

Abstract
The catalytic subunit of the herpes simplex virus 1 DNA polymerase (HSV-1 Pol) is essential for viral DNA synthesis and production of infectious virus in cell culture. While mutations that affect 5′–3′ polymerase activity have been evaluated in animal models of HSV-1 infection, mutations that affect other functions of HSV-1 Pol have not. In a previous report, we utilized bacterial artificial chromosome technology to generate defined HSV-1 pol mutants with lesions in the previously uncharacterized pre-NH2-terminal domain. We found that the extreme N-terminal 42 residues (deletion mutant polΔN43) were dispensable for replication in cell culture, while residues 44–49 (alanine-substitution mutant polA6) were required for efficient viral DNA synthesis and production of infectious virus. In this study, we sought to address the importance of these conserved elements in viral replication in a mouse corneal infection model. Mutant virus polΔN43 exhibited no meaningful defect in acute or latent infection despite strong conservation of residues 1–42 with HSV-2 Pol. The polA6 mutation caused a modest defect in replication at the site of inoculation, and was severely impaired for ganglionic replication, even at high inocula that permitted efficient corneal replication. Additionally, the polA6 mutation resulted in reduced latency establishment and subsequent reactivation. Moreover, we found that the polA6 replication defect in cultured cells was exacerbated in resting cells as compared to dividing cells. These results reveal an important role for the conserved motif at residues 44–49 of HSV-1 Pol for ganglionic viral replication.
Introduction
Herpes simplex virus 1 (HSV-1) infection following inoculation on the mouse cornea mirrors the pattern of disease progression observed in humans (reviewed by Efstathiou & Preston, 2005; Wagner & Bloom, 1997). Following HSV-1 replication on the cornea, viral particles enter nerve axon terminals and travel to neuronal cell bodies within the trigeminal ganglia (TG), wherein a second round of productive infection is initiated. Latent infections are established within the innervating sensory neurons, which harbour episomal viral DNA molecules and maintain a reservoir for the reactivation of infectious virus for the lifetime of the host.
Previous studies have established that certain viral DNA synthesis proteins that are not required for viral replication in cell culture are crucial for acute infection and reactivation in an animal host. For example, thymidine kinase-negative (TK−) mutants replicate like wild-type (WT) virus in dividing cells but replicate less well in resting cell cultures, likely due to reduced pools of thymine nucleotides (Field & Wildy, 1978; Jamieson et al., 1974). However, viral TK activity is absolutely essential for acute ganglionic replication and reactivation from latency of well-studied HSV-1 strains (Chen et al., 2004; Coen et al., 1989; Efstathiou et al., 1989; Tenser et al., 1989; Thompson & Sawtell, 2000). Similarly, viral ribonucleotide reductase-negative (RR−) mutants exhibit a modest defect in viral DNA synthesis in actively dividing cells, which is exacerbated during infection of resting cells, again likely due to reduced pools of nucleotides (Goldstein & Weller, 1988a, b; Jacobson et al., 1989; Nutter et al., 1985; Preston et al., 1988). Unlike HSV-1 TK, HSV-1 RR is required for viral replication on the mouse cornea as well as subsequent stages of infection in mice (Jacobson et al., 1989). In contrast, the catalytic subunit of the viral DNA polymerase (HSV-1 Pol) is absolutely essential for production of infectious virus in cell culture and in mice, presumably due to the role of 5′–3′ polymerase activity in production of progeny viral DNA molecules (Aron et al., 1975; Katz et al., 1990; Marcy et al., 1990; Purifoy et al., 1977). Drug resistant viruses containing point mutations in the pol gene that alter 5′–3′ polymerase activity exhibit varying degrees of attenuation during acute and latent infections in mice (Darby et al., 1984; Field & Coen, 1986; Larder & Darby, 1985; Pelosi et al., 1998). However, the roles of other HSV-1 Pol functions have yet to be evaluated in an animal model of infection.
The C-terminal half of HSV-1 Pol exhibits significant sequence and structural homology with other prokaryotic, eukaryotic, and viral DNA polymerases and is the best functionally characterized region of the protein (Fig. 1; Bernad et al., 1989; Liu et al., 2006; Wang et al., 1989). In contrast, the pre-NH2-terminal domain, which comprises the extreme N-terminal 141 residues, is unique to herpesvirus polymerases and a structural equivalent has yet to be identified in other published protein structures (Liu et al., 2006). Recently, using bacterial artificial chromosome (BAC) technology, we generated recombinant viruses with specific alterations at the 5′ end of the pol gene in order to assess the role of the previously uncharacterized pre-NH2-terminal domain of HSV-1 Pol (Fig. 1; Terrell & Coen, 2012). For one mutant, polΔN43, we deleted the extreme N-terminal 42 residues, which exhibit 76 % protein sequence identity between HSV-1 and -2 (Di Tommaso et al., 2011; Notredame et al., 2000). For a second mutant, polA6, we substituted HSV-1 Pol residues 44–49 (FYNPYL), which are strongly conserved within the human herpesvirus Pol family, with six alanines (Terrell & Coen, 2012). In cell culture polA6 exhibited an eightfold defect in viral yield with a similar decrease in viral DNA synthesis, while polΔN43 exhibited no defect in viral yield. Additionally, we demonstrated that the deletion and substitution mutations did not impact 5′–3′ polymerase activity of the corresponding mutant enzymes in vitro. Thus, the conserved motif FYNPYL plays a role in viral DNA synthesis processes during cellular infection that is distinct from 5′–3′ polymerase activity. We hypothesized that the conserved residues at the extreme N terminus and those functions outside 5′–3′ polymerase activity may be important for viral replication and reactivation in a mammalian host. In this present study, we sought to address this hypothesis by investigating the pre-NH2-terminal pol mutant viruses in a mouse model of infection.
Fig. 1.
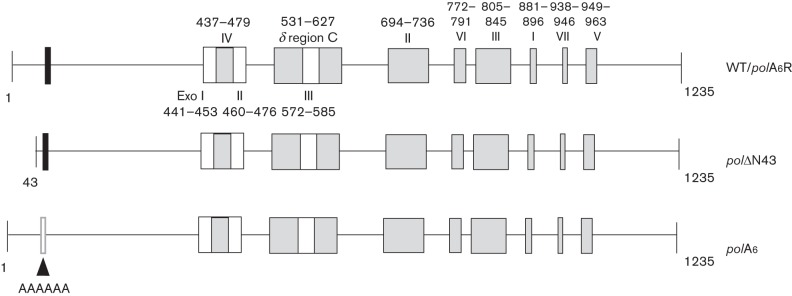
Locations of HSV-1 pre-NH2-terminal mutations within pol coding sequences. Coding sequences are depicted as lines, and conserved motifs and regions in the pre-NH2-terminal, polymerase and 3′–5′ exonuclease domains are shown as rectangles. The conserved motif at residues 44–49 (FYNPYL) in the pre-NH2-terminal domain is shown as a black rectangle, motifs conserved among 3′–5′ exonucleases (Exo I, II and III; Bernad et al., 1989; amino acid residues in HSV-1 Pol indicated below the top line) are shown as white boxes, and motifs conserved among α-like DNA polymerases (I–VII; Hwang et al., 1992; Wang et al., 1989) and a region conserved between HSV-1 Pol and DNA polymerase δ (δ-region C; Zhang et al., 1991) are shown as grey boxes (amino acid residues in HSV-1 Pol indicated above the top line). The top line shows the WT pol open reading frame which was restored in the background of polA6 to generate rescued derivative virus polA6R. The next line shows deletion mutant polΔN43, with a deletion of sequences encoding the extreme N-terminal 42 residues. The third line shows mutant polA6 in which the conserved motif at residues 44–49 was substituted with six alanines, as depicted by the white rectangle. The depicted mutations were incorporated at the pol locus of an infectious HSV-1 BAC clone as previously described (Terrell & Coen, 2012).
Results
Acute replication of pol mutant viruses on the mouse eye and TG
We sought to evaluate the role of conserved residues within the pre-NH2-terminal domain of HSV-1 Pol in a mouse model of infection and latency, hypothesizing that these residues could be important for acute and/or latent infection. The generation and characterization of recombinant pre-NH2-terminal domain pol mutant viruses (Fig. 1) has been described previously (Terrell & Coen, 2012). Deletion mutant polΔN43, which lacks the extreme N-terminal 42 residues that are highly conserved in HSV-1 and -2, displayed WT-like replication kinetics in cell culture. Mutant polA6, in which the conserved motif at HSV-1 Pol residues 44–49 was substituted with six alanines, exhibited decreased viral yield and DNA synthesis. Rescued derivative virus polA6R, in which the WT pol ORF was restored in the background of the mutant, recapitulated the WT phenotype and demonstrated that the replication defect was specifically attributable to the engineered mutation. Our analyses in the mouse model included these three viruses and the parental BAC-derived WT virus. CD-1 mice were infected on the eyes with 2×106 p.f.u. of virus following corneal scarification. At 1 day post-infection (p.i.), which is the time of peak replication on the eye (Coen et al., 1989), eyes were swabbed, and the eye swabs were titrated on the complementing cell line polB3, which inducibly expresses WT HSV-1 Pol (Hwang et al., 1997). The rescued derivative polA6R, as well as mutant polΔN43, generated infectious virus roughly as efficiently as WT (Fig. 2a; results shown in Fig. 2a, b represent pooled data from two independent experiments). However, mutant virus polA6 reproducibly exhibited a fivefold defect in viral replication at 1 day p.i., which was similar to the observed replication defect in cell culture (Terrell & Coen, 2012).
Fig. 2.
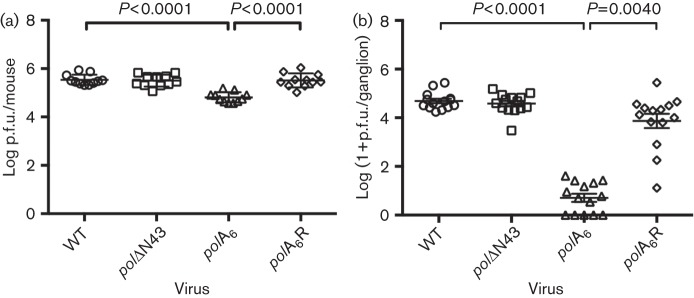
Acute replication of pre-NH2-terminal pol mutant viruses. CD-1 mice were infected on the eyes with 2×106 p.f.u. of the indicated BAC-derived viruses following corneal scarification. Data shown were pooled from two independent experiments and the means±sd for each dataset are plotted. Viral titres from eye swabs at 1 day p.i., n = 11 (a) and acutely infected TG at 3 days p.i., n = 14 (b) are shown. All viruses were analysed by one-way ANOVA (a) or Kruskal–Wallis test (b), and polA6 was compared to WT and polA6R using Bonferroni’s multiple comparisons post tests (a) or Dunn’s multiple comparisons post tests (b).
At 3 days p.i., which is the time of peak replication in the TG (Leib et al., 1989), mice were sacrificed and TG homogenates were titrated on polB3 cells. At this time, again, the rescued derivative virus polA6R and mutant polΔN43 exhibited acute ganglionic replication similar to that of WT (Fig. 2b). However, polA6 replication was drastically reduced with a ~1000-fold defect in infectious virus compared to its rescued derivative polA6R (P = 0.004; Fig. 2b). Although entire homogenates of ganglia from mice infected with polA6 were plated onto polB3 cells, a number of samples failed to yield detectable virus (Fig. 2b).
To determine whether acute ganglionic replication of mutant virus polA6 was merely delayed, in our second independent experiment, we extended the analysis of ganglionic replication through the first 7 days p.i. However, polA6 replication at 5 days p.i. remained three orders of magnitude lower than that of polA6R and WT (data not shown). By 7 days p.i., infectious virus became nearly undetectable in the TG for all viruses, including WT (data not shown). Therefore, polA6 exhibited severely impaired ganglionic replication throughout the acute phase of infection.
The acute ganglionic replication defect is not overcome by increasing the inoculum
Next, we asked whether the fivefold corneal replication defect at 1 day p.i. explained the severity of the acute ganglionic replication defect of mutant polA6. Accordingly, we tested whether increasing the inoculum dose could permit increased replication on the eye and thus overcome the severe ganglionic replication defect. We evaluated acute viral replication for equivalent doses of polA6 and polA6R (1×106 p.f.u.) as compared with 10-fold and 100-fold higher doses of polA6. At 1 day p.i., we observed the typical fivefold defect of polA6 for replication on the eye following the low dose inoculation, while at the highest dose of the mutant (1×108 p.f.u.), replication on the eye was similar to that of polA6R inoculated at the low dose (Fig. 3a). Nevertheless, infection with 1×108 p.f.u. of polA6 virus failed to overcome the ganglionic replication defect observed at 3 days p.i. (P<0.0001 versus polA6R) and did not even enhance viral replication in the TG as compared with 1×106 p.f.u. of polA6 (Fig. 3b). Additionally, we found that the replication defective phenotype persisted through 5 days p.i. in the TG for both low and high dose polA6 infections as compared with polA6R (Fig. 3c). Thus, regardless of the administered dose, mutant polA6 was unable to replicate efficiently in mouse TG in vivo.
Fig. 3.
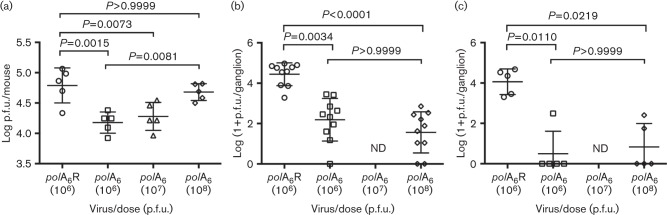
High dose polA6 infection cannot rescue ganglionic replication defect. Mouse eyes were infected with rescued derivative virus polA6R or mutant polA6 virus with the indicated dose in a 4 µl volume. Viral titres from eye swabs at 1 day p.i., n = 5 (a) and acutely infected TG at 3 days p.i., n = 10 (b) and 5 days p.i., n = 5 (c) are shown. All viruses were analysed by one-way ANOVA (a) or Kruskal–Wallis test (b, c), followed by Bonferroni’s multiple comparisons post tests (a) or Dunn’s multiple comparisons post tests (b, c) comparing polA6R to each mutant inoculum and the lowest mutant inoculum to the highest. ND, not determined.
Latency establishment and reactivation
We hypothesized that the acute replication defect exhibited by polA6 would compromise the efficiency of establishment of latency. To investigate this possibility, we quantified the number of viral DNA copies in latently infected TG using real-time PCR. Data presented in Fig. 4 were obtained from mice infected in the context of the two independent experiments presented in Fig. 2. Consistent with the robust level of viral replication during the acute phase of infection, both polΔN43 and polA6R viruses established latency as efficiently as WT virus (Fig. 4a). Interestingly, mutant virus polA6 reproducibly exhibited a sixfold decrease in latent viral DNA compared with its rescued derivative polA6R (P<0.0001; Fig. 4a).
Fig. 4.
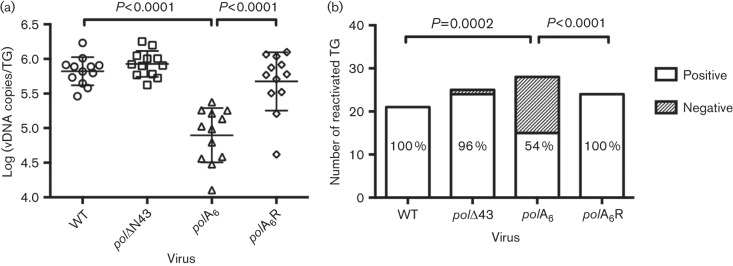
Latency establishment and reactivation of pol mutant viruses. CD-1 mice were infected with the indicated BAC-derived virus and latently infected TG were harvested at 30 days p.i. Data shown were pooled from two independent experiments. (a) Viral DNA (vDNA) was isolated from TG (n = 12), quantified via real-time PCR and normalized to mouse DNA. All viruses were analysed by one-way ANOVA, followed by Bonferonni’s multiple comparisons tests. WT was compared to each of the other viruses and polA6 was compared to polA6R. Non-significant P-values are not shown. (b) Virus reactivation from latently infected TG was evaluated using the dissociation method as described in Methods. The displayed P-values were obtained using Fisher’s exact tests. The percentage of reactivated TG for each virus is indicated in each column.
Previous reports have demonstrated that certain viral DNA synthesis proteins (e.g. TK and RR) that are dispensable for replication in cell culture are absolutely essential for reactivation, at least for laboratory HSV-1 strains (Chen et al., 2004; Coen et al., 1989; Efstathiou et al., 1989; Jacobson et al., 1989; Thompson & Sawtell, 2000). Therefore, we sought to determine whether the conserved HSV-1 Pol residues absent in mutants polΔN43 and polA6 were required for reactivation from latent infection. Reactivation assays were performed using the dissociation method in which latently infected TG are digested into single cell suspensions and plated individually onto polB3 monolayers in a six-well plate. Wells that did not yield infectious virus by 10 days post explant were harvested, freeze–thawed, sonicated and plated onto a fresh polB3 monolayer as a final test of reactivation. As expected, 100 % of WT- and polA6R-latently infected TG reactivated within 10 days post explant (Fig. 4b). Mutant virus polΔN43 displayed a comparable level of reactivation, with 22 of 23 (96 %) latently infected ganglia producing infectious virus (Fig. 4b), which demonstrated that the extreme N-terminal 42 residues are not required for reactivation. Reactivation of the polA6 virus was much less efficient (15/28; 54 %) and this reduction was statistically significant when compared with polA6R (Fig. 4b; P<0.0001). In addition to diminished reactivation efficiency, polA6 displayed slower reactivation kinetics than WT, polΔN43 and polA6R (data not shown). These results suggest that the conserved motif FYNPYL is necessary for efficient latency establishment, and either directly or indirectly for subsequent efficient reactivation of infectious virus.
Replication kinetics of polA6 in resting cells
Certain mutant viruses that exhibit decreased acute ganglionic replication also display replication defects in resting cell cultures (Bolovan et al., 1994; Brown et al., 1994; Field & Wildy, 1978; Goldstein & Weller, 1988a; Jacobson et al., 1989; Jamieson et al., 1974). In order to determine whether the metabolic state of the cell affected the in vitro growth defect observed with polA6, we analysed multi-cycle replication kinetics in resting and dividing cells. Cell cycle arrest was induced by maintaining confluent human foreskin fibroblast cells (HFFs) in medium containing 0.5 % FBS/DMEM for 96 h prior to infection. Western blot analysis (data not shown) of serum starved HFFs for retinoblastoma protein production and phosphorylation was similar to that reported previously from quiescent cell cultures (Ehmann et al., 2000; Song et al., 2000). Both resting and dividing HFFs were infected at an m.o.i. of 0.01 in triplicate, and whole-cell lysates were harvested at 48 h p.i. We included tk null virus dlsptk as a positive control in this experiment, as it has been established that such viruses exhibit impaired replication in resting cells (Field & Wildy, 1978; Jamieson et al., 1974). Accordingly, we found that dlsptk replication was similar to that of WT in actively dividing cells at 48 h p.i., and was reduced threefold in resting cell cultures (Table 1). As expected (Terrell & Coen, 2012), polA6 exhibited reduced viral yield in dividing cells with a sevenfold decrease in infectious virus compared with polA6R (Table 1). Interestingly, the polA6 replication defect was exacerbated in resting cell cultures to a 57-fold defect in viral yield at 48 h p.i. when compared to polA6R (Table 1). These results suggest that polA6 replication is further restricted in resting cells and this effect may contribute to the severity of the acute ganglionic replication defect observed in mice.
Table 1. Replication in dividing and resting HFFs.
| Titre (p.f.u. ml−1) | ||
| Virus | Dividing cells | Resting cells |
| WT | 2.1×108 | 9.1×107 |
| dlsptk | 2.5×108 | 3.1×107 |
| polA6 | 6.4×107 | 2.0×106 |
| polA6R | 4.3×108 | 1.1×108 |
Discussion
In this study, we addressed the roles of conserved residues within the pre-NH2-terminal domain of HSV-1 Pol in a mouse model of infection. We found that the extreme N-terminal 42 residues, which are deleted in mutant polΔN43, are dispensable for replication and reactivation from latent infection in mice following corneal inoculation. Thus, we have found no role for these residues either in cell culture (Terrell & Coen, 2012) or in vivo, at least in mice. This segment is conserved among a subset of simplexviruses within the subfamily Alphaherpesvirus that infect humans and monkeys (McGeoch et al., 2000), with viral DNA polymerases from Cercopithecine herpesvirus 1 (CeHV-1; B virus), CeHV-2 (simian agent 8; SA8), CeHV-16 (herpesvirus papio 2; HVP2), and Saimiriine herpesvirus 1 (SaHV-1) exhibiting 31–50 % protein sequence identity within the extreme N-terminal 42 residues of HSV-1 Pol (Di Tommaso et al., 2011; Notredame et al., 2000). There is even greater protein sequence identity (76 %) between this segment of HSV-1 and -2 Pol (Di Tommaso et al., 2011; Notredame et al., 2000). Perhaps conservation of this segment is just an accident of evolution that resulted in a longer pre-NH2-terminal domain for these viruses. Of note, however, is that pathogenesis of HSV infection in humans is very similar to that of B virus in its natural host, which is closely related to both HVP2 and SA8 (Elmore & Eberle, 2008). Thus, we cannot exclude the possibility that this conserved element might confer an advantage during viral replication that is specific to the biology of simplexviruses that was not detected in our mouse model of infection.
In contrast, we have demonstrated that a motif (FYNPYL) in the pre-NH2-terminal domain, which was replaced with a clustered alanine substitution in mutant polA6, has a significant role in both acute and latent infections in mice. The defect conferred by this mutation for viral replication on the mouse eye corresponded in magnitude with that in dividing cells in culture (Terrell & Coen, 2012). In contrast, replication at the secondary site of infection (TG) was much more drastically attenuated, and this defect could not be overcome by increasing the administered dose so that replication on the eye was efficient. We note that certain other HSV-1 mutants exhibit substantial defects in replication on the eye, but much less of a defect in acute ganglionic replication than does polA6 (Cai et al., 1993; Pelosi et al., 1998). We suggest therefore that the ganglionic replication defect reflects a specific block during polA6 infection. Restricted polA6 replication in resting cell cultures is consistent with this interpretation. Therefore, the function mediated by the conserved motif FYNPYL is of greater importance for viral replication in mouse TG and in resting cells. The motif FYNPYL is well conserved across avian and mammalian herpesviruses from all three herpesvirus subfamilies (Di Tommaso et al., 2011; Notredame et al., 2000), which underscores the importance of the as-yet-unidentified function of this conserved motif for viral replication.
Despite the severe impairment in ganglionic replication, the number of viral DNA copies in polA6-latently infected TG, a measure of establishment of latency, was only decreased by sixfold compared with polA6R. The decrease in latent viral DNA was more consistent with the fivefold corneal replication defect than the 1000-fold ganglionic replication defect. These observations are similar to those of a study of a TK− mutant derived from HSV-1 strain 17 (Thompson & Sawtell, 2000). However, examples exist in which mutant viruses that are defective for corneal replication sustain WT-like reactivation efficiencies and latent viral DNA loads (Perng et al., 1996; Thompson et al., 2009). Perhaps more surprisingly, and in considerable contrast to TK− mutants of laboratory strains, which fail to reactivate upon explant (Coen et al., 1989; Efstathiou et al., 1989; Thompson & Sawtell, 2000), the reactivation efficiency of polA6 was only twofold lower than that of polA6R despite the severe impairment in acute ganglionic replication for mutant polA6. It is especially remarkable that reactivation was this efficient given the decreased establishment of latency that we observed, on top of the severe acute ganglionic replication defect and the resting cell replication defect. One possible explanation for the relatively efficient reactivation of polA6 in vitro is that explant of ganglia alters neuronal physiology to overcome the defect of the virus. This may have precedent. Like polA6, an HSV-1 mutant with an insertion that truncates VP16 also displayed a severe defect for acute ganglionic replication while reactivating relatively efficiently from in vitro TG explants. A related HSV-1 mutant with an insertion in VP16 also reactivated efficiently from TG explants. However, this mutant failed to produce detectable infectious virus and lytic viral proteins in TG following hyperthermic stress in vivo (Thompson et al., 2009). Thus, reactivation of polA6 from latently infected TG in vivo might be highly impaired, akin to its acute ganglionic replication, in contrast to what was observed with the in vitro explant model.
Although we do not yet understand the biochemical basis for the polA6 replication defect, our results may provide some clues. Due to the nature of the mutation (see discussion in Terrell & Coen, 2012), we hypothesize that the defect is due to a disrupted protein–protein interaction. Our results indicate that this interaction would be important, yet non-essential in dividing cells in culture and on the eye in mice, but be more important in resting cells in culture and crucial for ganglionic replication in mice. One possible explanation is that a host protein that can partially compensate for the loss of function during polA6 infection in actively dividing cells, is reduced or absent in resting cells in culture and neurons in vivo. Alternatively, the mutation may have only diminished the binding affinity of HSV-1 Pol rather than completely abolishing a protein–protein interaction. In this scenario, the ability of the HSV-1 Pol enzyme to form a stable complex would be of greater importance in neurons and resting cells, perhaps due to limited expression of the protein binding partner. These possibilities are currently under investigation. The mechanism by which the conserved motif FYNPYL mediates efficient viral DNA synthesis and production of infectious virus may very well reflect a conserved replication mechanism for many herpesviruses.
Methods
Cells and viruses.
Vero cells (ATCC) and polB3 cells (kindly provided by Charles Hwang; Hwang et al., 1997) were maintained as previously described (Terrell & Coen, 2012). Human foreskin fibroblasts (HFFs; ATCC) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % FBS, 1 % penicillin/streptomycin, and 1 % amphotericin B. Viruses that were generated from an infectious BAC clone of KOS (I. Jurak, C. Cui, A. Pearson, A. Griffiths, P. A. Schaffer, D. M. Coen, unpublished results) included the virus derived from the WT BAC, polΔN43, polA6 and polA6R (Terrell & Coen, 2012). All BAC-derived viruses were propagated in and titrated on polB3 cells, which inducibly express WT HSV-1 Pol. Virus dlsptk, which contains a 360-bp deletion in the HSV-1 tk locus (outside the UL24 coding sequence) that abolishes enzymic activity (Coen et al., 1989), was amplified in Vero cells.
Assays of acute and latent infections in mice.
Animal care and experimentation procedures involving mice were approved by the Harvard Medical School Institutional Animal Care and Use Committee and were in compliance with federal guidelines. Seven-week-old CD-1 mice were anaesthetized with ketamine–xylazine, and 2×106 p.f.u., unless otherwise indicated, were administered to each eye in a volume of 4 µl via following corneal scarification as previously described (Pesola et al., 2005). Infectious virus was harvested from each mouse eye with pre-moistened cotton swabs that were resuspended in 1 ml of culture medium (DMEM supplemented with 5 % newborn calf serum, 1 % penicillin/streptomycin and 1 % amphotericin B) and stored at −80 °C until titration. Acutely infected TG were harvested and stored at −80 °C in 1 ml of culture medium. TG were thawed, dounce-homogenized, frozen, thawed and sonicated prior to titration. For any samples that were suspected to contain very low amounts of infectious virus, the entire lysate was plated onto confluent polB3 cells in six-well plates. Reactivation assays were performed by dissociating latently infected TG as previously described (Leib et al., 1991), and the cell suspension was added to individual wells on a six-well plate containing polB3 cell monolayers. Analysis of reactivation kinetics was performed similarly to that previously described (Balliet et al., 2007). Briefly, aliquots (150 µl) of the viral supernatant were harvested from each well and stored at −80 °C before plating on confluent 24-well plates containing polB3 cells for detection of infectious virus. For samples in which virus had not reactivated by 10 days post-dissociation, whole-cell lysates were freeze–thawed, sonicated and plated onto confluent polB3 cells (six-well plate), and monitored for an additional four days prior to fixing and staining.
Latent viral DNA detection via quantitative real-time PCR.
Latently infected TG were harvested from mock and HSV-1 infected mice at 30 days p.i. and processed for DNA isolation as previously described (Pesola et al., 2005). Real-time PCR assays were performed as previously described (Terrell & Coen, 2012) with primers that targeted the viral tk gene (Terrell & Coen, 2012) or the murine adipsin gene (Kramer et al., 2011) and resulted in ≥92 % mean PCR amplification efficiency in each assay. Viral and mouse DNA standards used for quantification of recovered DNA in latently infected TG were prepared as previously described (Pesola et al., 2005). R2 values for both viral and mouse DNA standard curves were ≥0.99.
Statistical analyses.
Statistical analyses were performed using GraphPad Prism (GraphPad Software). The analyses included one-way ANOVA with Bonferroni’s multiple comparisons post tests, Kruskal–Wallis analyses with Dunn’s multiple comparisons post tests, or Fisher's exact tests as indicated in the figure legends. For analysis of viral titres in vivo, 1 p.f.u. was added to all titres to avoid the undefined logarithm of zero.
Replication kinetics in resting cells.
Analysis of viral replication in actively dividing and resting cell cultures was performed as previously described (Field & Wildy, 1978; Jamieson et al., 1974) with modifications: HFFs (1×106) were seeded into six-well plates and, for dividing cells, were maintained in DMEM/10 % FBS for 24 h or less prior to infection. Resting cells were produced by maintaining cell cultures in DMEM/0.5 % FBS for 96 h prior to infection. Resting and dividing HFFs were infected at an m.o.i. of 0.01 in triplicate and whole-cell lysates were harvested at 48 h p.i. and titrated on polB3 cells.
Acknowledgements
We thank David Leib and Pam Rosato for helpful discussions, and Seamus McCarron at Harvard Medical School for technical assistance with real-time PCR assays. This work was supported by NIH award F31 AI084490 to S. L. T. and grant R01 AI019838 to D. M. C.
References
- Aron G. M., Purifoy D. J., Schaffer P. A. (1975). DNA synthesis and DNA polymerase activity of herpes simplex virus type 1 temperature-sensitive mutants. J Virol 16, 498–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balliet J. W., Kushnir A. S., Schaffer P. A. (2007). Construction and characterization of a herpes simplex virus type I recombinant expressing green fluorescent protein: acute phase replication and reactivation in mice. Virology 361, 372–383 10.1016/j.virol.2006.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernad A., Blanco L., Lázaro J. M., Martín G., Salas M. (1989). A conserved 3′→5′ exonuclease active site in prokaryotic and eukaryotic DNA polymerases. Cell 59, 219–228 10.1016/0092-8674(89)90883-0 [DOI] [PubMed] [Google Scholar]
- Bolovan C. A., Sawtell N. M., Thompson R. L. (1994). ICP34.5 mutants of herpes simplex virus type 1 strain 17syn+ are attenuated for neurovirulence in mice and for replication in confluent primary mouse embryo cell cultures. J Virol 68, 48–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown S. M., Harland J., MacLean A. R., Podlech J., Clements J. B. (1994). Cell type and cell state determine differential in vitro growth of non-neurovirulent ICP34.5-negative herpes simplex virus types 1 and 2. J Gen Virol 75, 2367–2377 10.1099/0022-1317-75-9-2367 [DOI] [PubMed] [Google Scholar]
- Cai W., Astor T. L., Liptak L. M., Cho C., Coen D. M., Schaffer P. A. (1993). The herpes simplex virus type 1 regulatory protein ICP0 enhances virus replication during acute infection and reactivation from latency. J Virol 67, 7501–7512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S. H., Pearson A., Coen D. M., Chen S. H. (2004). Failure of thymidine kinase-negative herpes simplex virus to reactivate from latency following efficient establishment. J Virol 78, 520–523 10.1128/JVI.78.1.520-523.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coen D. M., Kosz-Vnenchak M., Jacobson J. G., Leib D. A., Bogard C. L., Schaffer P. A., Tyler K. L., Knipe D. M. (1989). Thymidine kinase-negative herpes simplex virus mutants establish latency in mouse trigeminal ganglia but do not reactivate. Proc Natl Acad Sci U S A 86, 4736–4740 10.1073/pnas.86.12.4736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darby G., Churcher M. J., Larder B. A. (1984). Cooperative effects between two acyclovir resistance loci in herpes simplex virus. J Virol 50, 838–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Tommaso P., Moretti S., Xenarios I., Orobitg M., Montanyola A., Chang J. M., Taly J. F., Notredame C. (2011). T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res 39 (Suppl 2), W13–W17 10.1093/nar/gkr245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efstathiou S., Preston C. M. (2005). Towards an understanding of the molecular basis of herpes simplex virus latency. Virus Res 111, 108–119 10.1016/j.virusres.2005.04.017 [DOI] [PubMed] [Google Scholar]
- Efstathiou S., Kemp S., Darby G., Minson A. C. (1989). The role of herpes simplex virus type 1 thymidine kinase in pathogenesis. J Gen Virol 70, 869–879 10.1099/0022-1317-70-4-869 [DOI] [PubMed] [Google Scholar]
- Ehmann G. L., McLean T. I., Bachenheimer S. L. (2000). Herpes simplex virus type 1 infection imposes a G(1)/S block in asynchronously growing cells and prevents G(1) entry in quiescent cells. J Virol 267, 335–349 [DOI] [PubMed] [Google Scholar]
- Elmore D., Eberle R. (2008). Monkey B virus (Cercopithecine herpesvirus 1). Comp Med 58, 11–21 [PMC free article] [PubMed] [Google Scholar]
- Field H. J., Coen D. M. (1986). Pathogenicity of herpes simplex virus mutants containing drug resistance mutations in the viral DNA polymerase gene. J Virol 60, 286–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field H. J., Wildy P. (1978). The pathogenicity of thymidine kinase-deficient mutants of herpes simplex virus in mice. J Hyg (Lond) 81, 267–277 10.1017/S0022172400025109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein D. J., Weller S. K. (1988a). Factor(s) present in herpes simplex virus type 1-infected cells can compensate for the loss of the large subunit of the viral ribonucleotide reductase: characterization of an ICP6 deletion mutant. Virology 166, 41–51 10.1016/0042-6822(88)90144-4 [DOI] [PubMed] [Google Scholar]
- Goldstein D. J., Weller S. K. (1988b). Herpes simplex virus type 1-induced ribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: isolation and characterization of an ICP6 lacZ insertion mutant. J Virol 62, 196–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang C. B., Ruffner K. L., Coen D. M. (1992). A point mutation within a distinct conserved region of the herpes simplex virus DNA polymerase gene confers drug resistance. J Virol 66, 1774–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang Y. T., Liu B. Y., Coen D. M., Hwang C. B. (1997). Effects of mutations in the Exo III motif of the herpes simplex virus DNA polymerase gene on enzyme activities, viral replication, and replication fidelity. J Virol 71, 7791–7798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson J. G., Leib D. A., Goldstein D. J., Bogard C. L., Schaffer P. A., Weller S. K., Coen D. M. (1989). A herpes simplex virus ribonucleotide reductase deletion mutant is defective for productive acute and reactivatable latent infections of mice and for replication in mouse cells. Virology 173, 276–283 10.1016/0042-6822(89)90244-4 [DOI] [PubMed] [Google Scholar]
- Jamieson A. T., Gentry G. A., Subak-Sharpe J. H. (1974). Induction of both thymidine and deoxycytidine kinase activity by herpes viruses. J Gen Virol 24, 465–480 10.1099/0022-1317-24-3-465 [DOI] [PubMed] [Google Scholar]
- Katz J. P., Bodin E. T., Coen D. M. (1990). Quantitative polymerase chain reaction analysis of herpes simplex virus DNA in ganglia of mice infected with replication-incompetent mutants. J Virol 64, 4288–4295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer M. F., Jurak I., Pesola J. M., Boissel S., Knipe D. M., Coen D. M. (2011). Herpes simplex virus 1 microRNAs expressed abundantly during latent infection are not essential for latency in mouse trigeminal ganglia. Virology 417, 239–247 10.1016/j.virol.2011.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larder B. A., Darby G. (1985). Selection and characterisation of acyclovir-resistant herpes simplex virus type 1 mutants inducing altered DNA polymerase activities. Virology 146, 262–271 10.1016/0042-6822(85)90009-1 [DOI] [PubMed] [Google Scholar]
- Leib D. A., Bogard C. L., Kosz-Vnenchak M., Hicks K. A., Coen D. M., Knipe D. M., Schaffer P. A. (1989). A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J Virol 63, 2893–2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leib D. A., Nadeau K. C., Rundle S. A., Schaffer P. A. (1991). The promoter of the latency-associated transcripts of herpes simplex virus type 1 contains a functional cAMP-response element: role of the latency-associated transcripts and cAMP in reactivation of viral latency. Proc Natl Acad Sci U S A 88, 48–52 10.1073/pnas.88.1.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Knafels J. D., Chang J. S., Waszak G. A., Baldwin E. T., Deibel M. R., Jr, Thomsen D. R., Homa F. L., Wells P. A. & other authors (2006). Crystal structure of the herpes simplex virus 1 DNA polymerase. J Biol Chem 281, 18193–18200 10.1074/jbc.M602414200 [DOI] [PubMed] [Google Scholar]
- Marcy A. I., Olivo P. D., Challberg M. D., Coen D. M. (1990). Enzymatic activities of overexpressed herpes simplex virus DNA polymerase purified from recombinant baculovirus-infected insect cells. Nucleic Acids Res 18, 1207–1215 10.1093/nar/18.5.1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeoch D. J., Dolan A., Ralph A. C. (2000). Toward a comprehensive phylogeny for mammalian and avian herpesviruses. J Virol 74, 10401–10406 10.1128/JVI.74.22.10401-10406.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame C., Higgins D. G., Heringa J. (2000). T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol 302, 205–217 10.1006/jmbi.2000.4042 [DOI] [PubMed] [Google Scholar]
- Nutter L. M., Grill S. P., Cheng Y. C. (1985). Can ribonucleotide reductase be considered as an effective target for developing antiherpes simplex virus type II (HSV-2) compounds? Biochem Pharmacol 34, 777–780 10.1016/0006-2952(85)90757-9 [DOI] [PubMed] [Google Scholar]
- Pelosi E., Mulamba G. B., Coen D. M. (1998). Penciclovir and pathogenesis phenotypes of drug-resistant Herpes simplex virus mutants. Antiviral Res 37, 17–28 10.1016/S0166-3542(97)00054-5 [DOI] [PubMed] [Google Scholar]
- Perng G. C., Ghiasi H., Slanina S. M., Nesburn A. B., Wechsler S. L. (1996). High-dose ocular infection with a herpes simplex virus type 1 ICP34.5 deletion mutant produces no corneal disease or neurovirulence yet results in wild-type levels of spontaneous reactivation. J Virol 70, 2883–2893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesola J. M., Zhu J., Knipe D. M., Coen D. M. (2005). Herpes simplex virus 1 immediate-early and early gene expression during reactivation from latency under conditions that prevent infectious virus production. J Virol 79, 14516–14525 10.1128/JVI.79.23.14516-14525.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston V. G., Darling A. J., McDougall I. M. (1988). The herpes simplex virus type 1 temperature-sensitive mutant ts1222 has a single base pair deletion in the small subunit of ribonucleotide reductase. Virology 167, 458–467 [PubMed] [Google Scholar]
- Purifoy D. J., Lewis R. B., Powell K. L. (1977). Identification of the herpes simplex virus DNA polymerase gene. Nature 269, 621–623 10.1038/269621a0 [DOI] [PubMed] [Google Scholar]
- Song B., Liu J. J., Yeh K. C., Knipe D. M. (2000). Herpes simplex virus infection blocks events in the G1 phase of the cell cycle. Virology 267, 326–334 [DOI] [PubMed] [Google Scholar]
- Tenser R. B., Hay K. A., Edris W. A. (1989). Latency-associated transcript but not reactivatable virus is present in sensory ganglion neurons after inoculation of thymidine kinase-negative mutants of herpes simplex virus type 1. J Virol 63, 2861–2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrell S. L., Coen D. M. (2012). The pre-NH(2)-terminal domain of the herpes simplex virus 1 DNA polymerase catalytic subunit is required for efficient viral replication. J Virol 86, 11057–11065 10.1128/JVI.01034-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson R. L., Sawtell N. M. (2000). Replication of herpes simplex virus type 1 within trigeminal ganglia is required for high frequency but not high viral genome copy number latency. J Virol 74, 965–974 10.1128/JVI.74.2.965-974.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson R. L., Preston C. M., Sawtell N. M. (2009). De novo synthesis of VP16 coordinates the exit from HSV latency in vivo. PLoS Pathog 5, e1000352 10.1371/journal.ppat.1000352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner E. K., Bloom D. C. (1997). Experimental investigation of herpes simplex virus latency. Clin Microbiol Rev 10, 419–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T. S., Wong S. W., Korn D. (1989). Human DNA polymerase alpha: predicted functional domains and relationships with viral DNA polymerases. FASEB J 3, 14–21 [DOI] [PubMed] [Google Scholar]
- Zhang J., Chung D. W., Tan C. K., Downey K. M., Davie E. W., So A. G. (1991). Primary structure of the catalytic subunit of calf thymus DNA polymerase delta: sequence similarities with other DNA polymerases. Biochemistry 30, 11742–11750 10.1021/bi00115a002 [DOI] [PubMed] [Google Scholar]