


Abstract
Alternative transcript processing is an important mechanism for generating functional diversity in genes. However, little is known about the precise functions of individual isoforms. In fact, proteins (translated from transcript isoforms), not genes, are the function carriers. By integrating multiple human RNA-seq data sets, we carried out the first systematic prediction of isoform functions, enabling high-resolution functional annotation of human transcriptome. Unlike gene function prediction, isoform function prediction faces a unique challenge: the lack of the training data—all known functional annotations are at the gene level. To address this challenge, we modelled the gene–isoform relationships as multiple instance data and developed a novel label propagation method to predict functions. Our method achieved an average area under the receiver operating characteristic curve of 0.67 and assigned functions to 15 572 isoforms. Interestingly, we observed that different functions have different sensitivities to alternative isoform processing, and that the function diversity of isoforms from the same gene is positively correlated with their tissue expression diversity. Finally, we surveyed the literature to validate our predictions for a number of apoptotic genes. Strikingly, for the famous ‘TP53’ gene, we not only accurately identified the apoptosis regulation function of its five isoforms, but also correctly predicted the precise direction of the regulation.
INTRODUCTION
The generation of alternative products from a single gene locus is a common mechanism for increasing transcriptome and proteome complexity in eukaryotic cells. In particular, >90% of human genes undergo alternative splicing (1,2). Still, it remains unclear to what extent alternatively processed isoforms have divergent functions. Some studies have demonstrated that a large number of unconserved splicing events produce alternative isoforms at low abundance, and thus may be non-functional noise in the transcriptome (3,4). On the other hand, in many cases, alternatively spliced isoforms have distinct or even opposing functions (5). Moreover, many genomic variants relevant to inherited diseases change the ratio of alternatively spliced isoforms or generate disease-associated aberrant splicing products (6), suggesting the importance of maintaining a properly spliced transcriptome in healthy individuals.
Although recent years have seen an increase of studies on isoform-specific functions, most functional annotations for proteins are still only recorded at the gene level [e.g. in the Gene Ontology (7) database]. This is the case even when the original evidence was resolved at the isoform level. Owing to the limitations of current experimental techniques, there are very few data available for isoform functions, although such high-resolution data are crucial to understand protein functions. To fill this gap, this article reports the first systematic prediction of isoform functions by designing a novel multiple instance-based label propagation method and by integrating many genome-wide RNA-seq data sets.
In gene function prediction studies, protein sequence-based features (e.g. domain annotation and sequence similarity) and protein interactions are usually regarded as important characteristics and thus are widely used (8,9). However, existing encoding or annotation schemes severely limit the usefulness of such data for isoform function prediction, for four reasons. (i) Alternative splicing can regulate protein functions via the selective removal of structural domains (10,11). However, to assess protein functions on large scales, existing function prediction methods only use the number of shared domains to describe functional association between two genes (12). Without carefully investigating the detailed domain annotations, this method is insufficient to distinguish functionally distinct isoforms (13). (ii) Many alternatively spliced exons that regulate protein functions generate intrinsically disordered protein sequences (14,15), which have no influence on domain regions. (iii) Distinct isoform functions have been observed even in cases, where only a few amino acids change due to the alternative splicing (16–19). These subtle variances are difficult to capture with sequence-based features. (iv) The protein–protein interaction data frequently used in gene function studies are generally recorded at the gene level, without information about which isoform was actually tested in the experiments. Even in cases where a specific transcript has been annotated, most of the time it is the canonical isoform (i.e. the best studied one). This would lead to a systematic bias towards canonical isoforms when inferring isoform functions using protein interaction data.
RNA-seq technology can yield genome-wide unbiased expression profiles at the isoform level. We propose using the isoform co-expression networks derived from RNA-seq data to predict isoform functions. Given that several computational methods have been developed for isoform expression estimation (20–23) over the past several years, it is now feasible to profile the expression patterns of individual isoforms at high-throughput and in an unbiased manner, opening up great opportunities for elucidating cellular activities at the isoform level. Recent studies (15,24) indicate that isoform-level interactions are usually rewired by tissue-specific exons. As the function of a protein is largely determined by its interacting partners, such results further emphasize the importance of using expression data for isoform function prediction.
From an algorithmic viewpoint, the isoform function prediction problem is characterized by four major challenges:
The training data are unconventional. Most existing functional annotations are assigned to genes but not isoforms, yet each gene contains one or more isoforms. These type of data are exactly the ‘multiple instance (MI)-labelled’ data, in contrast to the ‘single instance-labelled data’ used by traditional machine learning methods, where the label (a discrete value representing one of two categories: +1 or −1) on a training instance gives complete information about its category. The labels on MI data are attached to sets of instances (or ‘bags’ in the jargon of MI learning), not to individual instances. The standard rule of MI-labelled data is that a bag is labelled positive only if at least one instance in the bag is positive, although we may not know which instances are positive, and the bag is labelled negative only if all instances within the bag are negative. In our context of functional annotations at the gene level, the isoforms are instances and each gene is a bag of isoforms. If a gene is labelled as having a function, then we know that at least one of its isoforms should have this function; on the other hand, if a gene is labelled as not having the function, then none of its isoforms have this function. However, formulating the existing functional annotation data as MI-labelled data is only a part of the solution because of the second challenge described below.
The isoform function prediction task is unconventional. In fact, we want to make two types of predictions. The first is ‘inheritance prediction’: given a gene with a function, we want to know which of its isoform(s) ‘inherit’ this function. The second is ‘de novo prediction’: we want to predict the functions of isoforms even for genes that are unknown to have these functions. Inheritance prediction can take full advantage of the current gene function annotations; however, it presents a novel prediction problem. The fact that a large number of genes are unannotated to many functions leads us to consider graph-based semi-supervised learning, also known as the label propagation (LP) method, due to its capacity for using unlabelled data. Nevertheless, few LP methods exist for MI-labelled networks (25,26), and none of these are suitable for inheritance predictions.
Integrating multiple isoform association networks with ML labels has never been done before. Many studies in the area of gene function prediction have demonstrated that combining multiple data sources can result in higher-quality function predictions (27). We believe that the same principle is valid for isoform function prediction. However, no method has been designed for the selection and integration of multiple MI-labelled networks.
There is a dearth of validation data for isoform function prediction. To assess the performance of our predictions, we need the functional annotations for isoforms.
To address the first two challenges (i, ii), we propose a new technique called instance-oriented MI label propagation (iMILP). iMILP enables both inheritance and de novo predictions by exploiting the benefits of unlabelled data. As previously mentioned, existing LP methods for MI-labelled data aim at classifying bags, not instances (25,26). In particular, they focus on identifying the single most positive instance in each positive bag, and do not attempt to optimize the scores of all instances within a bag during the learning process. In contrast, our iMILP method allows all qualified instances to inherit a positive bag’s label, via a ‘democratic’ learning process. To address challenge (iii), we recast the network selection problem as a feature selection problem, and introduce a wrapper strategy to solve the problem. Figure 1 illustrates the iMILP method and network selection and combination approach. To address challenge (iv), we validate predictions using the set of isoforms, whose host genes are annotated and contain only a single isoform. Although there may exist biases in this data set, it is the only large source of validation data on the isoform level that is available so far.
Figure 1.

Illustrations of (A–LP) the standard label propagation, with labels assigned to each node, (B–iMILP) the proposed instance-oriented MI label propagation, with labels assigned to bags of nodes and (C–iMILP + Network Selection) the method of integrating multiple networks before performing iMILP. Each node represents an instance, and nodes labelled as positive/negative/unknown are as grey/white/question-mark circles with solid lines. Bags of instances labelled as positive/negative are represented as the large grey/white ovals with dotted lines.
Therefore, we have performed the first systematic prediction of isoform functions by integrating 29 isoform co-expression networks constructed with RNA-seq data retrieved from the National Center for Biotechnology Information (NCBI) Sequence Read Archive database. Our iMILP method obtained an average area under the receiver operating characteristic curve (AUC) of 0.67, and assigned 70 392 function annotations to15 572 isoforms. Our results suggest that although many genes have isoforms carrying the same function, there is a substantial fraction of genes with functional variants. We also showed that functionally diverse isoforms usually have diverse expression patterns across tissues. An in-depth literature survey of isoforms related to apoptosis regulation confirms the majority of our predictions in this functional category.
MATERIALS AND METHODS
Isoform co-expression network construction
The messenger RNA (mRNA) isoform sequences were extracted from NCBI Reference Sequences (RefSeq, downloaded on January 2013) (28). We discarded all RefSeq records that were not manually reviewed. To construct the isoform co-expression networks, we retrieved 29 data sets of human full-length mRNA sequencing studies from the NCBI Sequence Read Archive database (29) (Supplementary Materials for the list of data sets). Each data set was required to have at least six experiments, and not to be a population study. The eXpress software (21), combined with Bowtie2 aligner (30), was used to infer isoform expression values. The RefSeq mRNA transcripts were used as transcriptome annotations. Only the ‘Coding DNA Sequence’ (CDS) sequences were considered to facilitate the calculation (see ‘Discussion’ section for more details). The mRNA level expression values were converted directly into protein isoform expressions. In cases where two or more RefSeq mRNA sequences correspond to the same protein sequence, they were regarded as belonging to a unique protein isoform, and their expression values were added.
In each RNA-seq data set, a protein isoform is retained for further analysis only if the coefficient of variation (the ratio of standard deviation to mean) of its expression profile is ≥0.3, and it is significantly expressed with the expression value ≥10 fragments per kilobase of exon per million fragments mapped (FPKM) in at least two experiments.
We then calculated the Pearson correlation coefficient (PCC) between the expression
profiles of each isoform pair meeting above criteria. To make the correlation estimates
comparable across data sets with different sample sizes, we applied Fisher’s
z transform (31). Given
a PCC estimate r, Fisher’s transformation score was calculated as
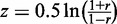 . The distributions of
z-scores vary from data set to data set, so we standardized the
z-scores to enforce zero mean and unit variance in each data set (32,33). By inverting the z-score, the corresponding
‘normalized’ correlation r' was calculated and used as an
edge weight in the co-expression networks. For fast computation, only co-expressed pairs
with normalized PCCs ≥0.5 were included in the isoform co-expression networks.
. The distributions of
z-scores vary from data set to data set, so we standardized the
z-scores to enforce zero mean and unit variance in each data set (32,33). By inverting the z-score, the corresponding
‘normalized’ correlation r' was calculated and used as an
edge weight in the co-expression networks. For fast computation, only co-expressed pairs
with normalized PCCs ≥0.5 were included in the isoform co-expression networks.
Functional annotation of genes
Gene Ontology (GO) data (7) and the UniProt Gene Ontology Annotation (UniProt-GOA) database (34) were used as the function categories and gene function annotations, respectively. By using the ID mapping information provided by the UniProt database, GO functions were assigned to each NCBI’s Gene ID, which includes one or more RefSeq transcripts. All GO annotations with the inferred from electronic annotation (IEA) evidence code were removed from consideration in our analysis because they have not been verified by human curators.
For a given GO term F, we labelled all its annotated genes as positive bags. The genes that are only annotated with its sibling GO terms are labelled as negative bags. The sibling GO terms of F are defined as those that share at least one direct parent with F and are not ancestors or descendants of F. We selected the functional categories by two criteria: (i) GO terms associated with >1000 genes (or <5 genes) were considered too general (or too specific) and thus ignored; (ii) If a GO term has >95% of its associated genes also annotated with its sibling GO terms, this GO term was not considered, as it is indistinguishable from its siblings. Finally, the remaining 4519 GO terms were selected and used in the function predictions.
Isoform function prediction
The proposed method consists of two components as shown in Figure 2. (i) The network selection and combination component chooses an optimal subset of networks relevant to the given GO category among all input isoform co-expression networks, then aggregates them into a single network, which is the input of the second component. (ii) The predictor component is a novel instance-oriented MI label propagation method. It takes the combined network as input and returns function predictions of isoforms. These two components will be explained in detail in the following two sections.
Figure 2.

Flowchart of the proposed method with two components: ‘network selection and combination’ and ‘predictor’. The network selection and combination component uses the wrapper strategy. The predictor component is our proposed iMILP method.
Instance-oriented MI label propagation method
All existing LP methods (25,26) for MI-labelled networks focus on classifying bags. They follow the rule that ‘knowing that one of the instances in the bag is positive is sufficient for predicting this bag as positive’. The consequence of this rule is that in a positive bag, all but the most positive instance are ignored. Therefore, these methods do not help when we need to answer an instance-level question such as ‘Which instances are positive in the positive bag?’ In our problem, we are more interested in knowing which isoforms (instances) inherit the function of the gene (bag) than which single isoform is the best representative of the gene’s function. We propose a novel iMILP method to make predictions at the instance level. Its label propagation rule is that ‘In the positive bag, a node (instance) that links to more nodes from positive bags receives a larger prediction score; any node that link to no other nodes from positive bags is demoted to have a prediction score of zero’. Applying this rule iteratively in the LP method clearly identifies all instances that are qualified to inherit the bag’s label. In the following, we first briefly review the standard LP algorithm, then describe how the iMILP algorithm adapts the LP approach to a network with MI labels.
Each node i in the network has a label yi
∈{+1, −1, 0}, representing a positive, negative or unknown instance,
respectively. LP methods usually keep track of a real-valued prediction score
fi∈[1, −1] for each node i,
which is often called a ‘soft label’. LP methods diffuse information from the
‘source nodes’ with unambiguous labels (yi
= +1 or −1) to direct neighbours by matrix multiplication:
f(1)
=
Lf(0),
where f(0) is a vector containing the initial
values of the soft labels for all nodes and its elements are usually assigned with the
labels yi. The matrix L is the normalized
Laplacian of the network (35). Propagating
the labels from source nodes to more distant nodes is accomplished by iteratively
performing the operation
f(t+1) =
Lf(t) (t
= 0,1,2, …), until f(t) converges,
where f(t) is the vector of soft
labels for all nodes at the t-th iteration. However, because the source
nodes are weakened by the diffusion, LP methods include the important step of restoring
all source nodes to their original extreme values before every diffusion step:
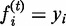 , where i is the index of
each source node. This practice was called ‘clamping’ the source nodes in the
pioneering work (36). The clamping step is
critical in that it provides source nodes with a ‘source of energy’ sufficient
to spread their labels to all reachable nodes in the network.
, where i is the index of
each source node. This practice was called ‘clamping’ the source nodes in the
pioneering work (36). The clamping step is
critical in that it provides source nodes with a ‘source of energy’ sufficient
to spread their labels to all reachable nodes in the network.
When the nodes have MI labels, things are different: the clamping step cannot be
performed for source nodes, as we have no nodes with labels y = +1 in the
positive bags. There do exist labels y = −1 for all nodes in the negative
bags. Therefore, to adapt the LP approach to the MI-labelled network, it is important to
redesign the ‘clamping’ step while keeping the diffusion step. The isoform
association network (with N isoforms) is represented as an adjacency
matrix 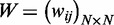 , where wij
denotes the intensity of association (normalized PCC) between isoforms i
and j. The normalized Laplacian of W is
, where wij
denotes the intensity of association (normalized PCC) between isoforms i
and j. The normalized Laplacian of W is
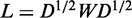 , where D is a diagonal
matrix with Dii =
Σjwij. We place all isoforms whose gene
(real bag) label is unknown (y = 0) into a single new pseudo bag
(called the ‘unlabelled bag’). Because these genes and their isoforms do not
provide any label constraints to the network, all their isoforms can be grouped into a
single pseudo bag without changing the result. We call this a pseudo bag because unlike
the other bags, it contains isoforms from more than one gene.
, where D is a diagonal
matrix with Dii =
Σjwij. We place all isoforms whose gene
(real bag) label is unknown (y = 0) into a single new pseudo bag
(called the ‘unlabelled bag’). Because these genes and their isoforms do not
provide any label constraints to the network, all their isoforms can be grouped into a
single pseudo bag without changing the result. We call this a pseudo bag because unlike
the other bags, it contains isoforms from more than one gene.
For ease of presentation, a variable name in bold font is a vector with continuous values. Having defined the network and terminology, our proposed iMILP algorithm is as follows:
Initialize the soft label f of each node (isoform) in a positive, negative or unlabelled bag (gene) as f = +1, −1 or 0, respectively.
- Clamp the soft labels of nodes as follows:
- For each node i in the positive bags,
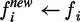 when
fi > ε
(ε is a positive number, close to 0), otherwise
when
fi > ε
(ε is a positive number, close to 0), otherwise
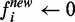 .
. - For each node i in the negative bags,
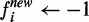 .
. - For each node i in the unlabelled bag, the soft label f remains unchanged:
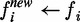 .
. - Within each bag (whether positive, negative or unlabelled), normalize the vector fnew containing the scores of all nodes in the bag, so that their squared sum is 1: f ← norm(fnew).
Diffuse labels: f ← Lf, where f is the vector of soft labels for all nodes.
Repeat step (ii) and (iii) until f converges.
We will now explain how the clamping step (step ii) works for the network with MI labels. Note that the clamping step is performed on the bag level. We first tune the soft labels within positive, negative or unlabelled bags in different ways, based on their definitions in the MI-labelling scheme, then normalize each bag for recharging bags.
In a positive bag, nodes with negative soft labels (or even more strictly, nodes with
f < ε) are ‘demoted’ to 0,
as indicated in step ii(a). The threshold ε should be
inversely related to the number of instances n in the positive bag. In
practice, we used ε =
0.01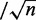 . This is a ‘democratic’ learning
process that retains all qualified instances of the bag. In contrast, existing MI +
LP learners (25,26) use an ‘authoritarian’ process that promotes
only one ‘witness node’ (the one with the maximum f value
after diffusion) in each positive bag, and ignores all others. In a negative bag,
following the MI-labelling rule, all nodes must be negative and receive equal
f scores −1, as shown in step ii(b). However, the negative
signals may dominate the network when there are many negative bags. This problem is
alleviated by the next step [step ii(d)] of normalizing the f scores in
the negative bags. In the single ‘unlabelled bag’, there are a large number of
isoforms whose host genes do not have labels. We keep their f scores
unchanged for they can serve as bridges for information to diffuse from the labelled bags
throughout the network. Although we do not clamp the nodes in the unlabelled bag, we still
need to normalize the bag to guarantee convergence after each diffusion step; otherwise,
the soft labels of these nodes can grow out of control. Therefore, the next normalization
step [step ii(d)] is important and has different purposes for different types of bags.
. This is a ‘democratic’ learning
process that retains all qualified instances of the bag. In contrast, existing MI +
LP learners (25,26) use an ‘authoritarian’ process that promotes
only one ‘witness node’ (the one with the maximum f value
after diffusion) in each positive bag, and ignores all others. In a negative bag,
following the MI-labelling rule, all nodes must be negative and receive equal
f scores −1, as shown in step ii(b). However, the negative
signals may dominate the network when there are many negative bags. This problem is
alleviated by the next step [step ii(d)] of normalizing the f scores in
the negative bags. In the single ‘unlabelled bag’, there are a large number of
isoforms whose host genes do not have labels. We keep their f scores
unchanged for they can serve as bridges for information to diffuse from the labelled bags
throughout the network. Although we do not clamp the nodes in the unlabelled bag, we still
need to normalize the bag to guarantee convergence after each diffusion step; otherwise,
the soft labels of these nodes can grow out of control. Therefore, the next normalization
step [step ii(d)] is important and has different purposes for different types of bags.
In the normalization step ii(d), we normalize all bags by constraining the squared sum of the soft labels in a bag equal to 1. This normalization has three implications for the solution: (i) all bags are equal, (ii) the soft labels f of nodes are proportional to their contributions to the bag and (iii) the larger the bag, the lower the f scores of its nodes. The first two effects are desirable. The third effect does not affect either inheritance or de novo predictions because soft labels only need to be comparable within a bag, not across bags. Figure 3 illustrates the diffusion and clamping steps using an example network with 15 nodes. It can be observed in the figure that f scores of the nodes inheriting labels of the positive/negative bags are replenished after each clamping step.
Figure 3.

Illustration of the iMILP approach with a 15-nodes example network. The initial network with its MI labels is shown in (A). Red dotted ovals represent positive bags of nodes, and green dotted ovals represent negative bags of nodes. (B) Each node is initialized with soft labels according to step i of the iMILP algorithm. After a series of alternating label propagation and clamping steps (C–F), the soft labels converge to (G), which gives the final prediction scores. The shade of colour in a node indicates the value of its soft label. The varying shades show how labels propagate in each diffusion step, and how the positive/negative bags are replenished in each clamping step, for preparing the diffusion in the next iteration. In (G), finally, both inheritance and de novo predictions are correctly made by the colour of these nodes.
After the soft labels f of all nodes converge, we need to make the final prediction for each node. For inheritance predictions, we assign positive labels to all nodes with non-zero f scores in the positive bags. The criterion should be more stringent for de novo predictions, which are less reliable. In practice, we have empirically set a threshold of 0.05 so that all nodes with f no less than this threshold in the pseudo unlabelled bag are predicted to be positive.
The algorithm is computationally efficient with a complexity of O(k|E|+k|V|), where k is the number of iterations (50 is usually enough for convergence), while |E| and |V| are the numbers of edges and nodes in the network, respectively. The source code is available on our website (http://zhoulab.usc.edu/IsoFP).
Network selection and combination algorithm
There exist several network selection and combination algorithms (8,37–39) for gene function prediction. However, all of them are designed for networks where labels are assigned to individual nodes. Therefore, they are not immediately suitable for networks with MI labels. To select informative networks for each GO category among all input networks, we cast the problem as a feature selection problem, where each network can be viewed as a feature. This viewpoint allows us to apply established feature selection strategies to the network selection problem. Specifically, we use the wrapper method, which is a widely used feature selection strategy (40). As shown in Figure 2, it uses the prediction performance of network subsets to guide a search for the best subset. To evaluate each subset, a performance score is obtained by applying the predictive model to selected networks with the K-fold regular cross-validation (details of the performance evaluation are in the following subsection). Our wrapper algorithm uses a greedy sequential forward strategy (41) to find the best subset of networks. The greedy search heuristics adds a new network to its currently selected subset only if doing so improves the prediction performance. The detailed procedure is presented below.
Input: M networks and a prediction performance measure
Output: a subset of networks G.
Step 1: Apply the predictor algorithm (iMILP) to each individual network in order to obtain their prediction performance scores. Then use these scores to sort the networks in the non-increasing order.
Step 2: Let G be a set containing the top-ranking networks. For each network i in the sorted order,
G′ = G∪{i} : add network i to the subset G;
Use the network subset G′ to predict isoform function and obtain its prediction performance score, perf(G′);
If perf(G′) > perf(G), G = G′; otherwise stop and return the selected networks.
After selecting a subset of m networks 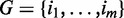 (where i is the index of each network), we used equal weights to combine
them into a single network as
(where i is the index of each network), we used equal weights to combine
them into a single network as 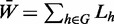 ,
where Lh is the normalized Laplacian of network
h.
,
where Lh is the normalized Laplacian of network
h.
Evaluation of the prediction performance
In this section, we explain in detail how the prediction performance evaluation is implemented for two different purposes: (i) as a part of the wrapper method, to score each subset combination of networks and (ii) to assess the proposed method as a whole. Below we present the two evaluation methods for these two purposes, respectively.
As aforementioned, the wrapper method needs to assign a quality score to each network subset. For this purpose, the prediction performance of the network subset selected is used as its quality score and can be estimated via the K-fold regular cross-validation. To make the large-scale evaluation of isoform-specific function predictions feasible, we take advantage of the functional annotations assigned to single-isoform genes because the only isoform in each single-isoform gene must carry the gene function. Therefore, for each GO term, we defined the positive isoforms as those from the positive single-isoform genes, and the negative isoforms as those from negative genes. We then performed the K-fold regular cross-validation, where all positive and negative genes (whatever single-isoform or multi-isoform genes) were randomly divided into K equal-size partitions, respectively. The test set uses all positive isoforms from positive single-isoform genes and all negative isoforms from negative genes in a partition, and the training set contains all labelled genes/bags of all other partitions. To dismiss possible random effects, this cross-validation procedure was repeated 10 times. For each round of cross-validation, we measured the AUC, which is the widely used performance measure in gene function prediction. Finally, we took the average AUC over all 10 rounds of the K-fold regular cross-validation. This average is reported as the quality score of the selected subset of networks for each GO term.
For the second purpose of assessing the performance of our proposed method as a whole, we used the K-fold nested cross-validation (42,43), which can provide a more unbiased estimate of the true performance than the K-fold regular cross-validation when a model selection step is involved (network subset selection in our case). The K-fold nested cross-validation includes the outer and inner cross-validation loops. The outer cross-validation loop is used to evaluate the prediction performance; and the inner cross-validation loop, which is actually a (K−1)-fold regular cross-validation, is used by the wrapper method to measure and select the best network subset (i.e. model selection), as described in the previous paragraph. The K-fold nested cross-validation was repeated 10 times, and the average AUC was reported as the final performance of our method for each GO term. In this study, we used K = 5 for the nested cross-validation, in which the inner loop is a 4-fold cross-validation for network selection.
Final predictions
We need to use all the labelled data for making the final predictions. Therefore, we can perform the following two steps: (i) apply the wrapper method with the K-fold regular cross-validation on all labelled data to select the best network subset and (ii) then train the predictive model with this best network subset on all labelled data for making final predictions. In practice, K = 5 is used for the final predictions.
Isoform function dissimilarity calculation
When a GO term is assigned to an isoform, all of its ancestors are assigned to this isoform accordingly because of the hierarchical relationships of GO terms. To remove unnecessary redundancy in the GO prediction results, we discarded every GO term being the ancestor of any other GO term assigned to the same isoform. Only isoforms that belong to the same gene and have predicted GO term(s) in the same GO branch were compared to investigate functional dissimilarity. Annotations in the three GO branches, biological process (BP), cellular component (CC) and molecular function (MF), were considered separately. The similarity score of two isoforms was estimated using G-SESAME method (44) and the dissimilarity score is simply defined as one minus the similarity score. The isoform functional divergence of a gene was calculated as the average dissimilarity score over all possible isoform pairs with GO annotations in the same branch.
RESULTS
Prediction performance of the iMILP method
The 29 RNA-seq data sets that we used to generate isoform co-expression networks cover a wide range of experimental and physiological conditions. We first applied our method to each single network. As shown in Figure 4A, no single network yielded an average AUC across all GO terms better than 0.53. The average AUC across all 29 single networks is only 0.48, even worse than a random guess (AUC 0.5). Therefore, we applied the wrapper method to select and combine a different subset of networks for each GO term. Our wrapper method resulted in a dramatically increased AUC score of 0.67, averaged across all GO terms (Figure 4A). This demonstrates the necessity of integrating multiple data sets for isoform function prediction.
Figure 4.

Prediction performance of the proposed method. (A) Average AUC score across all GO terms, for each individual isoform co-expression network (blue bars), and for the combined network using a different subset of networks for each GO term (red bar). (B) Average AUC scores over all terms within one of three GO branches (BP–biological process, CC–cellular component and MF–molecular function) and one of six ranges for the number of genes with the GO term (5–10, 11–20, 21–50, 51–100, 101–200 or 201–1000). GO terms annotating fewer genes are more specific. The bars show average AUC scores for each group.
GO annotations vary from highly specific functions that only involve a few genes, to some general categories with many associated genes, such as ‘cell cycle’. To investigate whether the performance of our label propagation method is influenced by the number of positive bags in the network, we divided the GO terms into 18 groups, following a standard procedure used in previous gene function prediction studies (8), then evaluated the performance of our algorithm in each group. These groups are based on the major GO branches (BP, CC and MF) and on sizes (the number of genes annotated with a GO term). Six size ranges were defined: [5,10], [11,20], [21,50], [51,100], [101,200], and [201,1000]. Figure 4B shows that the mean AUCs are higher than 0.6 in all GO groups. However, we also observed that the more genes annotated by a GO term, the (slightly) worse the prediction performance. This trend is consistent with previous gene function predictions (8). A possible explanation is that genes associated with GO terms of larger size are usually more heterogeneous, and thus it is harder to accurately predict the labels of their isoforms.
Functional annotations of isoforms
We applied our method to the entire training data set to generate the final function predictions of human isoforms. Our method selected and combined more than one networks for 75.0% of the GO terms (Figure 5A). The use of each network is shown in Figure 5B. We obtained 70 392 isoform-level function predictions, 13 621 of which were de novo function predictions, meaning the host genes are not annotated positively or negatively with predicted functions in the current GO database. Therefore, as a side product, our de novo predictions also contribute to functional annotation at the gene level. In addition, we predicted the functions of 8856 isoforms that have at least one annotation inherited from their host genes. In general, we believe that these inheritance predictions are more reliable than de novo predictions. Therefore, in the following analysis of the properties of isoform functions, we focused on the inheritance predictions.
Figure 5.

Network usage in the final predictions. (A) Given the number (K) of networks selected by the wrapper method to achieve the best AUC scores, each bar represents the number of GO terms that use the K networks for predictions. (B) The frequency of each network chosen in the final predictive models. The indexes of these networks are those of their corresponding RNA-seq data sets.
With the isoform-level annotation being resolved, we became interested in seeing which gene functions are usually shared by many isoforms of the same gene, and which functions are inherited by one or only a few isoforms. We proposed the concept of inheritance rate (IR): given a GO term and a multi-isoform gene annotated by this term, IR is calculated as the ratio between the number of isoforms assigned to the GO term and the total number of isoforms of this gene. A high IR suggests that this function of the gene is robust against alternative isoform processing; otherwise it is highly sensitive to this process. Among all GO terms annotated to at least 10 genes, the functions with the highest IR values are ‘nucleic acid transport’, ‘RNA splicing, via transesterification reactions’, ‘cellular protein localization’ and ‘hair follicle maturation’. The functions that are most sensitive to the regulation of isoforms are ‘regulation of membrane potential’, ‘actin cytoskeleton reorganization’, ‘taxis’ and ‘positive regulation of apoptotic process’.
Functional divergence among isoforms
Among the 7714 multi-isoform genes annotated in the RefSeq database, 2534 (791 or 1572) genes have at least two isoforms that have GO BP (CC or MF) terms assigned in our study. For each of these genes, we calculated the dissimilarity scores (see ‘Materials and Methods’ section) of all isoform pairs with annotations in the same GO branch, and then used their average as the gene level function dissimilarity score. We found that within each GO branch, a large number of genes have isoforms that share the same or similar functions (dissimilarity score between 0 and 0.1) (Figure 6A). Specifically, among BP, CC and MF annotations, 19.0 (482), 44.8 (354) and 30.7% (483) of the genes, respectively, have multiple isoforms annotated with identical functions. Nevertheless, there are also a considerable number of genes with functionally distinct isoforms. For example, 13.1% of genes have isoforms with a dissimilarity score based on their BP terms >0.5 (the proportions for CC and MF terms are 4.9 and 5.2%, respectively).
Figure 6.

Functional diversity of isoform-annotated genes. (A) The distribution of functional dissimilarities between isoforms of the same gene. The G-SESAME method was used to estimate the semantic dissimilarity scores of isoform functions. (B) Tissue expression diversity scores of genes with low or high functional diversity. Significant differences were observed in all the three GO categories. (P = 1.6 E-06, 3.2 E-02 and 9.7 E-03 for the BP, CC and MF GO tree branches, respectively. One-sided Wilcoxon tests were used to evaluate the P-values.)
We also investigated isoform properties that may be related to functional diversity. Recent studies (24,15) have found that isoforms with exons showing tissue-specific expression patterns can rewire the protein interaction network. Because protein functions are often exerted via protein–protein interactions, tissue-specific expression may lead to tissue-specific functions. We examined whether the tissue expression diversity of isoforms within a gene correlates with their functional diversity. We first calculated a tissue specificity score (45) for each isoform based on tissue expression data from the Illumina Human Body Map 2.0 project. For a gene with several isoforms, we defined the tissue diversity as the difference between the maximum and minimum specificity scores of its isoforms, to control the gene-level tissue specificity. Again for each GO branch separately (BP, CC or MF), we divided the genes into two groups with low and high functional diversity scores. To dismiss a possible bias introduced by the genes having different numbers of isoforms, we chose subsets from the low and high groups, where all genes have exactly two isoforms. As anticipated, we observed significantly higher tissue expression diversity in the groups with higher functional diversity, for all three GO domains (Figure 6B).
Literature validation of isoforms predicted to regulate apoptosis
The scarcity of literature on isoform-specific functions challenges any in-depth exploration of the functions predicted by our method. Nevertheless, because of the great abundance of cancer-related literature, there are considerable number of studies related to apoptosis-regulating isoforms. Apoptosis, the essential process that regulates cell death, is usually distressed in cancer cells. Intriguingly, several apoptosis genes are capable of regulating this process in the opposite direction via alternative isoforms. Below we provide a literature survey of four genes and their isoforms related to ‘regulation of apoptosis’.
The human tumour suppressor gene TP53 is well known for its role in inducing apoptosis and thus inhibiting tumorigenesis. According to its RefSeq annotation, 8 mRNA isoforms are ascribed to TP53, corresponding to seven unique protein products. The canonical isoform of TP53, p53α, is the full-length transcript composed of two transcription activation domains (TADs), a proline repeat domain (PXXP), a DNA binding domain (DBD), a nuclear localization signalling domain and an oligomerization domain (OD) (Figure 7). The other six protein products differ from the canonical form in TAD, PXXP, DBD and OD. Two of these, p53β and p53γ, are C-terminal isoforms produced by partial intron retention. Along with p53α, they demonstrate positive regulation of the apoptotic pathway (46–48). In p53β and p53γ, the OD is replaced by a short peptide of 10 and 15 residues, respectively. Isoform p53β is reported to enhance the transcriptional activity of p53α, and has weaker proapoptotic activity than p53α (46,47). A recent study found that mutant p53 breast cancer patients with p53γ expressed have a particularly good prognosis compared with those without this isoform expressed (48), indicating the capacity of p53γ to induce apoptosis. Interestingly, two other isoforms, Δ40p53α and Δ133p53α, are reported to have anti-apoptotic activity (49,46). Δ40p53α lacks the first TAD, but the DBD and OD are intact. It is able to interact with p53α, negatively regulating its transcriptional activity via competitive binding to specific DNA regions. In this way, it behaves as a suppressor of the full-length isoform p53α (49). Δ133p53α has even more amino acids deleted, losing both TADs and part of the OD. Thus, this isoform is an inhibitor of p53α (46).
Figure 7.

TP53 protein isoforms with functional annotations. The coding sequences (dark blue) are illustrated with splicing boundaries. Exonic regions are shown in proportion (boxes) but intronic regions are not (lines). The transcription activation domains (TAD) 1 and 2, proline rich domain (PXXP), DNA binding domain (DBD), nuclear localization signalling domain (NLS) and oligomerization domain (OD) are all annotated according to their locations in the canonical isoform, p53α. The biological function of each isoform is labelled as either positive (green arrow) or negative (red arrow) regulation of the apoptotic process.
All five isoforms described above were correctly predicted to have the GO term ‘regulation of apoptotic process’ (GO: 0042981) or any of its descendants (Table 1). We looked at whether the direction of the regulation could be also resolved at the isoform level. For each isoform, we checked our predictions for the two child terms ‘positive regulation of apoptotic process’ (GO: 0043065) and ‘negative regulation of apoptotic process’ (GO: 0043066). It should be emphasized that because we performed prediction for each GO term independently, the same isoform can be assigned both positive regulation and negative regulation by different predictions. Therefore, we have 10 predictions of regulation direction for the five isoforms. Surprisingly, even for TP53, a gene with many isoforms, our algorithm made the correct decisions in 9 of 10 predictions.
Table 1.
Prediction results on four apoptotic genes
| Gene | Isoform | Predicted as a regulator of apoptosis? | Positive regulation? |
Negative regulation? |
||
|---|---|---|---|---|---|---|
| Annotation | Prediction | Annotation | Prediction | |||
| TP53 | p53α | ○ | ○ | ○ | ✕ | ○ |
| p53β | ○ | ○ | ○ | ✕ | ✕ | |
| p53γ | ○ | ○ | ○ | ✕ | ✕ | |
| Δ40p53α | ○ | ✕ | ✕ | ○ | ○ | |
| Δ133p53α | ○ | ✕ | ✕ | ○ | ○ | |
| BCL2L1 | Bcl-xL | ○ | ✕ | ✕ | ○ | ○ |
| Bcl-xS | ○ | ○ | ✕ | ✕ | ✕ | |
| CFLAR | cFLIP-L | ○ | ○ | ✕ | ○ | ✕ |
| cFLIP-S | ○ | ✕ | ✕ | ○ | ○ | |
| DNAJA3 | Tid-1(L) | ○ | ○ | ✕ | ✕ | ✕ |
| Tid-1(S) | ○ | ✕ | ✕ | ○ | ○ | |
Positive and negative results are represented by circles and crosses, respectively.
We also surveyed the literature on three other apoptosis regulatory genes (Table 1), all of which have both apoptosis-inducing and apoptosis-suppressing isoforms. BCL2L1 has two isoforms: Bcl-xL inhibits and Bcl-xS promotes programmed cell death (50). CFLAR, the CASP8 and FADD-like apoptosis regulator, has two well-annotated isoforms cFLIP-L and cFLIP-S. Both isoforms are inhibitors of apoptotic proteins (51), and cFLIP-L is also a promoter of apoptosis (52). The third gene, DNAJA3, has two isoforms with opposite functions—one induces and the other represses the apoptotic process (53).
Our iMILP method successfully predicted the ‘regulation of apoptotic process’ function for all 11 isoforms (including the five TP53 isoforms). Note that this excellent recall rate (100%) cannot be attributed only to a preference for inheriting gene-level annotations in our predictions because there is clearly a selective yet precise (only one false positive) inheritance from genes to isoforms on the child GO terms ‘positive regulation of apoptotic process’ and ‘negative regulation of apoptotic process’. This result suggests that our method can achieve both high recall and high precision. On these four genes for which we could collect sufficient literature evidence, the overall accuracy of predictions on ‘positive regulation of apoptotic process’ and ‘negative regulation of apoptotic process’ are 72.7% (8/11) and 81.8% (9/11), respectively.
Furthermore, when we checked the predictions in detail, we found that BCL2L1 and CFLAR were not annotated with the GO term ‘positive regulation of apoptotic process’ or any of its descendants in the input data. They were only annotated with the sibling term ‘negative regulation of apoptotic process’, and thus were always treated as negative bags in our prediction. Our false-negative predictions of these isoforms were therefore caused by missannotations in the GO database. This discovery indicates that our evaluation underestimates the real power of the method.
In summary, given only isoform expression information, our method successfully annotated the positive or negative regulatory functions of apoptotic protein isoforms.
DISCUSSION
Reproducible and fast pipeline for estimating isoform expressions from RNA-seq data
We estimated isoform expression levels from RNA-seq data. Because this study uses a large number of RNA-seq data sets, it is important to choose the pipeline with the right trade-off between computation speed and estimation quality. Several transcript quantification tools exist, such as Cufflinks (20), eXpress (21), RSEM (22) and SLIDE (23). We compared the two most popular isoform abundance estimation pipelines, tophat+cufflinks and bowtie2+eXpress, in terms of runtime and reproducibility. Both pipelines generated reproducible isoform abundances in two replicates of three ENCODE human cell types (Supplementary Materials) (54). However, bowtie2+eXpress runs ∼10 times faster than tophat+cufflinks because bowtie2+eXpress works directly on the transcript sequences, without introducing genomic information. In addition, eXpress provides an online streaming mechanism, so that bowtie2 and eXpress can run at the same time using the pipe mechanism of Unix.
Also, because the function annotations were resolved at the protein level, it is not necessary to distinguish mRNAs that differ in the untranslated regions. Therefore, we reduced the computational cost using only the protein-coding sequence of each RefSeq mRNA in the expression evaluation.
Challenge in compilation of the negative data sets
There are no annotations of negative associations between a GO term and a gene. To generate the negative training data, we followed a popular method that is widely used in the literature on gene function prediction literatures (8): genes associated with the siblings of a given GO term are interpreted as negative annotations. However, because of the hierarchical structure of GO terms, any given GO term shares at least one parent with its siblings. This means that a GO term could be functionally similar to its siblings, especially for specific functions. Therefore, we should be cautious using the genes associated with the sibling terms as negative data. In fact, the number of genes shared between a GO term and its siblings can be used to estimate the functional similarity between the two GO terms. We found that among all GO terms with at least five and at most 1000 associated genes, 35.2% of them share most genes (≥95%) with their siblings. The GO terms with this level of overlap were not considered in our study (see ‘Materials and Methods’ section). This statistic also suggests that a substantial portion of the negative data compiled in this way may not really be negative, and that a better method is needed for further development of this framework. A recent study (55) proposed a more sophisticated method for choosing negative data, which could be incorporated into our future work.
Performance evaluation using single-isoform genes is not perfect
Owing to the lack of curated isoform function data, we used the annotations of single-isoform genes to assess the performance of our predictions. However, multiple isoforms of the same gene may share the same promoter regions and some consensus exons, which are not generally shared by a given pair of single-isoform genes. That is, the relationships between single-isoform genes and those between multiple isoforms of the same genes are different. Therefore, the proposed wrapper method directed by single-isoform genes may not be the best choice for inheritance predictions in multi-isoform genes, which aim at distinguishing the function difference between isoforms transcribed from the same locus. This issue can eventually be addressed by training our model on functional annotations with isoform-level resolution when they become available in the future. However, at this time, the proposed validation strategy is the best we can do.
Graph-based semi-supervised learning and multiple instance learning
These two learning models have been extensively studied in recent years. They were both proposed to solve the problems that have incompletely labelled data. To date, few studies have attempted to integrate the two learning schemes with the goal of fully using both unlabelled data and semi-labelled (or MI-labelled) data (25,26). All of them adopt the widely used MIL strategy of selecting the single instance with the maximum prediction score as the ‘witness instance’, and using only its score to fit the positive bag’s label. The approach works well for bag-level predictions, where one witness instance may be enough for learning, although it has some criticism even for this purpose (56). However, this strategy artificially forces the witness instance to have a much larger prediction score than other instances in the positive bag, weakening the roles of other instances and thus distorting instance-level inheritance predictions. In short, such strategy of declaring witness instances weakens the label propagation process, which actually has the power to subtly differentiate instances in a positive bag. In contrast, our proposed iMILP method ‘democratically’ treats all instances in a bag in the same manner. Empirically, iMILP converges quickly, although we do not yet have a theoretical convergence proof. In future work, we will provide a theoretical analysis of the iMILP method and examine its relationships with existing LP methods.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [NHLBI MAPGEN U01HL108634 and NIGMS R01GM105431] and the National Science Foundation [0747475 to X.J.Z.]; the National Science Council grant [NSC101-2627-B-005-002 to C.-C.L.]; the National Natural Science Foundation of China [61379092 and 11001256] and the Foundation for Members of Youth Innovation Promotion Association, CAS (to S.Z.). Funding for open access charge: NIH [NIGMS R01GM105431].
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 3.Pickrell JK, Pai AA, Gilad Y, Pritchard JK. Noisy splicing drives mRNA isoform diversity in human cells. PLoS Genet. 2010;6:e1001236. doi: 10.1371/journal.pgen.1001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Melamud E, Moult J. Stochastic noise in splicing machinery. Nucleic Acids Res. 2009;37:4873–4886. doi: 10.1093/nar/gkp471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Himeji D, Horiuchi T, Tsukamoto H, Hayashi K, Watanabe T, Harada M. Characterization of caspase-8L: a novel isoform of caspase-8 that behaves as an inhibitor of the caspase cascade. Blood. 2002;99:4070–4078. doi: 10.1182/blood.v99.11.4070. [DOI] [PubMed] [Google Scholar]
- 6.Pagani F, Baralle FE. Genomic variants in exons and introns: identifying the splicing spoilers. Nat. Rev. Genet. 2004;5:389–396. doi: 10.1038/nrg1327. [DOI] [PubMed] [Google Scholar]
- 7.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mostafavi S, Ray D, Warde-Farley D, Grouios C, Morris Q. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 2008;9(Suppl. 1):S4. doi: 10.1186/gb-2008-9-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clark WT, Radivojac P. Analysis of protein function and its prediction from amino acid sequence. Proteins. 2011;79:2086–2096. doi: 10.1002/prot.23029. [DOI] [PubMed] [Google Scholar]
- 10.Liu S, Altman RB. Large scale study of protein domain distribution in the context of alternative splicing. Nucleic Acids Res. 2003;31:4828–4835. doi: 10.1093/nar/gkg668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Resch A, Xing Y, Modrek B, Gorlick M, Riley R, Lee C. Assessing the impact of alternative splicing on domain interactions in the human proteome. J. Proteome Res. 2004;3:76–83. doi: 10.1021/pr034064v. [DOI] [PubMed] [Google Scholar]
- 12.Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38:W214–W220. doi: 10.1093/nar/gkq537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Severing EI, van Dijk AD, Morabito G, Busscher-Lange J, Immink RG, van Ham RC. Predicting the impact of alternative splicing on plant MADS domain protein function. PLoS One. 2012;7:e30524. doi: 10.1371/journal.pone.0030524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Romero PR, Zaidi S, Fang YY, Uversky VN, Radivojac P, Oldfield CJ, Cortese MS, Sickmeier M, LeGall T, Obradovic Z, et al. Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms. Proc. Natl Acad. Sci. USA. 2006;103:8390–8395. doi: 10.1073/pnas.0507916103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buljan M, Chalancon G, Eustermann S, Wagner GP, Fuxreiter M, Bateman A, Babu MM. Tissue-specific splicing of disordered segments that embed binding motifs rewires protein interaction networks. Mol. Cell. 2012;46:871–883. doi: 10.1016/j.molcel.2012.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vogan K, Underhill D, Gros P. An alternative splicing event in the Pax-3 paired domain identifies the linker region as a key determinant of paired domain DNA-binding activity. Mol. Cell. Biol. 1996;16:6677–6686. doi: 10.1128/mcb.16.12.6677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Merediz SAK, Schmidt M, Hoppe GJ, Alfken J, Meraro D, Levi BZ, Neubauer A, Wittiq B. Cloning of an interferon regulatory factor 2 isoform with different regulatory ability. Nucleic Acids Res. 2000;28:4219–4224. doi: 10.1093/nar/28.21.4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu CA, Lin WW, Obie C, Valle D. Molecular enzymology of mammalian delta 1-pyrroline-5-carboxylate synthase. Alternative splice donor utilization generates isoforms with different sensitivity to ornithine inhibition. J. Biol. Chem. 1999;274:6754–6762. doi: 10.1074/jbc.274.10.6754. [DOI] [PubMed] [Google Scholar]
- 19.Yan M, Wang LC, Hymowitz SG, Schilbach S, Lee J, Goddard A, de Vos AM, Gao WQ, Dixit VM. Two-amino acid molecular switch in an epithelial morphogen that regulates binding to two distinct receptors. Science. 2000;290:523–527. doi: 10.1126/science.290.5491.523. [DOI] [PubMed] [Google Scholar]
- 20.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roberts A, Pachter L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nat. Methods. 2013;10:71–73. doi: 10.1038/nmeth.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li JJ, Jiang CR, Brown JB, Huang H, Bickel PJ. Sparse linear modeling of next-generation mRNA sequencing (RNA-Seq) data for isoform discovery and abundance estimation. Proc. Natl Acad. Sci. USA. 2011;108:19867–19872. doi: 10.1073/pnas.1113972108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ellis JD, Barrios-Rodiles M, Colak R, Irimia M, Kim T, Calarco JA, Wang X, Pan Q, O’Hanlon D, Kim PM, et al. Tissue-specific alternative splicing remodels protein-protein interaction networks. Mol. Cell. 2012;46:884–892. doi: 10.1016/j.molcel.2012.05.037. [DOI] [PubMed] [Google Scholar]
- 25.Jia Y, Zhang C. Proceedings of the 23rd National Conference on Artificial Intelligence. CA: AAAI Press; 2008. Instance-level semisupervised multiple instance learning; pp. 640–645. [Google Scholar]
- 26.Wang C, Zhang L, Zhang HJ. Proceeding of the 1st ACM International Conference on Multimedia Information Retrieval-MIR’08. New York: ACM Press; 2008. Graph-based multiple-instance learning for object-based image retrieval; pp. 156–163. [Google Scholar]
- 27.Noble W, Ben-Hur A. Integrating information for protein function prediction. In: Lengauer T, editor. Bioinformatics-From Genomes to Therapies. Vol. 3. Weinheim: WILEY-VCH Verlag GmbH & Co. KGaA; 2007. pp. 1297–1314. [Google Scholar]
- 28.Pruitt KD, Tatusova T, Brown GR, Maglott DR. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 2012;40:D130–D135. doi: 10.1093/nar/gkr1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leinonen R, Sugawara H, Shumway M. The sequence read archive. Nucleic Acids Res. 2011;39:D19–D21. doi: 10.1093/nar/gkq1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Y, Schmidt B. Long read alignment based on maximal exact match seeds. Bioinformatics. 2012;28:i318–i324. doi: 10.1093/bioinformatics/bts414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anderson TW. An Introduction To Multivariate Statistical Analysis. 3rd edn. Hoboken, NJ: Wiley-Interscience; 2003. [Google Scholar]
- 32.Xu M, Kao MC, Nunez-Iglesias J, Nevins JR, West M, Zhou XJ. An integrative approach to characterize disease-specific pathways and their coordination: a case study in cancer. BMC Genomics. 2008;9(Suppl. 1):S12. doi: 10.1186/1471-2164-9-S1-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li W, Liu CC, Zhang T, Li H, Waterman MS, Zhou XJ. Integrative analysis of many weighted co-expression networks using tensor computation. PLoS Comput. Biol. 2011;7:e1001106. doi: 10.1371/journal.pcbi.1001106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barrell D, Dimmer E, Huntley RP, Binns D, O’Donovan C, Apweiler R. The GOA database in 2009–an integrated Gene Ontology Annotation resource. Nucleic Acids Res. 2009;37:D396–D403. doi: 10.1093/nar/gkn803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu W, Wang J, Chang SF. Robust and scalable graph-based semisupervised learning. Proc. IEEE. 2012;100:2624–2638. [Google Scholar]
- 36.Zhu X, Ghahramani Z. Learning from labeled and unlabeled data with label propagation. 2002 Technical Report CMU-CALD-02-107, Carnegie Mellon University. [Google Scholar]
- 37.Tsuda K, Shin H, Schölkopf B. Fast protein classification with multiple networks. Bioinformatics. 2005;21:ii59–ii65. doi: 10.1093/bioinformatics/bti1110. [DOI] [PubMed] [Google Scholar]
- 38.Kato T, Kashima H, Sugiyama M. Robust label propagation on multiple networks. IEEE Trans. Neural Netw. 2009;20:35–44. doi: 10.1109/TNN.2008.2003354. [DOI] [PubMed] [Google Scholar]
- 39.Mostafavi S, Morris Q. Fast integration of heterogeneous data sources for predicting gene function with limited annotation. Bioinformatics. 2010;26:1759–1765. doi: 10.1093/bioinformatics/btq262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saeys Y, Inza I, Larrañaga P. A review of feature selection techniques in bioinformatics. Bioinformatics. 2007;23:2507–2517. doi: 10.1093/bioinformatics/btm344. [DOI] [PubMed] [Google Scholar]
- 41.Shi Y, Cai Z, Xu L, Ren W, Goebel R, Lin G. Proceedings of IEEE Symposium on Computational Intelligence and Bioinformatics and Computational Biology (CIBCB) Toronto: 2006. A model-free greedy gene selection for microarray sample class prediction; pp. 1–8. [Google Scholar]
- 42.Varma S, Simon R. Bias in error estimation when using cross-validation for model selection. BMC Bioinformatics. 2006;7:91. doi: 10.1186/1471-2105-7-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruschhaupt M, Huber W, Poustka A, Mansmann U. A compendium to ensure computational reproducibility in high-dimensional classification tasks. Stat. Appl. Genet. Mol. Biol. 2004;3:1078. doi: 10.2202/1544-6115.1078. [DOI] [PubMed] [Google Scholar]
- 44.Wang JZ, Du Z, Payattakool R, Yu PS, Chen CF. A new method to measure the semantic similarity of GO terms. Bioinformatics. 2007;23:1274–1281. doi: 10.1093/bioinformatics/btm087. [DOI] [PubMed] [Google Scholar]
- 45.Yanai I, Benjamin H, Shmoish M, Chalifa-Caspi V, Shklar M, Ophir R, Bar-Even A, Horn-Saban S, Safran M, Domany E, et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics. 2005;21:650–659. doi: 10.1093/bioinformatics/bti042. [DOI] [PubMed] [Google Scholar]
- 46.Bourdon JC, Fernandes K, Murray-Zmijewski F, Liu G, Diot A, Xirodimas DP, Saville MK, Lane DP. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005;19:2122–2137. doi: 10.1101/gad.1339905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fujita K, Mondal AM, Horikawa I, Nguyen GH, Kumamoto K, Sohn JJ, Bowman ED, Mathe EA, Schetter AJ, Pine SR, et al. p53 isoforms Delta133p53 and p53beta are endogenous regulators of replicative cellular senescence. Nat. Cell Biol. 2009;11:1135–1142. doi: 10.1038/ncb1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bourdon JC, Khoury MP, Diot A, Baker L, Fernandes K, Aoubala M, Quinlan P, Purdie CA, Jordan LB, Prats AC, et al. p53 mutant breast cancer patients expressing p53γ have as good a prognosis as wild-type p53 breast cancer patients. Breast Cancer Res. 2011;13:R7. doi: 10.1186/bcr2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Courtois S, Verhaegh G, North S, Luciani MG, Lassus P, Hibner U, Oren M, Hainaut P. DeltaN-p53, a natural isoform of p53 lacking the first transactivation domain, counteracts growth suppression by wild-type p53. Oncogene. 2002;21:6722–6728. doi: 10.1038/sj.onc.1205874. [DOI] [PubMed] [Google Scholar]
- 50.Boise LH, González-García M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nuñez G, Thompson CB. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- 51.Krueger A, Schmitz I, Baumann S, Krammer PH, Kirchhoff S. Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J. Biol. Chem. 2001;276:20633–20640. doi: 10.1074/jbc.M101780200. [DOI] [PubMed] [Google Scholar]
- 52.Chang DW, Xing Z, Pan Y, Algeciras-Schimnich A, Barnhart BC, Yaish-Ohad S, Peter ME, Yang X. c-FLIP(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J. 2002;21:3704–3714. doi: 10.1093/emboj/cdf356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Syken J, De-Medina T, Münger K. TID1, a human homolog of the Drosophila tumor suppressor l(2)tid, encodes two mitochondrial modulators of apoptosis with opposing functions. Proc. Natl Acad. Sci. USA. 1999;96:8499–8504. doi: 10.1073/pnas.96.15.8499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Youngs N, Penfold-Brown D, Drew K, Shasha D, Bonneau R. Parametric Bayesian priors and better choice of negative examples improve protein function prediction. Bioinformatics. 2013;29:1190–1198. doi: 10.1093/bioinformatics/btt110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ngo TD, Le DD, Satoh S. Proceeding of the International Conference on Image Analysis and Processing (ICIAP) Ravenna, Italy: 2011. Improving image categorization by using multiple instance learning with spatial relation; pp. 108–117. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.