

Abstract
Considerable progress has been made in the past few years in the fight against Alzheimer’s disease (AD) and Parkinson’s disease (PD). Neuropathological studies in human brains and experimental in vivo and in vitro models support the notion that synapses are affected even at the earliest stages of the neurodegenerative process. The objective of this manuscript is to review some of the mechanisms of synaptic damage in AD and PD. Some lines of evidence support the notion that oligomeric neurotoxic species of amyloid β, α-synuclein, and Tau might contribute to the pathogenesis of synaptic failure at early stages of the diseases. The mechanisms leading to synaptic damage by oligomers might involve dysregulation of glutamate receptors and scaffold molecules that results in alterations in the axonal transport of synaptic vesicles and mitochondria that later on lead to dendritic and spine alterations, axonal dystrophy, and eventually neuronal loss. However, while some studies support a role of oligomers, there is an ongoing debate as to the exact nature of the toxic species. Given the efforts toward earlier clinical and preclinical diagnosis of these disorders, understanding the molecular and cellular mechanisms of synaptic degeneration is crucial toward developing specific biomarkers and new therapies targeting the synaptic apparatus of vulnerable neurons.
INTRODUCTION
The past few years have witnessed considerable progress in the fight against Alzheimer’s disease (AD), with the introduction of the revised clinical [1] and neuropathological [2] criteria for the diagnosis of AD, identification of new biomarkers [3–5], better characterization of the poly-genetic aspects of AD [6, 7], and a more clear understanding of the contribution of neurotoxic aggregates of amyloid β (Aβ) [8–10] and microtubule associated protein τ (Tau) [11, 12], to the pathogenesis of neurodegeneration in AD. Likewise, in disorders with parkinsonism and dementia such as Parkinson’s disease (PD), PD with dementia (PDD) and dementia with Lewy bodies (DLB) (jointly denominated Lewy body disease [LBD]) [13] dramatic progress has been made in identifying new genes involved in familial [14] and sporadic [15] forms, several of them possibly converging on the α-synuclein (α-syn) pathway [16, 17].
In 2013 an estimated 5.2 million Americans of all ages have AD and 1 million have PD [18]. This year an estimated 450,000 people in the US will die with AD, making AD the sixth-leading cause of death in the US [18]. Without a cure, the number of cases of AD, as defined by the 1984 and DSM-IV criteria, will double by the year 2050, with western states experiencing the highest rates [18]. The new criteria published in 2011 proposed three stages of the disease, namely preclinical AD, mild cognitive impairment (MCI) due to AD, and dementia due to AD [1]. The 2011 criteria proposes that AD begins before the development of symptoms and that new positron emission tomography (PET) and cerebral spinal cord fluid (CSF) biomarkers are able to identify brain alterations before the onset neurological alterations [1]. However, the predictive value of such biomarkers is not yet proven in sporadic preclinical cases [19]. If AD can be detected earlier, as defined by the 2011 criteria, the number of people reported to have AD will be much larger than 5 million.
In 2011 a workgroup of experts was organized to revise the 1997 neuropathological criteria for the diagnosis of AD and related disorders [2]. The 1997 criteria required a history of dementia [20], while the new criteria disentangle the clinico-pathologic term “Alzheimer’s disease” from AD neuropathologic change [2]. Using the new criteria, AD neuropathologic change would be ranked along three parameters (Amyloid, Braak, CERAD) to obtain an “ABC score”. For this purpose a modified version of Thal phases of Aβ plaque accumulation was proposed [21], adapted to a four-point scale, continued use of the staging scheme for neurofibrillary tangles as described by Braak [22], reduced to four stages that improves inter-rater reliability, and continued use of CERAD protocol for neuritic plaque scoring [23]. The new criteria provided guidance on clinico-pathologic correlations for pathologists reporting autopsy findings based on the literature and analysis of the National Alzheimer’s Coordinating Center (NACC) database. The new criteria also emphasized the importance of assessing non-AD brain lesions in recognition of commonly co-morbid conditions in cognitively impaired elderly. Among the co-morbid conditions, synucleinopathies such as PD, PDD and DLB, are important given that over 75% of patients with AD display LB’s in the amygdala [24, 25] and about 25% of patients with AD develop parkinsonism [26].
The main purpose of this manuscript is to review evidence supporting the synapse failure hypothesis of AD and LDB and the role of Aβ, α-syn, and Tau accumulation in the pathogenesis of this process. We conclude that synaptic dysfunction occurs early, followed by pre-synaptic and spine loss, axonal dystrophy and eventually neuronal loss. We focus on synapses because Aβ is released at the synaptic terminal [27] and α-syn localizes to the synaptic vesicles [28] where they can effect synaptic transmission. However, a number of other cellular substrates play an equal important role (e.g.: neuro-inflammation, vascular, glial) and deserve close consideration. For example, a recent GWAS study highlighted the association of AD with innate immune response [29–33].
SYNAPTIC DAMAGE AND Aβ IN EARLY ALZHEIMER’S DISEASE
For several years the classical definition of neurodegeneration in disorders such as AD and PD was limited to the finding of selective neuronal loss and astrogliosis. This concept has now been expanded to include synaptic loss and neuro-inflammation. Synaptic damage can be detected at the earliest stages of AD. Patients with MCI demonstrate loss of pre-synaptic proteins such as synaptophysin, VAMP2, and SNAP25 and post-synaptic markers such as PSD95 and Shank1 [34]. Likewise ultrastructural [35] and confocal microscopy studies [36] have shown progressive alterations of synapses in early stages of AD and in APP tg models [37]. This has been confirmed in experimental APP transgenic models [38], as well as after acute injection of Aβ oligomers [39]. These studies have shown more severe loss of glutaminergic terminals but not GABAergic terminals in the hippocampus [40, 41]. Consistent with the neuropathological and structural studies, recent gene array investigations have shown that in early AD there is altered expression of genes involved in synaptic vesicle trafficking and release, neurotransmitter receptors and receptor trafficking, postsynaptic density scaffolding, cell adhesion regulating synaptic stability, and neuromodulatory systems [42–45]. The memory impairment in patients with AD is related to synaptic loss in the neocortex and limbic system [46–48]. In contrast, cognitive impairment does not correlated with Aβ plaques in the brain. The loss of synapses in AD is greater than the extent of the neuronal loss in the cortex. This suggests that synaptic damage precedes the loss of neuronal cell bodies. This is why synapses are a good correlate to cognitive deficits [43, 46, 47, 49–52]. The remaining synapses appear to be enlarged representing a possible compensatory mechanism [47, 53, 54].
The mechanisms of synaptic loss in AD might involve axonal transport defects, oxidative stress, mitochondrial damage, and neuroinflammation among others [55]. Increasing levels of Aβ1–42, the proteolytic product of APP metabolism are also suspected to be centrally involved in the pathogenesis of synaptic damage in AD [56–60] (Fig 1A). Accumulation of Aβ in AD is the result of an imbalance in the mechanisms of synthesis, aggregation, and clearance (Fig 1B). Increased synthesis and aggregation has a prominent role in familial AD, and altered clearance including degradation and autophagy has a role in sporadic AD [61, 62]. The mechanisms through which accumulation of Aβ and other APP metabolites might lead to synaptic damage and neurodegeneration are under investigation. More specifically, the potential role of neurotoxic Aβ oligomers has emerged as a topic of considerable interest in recent years [63–66] (Fig 1).
Figure 1.

Schematic diagram showing the processing of APP, the formation of Aβ oligomers, and its interaction with Tau in the mechanisms of synapse loss. A) APP is cleaved by β and γ-secretases to form sAPPβ. Monomeric Aβ42 can form Aβ oligomers that can be cleared by ApoE and proteases such as neprilysin and IDE. Both Aβ monomers and oligomers progress to fibrils and plaques, while Aβ oligomers interact with surface receptors that in turn activate various kinases to alter Tau, leading to loss of axonal transport of neurotrophic factors and impaired mitochondrial function, culminating in neurotoxicity. B) Aβ production is dependent on both Aβ clearance, aggregation and synthesis.
Monomeric Aβ can aggregate to form amyloid fibrils, protofibrils, annular structures [67], Aβ-derived diffusible ligands (ADDLs) [68] and smaller order oligomeric species (for reviews, see [69–74]). Oligomers of Aβ can organize into dimers, trimers, tetramers, and higher order arrays that can form annular structures [75]. Smaller oligomers are divided into those generated from synthetic peptides and those purified from cells, transgenic (tg) mice, or AD human brains [8, 69, 76]. However, it is worth noting that there is great heterogeneity in the Aβ arrays accumulating in the brain of AD patients, and more recent studies have highlighted that there is uncertainty around the pathological significance of some of these oligomeric species [76].
An example of a naturally occurring oligomer specie is Aβ*56 derived from the brains of APP tg mice, which has been shown to promote age-dependent memory deficits [77]. Aβ*56 and Aβ trimers secreted by cultured cells could turn out to share common synaptotoxic properties [69]. The Aβ dimers, trimers, and higher order oligomers secreted by cultured neurons inhibit LTP, damage spines, and interfere with activity-regulated cytoskeleton associated protein (Arc) location [64, 65, 69, 78, 79]. Additional studies have shown that Aβ dimers extracted from human CSF disrupt synaptic plasticity and inhibit hippocampal LTP in vivo [80] (Fig 1). Together, these studies indicate that Aβ oligomers, ranging in size from 2–12 subunits, might be responsible for the synaptic damage and memory deficits [81]. A number of recent studies have begun to investigate the possibility that Aβ oligomers might interfere with synaptic function by altering synaptic proteins such as post-synaptic density-95 (PSD95) [82–85], Shank1 [34], and glutamate receptors [86].
Although the neurotoxic effects of the Aβ have been widely studied in experimental models, less is known about the characteristics of the oligomers across the spectrum of AD and how this correlates with cognition and synaptic proteins. We have previously utilized immunoblot analysis to investigate the relationship between levels of Aβ oligomers and synaptic proteins in fractions from the brains of AD patients and APP tg mice. Our studies show that Aβ oligomers, in particular dimers and pentamers, progressively accumulate in the brains of AD patients, as well as in APP tg mice. This was accompanied by reductions in the levels of synaptic scaffold proteins such as PSD95, Shank1 and Shank3 [34].
While accumulation of Aβ oligomers at the synaptic site have been proposed to be an important trigger in the pathogenesis of AD, this hypothesis has been challenged by the lack of a unified description of the toxic oligomer [76] and by recent negative results from clinical trials using Aβ vaccines [87]. Alternative explanation as to why the vaccine trials showed little or no efficacy include that patients treated were at late stages of the disease and that the antibodies did not target specific Aβ oligomers [88].
DOWNSTREAM MECHANISMS OF SYNAPTIC DEGENERATION IN ALZHEIMER’S DISEASE
As described in the previous section, synaptic degeneration occurs early in the progression of AD involving neocortical and limbic system circuitries. Upstream of the cascade is the accumulation of Aβ oligomers at the synaptic sites. The process of synaptic damage could involve a multistep process beginning with dysregulation of glutamate receptors [89, 90] and scaffold molecules such as PSD95 and Shank1 [34] that results in alterations in the axonal transport of synaptic vesicles and mitochondria that later lead to dendritic and spine alterations, as well as axonal dystrophy (Fig 2). Therefore, in the early stages synaptic and network dysfunction [91, 92] might be the norm and actual loss of pre-synaptic terminals and dendritic spines will occur later as synapses become more damaged (Fig 2). Some studies have suggested that at the early stages of the disease progression, aberrant synaptic sprouting might occur as a compensatory mechanism [44]. This is followed by axonal degeneration, while neuronal loss occurs in the later stages of the disease.
Figure 2.

Schematic representation of progression of the mechanisms of synaptic damage in AD and synucleinopathies. At the earliest stages, oligomers interfere with the transport of synaptic vesicle proteins and glutamate receptors resulting in functional deficits that are potentially reversible. Later on signaling pathways and Tau are engage in association with axonal transport defects of trophic factors and mitochondria that in turn lead to synaptic loss and oxidative stress. This is followed by axonal alterations and eventually neuronal loss, resulting in irreversible damage different neurotoxic insults, their effect on neuronal function, and stage with in the disease progression.
Downstream to the accumulation of Aβ oligomers at the synaptic sites there are a number of receptors and signaling cascades involved that converge on abnormal Tau phosphorylation, aggregation and mis-localization from the pre-synaptic to the axonal site (Fig 2). Several Aβ oligomers receptors have been identified including mGluR5 [93], ephrin (ephR2) [94], prion protein (PrP) [95] and others. Extracellular Aβ oligomers bound to lipid-anchored PrP(C) activates intracellular Fyn kinase to perturb synapses (Fig 1) [96–98]. Moreover, recent studies have shown that co-expression of the metabotropic glutamate receptor, mGluR5, allowed PrP(C)-bound Aβ to activate Fyn. PrP(C) and mGluR5 interact physically, and cytoplasmic Fyn forms a complex with mGluR5, resulting in eEF2 phosphorylation and dendritic spine loss [99].
Once bound to synapses, Aβ oligomers can dysregulate the activity and reduce the surface expression of both N-methyl-D-aspartate (NMDA) and 2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl)-propanoic acid (AMPA) types of glutamate receptors, impairing signaling pathways involved in synaptic plasticity [100, 101]. Another interesting finding is that the glutamate receptor system involved in synaptic loss in AD is represented by extracellular NMDARs (eNMDARs). Recent studies have shown that Aβ induces the release of astrocytic glutamate, which in turn activates extrasynaptic NMDA receptors on neurons [102]. This eNMDAR activity leads to synaptic transmission, tau phosphorylation, and caspase-3 activation, each of which is implicated in spine loss. Nitromemantine, which blocks eNMDARs activity, protects synapses from Aβ oligomer toxicity [102] (Fig 1).
Finally, recent work has also found that Aβ oligomers are ligands with nanomolar affinity to paired immunoglobulin-like receptor B (PirB) in murines and its human ortholog, leukocyte immunoglobulin-like receptor B2 (LilrB2). The extracellular domains of PirB and LilrB2 mediate this role, leading to cofilin signaling. The synapto-toxic effects of Aβ oligomers require PirB, and in a transgenic model of AD, PirB not only contributed to memory deficits present in adult mice, but also mediated loss of synaptic plasticity the cortex [103].
Downstream to Aβ, recent studies suggests an emerging role for Tau at the synapse (Fig 2). Even in the absence of tangles, mice over-expressing human Tau display significant synaptic degeneration, suggesting that soluble, oligomeric Tau is the synaptotoxic species [104]. Aggregated or hyperphosphorylated Tau is able to interact with post-synaptic signaling complexes, regulating glutamatergic receptor content in dendritic spines [105], and influencing axonal mitochondrial transport [106, 107]. Interestingly, reducing Tau by genetic means [106] or with immunotherapy reduces behavioral deficits, synaptic dysfunction and network degeneration [108] (Fig 2).
Recent studies suggest that the abnormal localization of Tau to the dendrites might play a role in AD [109]. In support of this possibility, a study showed that Tau translocation to dendrites is mediated by spastin, a microtubule (MT)-degrading enzyme. Spastin is recruited by MT polyglutamylation, induced by Tau mis-sorting, triggering translocalization of TTLL6 (Tubulin-Tyrosine-Ligase-Like-6) into dendrites. Consequences of this translocation include spine loss, as well as mitochondria and neurofilament mislocalization. Adding Tau to Tau-deficient neurons reestablishes Aβ-induced toxicity, which requires phosphorylation of Tau’s KXGS motifs. Transgenic mice overexpressing Tau show TTLL6 translocalization into dendrites and decreased MT stability [110].
Another interesting downstream pathway includes α1-takusan, which was previously identified as a protein that enhances synaptic activity via interaction with PSD-95 and mitigates oligomeric Aβ-induced synaptic loss. In contrast, knockdown of takusan results in enhanced synaptic damage. α1-Takusan interacts with Tau either directly or indirectly, and prevents Aβ-induced Tau aggregation and mitochondrial pathology. α1-Takusan protects synapses from Aβ-induced insult via interaction with PSD-95 and Tau [111].
In addition to the indirect interactions between Aβ and Tau mediated by receptors and signaling pathways, recent studies suggest that monomeric and oligomeric Aβ directly interacts with tau in neurons affected by AD. These interactions progressively increased with the disease process damaging synapses, leading to cognitive decline in AD patients [112].
In between the Aβ oligomer receptors and Tau, a number of studies suggest the role for abnormal activation of signaling cascades including GSK3β, CDK5 and Fyn kinase (Fig 1). Pharmacological interventions targeting the interactions between Aβ oligomer and receptors as well as blocking these kinases have been proposed for the treatment of AD. A number of reviews deal with this subject in greater detail [113–116].
SYNUCLEIN ACCUMULATION IN SYNAPTIC DEGENERATION IN LEWY BODY DISEASE
Lewy body diseases (LBDs) form a heterogeneous group of disorders including PD, PDD and DLB [13]They are often referred to as synucleinopathies as the accumulation of the presynaptic protein α-syn is what characterizes LBDs. α-Syn is a highly abundant protein at the pre-synaptic terminals [117–119], where it is associated with the distal reserve pool of synaptic vesicles [120–122] and has a role in the regulation of neurotransmitter release, synaptic function and plasticity [123, 124] (Fig 3). Various mutations (A53T, A30P, and E46K) [125–127] and multiplications of [128] within the gene encoding of α-syn (SNCA) leads to dominant familial parkinsonism (Fig 3A). Furthermore, certain polymorphisms in SNCA are associated with elevated risk levels for sporadic PD [129]. An increasing group of evidence from animal models, as well as data from genetic, biochemical and biophysical studies support the hypothesis that the processes of α-syn oligomerization [130, 131] and fibril growth [132, 133] have central roles in the pathogenesis of PD and other synucleinopathies [134–136] (Fig 3A).
Figure 3.
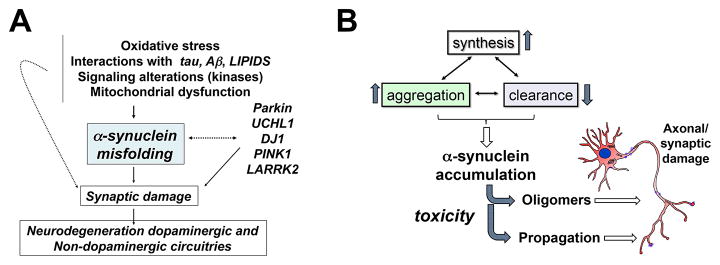
Role of α-syn in the mechanisms of synapse loss in Lewy body disease. A) Neurotoxic events and genetic predisposition lead to α-syn misfolding and aggregation that in turn leads to synaptic damage and ultimately neurodegeneration of dopaminergic and non-dopaminergic circuits. B) Increased synthesis and aggregation, and/or decreased clearance of α-syn leads to α-syn accumulation causing toxicity via oligomers and propagation of the toxic species resulting in in axonal and synaptic damage.
α-Syn aggregates that might play a role in LBD include oligomers, protofibrils, and fibrils. The Lewy bodies, which are the hallmark of LBD, contain mostly fibrillar forms of α-syn [137, 138]. As is the case with Aβ for AD [76], there is no consensus as to the precise α-syn aggregates that are responsible for the synaptic damage in LBD [135]. However, there is indirect evidence supporting the existence of oligomeric α-syn intermediates in vivo under pathophysiological conditions[120, 131, 139–141].
A number of oligomeric intermediates of different morphologies, including spherical, chain-like, and annular oligomers have been described prior to fibril formation by α-syn [142]. Studies suggest that α-syn oligomers can be divided into small (~2–5 mers), medium (~5–15 mers), and large (~15–150 mers)[143, 144]. It is unclear which of these aggregates are physiological and which ones represent toxic species. A couple of studies have reported a stable native α-syn tetramer [145, 146] while other studies suggest the monomer as the native structure [147]. A more recent study suggests that most of the native α-syn is a monomer with a small fraction as trimers and tetramers that prevent non-membrane-bound monomers from aggregating [148]. In vitro studies suggest that the oligomers that undergo a conformational change leading to PK-resistant species might be more toxic [143].
The levels of α-syn is regulated through the balance between rates of α-syn synthesis, aggregation, and clearance [149] (Fig 3B). Dysfunction of one or more of the pathways that balance these rates can lead to anomalous and, therefore, toxic levels of α-syn. For example, in certain forms of familial parkinsonism, multiplication of SNCA leads to elevated accumulation of α-syn due to the increase in the protein expression levels [128], whereas in other forms, SNCA mutations enhance the propensity of α-syn to aggregate [130]. A genome-wide association study (GWAS) linked certain variations in the SNCA gene to higher risk for developing PD [15]. A representative example of such a polymorphism is known as Rep1. Rep1 occurs in the promoter region of SNCA and could increase the susceptibility to PD by increasing the expression of α-syn [150]. Clearance of α-syn monomers and aggregates takes place via direct proteolysis (i.e., by neurosin or matrix metalloprotease 9 (MMP9)) [151], binding to molecular chaperones (for example, heat shock proteins (HSPs)) [152], the proteasome [153–155], and autophagy (related to the activity of the lysosome) [149, 156–158]. In some isolated forms of PD and DLB, inability of the autophagy pathways to eliminate oligomers might facilitate α-syn-mediated toxicity [157]. It has been shown that chaperone-mediated autophagy [121] of mutant α-syn is impaired. In PD and DLB, regulation of the levels of key autophagy molecules such as ATG7, a ubiquitin-like modifier-activating enzyme, and mTOR, a serine–threonine-protein kinase, are impaired [159].
Accumulation of protease K (PK)- resistant α-syn aggregates at the synaptic site results in early degeneration in selected circuitries in PD, PDD and DLB [160]. Degeneration in DLB cases is more closely associated with synaptic accumulation of PK-resistant α-syn than to Lewy bodies [161]. By confocal microscopy there is an average 30–40% loss of synapses in the frontal and temporal cortex in patients with DLB. Synapse loss in the frontal-temporal cortex in DLB patients correlates well with the cognitive impairment [162].
In addition to the direct damage to the synaptic membrane, α-syn oligomers might also trigger synaptoxicity by damaging mitochondria [163], lysosomes [164], or disrupting microtubules [165]. Moreover, a recent study showed that α-syn aggregates might interfere with the axonal transport of synaptic proteins, such as synapsin-1 [123]. Therefore in the early stages of the process of synaptic degeneration in synucleonopathies, there is a failure of synaptic function due to altered transport of vesicles, synaptic proteins, and mitochondria (Fig 2). This then leads to pre-synaptic terminal loss, dendritic damage, axonal dystrophy and eventually degeneration of selective neuronal populations within the striato-nigral and cortico-limbic systems among others (Fig 2).
Similarly to what has been described in the previous sections for AD and Aβ oligomers [93], in PD/DLB α-syn accumulating at the synapses might interact with several receptors including mGluR5 [166]. Activation of glutamate receptors could lead to excitotoxicity and activation of signaling pathways that target Tau aggregation and phosphorylation [167–170] (Fig 2). However it is possible that downstream of α-syn and signaling pathways, targets other than Tau might also be involved because knocking down Tau in α-syn tg mice does not completely rescue the deficits associated [171].
In addition to the role of α-syn accumulating at the synaptic site, recent evidence suggests that under pathological conditions, toxic α-syn oligomers could be released from neurons [172–174] (Fig 3B). Failure of the intracellular clearance pathways, such as autophagy, might contribute to the pathological release of α-syn [157, 175]. Extracellular α-syn aggregates can then transfer from neuron to neuron or from neuron to glial cell [176] where they can nucleate further intracellular aggregation and/or trigger neuro-inflammation and exacerbate the synaptic pathology and neuronal loss [157, 177]. We have recently found that extracellular α-syn released from neuronal cells is an endogenous agonist for Toll-like receptor 2 (TLR2), which activates inflammatory responses in microglia. The TLR2 ligand activity of α-syn is conformation-sensitive; only specific types of oligomer can interact with and activate TLR2. This paracrine interaction between neuron-released oligomeric α-syn and TLR2 in microglia suggests that both of these proteins are novel therapeutic targets for modification of neuroinflammation in PD and related neurological diseases [178]. It is likely that similar to Aβ oligomers, there are several other neuronal and glial receptors for oligomeric α-syn.
Supporting a potential role of extracellular α-syn in the synaptopathology in LBD, previous studies have shown accumulation of α-syn in fetal grafted neurons in patients with PD [179], as well as in grafted neuronal precursor cells in the hippocampus [157] and basal ganglia [180] in mouse models. Interestingly, α-syn has also been shown to ectopically accumulate in oligodendroglial cells in multiple system atrophy (another synucleinopathy) [181] and in astroglial cells in PD [176, 181]. Moreover, the ascending distribution of the Lewy body pathology in LBD, as described by Braak [182] and recent studies published by the group of Dr. V. Lee showing prion-like propagation of α-syn after intra-cerebral injection of seeds [183], which has been interpreted to support the dissemination of α-syn from subcortical to cortical brain regions. Hardy has recently reviewed this topic and identified the caveats of using terminology such as “prion” to describe PD and suggests the use of the term “templating” [184].
In summary, Aβ, α-syn, and Tau aggregates might play a role in synaptic damage in AD and LBD. At the earliest stages, it has been proposed that oligomers might interfere with the transport of synaptic vesicle proteins and glutamate receptors resulting in functional deficits that are potentially reversible; however, further investigation as to the precise nature of the toxic oligomers is necessary. Later on signaling pathways and Tau might be engaged in association with axonal transport defects of trophic factors and mitochondria that in turn lead to synaptic loss and oxidative stress. This could be followed by axonal alterations and eventually neuronal loss and neuroinflammation, resulting in irreversible damage (Fig 2). Given the efforts toward earlier and preclinical diagnosis of AD, PD, and related disorders, understanding the molecular and cellular mechanisms of synaptic degeneration is crucial to developing specific biomarkers and new therapies targeting the synaptic apparatus of vulnerable neurons.
Acknowledgments
Grant support: National Institute of Health through the following grants: AG5131, AG18440, AG022074, and NS044233 (EM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jack CR, Jr, Albert MS, Knopman DS, McKhann GM, Sperling RA, Carrillo MC, et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:257–62. doi: 10.1016/j.jalz.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012;8:1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galasko D, Golde TE. Biomarkers for Alzheimer’s disease in plasma, serum and blood - conceptual and practical problems. Alzheimers Res Ther. 2013;5:10. doi: 10.1186/alzrt164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson KA, Minoshima S, Bohnen NI, Donohoe KJ, Foster NL, Herscovitch P, et al. Appropriate use criteria for amyloid PET: a report of the Amyloid Imaging Task Force, the Society of Nuclear Medicine and Molecular Imaging, and the Alzheimer’s Association. Alzheimers Dement. 2013;9:e-1–16. doi: 10.1016/j.jalz.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kang JH, Korecka M, Toledo JB, Trojanowski JQ, Shaw LM. Clinical Utility and Analytical Challenges in Measurement of Cerebrospinal Fluid Amyloid-beta1–42 and tau Proteins as Alzheimer Disease Biomarkers. Clin Chem. 2013 doi: 10.1373/clinchem.2013.202937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bettens K, Sleegers K, Van Broeckhoven C. Genetic insights in Alzheimer’s disease. Lancet Neurol. 2013;12:92–104. doi: 10.1016/S1474-4422(12)70259-4. [DOI] [PubMed] [Google Scholar]
- 7.Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harb Perspect Med. 2012:2. doi: 10.1101/cshperspect.a006296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Masters CL, Selkoe DJ. Biochemistry of amyloid beta-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006262. doi: 10.1101/cshperspect.a006262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stefani M. Structural features and cytotoxicity of amyloid oligomers: implications in Alzheimer’s disease and other diseases with amyloid deposits. Prog Neurobiol. 2012;99:226–45. doi: 10.1016/j.pneurobio.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Sarell CJ, Stockley PG, Radford SE. Assessing the causes and consequences of co-polymerization in amyloid formation. Prion. 2013:7. doi: 10.4161/pri.26415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chesser AS, Pritchard SM, Johnson GV. Tau Clearance Mechanisms and Their Possible Role in the Pathogenesis of Alzheimer Disease. Front Neurol. 2013;4:122. doi: 10.3389/fneur.2013.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cardenas AM, Ardiles AO, Barraza N, Baez-Matus X, Caviedes P. Role of tau protein in neuronal damage in Alzheimer’s disease and Down syndrome. Arch Med Res. 2012;43:645–54. doi: 10.1016/j.arcmed.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 13.McKeith IG. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. J Alzheimers Dis. 2006;9:417–23. doi: 10.3233/jad-2006-9s347. [DOI] [PubMed] [Google Scholar]
- 14.Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet. 2006;7:306–18. doi: 10.1038/nrg1831. [DOI] [PubMed] [Google Scholar]
- 15.Edwards TL, Scott WK, Almonte C, Burt A, Powell EH, Beecham GW, et al. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet. 2010;74:97–109. doi: 10.1111/j.1469-1809.2009.00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ozansoy M, Basak AN. The central theme of Parkinson’s disease: alpha-synuclein. Mol Neurobiol. 2013;47:460–5. doi: 10.1007/s12035-012-8369-3. [DOI] [PubMed] [Google Scholar]
- 17.Hardy J. Genetic analysis of pathways to Parkinson disease. Neuron. 2010;68:201–6. doi: 10.1016/j.neuron.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Association As. 2013 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dementia. 2013;9:1–71. doi: 10.1016/j.jalz.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 19.Jack CR, Jr, Wiste HJ, Weigand SD, Knopman DS, Lowe V, Vemuri P, et al. Amyloid-first and neurodegeneration-first profiles characterize incident amyloid PET positivity. Neurology. 2013;81:1732–40. doi: 10.1212/01.wnl.0000435556.21319.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hyman BT, Trojanowski JQ. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropath Exp Neurol. 1997;56:1095–7. doi: 10.1097/00005072-199710000-00002. [DOI] [PubMed] [Google Scholar]
- 21.Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 22.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 23.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–86. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 24.Hamilton RL. Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol. 2000;10:378–84. doi: 10.1111/j.1750-3639.2000.tb00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Halliday GM, Holton JL, Revesz T, Dickson DW. Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol. 2011;122:187–204. doi: 10.1007/s00401-011-0852-9. [DOI] [PubMed] [Google Scholar]
- 26.Hansen L, Masliah E, Terry R. A neuropathologic subset of Alzheimer’s disease with concomitant Lewy body disease and spongiform change. Acta Neuropathol. 1989;78:194–201. doi: 10.1007/BF00688209. [DOI] [PubMed] [Google Scholar]
- 27.Wei W, Nguyen LN, Kessels HW, Hagiwara H, Sisodia S, Malinow R. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat Neurosci. 2010;13:190–6. doi: 10.1038/nn.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iwai A, Masliah E, Yoshimoto M, De Silva R, Ge N, Kittel A, et al. The precursor protein of non-Aβ component of Alzheimer’s disease amyloid (NACP) is a presynaptic protein of the central nervous system. Neuron. 1994;14:467–75. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- 29.Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43:429–35. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43:436–41. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368:117–27. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Swarup V, Geschwind DH. Alzheimer’s disease: From big data to mechanism. Nature. 2013;500:34–5. doi: 10.1038/nature12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ertekin-Taner N. Alzheimer disease: the quest for Alzheimer disease genes--focus on CSF tau. Nat Rev Neurol. 2013;9:368–70. doi: 10.1038/nrneurol.2013.117. [DOI] [PubMed] [Google Scholar]
- 34.Pham E, Crews L, Ubhi K, Hansen L, Adame A, Cartier A, et al. Progressive accumulation of amyloid-beta oligomers in Alzheimer’s disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. FEBS J. 2010;277:3051–67. doi: 10.1111/j.1742-4658.2010.07719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scheff SW, Price DA, Schmitt FA, Roberts KN, Ikonomovic MD, Mufson EJ. Synapse stability in the precuneus early in the progression of Alzheimer’s disease. J Alzheimers Dis. 2013;35:599–609. doi: 10.3233/JAD-122353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Masliah E, Hansen L, DeTeresa R, Terry R. Confocal laser imaging of synapse-plaque relationships in Alzheimer disease. J Neuropathol Exp Neurol. 1990;49:335. [Google Scholar]
- 37.Alonso-Nanclares L, Merino-Serrais P, Gonzalez S, DeFelipe J. Synaptic changes in the dentate gyrus of APP/PS1 transgenic mice revealed by electron microscopy. J Neuropath Exp Neurol. 2013;72:386–95. doi: 10.1097/NEN.0b013e31828d41ec. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, et al. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–8. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Freir DB, Fedriani R, Scully D, Smith IM, Selkoe DJ, Walsh DM, et al. Abeta oligomers inhibit synapse remodelling necessary for memory consolidation. Neurobiol Aging. 2011;32:2211–8. doi: 10.1016/j.neurobiolaging.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Canas PM, Simoes AP, Rodrigues RJ, Cunha RA. Predominant loss of glutamatergic terminal markers in a beta-amyloid peptide model of Alzheimer’s disease. Neuropharmacology. 2014;76(Pt A):51–6. doi: 10.1016/j.neuropharm.2013.08.026. [DOI] [PubMed] [Google Scholar]
- 41.Mitew S, Kirkcaldie MT, Dickson TC, Vickers JC. Altered synapses and gliotransmission in Alzheimer’s disease and AD model mice. Neurobiol Aging. 2013;34:2341–51. doi: 10.1016/j.neurobiolaging.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 42.Mufson EJ, Binder L, Counts SE, DeKosky ST, de Toledo-Morrell L, Ginsberg SD, et al. Mild cognitive impairment: pathology and mechanisms. Acta Neuropathol. 2012;123:13–30. doi: 10.1007/s00401-011-0884-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007;68:1501–8. doi: 10.1212/01.wnl.0000260698.46517.8f. [DOI] [PubMed] [Google Scholar]
- 44.Berchtold NC, Coleman PD, Cribbs DH, Rogers J, Gillen DL, Cotman CW. Synaptic genes are extensively downregulated across multiple brain regions in normal human aging and Alzheimer’s disease. Neurobiol Aging. 2013;34:1653–61. doi: 10.1016/j.neurobiolaging.2012.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang RY, Nouwens AS, Dodd PR, Etheridge N. The synaptic proteome in Alzheimer’s disease. Alzheimers Dement. 2013;9:499–511. doi: 10.1016/j.jalz.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 46.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–80. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 47.DeKosky S, Scheff S. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–64. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 48.DeKosky ST, Scheff SW, Styren SD. Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration. 1996;5:417–21. doi: 10.1006/neur.1996.0056. [DOI] [PubMed] [Google Scholar]
- 49.Scheff SW, DeKosky ST, Price DA. Quantitative assessment of cortical synaptic density in Alzheimer’s disease. Neurobiol Aging. 1990;11:29–37. doi: 10.1016/0197-4580(90)90059-9. [DOI] [PubMed] [Google Scholar]
- 50.Scheff S, Sparks D, Price D. Quantitative assessment of synaptic density in the entorhinal cortex in Alzheimer’s disease. FASEB J. 1993;34:356–61. doi: 10.1002/ana.410340309. [DOI] [PubMed] [Google Scholar]
- 51.Masliah E, Terry R. The role of synaptic pathology in the mechanisms of dementia in Alzheimer’s disease. Clin Neurosci. 1994;1:192–8. [Google Scholar]
- 52.Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, et al. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–31. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- 53.Scheff S, DeKosky S, Price D. Quantitative assessment of cortical synaptic density in Alzheimer’s disease. Neurobiol Aging. 1990;11:29–37. doi: 10.1016/0197-4580(90)90059-9. [DOI] [PubMed] [Google Scholar]
- 54.Scheff S, Price D. Synapse loss in the temporal lobe in Alzheimer’s disease. Ann Neurol. 1993;33:190–9. doi: 10.1002/ana.410330209. [DOI] [PubMed] [Google Scholar]
- 55.Krstic D, Knuesel I. Deciphering the mechanism underlying late-onset Alzheimer disease. Nat Rev Neurol. 2013;9:25–34. doi: 10.1038/nrneurol.2012.236. [DOI] [PubMed] [Google Scholar]
- 56.Masliah E, Crews L. Genetically engineered mouse models of neurodegenerative disorders. In: Uversky VN, editor. Protein Misfolding, Aggregation and Conformational Diseases. New York: Kluwer Academic/Plenum; 2006. [Google Scholar]
- 57.Sisodia SS, Price DL. Role of the beta-amyloid protein in Alzheimer’s disease. FASEB J. 1995;9:366–70. doi: 10.1096/fasebj.9.5.7896005. [DOI] [PubMed] [Google Scholar]
- 58.Selkoe D. Amyloid b protein precursor and the pathogenesis of Alzheimer’s disease. Cell. 1989;58:611–2. doi: 10.1016/0092-8674(89)90093-7. [DOI] [PubMed] [Google Scholar]
- 59.Selkoe D. Amyloid b-protein deposition as a seminal pathogenic event in AD: an hypothesis. Neurobiol Aging. 1990;11:299. [Google Scholar]
- 60.Selkoe D. Physiological production of the b-amyloid protein and the mechanisms of Alzheimer’s disease. Trends Neurosci. 1993;16:403–9. doi: 10.1016/0166-2236(93)90008-a. [DOI] [PubMed] [Google Scholar]
- 61.Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, McConlogue L, et al. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7:612–8. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- 62.Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120:4081–91. doi: 10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- 63.Klein WL. ADDLs & protofibrils--the missing links? Neurobiol Aging. 2002;23:231–5. doi: 10.1016/s0197-4580(01)00312-8. [DOI] [PubMed] [Google Scholar]
- 64.Klein WL, Krafft GA, Finch CE. Targeting small Abeta oligomers: the solution to an Alzheimer’s disease conundrum? Trends Neurosci. 2001;24:219–24. doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- 65.Walsh DM, Selkoe DJ. Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein Pept Lett. 2004;11:213–28. doi: 10.2174/0929866043407174. [DOI] [PubMed] [Google Scholar]
- 66.Glabe CC. Amyloid accumulation and pathogensis of Alzheimer’s disease: significance of monomeric, oligomeric and fibrillar Abeta. Subcell Biochem. 2005;38:167–77. doi: 10.1007/0-387-23226-5_8. [DOI] [PubMed] [Google Scholar]
- 67.Glabe CG. Structural classification of toxic amyloid oligomers. J Biol Chem. 2008;283:29639–43. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, et al. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–53. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–13. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–98. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 71.Teplow DB. Structural and kinetic features of amyloid beta-protein fibrillogenesis. Amyloid. 1998;5:121–42. doi: 10.3109/13506129808995290. [DOI] [PubMed] [Google Scholar]
- 72.Roychaudhuri R, Yang M, Hoshi MM, Teplow DB. Amyloid beta-protein assembly and Alzheimer disease. J Biol Chem. 2009;284:4749–53. doi: 10.1074/jbc.R800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Klein WL. Synaptotoxic amyloid-beta oligomers: a molecular basis for the cause, diagnosis, and treatment of Alzheimer’s disease? J Alzheimers Dis. 2013;33 (Suppl 1):S49–65. doi: 10.3233/JAD-2012-129039. [DOI] [PubMed] [Google Scholar]
- 74.Mucke L, Selkoe DJ. Neurotoxicity of amyloid beta-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med. 2012;2:a006338. doi: 10.1101/cshperspect.a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tsigelny IF, Sharikov Y, Kouznetsova VL, Greenberg JP, Wrasidlo W, Gonzalez T, et al. Structural Diversity of Alzheimer’s Disease Amyloid-beta Dimers and Their Role in Oligomerization and Fibril Formation. J Alzheimers Dis. 2013 doi: 10.3233/JAD-131589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Benilova I, Karran E, De Strooper B. The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci. 2012;15:349–57. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- 77.Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–7. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 78.Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006;572:477–92. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kerrigan TL, Randall AD. A new player in the “synaptopathy” of Alzheimer’s disease - arc/arg 3. 1. Front Neurol. 2013;4:9. doi: 10.3389/fneur.2013.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, Wallin A, et al. Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci. 2008;28:4231–7. doi: 10.1523/JNEUROSCI.5161-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, et al. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, Cole GM. Synaptic changes in Alzheimer’s disease: increased amyloid-beta and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am J Pathol. 2004;165:1809–17. doi: 10.1016/s0002-9440(10)63436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Simon AM, Schiapparelli L, Salazar-Colocho P, Cuadrado-Tejedor M, Escribano L, Lopez de Maturana R, et al. Overexpression of wild-type human APP in mice causes cognitive deficits and pathological features unrelated to Abeta levels. Neurobiol Dis. 2009;33:369–78. doi: 10.1016/j.nbd.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 84.Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, et al. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A. 2009;106:4012–7. doi: 10.1073/pnas.0811698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sultana R, Banks WA, Butterfield DA. Decreased levels of PSD95 and two associated proteins and increased levels of BCl2 and caspase 3 in hippocampus from subjects with amnestic mild cognitive impairment: Insights into their potential roles for loss of synapses and memory, accumulation of Abeta, and neurodegeneration in a prodromal stage of Alzheimer’s disease. J Neurosci Res. 2010;88:469–77. doi: 10.1002/jnr.22227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Almeida CG, Tampellini D, Takahashi RH, Greengard P, Lin MT, Snyder EM, et al. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis. 2005;20:187–98. doi: 10.1016/j.nbd.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 87.Prins ND, Scheltens P. Treating Alzheimer’s disease with monoclonal antibodies: current status and outlook for the future. Alzheimers Res Ther. 2013;5:56. doi: 10.1186/alzrt220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lemere CA, Masliah E. Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat Rev Neurol. 2010;6:108–19. doi: 10.1038/nrneurol.2009.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mota SI, Ferreira IL, Rego AC. Dysfunctional synapse in Alzheimer’s disease - A focus on NMDA receptors. Neuropharmacology. 2014;76(Pt A):16–26. doi: 10.1016/j.neuropharm.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 90.Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, et al. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–43. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Palop JJ, Chin J, Mucke L. A network dysfunction perspective on neurodegenerative diseases. Nature. 2006;443:768–73. doi: 10.1038/nature05289. [DOI] [PubMed] [Google Scholar]
- 92.Palop JJ, Mucke L, Roberson ED. Quantifying biomarkers of cognitive dysfunction and neuronal network hyperexcitability in mouse models of Alzheimer’s disease: depletion of calcium-dependent proteins and inhibitory hippocampal remodeling. Methods Mol Biol. 2011;670:245–62. doi: 10.1007/978-1-60761-744-0_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, Klein WL, et al. Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron. 2010;66:739–54. doi: 10.1016/j.neuron.2010.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cisse M, Halabisky B, Harris J, Devidze N, Dubal DB, Sun B, et al. Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature. 2011;469:47–52. doi: 10.1038/nature09635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–32. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chin J, Palop JJ, Puolivali J, Massaro C, Bien-Ly N, Gerstein H, et al. Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer’s disease. J Neurosci. 2005;25:9694–703. doi: 10.1523/JNEUROSCI.2980-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chin J, Palop JJ, Yu GQ, Kojima N, Masliah E, Mucke L. Fyn kinase modulates synaptotoxicity, but not aberrant sprouting, in human amyloid precursor protein transgenic mice. J Neurosci. 2004;24:4692–7. doi: 10.1523/JNEUROSCI.0277-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Um JW, Strittmatter SM. Amyloid-beta induced signaling by cellular prion protein and Fyn kinase in Alzheimer disease. Prion. 2013;7:37–41. doi: 10.4161/pri.22212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Um JW, Kaufman AC, Kostylev M, Heiss JK, Stagi M, Takahashi H, et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron. 2013;79:887–902. doi: 10.1016/j.neuron.2013.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Paula-Lima AC, Brito-Moreira J, Ferreira ST. Deregulation of excitatory neurotransmission underlying synapse failure in Alzheimer’s disease. J Neurochem. 2013;126:191–202. doi: 10.1111/jnc.12304. [DOI] [PubMed] [Google Scholar]
- 101.Sivanesan S, Tan A, Rajadas J. Pathogenesis of Abeta oligomers in synaptic failure. Current Alzheimer research. 2013;10:316–23. doi: 10.2174/1567205011310030011. [DOI] [PubMed] [Google Scholar]
- 102.Talantova M, Sanz-Blasco S, Zhang X, Xia P, Akhtar MW, Okamoto S, et al. Abeta induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci U S A. 2013;110:E2518–27. doi: 10.1073/pnas.1306832110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kim T, Vidal GS, Djurisic M, William CM, Birnbaum ME, Garcia KC, et al. Human LilrB2 is a beta-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer’s model. Science. 2013;341:1399–404. doi: 10.1126/science.1242077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pooler AM, Noble W, Hanger DP. A role for tau at the synapse in Alzheimer’s disease pathogenesis. Neuropharmacology. 2014;76(Pt A):1–8. doi: 10.1016/j.neuropharm.2013.09.018. [DOI] [PubMed] [Google Scholar]
- 105.Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell. 2010;142:387–97. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 106.Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, et al. Tau reduction prevents Abeta-induced defects in axonal transport. Science. 2010;330:198. doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shahpasand K, Uemura I, Saito T, Asano T, Hata K, Shibata K, et al. Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer’s disease. J Neurosci. 2012;32:2430–41. doi: 10.1523/JNEUROSCI.5927-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci. 2007;27:9115–29. doi: 10.1523/JNEUROSCI.2361-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zempel H, Thies E, Mandelkow E, Mandelkow EM. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010;30:11938–50. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zempel H, Luedtke J, Kumar Y, Biernat J, Dawson H, Mandelkow E, et al. Amyloid-beta oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. EMBO J. 2013;32:2920–37. doi: 10.1038/emboj.2013.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nakanishi N, Ryan SD, Zhang X, Khan A, Holland T, Cho EG, et al. Synaptic protein alpha1-takusan mitigates amyloid-beta-induced synaptic loss via interaction with tau and postsynaptic density-95 at postsynaptic sites. J Neurosci. 2013;33:14170–83. doi: 10.1523/JNEUROSCI.4646-10.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Manczak M, Reddy PH. Abnormal interaction of oligomeric amyloid-beta with phosphorylated tau: implications to synaptic dysfunction and neuronal damage. J Alzheimers Dis. 2013;36:285–95. doi: 10.3233/JAD-130275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Citron M. Alzheimer’s disease: strategies for disease modification. Nature reviews Drug discovery. 2010;9:387–98. doi: 10.1038/nrd2896. [DOI] [PubMed] [Google Scholar]
- 114.Nistico R, Pignatelli M, Piccinin S, Mercuri NB, Collingridge G. Targeting synaptic dysfunction in Alzheimer’s disease therapy. Mol Neurobiol. 2012;46:572–87. doi: 10.1007/s12035-012-8324-3. [DOI] [PubMed] [Google Scholar]
- 115.Salomone S, Caraci F, Leggio GM, Fedotova J, Drago F. New pharmacological strategies for treatment of Alzheimer’s disease: focus on disease modifying drugs. Br J Clin Pharmacol. 2012;73:504–17. doi: 10.1111/j.1365-2125.2011.04134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yang K, Belrose J, Trepanier CH, Lei G, Jackson MF, MacDonald JF. Fyn, a potential target for Alzheimer’s disease. J Alzheimers Dis. 2011;27:243–52. doi: 10.3233/JAD-2011-110353. [DOI] [PubMed] [Google Scholar]
- 117.Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, et al. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–75. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- 118.Jakes R, Spillantini MG, Goedert M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994;345:27–32. doi: 10.1016/0014-5793(94)00395-5. [DOI] [PubMed] [Google Scholar]
- 119.Withers GS, George JM, Banker GA, Clayton DF. Delayed localization of synelfin (synuclein, NACP) to presynaptic terminals in cultured rat hippocampal neurons. Brain Res Dev Brain Res. 1997;99:87–94. doi: 10.1016/s0165-3806(96)00210-6. [DOI] [PubMed] [Google Scholar]
- 120.Kahle PJ, Neumann M, Ozmen L, Muller V, Jacobsen H, Schindzielorz A, et al. Subcellular localization of wild-type and Parkinson’s disease-associated mutant alpha -synuclein in human and transgenic mouse brain. J Neurosci. 2000;20:6365–73. doi: 10.1523/JNEUROSCI.20-17-06365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lee SJ, Jeon H, Kandror KV. Alpha-synuclein is localized in a subpopulation of rat brain synaptic vesicles. Acta Neurobiol Exp. 2008;68:509–15. doi: 10.55782/ane-2008-1717. [DOI] [PubMed] [Google Scholar]
- 122.Zhang L, Zhang C, Zhu Y, Cai Q, Chan P, Ueda K, et al. Semi-quantitative analysis of alpha-synuclein in subcellular pools of rat brain neurons: an immunogold electron microscopic study using a C-terminal specific monoclonal antibody. Brain res. 2008;1244:40–52. doi: 10.1016/j.brainres.2008.08.067. [DOI] [PubMed] [Google Scholar]
- 123.Scott DA, Tabarean I, Tang Y, Cartier A, Masliah E, Roy S. A pathologic cascade leading to synaptic dysfunction in alpha-synuclein-induced neurodegeneration. J Neurosci. 2010;30:8083–95. doi: 10.1523/JNEUROSCI.1091-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–7. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998;18:106–8. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 126.Polymeropoulos M, Lavedan C, Leroy E, Ide S, Dehejia A, Dutra A, et al. Mutation in the a-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–7. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 127.Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–73. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 128.Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004;364:1167–9. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- 129.Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41:1308–12. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med. 1998;4:1318–20. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- 131.Tsigelny IF, Sharikov Y, Miller MA, Masliah E. Mechanism of alpha-synuclein oligomerization and membrane interaction: theoretical approach to unstructured proteins studies. Nanomedicine. 2008;4:350–7. doi: 10.1016/j.nano.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Oueslati A, Fournier M, Lashuel HA. Role of post-translational modifications in modulating the structure, function and toxicity of alpha-synuclein: implications for Parkinson’s disease pathogenesis and therapies. Prog Brain Res. 2010;183:115–45. doi: 10.1016/S0079-6123(10)83007-9. [DOI] [PubMed] [Google Scholar]
- 133.Taschenberger G, Garrido M, Tereshchenko Y, Bahr M, Zweckstetter M, Kugler S. Aggregation of alphaSynuclein promotes progressive in vivo neurotoxicity in adult rat dopaminergic neurons. Acta Neuropathol. 2012;123:671–83. doi: 10.1007/s00401-011-0926-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci U S A. 2011;108:4194–9. doi: 10.1073/pnas.1100976108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14:38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lee VM, Trojanowski JQ. Mechanisms of Parkinson’s disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron. 2006;52:33–8. doi: 10.1016/j.neuron.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 137.Kosaka K. Diffuse Lewy body disease in Japan. JNeurol. 1990;237:197–204. doi: 10.1007/BF00314594. [DOI] [PubMed] [Google Scholar]
- 138.Dickson DW, Crystal H, Mattiace LA, Kress Y, Schwagerl A, Ksiezak-Reding H, et al. Diffuse Lewy body disease: light and electron microscopic immunocytochemistry of senile plaques. Acta Neuropathol. 1989;78:572–84. doi: 10.1007/BF00691284. [DOI] [PubMed] [Google Scholar]
- 139.Kahle PJ, Neumann M, Ozmen L, Muller V, Odoy S, Okamoto N, et al. Selective insolubility of alpha-synuclein in human Lewy body diseases is recapitulated in a transgenic mouse model. Am J Pathol. 2001;159:2215–25. doi: 10.1016/s0002-9440(10)63072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, et al. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol. 1998;152:879–84. [PMC free article] [PubMed] [Google Scholar]
- 141.Lee MK, Stirling W, Xu Y, Xu X, Qui D, Mandir AS, et al. Human alpha-synuclein-harboring familial Parkinson’s disease-linked Ala-53 --> Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc Natl Acad Sci U S A. 2002;99:8968–73. doi: 10.1073/pnas.132197599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Horvath I, Weise CF, Andersson EK, Chorell E, Sellstedt M, Bengtsson C, et al. Mechanisms of Protein Oligomerization: Inhibitor of Functional Amyloids Templates alpha-Synuclein Fibrillation. J Am Chem Soc. 2012;134:3439–44. doi: 10.1021/ja209829m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cremades N, Cohen SI, Deas E, Abramov AY, Chen AY, Orte A, et al. Direct Observation of the Interconversion of Normal and Toxic Forms of alpha-Synuclein. Cell. 2012;149:1048–59. doi: 10.1016/j.cell.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A, et al. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J Neurosci. 2007;27:9220–32. doi: 10.1523/JNEUROSCI.2617-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LT, Liao J, et al. A soluble alpha-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci U S A. 2011;108:17797–802. doi: 10.1073/pnas.1113260108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bartels T, Choi JG, Selkoe DJ. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–10. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Fauvet B, Mbefo MK, Fares MB, Desobry C, Michael S, Ardah MT, et al. Alpha-synuclein in the central nervous system and from erythrocytes, mammalian cells and E. coli exists predominantly as a disordered monomer. J Biol Chem. 2012;287:15345–64. doi: 10.1074/jbc.M111.318949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gurry T, Ullman O, Fisher CK, Perovic I, Pochapsky T, Stultz CM. The dynamic structure of alpha-synuclein multimers. J Am Chem Soc. 2013;135:3865–72. doi: 10.1021/ja310518p. [DOI] [PubMed] [Google Scholar]
- 149.Kragh CL, Ubhi K, Wyss-Corey T, Masliah E. Autophagy in dementias. Brain Pathol. 2012;22:99–109. doi: 10.1111/j.1750-3639.2011.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Maraganore DM, de Andrade M, Elbaz A, Farrer MJ, Ioannidis JP, Kruger R, et al. Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA. 2006;296:661–70. doi: 10.1001/jama.296.6.661. [DOI] [PubMed] [Google Scholar]
- 151.Iwata A, Maruyama M, Akagi T, Hashikawa T, Kanazawa I, Tsuji S, et al. Alpha-synuclein degradation by serine protease neurosin: implication for pathogenesis of synucleinopathies. Hum Mol Genet. 2003;12:2625–35. doi: 10.1093/hmg/ddg283. [DOI] [PubMed] [Google Scholar]
- 152.Klucken J, Shin Y, Masliah E, Hyman BT, McLean PJ. Hsp70 Reduces alpha-Synuclein Aggregation and Toxicity. J Biol Chem. 2004;279:25497–502. doi: 10.1074/jbc.M400255200. [DOI] [PubMed] [Google Scholar]
- 153.McNaught KS, Mytilineou C, Jnobaptiste R, Yabut J, Shashidharan P, Jennert P, et al. Impairment of the ubiquitin-proteasome system causes dopaminergic cell death and inclusion body formation in ventral mesencephalic cultures. J Neurochem. 2002;81:301–6. doi: 10.1046/j.1471-4159.2002.00821.x. [DOI] [PubMed] [Google Scholar]
- 154.McNaught KS, Bjorklund LM, Belizaire R, Isacson O, Jenner P, Olanow CW. Proteasome inhibition causes nigral degeneration with inclusion bodies in rats. Neuroreport. 2002;13:1437–41. doi: 10.1097/00001756-200208070-00018. [DOI] [PubMed] [Google Scholar]
- 155.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–13. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 156.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–5. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 157.Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009;106:13010–5. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, et al. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci. 2009;29:13578–88. doi: 10.1523/JNEUROSCI.4390-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Crews L, Spencer B, Desplats P, Patrick C, Paulino A, Rockenstein E, et al. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One. 2010;5:e9313. doi: 10.1371/journal.pone.0009313. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 160.Schulz-Schaeffer WJ. The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010;120:131–43. doi: 10.1007/s00401-010-0711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kramer ML, Schulz-Schaeffer WJ. Presynaptic alpha-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J Neurosci. 2007;27:1405–10. doi: 10.1523/JNEUROSCI.4564-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Masliah E, Mallory M, DeTeresa R, Alford M, Hansen L. Differing patterns of aberrant neuronal sprouting in Alzheimer’s disease with and without Lewy bodies. Brain Res. 1993;617:258–66. doi: 10.1016/0006-8993(93)91093-8. [DOI] [PubMed] [Google Scholar]
- 163.Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, et al. alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol. 2000;157:401–10. doi: 10.1016/s0002-9440(10)64553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Manzoni C, Lewis PA. Dysfunction of the autophagy/lysosomal degradation pathway is a shared feature of the genetic synucleinopathies. FASEB J. 2013;27:3424–9. doi: 10.1096/fj.12-223842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Alim MA, Ma QL, Takeda K, Aizawa T, Matsubara M, Nakamura M, et al. Demonstration of a role for alpha-synuclein as a functional microtubule-associated protein. J Alzheimers Dis. 2004;6:435–42. doi: 10.3233/jad-2004-6412. discussion 43–9. [DOI] [PubMed] [Google Scholar]
- 166.Price DL, Rockenstein E, Ubhi K, Phung V, MacLean-Lewis N, Askay D, et al. Alterations in mGluR5 expression and signaling in Lewy body disease and in transgenic models of alpha-synucleinopathy--implications for excitotoxicity. PLoS One. 2010;5:e14020. doi: 10.1371/journal.pone.0014020. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 167.Kaul T, Credle J, Haggerty T, Oaks AW, Masliah E, Sidhu A. Region-specific tauopathy and synucleinopathy in brain of the alpha-synuclein overexpressing mouse model of Parkinson’s disease. BMC Neurosci. 2011;12:79. doi: 10.1186/1471-2202-12-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Haggerty T, Credle J, Rodriguez O, Wills J, Oaks AW, Masliah E, et al. Hyperphosphorylated Tau in an alpha-synuclein-overexpressing transgenic model of Parkinson’s disease. Eur J Neurosci. 2011;33:1598–610. doi: 10.1111/j.1460-9568.2011.07660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wills J, Jones J, Haggerty T, Duka V, Joyce JN, Sidhu A. Elevated tauopathy and alpha-synuclein pathology in postmortem Parkinson’s disease brains with and without dementia. Exp Neurol. 2010;225:210–8. doi: 10.1016/j.expneurol.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Duka V, Lee JH, Credle J, Wills J, Oaks A, Smolinsky C, et al. Identification of the sites of tau hyperphosphorylation and activation of tau kinases in synucleinopathies and Alzheimer’s diseases. PLoS One. 2013;8:e75025. doi: 10.1371/journal.pone.0075025. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 171.Morris M, Koyama A, Masliah E, Mucke L. Tau reduction does not prevent motor deficits in two mouse models of Parkinson’s disease. PLoS One. 2011;6:e29257. doi: 10.1371/journal.pone.0029257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Jang A, Lee HJ, Suk JE, Jung JW, Kim KP, Lee SJ. Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J Neurochem. 2010;113:1263–74. doi: 10.1111/j.1471-4159.2010.06695.x. [DOI] [PubMed] [Google Scholar]
- 173.Danzer KM, Ruf WP, Putcha P, Joyner D, Hashimoto T, Glabe C, et al. Heat-shock protein 70 modulates toxic extracellular alpha-synuclein oligomers and rescues trans-synaptic toxicity. FASEB J. 2011;25:326–36. doi: 10.1096/fj.10-164624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Alvarez-Erviti L, Couch Y, Richardson J, Cooper JM, Wood MJ. Alpha-synuclein release by neurons activates the inflammatory response in a microglial cell line. Neurosci Res. 2011;69:337–42. doi: 10.1016/j.neures.2010.12.020. [DOI] [PubMed] [Google Scholar]
- 175.Lee HJ, Cho ED, Lee KW, Kim JH, Cho SG, Lee SJ. Autophagic failure promotes the exocytosis and intercellular transfer of alpha-synuclein. Exp Mol Med. 2013;45:e22. doi: 10.1038/emm.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, et al. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem. 2010;285:9262–72. doi: 10.1074/jbc.M109.081125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, et al. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. 2013;4:1562. doi: 10.1038/ncomms2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Crews L, Mizuno H, Desplats P, Rockenstein E, Adame A, Patrick C, et al. Alpha-synuclein alters Notch-1 expression and neurogenesis in mouse embryonic stem cells and in the hippocampus of transgenic mice. J Neurosci. 2008;28:4250–60. doi: 10.1523/JNEUROSCI.0066-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hansen C, Angot E, Bergstrom AL, Steiner JA, Pieri L, Paul G, et al. alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest. 2011;121:715–25. doi: 10.1172/JCI43366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wakabayashi K, Hayashi S, Kakita A, Yamada M, Toyoshima Y, Yoshimoto M, et al. Accumulation of α synuclein/NACP is a cytopathological feature common to Lewy body disease and multiple system atrophy. Acta Neuropathol. 1998;96:445–52. doi: 10.1007/s004010050918. [DOI] [PubMed] [Google Scholar]
- 182.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 183.Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–53. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hardy J, Revesz T. The spread of neurodegenerative disease. N Engl J Med. 2012;366:2126–8. doi: 10.1056/NEJMcibr1202401. [DOI] [PubMed] [Google Scholar]