

Abstract
Pseudomonas aeruginosa is one of the most dreaded opportunistic pathogens accounting for 10 % of hospital-acquired infections, with a 50 % mortality rate in chronically ill patients. The increased prevalence of drug-resistant isolates is a major cause of concern. Resistance in P. aeruginosa is mediated by various mechanisms, some of which are shared among different classes of antibiotics and which raise the possibility of cross-resistance. The goal of this study was to explore the effect of subinhibitory concentrations (SICs) of clinically relevant antibiotics and the role of a global antibiotic resistance and virulence regulator, AmpR, in developing cross-resistance. We investigated the induction of transient cross-resistance in P. aeruginosa PAO1 upon exposure to SICs of antibiotics. Pre-exposure to carbapenems, specifically imipenem, even at 3 ng ml−1, adversely affected the efficacy of clinically used penicillins and cephalosporins. The high β-lactam resistance was due to elevated expression of both ampC and ampR, encoding a chromosomal β-lactamase and its regulator, respectively. Differences in the susceptibility of ampR and ampC mutants suggested non-AmpC-mediated regulation of β-lactam resistance by AmpR. The increased susceptibility of P. aeruginosa in the absence of ampR to various antibiotics upon SIC exposure suggests that AmpR plays a major role in the cross-resistance. AmpR was shown previously to be involved in resistance to quinolones by regulating MexEF–OprN efflux pump. The data here further indicate the role of AmpR in cross-resistance between quinolones and aminoglycosides. This was confirmed using quantitative PCR, where expression of the mexEF efflux pump was further induced by ciprofloxacin and tobramycin, its substrate and a non-substrate, respectively, in the absence of ampR. The data presented here highlight the intricate cross-regulation of antibiotic resistance pathways at SICs of antibiotics and the need for careful assessment of the order of antibiotic regimens as this may have dire consequences. Targeting a global regulator such as AmpR that connects diverse pathways is a feasible therapeutic approach to combat P. aeruginosa pathogenesis.
Introduction
Treatment of Pseudomonas aeruginosa infections poses a clinical challenge due to the extensive spread of multidrug-resistant isolates (Barbier & Wolff, 2010; Curcio, 2013). The alarming development of resistance to almost all clinically relevant antibiotics has led to P. aeruginosa being classified as one of the ESKAPE pathogens, which account for 40 % of all nosocomial infections (Rice, 2010; Pendleton et al., 2013). P. aeruginosa has adapted to prevail and infect widely, in part due to its high degree of resistance to antibiotics, and its expression of a large arsenal of virulence factors posing a serious threat in the clinical setting (Kerr & Snelling, 2009).
The major mechanisms contributing to antibiotic resistance in P. aeruginosa include an impermeable outer membrane, expression of efflux pumps, target alteration and production of drug-inactivating enzymes (Livermore, 2002; Zavascki et al., 2010; Alvarez-Ortega et al., 2011). The highly impermeable membrane provides P. aeruginosa with natural resistance to many antimicrobials (Angus et al., 1982; Nikaido, 2003). To deal with the antimicrobials that cross the cell-wall barrier, the P. aeruginosa PAO1 genome encodes 10 resistance–nodulation–division systems that include MexAB–OprM, MexCD–OprJ, MexEF–OprN and MexXY–OprM contributing to antibiotic resistance (Stover et al., 2000; Lister et al., 2009). Upregulation of the MexAB–OprM efflux pump confers resistance to many different classes of antibiotics including fluoroquinolones, β-lactams, sulfonamides, chloramphenicol and trimethoprim (Li et al., 1995). The MexCD–OprJ pump predominantly expels quinolones, macrolides, tetracycline, chloramphenicol and fourth-generation cephalosporins such as cefepime (FEP) and cefpirome (Masuda et al., 1996; Poole et al., 1996a). The MexEF–OprN pump is involved in efflux of fluoroquinolones, chloramphenicol and trimethoprim (Köhler et al., 1997). The MexXY–OprM pump is involved in the efflux of aminoglycosides, quinolones, tetracyclines and some β-lactams (Morita et al., 2012). Importantly, it is the only pump involved in the efflux of aminoglycosides. It is evident that the efflux pumps share substrates and may involve a coordinated expression to deal with the onslaught of antibiotics.
Another major determining factor contributing to antibiotic resistance in P. aeruginosa is the overexpression of antibiotic-hydrolysing enzymes such as β-lactamases that are either chromosomally encoded or acquired (Hennessey, 1967; Lindberg & Normark, 1986; Jacoby, 2009; Castillo-Vera et al., 2012; Yong et al., 2012). The β-lactamases degrade β-lactam antibiotics (Normark et al., 1986; Hanson & Sanders, 1999; Gupta, 2008; Zhao & Hu, 2010; Bonnin et al., 2013). The major chromosomally encoded β-lactamase in P. aeruginosa is AmpC, whose expression is positively regulated by a LysR-type transcriptional regulator, AmpR (Lodge et al., 1990, 1993). In addition to AmpC, P. aeruginosa expresses a second chromosomal β-lactamase, PoxB (Girlich et al., 2004; Kong et al., 2005a), and can also acquire extended-spectrum metallo-β-lactamases on mobile genetic elements and plasmids (Bradford, 2001; Poirel et al., 2012; Zhu et al., 2013). In recent years, it has become apparent that P. aeruginosa co-regulates antibiotic resistance and virulence (Gooderham & Hancock, 2009; Yeung et al., 2011; Balasubramanian et al., 2013b). Our studies show that AmpR plays a major role in this co-regulation (Kong et al., 2005b; Balasubramanian et al., 2011, 2012). In addition to β-lactam resistance, AmpR regulates fluoroquinolone resistance, expression of many different virulence factors, QS-regulated phenotypes and biofilm formation (Balasubramanian et al., 2011, 2012, 2013a).
The current treatment regimen for P. aeruginosa infections involves fluoroquinolones, aminoglycosides and β-lactams alone or in combination (Cystic Fibrosis Foundation, 2011; Vardakas et al., 2013). The major β-lactams used extensively in clinical settings include carbapenems [imipenem (IPM) or meropenem (MEM)], third-generation cephalosporins [ceftazidime (CAZ)], Gram-negative-specific drugs [monobactam aztreonam (ATM)] and penicillin derivatives [piperacillin (PIP), either alone or in combination with tazobactam (TZP)] (Giamarellou & Kanellakopoulou, 2008; Page & Heim, 2009). The use of combination therapy for treatment of P. aeruginosa infections has been controversial. Many studies have dismissed the efficacy and advantages of combination therapy over monotherapy (Boyd & Nailor, 2011; Johnson et al., 2011; Tamma et al., 2012). In fact, adverse effects of combination therapy have been reported frequently (Paul et al., 2004, 2006). In addition, P. aeruginosa isolates from patients undergoing combination therapy have a higher rate of resistance, and, with simultaneous exposure, this allows the co-evolution of resistance to multiple classes of antibiotics (McGowan, 2006). However, compelling data on the interplay between antibiotics in the development of resistance is missing. We hypothesize that use of the same mechanisms involving efflux pumps or regulators such as AmpR may contribute to the development of cross-resistance to multiple classes of antibiotics.
Pathogens have adapted to respond to a gradient of antibiotic concentration in the environment as well as within the host during therapy (Baquero & Negri, 1997; Baquero et al., 2008). Whilst much research is focused on studying bacterial resistance to high antibiotic doses, the response to subinhibitory concentrations (SICs) remains largely unexplored. During the last decade, whole-genome studies have enabled identification of the effects of SICs of antibiotics on cellular mechanisms other than their direct targets (Davies et al., 2006). In this study, we explored the role of pre-exposure to SICs of clinically relevant antibiotics on the induction of transient cross-resistance in P. aeruginosa. Our studies also provide insights into the role of AmpR in regulating antibiotic cross-resistance in response to SICs of antibiotics.
Methods
Bacterial strains, growth conditions and antibiotics.
The prototypic P. aeruginosa strain PAO1 and its isogenic ampR deletion mutant, PAOΔampR, used in this study have been described previously (Stover et al., 2000; Balasubramanian et al., 2012). Luria–Bertani broth (Fisher Scientific) was used for routine cultivation of strains and was supplemented with 1.5 % agar when needed. Cation-adjusted Mueller–Hinton (CAMH) broth (Difco) was used for MIC studies and chequerboard assays. All antibiotics were obtained from Sigma-Aldrich. The antibiotics used in the study were: penicillins (amoxicillin, AMX; ampicillin/sulbactam, SAM; PIP; TZP; ticarcillin, TIC; ticarcillin/clavulanic acid, TIM), cephalosporins (CAZ; FEP; cefotaxime, CTX), carbapenems (IPM; MEM; doripenem, DOR; ertapenem, ETP), monobactam (ATM), quinolones (ciprofloxacin, CIP; ofloxacin, OFX; levofloxacin, LVX) and aminoglycosides (tobramycin, TOB; amikacin, AMK).
Construction of PAOΔampC.
An in-frame deletion of ampC was constructed using overlapextension PCR and homologous recombination as described previously (Balasubramanian et al., 2012). Briefly, sequences upstream (855 bp) and downstream (875 bp) of the target deletion were amplified using primer pairs DZampCUF1 (5′-GGAATTCAAGACGATGCTCCGGGTCAGTG-3′) and DZampCUR1 (5′-GATACCAGATTCCCCTGCCTGTCTAGCTAGCTAGAATGCTC-3′), and DZampCDF2 (5′-CTAGCTAGCTAGAATGCTCAAGCGCGCTCGCGAGGGCGACGGA-3′) and DZampCDR2 (5′-CGGGATCCGACCCTGCATACCATCAAGG-3′), respectively. The two amplicons were then ligated through PCR and cloned into the suicide vector pEXG2 (Rietsch et al., 2005). The resultant plasmid was moved into PAO1 for homologous recombination with the genomic DNA. Clones were screened for gentamicin sensitivity (75 µg ml−1) and sucrose resistance (8 % sucrose) corresponding to a double cross-over recombination event and replacement of the target gene with the deletion product. The presence of the deletion in PAOΔampC (PKM201) was confirmed by PCR amplification and sequencing of the deletion product (data not shown).
MICs.
The MICs of clinically relevant antibiotics were determined for P. aeruginosa strains using E-strips following the manufacturer’s protocol (bioMérieux). E-test MICs are widely regarded as being reliable and the results are identical to the broth microdilution method (Arendrup et al., 2001; Pankuch et al., 2006; Amsler et al., 2010) recommended by the Clinical and Laboratory Standards Institute (CLSI, 2006). To determine whether exposure to SICs of the antibiotics altered the P. aeruginosa susceptibility profile, exponential-phase cells (OD600 of 0.6) were divided into two pools. One pool was exposed to SICs (≤0.25 MIC) of antibiotic (one antibiotic at a time) for 1 h at 37 °C and plated on CAMH plates containing the same antibiotic. The second pool was not pre-exposed to antibiotic and was plated on plain CAMH plates. E-test assays were then performed with a panel of antibiotics to compare unexposed and pre-exposed MICs. The SICs of the various antibiotics used were: 0.1 µg ml−1 (IPM, CAZ and TOB), 0.05 µg ml−1 (CIP and MEM) and 0.2 µg ml−1 (PIP). All the assays were performed at least in duplicate. In general, ≥2.5-fold difference in the MIC profile was considered significant.
Chequerboard assays.
Interactions between antibiotics were determined using chequerboard assays in 96-well plates (Sopirala et al., 2010). Two-dimensional chequerboard assays were performed with twofold serial dilutions of each antibiotic, along the rows (for antibiotic 1) and columns (for antibiotic 2) in a 96-well plate (BD Labware). The bacterial cells were diluted according to CLSI recommendations (CLSI, 2006) and 100 µl dilution (5×105 c.f.u. ml−1) was added to each well containing antibiotics, for a total reaction volume of 200 µl per well. Results were observed after 16–18 h static incubation at 37 °C. The first clear well containing both antibiotics was used to calculate the fractional inhibitory concentration (FIC) as follows: FIC of antibiotic A (FICA) = MIC of antibiotic A in combination/MIC of antibiotic A alone; FIC of antibiotic B (FICB) = MIC of antibiotic B in combination/MIC of antibiotic B alone; FIC index (FICi) = FICA+FICB. FICi data were interpreted as follows: <0.5, synergy; >0.5–4.0, indifference; >4.0, antagonism (Odds, 2003). Chequerboard assays were performed with the following antibiotics: IPM (0.003–0.2 µg ml−1 and 0.05–3.2 µg ml−1) with CAZ (0.8–51.2 µg ml−1), PIP (12.5–800 µg ml−1), TIC (12.5–800 µg ml−1) and ATM (0.5–32 µg ml−1). The chequerboard assays were performed in triplicate.
RNA isolation, cDNA synthesis and quantitative real-time PCR (qPCR).
Total RNA was isolated from PAO1 and PAOΔampR grown to mid-exponential phase with or without pre-exposure to SICs of antibiotics (as described above). RNA isolation, cDNA synthesis and qPCR assays were performed as described previously (Balasubramanian et al., 2012). The gene-specific primers used for qPCR were: ampC: forward, 5′-CGCCGTACAACCGGTGAT-3′; reverse, 5′-CGGCCGTCCTCTTTCGA-3′; ampR: forward, 5′-CATTGGCCTTCATCACCGGTTGTA-3′; reverse, 5′-GGTTTCTCATGCAGCCAACGACAA-3′; poxB: forward, 5′-AATCGGCCAGGTTGTGGATAA-3′; reverse, 5′-GGAGCAGAAAGCGGGTCTGT-3′; and mexE: forward, 5′-AAGTCATCGAACAACCGCTGAACG-3′; reverse, 5′-TTCTTCACCAGTGCGCCTTCAT-3′. Ten nanograms of cDNA was used per reaction well in the qPCR assays. As an internal control, the clpX gene (PA1802) was included to ensure equal amounts of RNA were used in all samples. qPCR assays for each gene were performed at least in biological duplicates, each with technical triplicates. Melting curves were determined to ensure primer specificity. Gene expression values were normalized to the non-antibiotic-treated PAO1 values and are represented as means±se.
Statistical analysis.
All data were analysed for statistical significance using Student’s t-test on GraphPad statistical analysis software.
Results and Discussion
Effect of exposure to SICs of antibiotics on the P. aeruginosa PAO1 susceptibility profile
In this study, the effects of pre-exposure to SICs of various clinically relevant antibiotics on the P. aeruginosa PAO1 susceptibility profile were determined (Table 1). After pre-exposure, the cells were plated on CAMH plates with and without SICs of antibiotics. The MIC profile was determined by E-test (Table 1). The effect on P. aeruginosa PAO1 susceptibility was dependent on the class of antibiotic used for the SIC exposure (Table 1).
Table 1. Resistance profile of P. aeruginosa PAO1 after SIC antibiotic exposure.
Results are shown as MIC values (µg ml−1).
| Class | Antibiotic | None* | SIC antibiotic exposure | ||||
| IPM (0.1 µg ml−1) | CAZ (0.1 µg ml−1) | PIP (0.2 µg ml−1) | CIP (0.05 µg ml−1) | TOB (0.1 µg ml−1) | |||
| Penicillin | AMX | >256 | >256 | >256 | >256 | >256 | >256 |
| SAM | 96 | 96 | 64 | 256 | 32 | 256 | |
| TZP | 4 | 128 | 3/2 | 4 | 1.5 | 4 | |
| TIM | 24 | 256 | 16 | 24 | 8 | 16 | |
| Cephalosporin | CAZ | 1.5 | 8 | 1 | 2 | 0.75 | 1 |
| FEP | 2.0 | 6 | 1.5 | 2 | 1 | 3 | |
| Carbapenem | IPM | 1.5 | 3 | 1.5 | 1.5 | 0.75 | 1.5 |
| DOR | 0.38 | 0.5 | 0.19 | 0.25 | 0.19 | 0.5 | |
| MEM | 0.38 | 0.25 | 0.25 | 0.5 | 0.75 | 0.5 | |
| ETP | >32 | nd | nd | >32 | 6 | >32 | |
| Monobactam | ATM | 3 | 6/8 | 1.5 | 3 | 1 | 3 |
| Quinolone | OFX | 1 | 1.5 | 1 | 1.5 | 0.5 | 1.5 |
| CIP | 0.25 | 0.25 | 0.25 | 0.25 | 0.125 | 0.38 | |
| LVX | 0.5 | 0.5 | 0.5 | 0.5 | 0.25 | 0.75 | |
| Aminoglycoside | AMK | 4 | 6 | 4 | 4 | 4 | 4 |
| TOB | 1 | 0.75 | 0.75 | 0.75 | 0.75 | 0.75 | |
nd, Not determined.
MIC of PAO1 with no antibiotic exposure.
Pre-exposure to a SIC of the β-lactam PIP (0.2 µg ml−1) and the aminoglycoside TOB (0.1 µg ml−1) resulted in a significant increase in resistance only to SAM (2.7-fold) and not to the other antibiotics tested (Table 1). In contrast, when PAO1 was pre-exposed to a SIC of the quinolone CIP (0.05 µg ml−1), there was increased susceptibility to almost all the classes of antibiotics tested (SAM, 3-fold; TZP, 2.7-fold; TIM, 3-fold; ETP, 5.3-fold; ATM, 3-fold). SIC exposure to the cephalosporin CAZ (0.1 µg ml−1) marginally enhanced PAO1 susceptibility to all classes of antibiotics except quinolones (Table 1). Although the difference in MICs was only between 1.5- and 2.0-fold, which is typically not considered significant, the trend was consistent over the different classes of antibiotics (Table 1). Such marginal differences are suggestive of creeping baseline MICs, contributing to breakthrough resistance in the clinical setting (Fernández et al., 2011).
Exposure to a SIC of IPM (0.1 µg ml−1) significantly enhanced PAO1 cross-resistance to penicillins (TZP, 32-fold; TIM, 10.7-fold), cephalosporins (CAZ, 5.3-fold; FEP, 3-fold) and monobactams (ATM, 2.7-fold) but had little or no effect on other carbapenems (DOR, MEM and ETP), quinolones and aminoglycosides (Table 1). The gain in resistance to the penicillins was significant enough to cross their MIC breakpoints and classify the strain as clinically resistant, in accordance with CLSI standards (TZP and TIM: ≥128 µg ml−1; CLSI, 2006). This is in agreement with previous findings where exposure to a SIC of IPM led to clinical resistance in P. aeruginosa (Livermore, 1987). To determine whether the trend was true of other carbapenems, PAO1 was exposed to a SIC of MEM (0.05 µg ml−1) before determining the MIC (Table 2). Compared with unexposed PAO1, SIC MEM-exposed cells displayed a marginal increase in resistance towards TZM (3-fold) and CAZ (2-fold; Table 2) but showed no effect on the rest of the antibiotics (data not shown). Thus, it seems that, although carbapenems in general have the potential to induce resistance to other antibiotics, the extent varies and IPM is a much more effective inducer of transient cross-resistance compared to MEM (Table 2).
Table 2. MICs of penicillins and cephalosporins in response to SIC carbapenem exposure.
Results are shown as MIC values (µg ml−1).
| Class | Antibiotic | None* | SIC antibiotic exposure | |
| IPM (0.1 µg ml−1)† | MEM (0.05 µg ml−1) | |||
| Penicillin | TZP | 4 | 128 | 12 |
| TIM | 24 | 256 | 32 | |
| Cephalosporin | CAZ | 1.5 | 8 | 3 |
| FEP | 2 | 6 | 2 | |
MIC of PAO1 with no antibiotic exposure.
IPM-exposed PAO1 MIC values are from Table 1 for comparison.
The susceptibility profile of the pre-exposed and unexposed cells was similar on plates containing no antibiotic (data not shown). Also, the difference seen in the profile of the pre-exposed cells on plates contacting antibiotics (Table 1) was lost when subcultured on plates without any antibiotics (data not shown). These results showed that the changes conferred are transient and that the altered susceptibility is observed only in the presence of SICs of antibiotics. This implies the existence of a dynamic interplay between different classes of antibiotics at SICs in P. aeruginosa. The findings also necessitate a careful assessment of both combinatorial therapy and the antibiotic treatment history of a patient, because trailing concentrations of one antibiotic such as IPM can provide resistance to other subsequently used antibiotics. The role of carbapenems in the transient cross-resistance was explored further.
A concentration as low as 3 ng IPM ml−1 can induce β-lactam cross-resistance
The data from cells exposed to a SIC of IPM suggested a widespread antagonistic effect with other antibiotics (Table 1). In order to determine the extent of combinatorial cross-resistance, chequerboard assays were performed for IPM (0.05–3.2 µg ml−1) with PIP, TIC, CAZ and ATM as described in Methods. The FIC for each antibiotic and the FICi for each combination were calculated (Fig. 1). Any two drugs were considered synergistic, indifferent (or non-synergistic) or antagonistic if the FICi scores were <0.5, >0.5 but <4.0 or>4.0, respectively (Odds, 2003).
Fig. 1.

IPM chequerboard assays. The effects of combinatorial cross-resistance of IPM with PIP (a), TIC (b), CAZ (c) and ATM (d) on P. aeruginosa PAO1 was determined. The FIC and FICi values were calculated as described in Methods. The antibiotics were used at the following concentrations: IPM, 0.05–3.2 µg ml−1; PIP, 12.5–800 µg ml−1: TIC, 12.5–800 µg ml−1; CAZ, 0.8–51.2 µg ml−1; ATM, 0.5–32 µg ml−1. All assays were performed in triplicate.
The FICi for the IPM/PIP combination was 8.06, suggesting that the antibiotic pair was antagonistic (Fig. 1). When IPM was combined with TIC, CAZ or ATM, the FICi was 2.06 in each case, suggesting indifference. However, a closer assessment of the FIC for individual antibiotics in the combinations tested revealed that the FICi was heavily skewed in one direction. For example, the FIC of PIP in the presence of IPM was 8, whereas when reversed, the score was 0.06, suggesting an antagonistic and synergistic effect, respectively (Fig. 1). Similarly, the FIC of TIC, CAZ and ATM with IPM (FIC = 2.0) suggested a non-synergistic interaction, whereas the FIC of IPM with these three antibiotics was 0.06, indicating synergy (Fig. 1). Thus, the chequerboard data suggested that IPM became more potent at killing cells at lower concentrations in the presence of other antibiotics like PIP, TIC, CAZ and ATM. More importantly, the other antibiotics became less effective (high MIC) in the presence of low doses of IPM (Fig. 1). These results highlight the significance of the order of antibiotic treatment. A better treatment output can be expected if PIP, CAZ or TIC therapy is followed by IPM, whereas the reverse order could lead to high resistance to PIP, CAZ or TIC at diminishing IPM concentrations.
The susceptibility to various antibiotics in the presence of a SIC of IPM was concentration dependent (Fig. 1), the effect being more pronounced at 0.1 µg ml−1 and starting to drop at 0.4 µg ml−1 as it approached the MIC for IPM. To further explore the lowest concentration limit, another chequerboard assay was carried out with 0.003–0.2 µg IPM ml−1, whilst the other β-lactams were kept at the same concentrations as before. The results clearly demonstrated that, even at concentrations as low as 3–6 ng ml−1, IPM was able to alter the resistance to PIP, CAZ, TIC and ATM (Fig. 2).
Fig. 2.
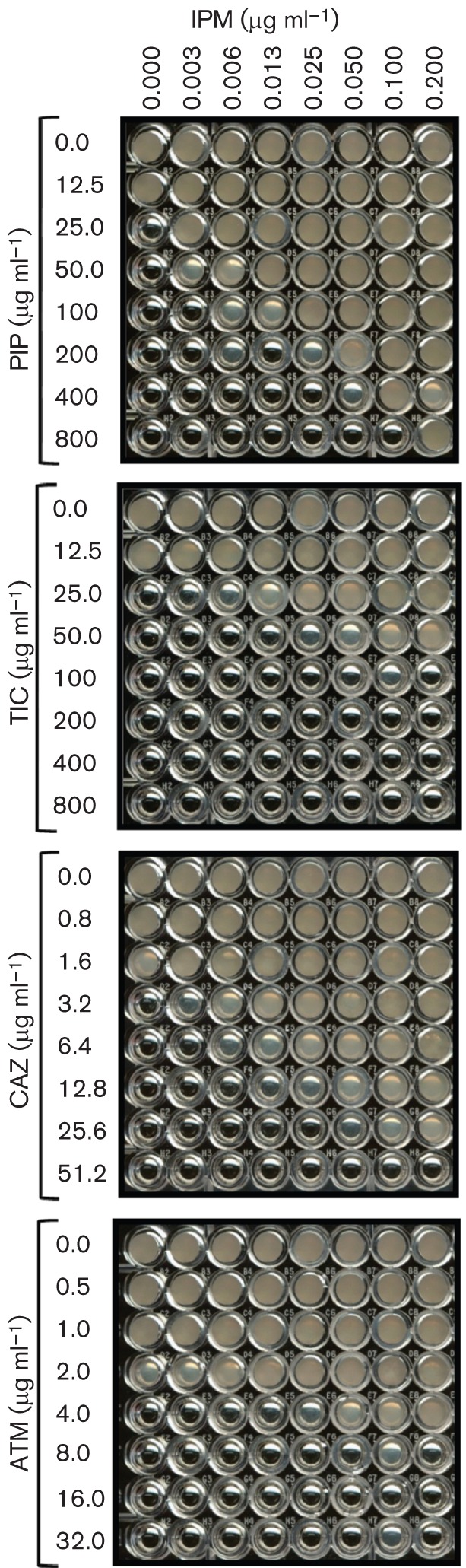
Low-range IPM chequerboard assays. The effects of co-exposure of very low concentrations of IPM with PIP (a), TIC (b), CAZ (c) and ATM (d) on P. aeruginosa PAO1 was determined. The antibiotics were used at the following concentrations: IPM, 0.003–0.2 µg ml−1; PIP, 12.5–800 µg ml−1; TIC, 12.5–800 µg ml−1; CAZ, 0.8–51.2 µg ml−1; ATM, 0.5–32 µg ml−1. All assays were performed at least in triplicate.
IPM is often used to treat multidrug-resistant P. aeruginosa infections (Page & Heim, 2009). SICs of IPM are known to confer clinical resistance (Livermore, 1987). However, this is the first study to reveal that a concentration as low as 3 ng IPM ml−1 can enhance resistance to clinically used penicillins and cephalosporins (Table 1, Fig. 2). The biphasic response that shows low-dose stimulation and high-dose inhibition is termed hormesis (Stebbing, 1982; Davies et al., 2006). An earlier study also linked the use of IPM with the risk of emergence of antibiotic resistance (Carmeli et al., 1999). A SIC of IPM has also been shown to induce genes coding for alginate synthesis and biofilm formation, hallmarks of chronic infection (Bagge et al., 2004). Together, these findings indicate that the use of IPM as an anti-pseudomonal drug comes with a high cost in terms of increased resistance to other clinically used antibiotics, and favours chronic infection at its lingering low concentrations.
Induction of ampC and ampR expression by SICs of clinically used antibiotics
In P. aeruginosa, AmpC β-lactamase is the primary mediator of β-lactam resistance, whose expression is modulated by the AmpR in response to the inducers (Lodge et al., 1990, 1993). In order to determine the most proficient inducer of ampC, mRNA levels were quantified using qPCR after exposure of PAO1 to SICs of various antibiotics (Fig. 3). As expected, the most potent inducer of ampC expression was IPM [relative quantity (RQ): 221±16, P = 0.003], corroborating previous findings (Livermore & Yang, 1987) and further supporting the enhanced resistance seen in the MIC and chequerboard assays (Table 1, Fig. 1). A similar induction of ampC expression in response to a SIC of IPM was also seen in P. aeruginosa biofilm (Bagge et al., 2004). The qPCR assays also identified MEM as another moderately strong inducer of ampC (RQ: 58±3, P = 0.001). However, the poor induction of ampC with SICs of other β-lactams (CAZ and PIP) was similar to the non-β-lactams (CIP and TOB; Fig. 3).
Fig. 3.
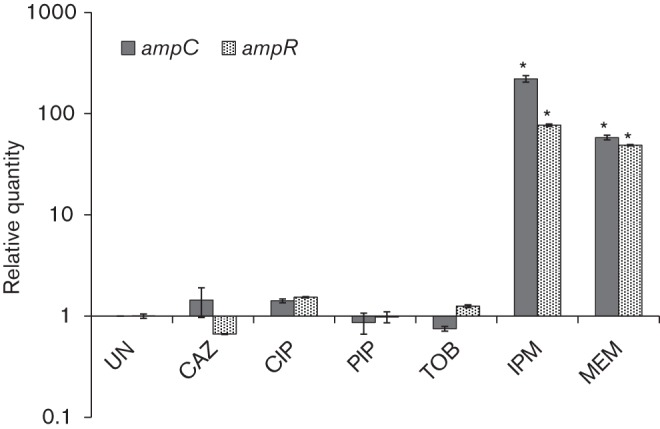
Induction of ampR and ampC expression. qPCR was used to quantify the relative quantities of ampR and ampC mRNA after P. aeruginosa PAO1 cells were treated with SICs of various antibiotics. Data were normalized to expression in the untreated cells. The clpX gene (PA1802) was used as the housekeeping control. *P≤0.003 compared with expression in the uninduced (UN) condition.
To determine whether the induction of ampC is a function of increased expression of ampR, its transcript levels were also quantified. Pre-exposure to SICs of both IPM (RQ: 77±2, P<0.0001) and MEM (RQ: 49±1, P<0.0001) were found to induce the expression of ampR, although expression was lower compared with ampC expression (Fig. 3). Induction of ampR expression in P. aeruginosa PAO1 at SIC exposure to antibiotics has not been shown before. Although ampR expression is induced by a β-lactam in the mucoid P. aeruginosa variant PDO300, this is due to its regulation by the alginate master regulator AlgT/U (Balasubramanian et al., 2011). A previous transcriptional fusion study from our group showed that ampR expression in P. aeruginosa PAO1 is not significantly affected in the presence of inducer (Kong et al., 2005b). Even Citrobacter freundii ampR is known to be expressed constitutively in a heterologous host, irrespective of the inducer (Lindquist et al., 1989). However, the above studies used benzylpenicillin and 6-aminopenicillanic acid as inducer, respectively. We showed here that IPM was far more effective than the other β-lactams at inducing ampC and ampR expression (Fig. 3). What makes carbapenems and specifically IPM a strong inducer, even at very low concentrations, is still not known and needs to be explored further.
Absence of AmpR leads to enhanced susceptibility following SIC antibiotic exposure
The role of AmpR in antibiotic resistance is well established (Balasubramanian et al., 2011, 2012; Cabot et al., 2012). To determine the role of AmpR in transient cross-resistance, specifically in the presence of IPM, an E-test was performed on PAOΔampR with and without exposure to SICs of antibiotics (Table 3).
Table 3. Susceptibility profile of P. aeruginosa PAOΔampR and PAOΔampC after SIC antibiotic exposure.
Results are shown as MIC values (µg ml−1).
| Class | Antibiotic | None* | None† | SIC antibiotic exposure | ||||||||||||
| IPM (0.1 µg ml−1) | MEM (0.05 µg ml−1) | CAZ (0.1 µg ml−1) | PIP (0.2 µg ml−1) | CIP (0.05 µg ml−1) | TOB (0.1 µg ml−1) | |||||||||||
| ampR | ampC | ampR | ampC | ampR | ampC | ampR | ampC | ampR | ampC | ampR | ampC | ampR | ampC | |||
| Penicillin | AMX | >256 | 12 | 12 | 12 | 12 | 12 | 12 | 12 | 12 | 8 | 24 | 8 | 6 | 12 | 16 |
| SAM | 96 | 12 | 16 | 3 | 12 | 8 | 8 | 6 | 16 | 8 | 32 | 6 | 6 | 4 | 24 | |
| TZP | (4) | 3 | 4 | 1 | 1.5 | 2 | 3 | 1.5 | 3 | 3/2 | 4 | 2 | 1.5 | 2 | 3 | |
| TIM | 24 | 24 | 48 | 8 | 16 | 6 | 12 | 4 | 16 | 8 | 24 | 4 | 6 | 6 | 12 | |
| Cephalosporin | CAZ | 1.5 | 1.5 | 2 | 0.38 | 1 | 1 | 1 | 1 | 1.5 | 1.5 | 2 | 1 | 0.75 | 1 | 1.5 |
| FEP | 2 | 1.5 | 2 | 0.5 | 1.5 | 1 | 0.75 | 0.75 | 1.5 | 1.5 | 3 | 1 | 1 | 2 | 3 | |
| Carbapenem | IPM | 1.5 | 0.38 | 0.5 | 0.38 | 0.38 | 0.38 | 0.25 | 0.38 | 0.25 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| DOR | 0.38 | 0.25 | 0.125 | 0.25 | 0.125 | 0.19 | 0.064 | 0.25 | 0.094 | 0.25 | 0.19 | 0.38 | 0.064 | 0.5 | 0.125 | |
| MEM | 0.38 | 0.5 | 0.3 | 0.25 | 0.25 | 0.5 | 0.19 | 0.25 | 0.125 | 0.5 | 0.25 | 0.38 | 1 | 0.5 | 1.5 | |
| ETP | >32 | 6 | 8 | nd | nd | nd | nd | nd | nd | 8 | 8 | 6 | ND | 8 | ND | |
| Monobactam | ATM | 3 | 1.5 | 3 | 0.5 | 1.5 | 1.5 | 3 | 0.75 | 1.5 | 1.5 | 3 | 1 | 1 | 1 | 3 |
| Quinolone | CIP | 0.25 | 1.5 | 0.25 | 1.5 | 0.25 | 1.5 | 0.25 | 1.5 | 0.25 | 1.5 | 0.38 | 1.5 | 0.125 | 1.5 | 0.38 |
| LVX | 0.5 | 6 | 0.5 | 6 | 0.5 | 3 | 0.5 | 4 | 0.5 | 4 | 0.5 | 4 | 0.25 | 4 | 0.75 | |
| Aminoglycoside | AMK | 4 | 3 | 4 | 3 | 4 | 3 | 3 | 2 | 4 | 3 | 4 | 3 | 6 | 3 | 4 |
| TOB | 1 | 0.5 | 1 | 0.38 | 0.75 | 0.38 | 0.5 | 0.38 | 0.75 | 0.38 | 0.75 | 0.25 | 0.75 | 0.38 | 0.75 | |
nd, Not determined.
*PAO1 MIC values are from Table 1 for comparison.
†PAOΔampR and PAOΔampC with no antibiotic exposure.
In the absence of SIC exposure, the loss of ampR resulted in increased sensitivity to most of the β-lactams that were tested such as penicillins (AMX and SAM), carbapenems (IPM and ETP) and monobactam (ATM) but not the cephalosporins (CAZ and FEP; Table 3). The increased sensitivity could be due to loss of ampC expression in PAOΔampR.
Pre-exposure to SICs of antibiotics made PAOΔampR more susceptible to penicillins, cephalosporins and monobactam (Table 3). Specifically, exposure to a SIC of IPM increased the susceptibility of PAOΔampR to many penicillins (SAM, fourfold; TZP, threefold; TIM, threefold), cephalosporins (CAZ, fourfold; FEP, threefold) and monobactam (ATM, threefold). The data indicated that AmpR plays a key role in subinhibitory IPM-mediated β-lactam resistance in P. aeruginosa (Table 3).
Apart from β-lactams, loss of ampR also resulted in increased susceptibility to the aminoglycosides AMK and TOB (Table 3). The sensitivity to aminoglycosides was enhanced upon pre-exposure to various β-lactams and non-β-lactams. The change in TOB susceptibility of PAOΔampR upon SIC antibiotic exposure was also confirmed by the broth dilution method (data not shown). Regulation of aminoglycoside resistance by AmpR is not AmpC-mediated because the resistance profile of PAOΔampC remained unaltered (Table 3). The data clearly suggest that AmpR plays a critical role in regulating antibiotic cross-resistance in P. aeruginosa.
Loss of ampR, in addition to rendering P. aeruginosa sensitive to many β-lactam antibiotics, also leads to reduced production of acute virulence factors (Balasubramanian et al., 2012, 2013a). This finding further augments the suggestion that AmpR is a viable drug candidate as it would make P. aeruginosa less virulent, and sensitive to β-lactams and aminoglycosides, without confounding their efficacy at SIC pre-exposure to antibiotics.
AmpR-mediated cross-resistance to β-lactams is partially contributed by AmpC
Largely, AmpR regulates β-lactam resistance through AmpC as both PAOΔampR and PAOΔampC became sensitive to β-lactams compared with PAO1 (Table 3). To investigate whether AmpR could regulate β-lactam resistance an in AmpC-independent manner, the MIC profile was compared between PAOΔampR and PAOΔampC with and without exposure to SICs of antibiotics. Compared with PAOΔampC, pre-exposure to various β-lactams and TOB made PAOΔampR more susceptible to many penicillins (SAM, TZP and TIM), cephalosporins (CAZ and FEP) and monobactam (ATM). The difference in susceptibility showed that AmpR also regulates β-lactam resistance by AmpC-independent mechanisms. One such mechanism could involve MexR, the regulator of the MexAB–OprM pump involved in the efflux of β-lactam antibiotics (Poole et al., 1996b). This is further supported by previous qPCR studies that show positive regulation of mexR expression by AmpR (Balasubramanian et al., 2012). This would argue that, in the absence of ampR, the expression of the MexAB–OprM pump should confer increased resistance. However, the data from our laboratory show that deleting ampC in PAO1 (Table 3) or in PAOΔampR (D. Zincke, unpublished data) diminishes β-lactam resistance, in spite of having a functional MexAB system. This observation suggests that the MexAB pump by itself is not enough to confer β-lactam resistance. Although induction of ampC by AmpR seems to be the primary mechanism that leads to enhanced resistance, we are far from understanding the role of various converging pathways in the regulation of antibiotic resistance.
AmpR is involved in cross-resistance to quinolones
The MIC data comparison showed that the increase in resistance to quinolones (CIP and LVX) in PAOΔampR was AmpC-independent because PAOΔampC had no effect on resistance to quinolones (Table 3). This finding is in agreement with a previously shown role of AmpR in quinolone resistance through regulation of mexEF–oprN encoding the efflux pump and its regulator mexT, in the absence of antibiotic induction (Köhler et al., 1999; Balasubramanian et al., 2012). To investigate the role of AmpR in the expression of mexEF–oprN efflux pump and cross-induction by clinically used antibiotics, we checked the expression of mexE, the first gene of the mexEF–oprN operon, in PAO1 and PAOΔampR after exposure to SICs of antibiotics (Fig. 4).
Fig. 4.
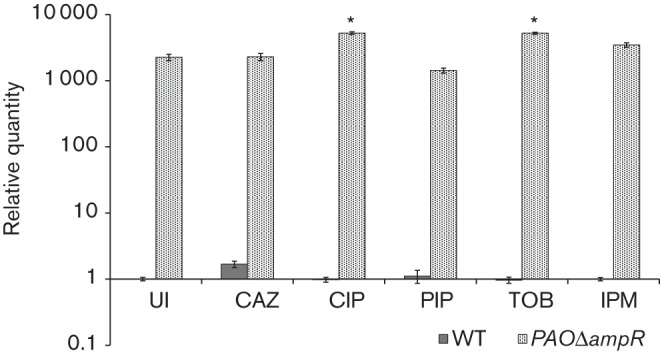
mexE expression in response to SICs. Expression of mexE was determined in PAO1 (WT) and PAOΔampR cells, with and without SIC treatment with various antibiotics. Data were normalized to expression in the PAO1 untreated cells. The clpX gene (PA1802) was used as the housekeeping control. UI, uninduced. *P≤0.0008 compared with the WT untreated (UI) strain.
In the absence of any pre-exposure to antibiotics, PAOΔampR had a high mexE expression compared with PAO1 (RQ in PAOΔampR: 2253±251, P = 0.0001; Fig. 4), which is in agreement with previous findings (Balasubramanian et al., 2012). However, mexE expression could not be induced in PAO1 with any of the antibiotics including the MexEF–OprN substrate CIP (Fig. 4). The absence of induction was not surprising, as PAO1 has a functional AmpR that represses mexE expression (Balasubramanian et al., 2012). In addition, PAO1 harbours an 8 bp insertion in mexT encoding the positive regulator of mexEF–oprN transcription, rendering it non-inducible (Maseda et al., 2000).
In the presence of a SIC of CIP, a MexEF–OprN substrate, expression of mexE in PAOΔampR was further induced twofold (RQ: untreated 2253±251, CIP treated 5241±246, P = 0.0001; Fig. 4). This suggested that there is an additional level of transcriptional control in the presence of CIP, which, unlike AmpR, positively regulates mexE transcription. In addition, there was a twofold induction of mexE expression upon treatment with a SIC of the aminoglycoside TOB, a non-MexEF substrate (RQ in PAOΔampR: untreated 2253±251, TOB treated 5207±192, P = 0.0008; Fig. 4). The mechanism resulting in the induction of a quinolone-specific efflux pump due to exposure to a SIC of an aminoglycoside is not clear.
The efflux pumps MexAB, MexEF and MexXY are predominantly involved in efflux of β-lactams, fluoroquinolones and aminoglycosides, respectively. However, cross-induction of these by different pump substrates is not uncommon. Expression of mexXY is induced by the quinolone OFX in the absence of mexAB (Masuda et al., 2000). Our study showed that expression of mexE is induced by an aminoglycoside in the absence of ampR (Fig. 4), where mexAB expression increases significantly (Balasubramanian et al., 2012). Together, these findings suggest that all three efflux pumps share regulation, and AmpR seems to play an important role in this process.
Conclusions
The development and persistence of antibiotic resistance is a complicated and multifactorial phenomenon necessitating dedicated research to understand and control this problem. Both high and low concentrations of antibiotics have been identified to result in the development of resistance (reviewed by Andersson & Hughes, 2011). Our data showed that exposure to SICs of antibiotics has a widespread role in inducing transient cross-resistance and expression of antibiotic resistance genes. Residual amounts of IPM could be enough to provide resistance to subsequent β-lactam treatment. Whilst the increase in resistance upon SIC exposure to carbapenems is evident and expected, the increase in susceptibility with CIP was unexpected. The unexpected SIC sensitization to CIP warrants further investigation.
We also showed that AmpR has a wider role in regulating antibiotic resistance than previously thought. The data confirmed that AmpR plays a major role in the development of transient cross-resistance upon SIC induction. Additionally, AmpR was shown to regulate β-lactam resistance in an AmpC-dependent and -independent manner as well as non-β-lactam resistance such as that of quinolones and aminoglycosides. Together with our transcriptomic studies that established AmpR as a major regulator of virulence (Balasubramanian et al., 2012, 2013a), AmpR may be a suitable drug target for combating P. aeruginosa infections.
In conclusion, a strict and guided use of antibiotics and monitoring patient treatment history along with a careful assessment of the usefulness of therapeutic approaches is needed. With fewer new antibiotics being discovered, the focus should also be on developing new therapeutic strategies involving important players of resistance and virulence, such as AmpR. Although PAOΔampR displays fluoroquinolone resistance, targeting AmpR is still a good proposition because strains will become susceptible to β-lactams and have reduced virulence factor production.
Acknowledgements
This study was supported in part by the National Institutes of Health – Minority Biomedical Research Support SCORE (SC1AI081376; to K. M. and H. K.), Research Initiative for Scientific Enhancement graduate student fellowship (NIH/NIGMS R25 GM61347; to D. Z.), National Science Foundation under grant no. IIP-1237818 (PFI-AIR: CREST-I/UCRC-Industry Ecosystem to Pipeline Research), Herbert Wertheim College of Medicine (to H. K.) and Florida International University Dissertation Year Fellowship (to D. B.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Abbreviations:
- AMK
amikacin
- AMX
amoxicillin
- ATM
aztreonam
- CAZ
ceftazidime
- CIP
ciprofloxacin
- CLSI
Clinical and Laboratory Standards Institute
- DOR
doripenem
- ETP
ertapenem
- FEP
cefepime
- FIC
fractional inhibitory concentration
- FICi
FIC index
- IPM
imipenem
- LVX
levofloxacin
- MEM
meropenem
- OFX
ofloxacin
- PIP
piperacillin
- RQ
relative quantity
- SAM
ampicillin/sulbactam
- SIC
subinhibitory concentration
- TIC
ticarcillin
- TIM
ticarcillin/clavulanic acid
- TOB
tobramycin
- TZP
piperacillin/tazobactam
References
- Alvarez-Ortega C., Wiegand I., Olivares J., Hancock R. E., Martínez J. L. (2011). The intrinsic resistome of Pseudomonas aeruginosa to β-lactams. Virulence 2, 144–146 10.4161/viru.2.2.15014 [DOI] [PubMed] [Google Scholar]
- Amsler K., Santoro C., Foleno B., Bush K., Flamm R. (2010). Comparison of broth microdilution, agar dilution, and Etest for susceptibility testing of doripenem against Gram-negative and Gram-positive pathogens. J Clin Microbiol 48, 3353–3357 10.1128/JCM.00494-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson D. I., Hughes D. (2011). Persistence of antibiotic resistance in bacterial populations. FEMS Microbiol Rev 35, 901–911 10.1111/j.1574-6976.2011.00289.x [DOI] [PubMed] [Google Scholar]
- Angus B. L., Carey A. M., Caron D. A., Kropinski A. M., Hancock R. E. (1982). Outer membrane permeability in Pseudomonas aeruginosa: comparison of a wild-type with an antibiotic-supersusceptible mutant. Antimicrob Agents Chemother 21, 299–309 10.1128/AAC.21.2.299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendrup M., Lundgren B., Jensen I. M., Hansen B. S., Frimodt-Møller N. (2001). Comparison of Etest and a tablet diffusion test with the NCCLS broth microdilution method for fluconazole and amphotericin B susceptibility testing of Candida isolates. J Antimicrob Chemother 47, 521–526 10.1093/jac/47.5.521 [DOI] [PubMed] [Google Scholar]
- Bagge N., Schuster M., Hentzer M., Ciofu O., Givskov M., Greenberg E. P., Høiby N. (2004). Pseudomonas aeruginosa biofilms exposed to imipenem exhibit changes in global gene expression and β-lactamase and alginate production. Antimicrob Agents Chemother 48, 1175–1187 10.1128/AAC.48.4.1175-1187.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian D., Kong K. F., Jayawardena S. R., Leal S. M., Sautter R. T., Mathee K. (2011). Co-regulation of β-lactam resistance, alginate production and quorum sensing in Pseudomonas aeruginosa. J Med Microbiol 60, 147–156 10.1099/jmm.0.021600-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian D., Schneper L., Merighi M., Smith R., Narasimhan G., Lory S., Mathee K. (2012). The regulatory repertoire of Pseudomonas aeruginosa AmpC β-lactamase regulator AmpR includes virulence genes. PLoS ONE 7, e34067 10.1371/journal.pone.0034067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian D., Kumari H., Jaric M., Fernandez M., Turner K. H., Dove S. L., Narasimhan G., Lory S., Mathee K. (2013a). Deep sequencing analyses expands the Pseudomonas aeruginosa AmpR regulon to include small RNA-mediated regulation of iron acquisition, heat shock and oxidative stress response. Nucleic Acids Res. 10.1093/nar/gkt942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian D., Schneper L., Kumari H., Mathee K. (2013b). A dynamic and intricate regulatory network determines Pseudomonas aeruginosa virulence. Nucleic Acids Res 41, 1–20 10.1093/nar/gks1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquero F., Negri M. C. (1997). Selective compartments for resistant microorganisms in antibiotic gradients. BioEssays 19, 731–736 10.1002/bies.950190814 [DOI] [PubMed] [Google Scholar]
- Baquero F., Martínez J. L., Cantón R. (2008). Antibiotics and antibiotic resistance in water environments. Curr Opin Biotechnol 19, 260–265 10.1016/j.copbio.2008.05.006 [DOI] [PubMed] [Google Scholar]
- Barbier F., Wolff M. (2010). [Multi-drug resistant Pseudomonas aeruginosa: towards a therapeutic dead end?]. Med Sci (Paris) 26, 960–968 (in French). 10.1051/medsci/20102611960 [DOI] [PubMed] [Google Scholar]
- Bonnin R. A., Poirel L., Nordmann P., Eikmeyer F. G., Wibberg D., Pühler A., Schlüter A. (2013). Complete sequence of broad-host-range plasmid pNOR-2000 harbouring the metallo-β-lactamase gene blaVIM-2 from Pseudomonas aeruginosa. J Antimicrob Chemother 68, 1060–1065 10.1093/jac/dks526 [DOI] [PubMed] [Google Scholar]
- Boyd N., Nailor M. D. (2011). Combination antibiotic therapy for empiric and definitive treatment of gram-negative infections: insights from the Society of Infectious Diseases Pharmacists. Pharmacotherapy 31, 1073–1084 10.1592/phco.31.11.1073 [DOI] [PubMed] [Google Scholar]
- Bradford P. A. (2001). Extended-spectrum β-lactamases in the 21st century: characterization, epidemiology, and detection of this important resistance threat. Clin Microbiol Rev 14, 933–951 10.1128/CMR.14.4.933-951.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabot G., Ocampo-Sosa A. A., Domínguez M. A., Gago J. F., Juan C., Tubau F., Rodríguez C., Moyà B., Peña C. & other authors (2012). Genetic markers of widespread extensively drug-resistant Pseudomonas aeruginosa high-risk clones. Antimicrob Agents Chemother 56, 6349–6357 10.1128/AAC.01388-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeli Y., Troillet N., Eliopoulos G. M., Samore M. H. (1999). Emergence of antibiotic-resistant Pseudomonas aeruginosa: comparison of risks associated with different antipseudomonal agents. Antimicrob Agents Chemother 43, 1379–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo-Vera J., Ribas-Aparicio R. M., Nicolau C. J., Oliver A., Osorio-Carranza L., Aparicio-Ozores G. (2012). Unusual diversity of acquired β-lactamases in multidrug-resistant Pseudomonas aeruginosa isolates in a Mexican hospital. Microb Drug Resist 18, 471–478 10.1089/mdr.2011.0183 [DOI] [PubMed] [Google Scholar]
- CLSI (2006). Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; Approved standard M7-A7, 7th edn. Wayne, PA: Clinical and Laboratory Standards Institute [Google Scholar]
- Curcio D. (2013). Multidrug-resistant Gram-negative bacterial infections: are you ready for the challenge? Curr Clin Pharmacol 8, 1–12 [DOI] [PubMed] [Google Scholar]
- Cystic Fibrosis Foundation (2011). Patient Registry Annual Data Report. Bethesda: Maryland Cystic Fibrosis Foundation [Google Scholar]
- Davies J., Spiegelman G. B., Yim G. (2006). The world of subinhibitory antibiotic concentrations. Curr Opin Microbiol 9, 445–453 10.1016/j.mib.2006.08.006 [DOI] [PubMed] [Google Scholar]
- Fernández L., Breidenstein E. B., Hancock R. E. (2011). Creeping baselines and adaptive resistance to antibiotics. Drug Resist Updat 14, 1–21 10.1016/j.drup.2011.01.001 [DOI] [PubMed] [Google Scholar]
- Giamarellou H., Kanellakopoulou K. (2008). Current therapies for Pseudomonas aeruginosa. Crit Care Clin 24, 261–278, viii 10.1016/j.ccc.2007.12.004 [DOI] [PubMed] [Google Scholar]
- Girlich D., Naas T., Nordmann P. (2004). Biochemical characterization of the naturally occurring oxacillinase OXA-50 of Pseudomonas aeruginosa. Antimicrob Agents Chemother 48, 2043–2048 10.1128/AAC.48.6.2043-2048.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooderham W. J., Hancock R. E. (2009). Regulation of virulence and antibiotic resistance by two-component regulatory systems in Pseudomonas aeruginosa. FEMS Microbiol Rev 33, 279–294 10.1111/j.1574-6976.2008.00135.x [DOI] [PubMed] [Google Scholar]
- Gupta V. (2008). Metallo β lactamases in Pseudomonas aeruginosa and Acinetobacter species. Expert Opin Investig Drugs 17, 131–143 10.1517/13543784.17.2.131 [DOI] [PubMed] [Google Scholar]
- Hanson N. D., Sanders C. C. (1999). Regulation of inducible AmpC β-lactamase expression among Enterobacteriaceae. Curr Pharm Des 5, 881–894 [PubMed] [Google Scholar]
- Hennessey T. D. (1967). Inducible β-lactamase in Enterobacter. J Gen Microbiol 49, 277–285 10.1099/00221287-49-2-277 [DOI] [PubMed] [Google Scholar]
- Jacoby G. A. (2009). AmpC β-lactamases. Clin Microbiol Rev 22, 161–182 10.1128/CMR.00036-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson S. J., Ernst E. J., Moores K. G. (2011). Is double coverage of Gram-negative organisms necessary? Am J Health Syst Pharm 68, 119–124 10.2146/ajhp090360 [DOI] [PubMed] [Google Scholar]
- Kerr K. G., Snelling A. M. (2009). Pseudomonas aeruginosa: a formidable and ever-present adversary. J Hosp Infect 73, 338–344 10.1016/j.jhin.2009.04.020 [DOI] [PubMed] [Google Scholar]
- Köhler T., Michéa-Hamzehpour M., Henze U., Gotoh N., Curty L. K., Pechère J.-C. (1997). Characterization of MexE-MexF-OprN, a positively regulated multidrug efflux system of Pseudomonas aeruginosa. Mol Microbiol 23, 345–354 10.1046/j.1365-2958.1997.2281594.x [DOI] [PubMed] [Google Scholar]
- Köhler T., Epp S. F., Curty L. K., Pechère J. C. (1999). Characterization of MexT, the regulator of the MexE-MexF-OprN multidrug efflux system of Pseudomonas aeruginosa. J Bacteriol 181, 6300–6305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong K. F., Jayawardena S. R., Del Puerto A., Wiehlmann L., Laabs U., Tümmler B., Mathee K. (2005a). Characterization of poxB, a chromosomal-encoded Pseudomonas aeruginosa oxacillinase. Gene 358, 82–92 10.1016/j.gene.2005.05.027 [DOI] [PubMed] [Google Scholar]
- Kong K. F., Jayawardena S. R., Indulkar S. D., Del Puerto A., Koh C. L., Høiby N., Mathee K. (2005b). Pseudomonas aeruginosa AmpR is a global transcriptional factor that regulates expression of AmpC and PoxB β-lactamases, proteases, quorum sensing, and other virulence factors. Antimicrob Agents Chemother 49, 4567–4575 10.1128/AAC.49.11.4567-4575.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. Z., Nikaido H., Poole K. (1995). Role of mexA-mexB-oprM in antibiotic efflux in Pseudomonas aeruginosa. Antimicrob Agents Chemother 39, 1948–1953 10.1128/AAC.39.9.1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg F., Normark S. (1986). Contribution of chromosomal β-lactamases to β-lactam resistance in enterobacteria. Rev Infect Dis 8 (Suppl. 3), S292–S304 10.1093/clinids/8.Supplement_3.S292 [DOI] [PubMed] [Google Scholar]
- Lindquist S., Lindberg F., Normark S. (1989). Binding of the Citrobacter freundii AmpR regulator to a single DNA site provides both autoregulation and activation of the inducible ampC β-lactamase gene. J Bacteriol 171, 3746–3753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister P. D., Wolter D. J., Hanson N. D. (2009). Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin Microbiol Rev 22, 582–610 10.1128/CMR.00040-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livermore D. M. (1987). Clinical significance of β-lactamase induction and stable derepression in Gram-negative rods. Eur J Clin Microbiol 6, 439–445 10.1007/BF02013107 [DOI] [PubMed] [Google Scholar]
- Livermore D. M. (2002). Multiple mechanisms of antimicrobial resistance in Pseudomonas aeruginosa: our worst nightmare? Clin Infect Dis 34, 634–640 10.1086/338782 [DOI] [PubMed] [Google Scholar]
- Livermore D. M., Yang Y. J. (1987). β-Lactamase lability and inducer power of newer β-lactam antibiotics in relation to their activity against β-lactamase-inducibility mutants of Pseudomonas aeruginosa. J Infect Dis 155, 775–782 10.1093/infdis/155.4.775 [DOI] [PubMed] [Google Scholar]
- Lodge J. M., Minchin S. D., Piddock L. J., Busby S. J. (1990). Cloning, sequencing and analysis of the structural gene and regulatory region of the Pseudomonas aeruginosa chromosomal ampC β-lactamase. Biochem J 272, 627–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge J., Busby S., Piddock L. (1993). Investigation of the Pseudomonas aeruginosa ampR gene and its role at the chromosomal ampC β-lactamase promoter. FEMS Microbiol Lett 111, 315–320 [DOI] [PubMed] [Google Scholar]
- Maseda H., Saito K., Nakajima A., Nakae T. (2000). Variation of the mexT gene, a regulator of the MexEF-oprN efflux pump expression in wild-type strains of Pseudomonas aeruginosa. FEMS Microbiol Lett 192, 107–112 10.1111/j.1574-6968.2000.tb09367.x [DOI] [PubMed] [Google Scholar]
- Masuda N., Gotoh N., Ohya S., Nishino T. (1996). Quantitative correlation between susceptibility and OprJ production in NfxB mutants of Pseudomonas aeruginosa. Antimicrob Agents Chemother 40, 909–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda N., Sakagawa E., Ohya S., Gotoh N., Tsujimoto H., Nishino T. (2000). Substrate specificities of MexAB-OprM, MexCD-OprJ, and MexXY-oprM efflux pumps in Pseudomonas aeruginosa. Antimicrob Agents Chemother 44, 3322–3327 10.1128/AAC.44.12.3322-3327.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan J. E., Jr (2006). Resistance in nonfermenting Gram-negative bacteria: multidrug resistance to the maximum. Am J Med 119 (Suppl. 1), S29–S36, discussion S62–S70 10.1016/j.amjmed.2006.03.014 [DOI] [PubMed] [Google Scholar]
- Morita Y., Tomida J., Kawamura Y. (2012). MexXY multidrug efflux system of Pseudomonas aeruginosa. Front Microbiol 3, 408 10.3389/fmicb.2012.00408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H. (2003). Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev 67, 593–656 10.1128/MMBR.67.4.593-656.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normark S., Lindquist S., Lindberg F. (1986). Chromosomal β-lactam resistance in enterobacteria. Scand J Infect Dis Suppl 49, 38–45 [PubMed] [Google Scholar]
- Odds F. C. (2003). Synergy, antagonism, and what the chequerboard puts between them. J Antimicrob Chemother 52, 1 10.1093/jac/dkg301 [DOI] [PubMed] [Google Scholar]
- Page M. G., Heim J. (2009). Prospects for the next anti-Pseudomonas drug. Curr Opin Pharmacol 9, 558–565 10.1016/j.coph.2009.08.006 [DOI] [PubMed] [Google Scholar]
- Pankuch G. A., Lin G., Hoellman D. B., Good C. E., Jacobs M. R., Appelbaum P. C. (2006). Activity of retapamulin against Streptococcus pyogenes and Staphylococcus aureus evaluated by agar dilution, microdilution, E-test, and disk diffusion methodologies. Antimicrob Agents Chemother 50, 1727–1730 10.1128/AAC.50.5.1727-1730.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul M., Benuri-Silbiger I., Soares-Weiser K., Leibovici L. (2004). β Lactam monotherapy versus β lactam-aminoglycoside combination therapy for sepsis in immunocompetent patients: systematic review and meta-analysis of randomised trials. BMJ 328, 668 10.1136/bmj.38028.520995.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul M., Silbiger I., Grozinsky S., Soares-Weiser K., Leibovici L. (2006). β Lactam antibiotic monotherapy versus β lactam-aminoglycoside antibiotic combination therapy for sepsis. Cochrane Database Syst Rev 1, CD003344. [DOI] [PubMed] [Google Scholar]
- Pendleton J. N., Gorman S. P., Gilmore B. F. (2013). Clinical relevance of the ESKAPE pathogens. Expert Rev Anti Infect Ther 11, 297–308 10.1586/eri.13.12 [DOI] [PubMed] [Google Scholar]
- Poirel L., Bonnin R. A., Nordmann P. (2012). Genetic support and diversity of acquired extended-spectrum β-lactamases in Gram-negative rods. Infect Genet Evol 12, 883–893 10.1016/j.meegid.2012.02.008 [DOI] [PubMed] [Google Scholar]
- Poole K., Gotoh N., Tsujimoto H., Zhao Q., Wada A., Yamasaki T., Neshat S., Yamagishi J., Li X. Z., Nishino T. (1996a). Overexpression of the mexC-mexD-oprJ efflux operon in nfxB-type multidrug-resistant strains of Pseudomonas aeruginosa. Mol Microbiol 21, 713–725 10.1046/j.1365-2958.1996.281397.x [DOI] [PubMed] [Google Scholar]
- Poole K., Tetro K., Zhao Q., Neshat S., Heinrichs D. E., Bianco N. (1996b). Expression of the multidrug resistance operon mexA-mexB-oprM in Pseudomonas aeruginosa: mexR encodes a regulator of operon expression. Antimicrob Agents Chemother 40, 2021–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice L. B. (2010). Progress and challenges in implementing the research on ESKAPE pathogens. Infect Control Hosp Epidemiol 31 (Suppl 1), S7–S10 10.1086/655995 [DOI] [PubMed] [Google Scholar]
- Rietsch A., Vallet-Gely I., Dove S. L., Mekalanos J. J. (2005). ExsE, a secreted regulator of type III secretion genes in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 102, 8006–8011 10.1073/pnas.0503005102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sopirala M. M., Mangino J. E., Gebreyes W. A., Biller B., Bannerman T., Balada-Llasat J. M., Pancholi P. (2010). Synergy testing by Etest, microdilution checkerboard, and time-kill methods for pan-drug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 54, 4678–4683 10.1128/AAC.00497-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stebbing A. R. (1982). Hormesis – the stimulation of growth by low levels of inhibitors. Sci Total Environ 22, 213–234 10.1016/0048-9697(82)90066-3 [DOI] [PubMed] [Google Scholar]
- Stover C. K., Pham X. Q., Erwin A. L., Mizoguchi S. D., Warrener P., Hickey M. J., Brinkman F. S., Hufnagle W. O., Kowalik D. J. & other authors (2000). Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406, 959–964 10.1038/35023079 [DOI] [PubMed] [Google Scholar]
- Tamma P. D., Cosgrove S. E., Maragakis L. L. (2012). Combination therapy for treatment of infections with Gram-negative bacteria. Clin Microbiol Rev 25, 450–470 10.1128/CMR.05041-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardakas K. Z., Tansarli G. S., Bliziotis I. A., Falagas M. E. (2013). β-Lactam plus aminoglycoside or fluoroquinolone combination versus β-lactam monotherapy for Pseudomonas aeruginosa infections: a meta-analysis. Int J Antimicrob Agents 41, 301–310 10.1016/j.ijantimicag.2012.12.006 [DOI] [PubMed] [Google Scholar]
- Yeung A. T., Bains M., Hancock R. E. (2011). The sensor kinase CbrA is a global regulator that modulates metabolism, virulence, and antibiotic resistance in Pseudomonas aeruginosa. J Bacteriol 193, 918–931 10.1128/JB.00911-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong D., Toleman M. A., Bell J., Ritchie B., Pratt R., Ryley H., Walsh T. R. (2012). Genetic and biochemical characterization of an acquired subgroup B3 metallo-β-lactamase gene, blaAIM-1, and its unique genetic context in Pseudomonas aeruginosa from Australia. Antimicrob Agents Chemother 56, 6154–6159 10.1128/AAC.05654-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavascki A. P., Carvalhaes C. G., Picão R. C., Gales A. C. (2010). Multidrug-resistant Pseudomonas aeruginosa and Acinetobacter baumannii: resistance mechanisms and implications for therapy. Expert Rev Anti Infect Ther 8, 71–93 10.1586/eri.09.108 [DOI] [PubMed] [Google Scholar]
- Zhao W. H., Hu Z. Q. (2010). β-Lactamases identified in clinical isolates of Pseudomonas aeruginosa. Crit Rev Microbiol 36, 245–258 10.3109/1040841X.2010.481763 [DOI] [PubMed] [Google Scholar]
- Zhu B., Zhang P., Huang Z., Yan H. Q., Wu A. H., Zhang G. W., Mao Q. (2013). Study on drug resistance of Pseudomonas aeruginosa plasmid-mediated AmpC β-lactamase. Mol Med Rep 7, 664–668 [DOI] [PubMed] [Google Scholar]