

Abstract
Objectives
ACAT2 is the exclusive cholesterol-esterifying enzyme in hepatocytes and enterocytes. Hepatic ABCA1 transfers unesterified cholesterol (UC) to apoAI, thus generating HDL. By changing the hepatic UC pool available for ABCA1, ACAT2 may affect HDL metabolism. The aim of this study was to reveal whether hepatic ACAT2 influences HDL metabolism.
Design
WT and LXRα/β double knockout (DOKO) mice were fed a western-type diet for 8 weeks. Animals were i.p. injected with an antisense oligonucleotide targeted to hepatic ACAT2 (ASO6), or with an ASO control. Injections started 4 weeks after, or concomitantly with, the beginning of the diet.
Results
ASO6 reduced liver cholesteryl esters, while not inducing UC accumulation. ASO6 increased hepatic ABCA1 protein independently of the diet conditions. ASO6 affected HDL lipids (increased UC) only in DOKO, while it increased apoE-containing HDL in both genotypes. In WT mice ASO6 led to the appearance of large HDL enriched in apoAI and apoE.
Conclusions
The use of ASO6 revealed a new pathway by which the liver may contribute to HDL metabolism in mice. ACAT2 seems to be a hepatic player affecting the cholesterol fluxes fated to VLDL or to HDL, the latter via up-regulation of ABCA1.
Introduction
For many years, the inhibition of intracellular cholesterol esterification has been considered as a potential strategy to prevent atherosclerosis [1]. Acyl-coenzyme A:cholesterol acyltransferase (ACAT) is an enzyme located in the endoplasmic reticulum that catalyses the synthesis of cholesteryl esters (CE) by conjugating cholesterol to long-chain fatty acids; mainly oleic and palmitic acids. It is now clear that the two enzymes ACAT1 and ACAT2, which are encoded by the Soat1 and Soat2 genes respectively, localize in different cell types and have separate physiological functions (for review see [2], [3]). ACAT1 is ubiquitously expressed and provides essential housekeeping functions to prevent the toxicity induced by increasing amounts of unesterified cholesterol (UC) in cells. Conversely, ACAT2 is exclusively expressed in hepatocytes and enterocytes, and it synthesizes CE that can be incorporated into apoB-containing lipoproteins (VLDL and chylomicrons). In mice deletion of Soat1 or Soat2 genes revealed a diverse role for the different ACAT enzymes in atherosclerosis. In Soat1 knockout animals, ACAT1 deficiency led to a marked alteration in cholesterol metabolism resulting in massive accumulation of UC, which caused numerous skin and brain lesions, and worsened atherosclerosis. Conversely, the deletion of the Soat2 gene has been consistently atheroprotective [4]–[6]. Previous studies also suggest a clear atherogenic potential of ACAT2-derived CE also in humans. In both women and men, the Atherosclerosis Risk in Communities (ARIC) study revealed an association between the carotid intima-media thickness and the ACAT2-derived CE in lipoproteins [7]. Also, the Uppsala Longitudinal Study of Adult Men (ULSAM) showed an association between the ACAT2-derived CE in lipoproteins and CVD mortality [8]. Finally, ACAT2-derived CE in lipoproteins were able to predict coronary artery disease in patients with symptoms of acute coronary syndrome [9]. Athero-protection was also achieved in mice using anti-sense oligonucleotide targeted to hepatic Soat2 mRNA (ASO6) and resulting in a decreased ACAT2 activity in the liver [10]. These studies did not address whether a decreased hepatic ACAT2 activity modifies HDL metabolism. In addition, inconsistent results on HDL cholesterol levels were obtained in mice in which ACAT2 activity was reduced by genetic manipulation [4], [6], [11]–[14]. We hypothesized that a low ACAT2 activity in the liver would result in a greater UC mass that in turn could be secreted into nascent HDL by ABCA1. Hence, we reduced hepatic ACAT2 activity by injection of ASO6 in male C57BL/6 and in Liver X Receptor (LXR) α/β double knockout (DOKO) mice. In DOKO mice the link between ACAT2 activity and HDL metabolism should be more evident. This mouse model is characterized by a reduced bile acid (BA) synthesis, which is paralleled by a maintained ABCA1 expression in the liver. Considering the possible fate of cholesterol in the hepatocytes (i.e. the esterification by ACAT2 to be incorporated in VLDL or stored, the conversion into BA, and the efflux into nascent HDL) a reduction of the ACAT2 activity in DOKO mice, may indeed lead to a greater UC mass available for secretion into nascent HDL. Finally, we also investigated whether the metabolic response following a decreased hepatic ACAT2 activity was influenced by the time frame in which the animals were fed a western type of diet.
Our results identify hepatic ACAT2 as an important player for HDL metabolism since the activity of this enzyme may influence the intracellular cholesterol fated to secretion either into VLDL or into HDL particles; the latter via an increased expression of the ABCA1 protein.
Material and Methods
Animals and experimental design
Ten-week-old male wild-type (WT), and LXRα and LXR β double knockout (DOKO) mice, on a pure C57BL/6 genetic background (as previously described [15]), were housed on a regular 12-h light/12-h dark cycle with free access to water and food. Until the onset of the studies, mice were fed a chow diet (Special Diet Services; SDS, NOVA-SCB, Sweden – Rat and Mouse n.1 Maintenance, 9.5 mm pellet; RM1 (P)). During the two experiments animals were fed a western-type diet, containing 10% saturated fat and 0.2% cholesterol (w/w) (Harlan Laboratories - TD.07346, 4% PO, 2% SBO, 0.2% Cholesterol Diet - 1/2" pellet). In the first experiment (4-weeks), the diet was started 4 weeks prior to and maintained during the following 4 weeks of treatment with biweekly i.p. injection (25 MPK) of either an anti-sense oligonucleotide specifically targeted to hepatic ACAT2 gene (ASO 6) [10] in half of the animals, or with a control anti-sense oligonucleotide (ASOctrl) in the other half. In the second experiment (0-weeks) WT and DOKO mice were given the western-type diet for 10 weeks and concomitantly treated with the same doses of ASO6 and ASOctrl used in the 4-weeks experiment. In all the experiments, animals were fasted 4 hours prior to euthanasia, which was performed by inhalation of carbon dioxide. Blood, tissues and organs were then collected, and stored at −80°C until the analyses. The Swedish Board of Agriculture, Ethical committee on Animal Experiments, Stockholm South approved all the animal studies (Permit number: S17-07, 2007-03-30).
Antisense Oligonucleotides (ASOs)
The 5-10-5 methoxy ethyl chimeric 20-mer oligonucleotides with fully modified phosphorothioate backbones were kindly donated by ISIS Pharmaceuticals (Carlsbad, CA, USA). ASO6 contained a sequence-targeting mouse Soat2. The ASOctrl l was not complementary to the ACAT2 sequence and did not hybridize with any specific gene target. The sequences of these ASOs were as follows:
ASO6: 5′-TTCGGAAATGTTGCACCTCC-3′;
ASOctrl: 5′-CCTTCCCTGAAGGTTCCTCC-3′.
Isolation of liver microsomes and ACAT2 activity assay
Liver samples (50 to 150 mg) were homogenized in 3 mL ice-cold buffer containing 0.1 mol/L K2HPO4, 0.25 mol/L sucrose, and 1 mmol/L EDTA, pH 7.4. A protease inhibitor cocktail (Sigma) was added to the buffer before homogenization. The homogenate was then centrifuged for 15 min at 12 000 g (4°C) to remove cell debris. The resulting supernatant was centrifuged for 60 min at 100 000 g. The microsomal pellet from this spin was re-suspended in 0.1 mol/L K2HPO4 at pH 7.4 and immediately frozen at –80°C. Total ACAT enzymatic activity was determined in hepatic microsomes as previously described [16], except that pre-incubation included a cholesterol-saturated solution of β-hydroxypropyl cyclodextrin for 30 min before addition of 14[C] oleoyl Co-A (PerkinElmer, Uppland Väsby, Sweden). In separate tubes, pyripyropene A, a specific ACAT2 inhibitor [17], was included in the preincubation at a concentration of 5 μmol/L to differentiate ACAT1 (uninhibited) and ACAT2 (total–ACAT1) activities. Pyripyropene A was a kind gift of Prof. Hiroshi Tomoda, Dept. of Microbial Chemistry, Graduate School of Pharmaceutical Sciences, Kitasato University, Japan.
Preparation of liver plasma membrane and western blot analysis
Liver samples (∼ 200 mg) were homogenized in 3 mL ice-cold buffer containing 20 mmol/LTris-HCl, 0.25 M sucrose, and 2 mmol/L MgCl2. A protease inhibitor cocktail (complete MINI Roche Diagnostics GmbH, Mannheim, Germany) was added to the buffer before homogenization. The homogenate was then centrifuged for 10 min at 2000×g (4°C) to remove fat. The resulting intermediate phase was centrifuged for 45 min at 32000 rpm using a Beckman Ultracentrifuge XL-70. The pellet was re-suspended in 100 μl of Lysis Buffer (80 mmol/L NaCl, 50 mmol/L Tris-HCl, 2 mmol/L CaCl2, 1% TritonX-100 and protease inhibitor cocktail), and immediately frozen at –80°C. After protein determination (DC™ protein assay, Bio-Rad Laboratories, Hercules, USA) membranes were pooled group-wise. Reduced pooled membranes (10, 20, 30 μg or 40, 60, 80 μg protein) were separated on a NuPage 3–8% Tris-Acetate gel and then transferred onto nitrocellulose membranes (Invitrogen, Carlsbad, USA). After blocking in 5% non-fat dry milk in PBS-T (PBS with 0.1% Tween-20), the nitrocellulose membranes were incubated overnight at 4°C with the primary antibody (Ab) specific for the protein of interest in 5% non-fat milk powder in PBS-T. ABCA1 was detected with mouse monoclonal Ab (1∶1000, ab18180, Abcam LtD, Cambridge, UK) and as secondary antibody a peroxidase-conjugated goat anti-mouse antibody was used (1∶20000; Pierce Biotechnology, Inc., Rockford). SR-BI was detected with rabbit polyclonal Ab (1∶3000, ab396, Abcam LtD, Cambridge, UK) and a peroxidase-conjugated donkey anti-rabbit antibody (1∶60000; GE Healthcare, UK). LDLr protein was detected with rabbit monoclonal Ab (1∶3000, ab52818, Abcam LtD, Cambridge, UK) and a peroxidase-conjugated donkey anti-rabbit antibody (1∶60000; GE Healthcare, UK). The specific bands were detected using SuperSignal chemiluminescence kit (Pierce Biotechnology, Inc., Rockford) and Bio-Rad Universal Hood II and quantified by Bio-Rad Quantity One software (Bio-Rad Laboratories, Hercules, USA). Signals were plotted by μg-loaded protein and the slope of the curves was calculated by method of least square. The slope of the ASOctrl group was set equal to 100%.
Serum and liver lipid analysis
Serum lipoproteins were fractionated by size from 12 μL of individual serum samples using a Superose 6 PC 3.2/30 column (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) as previously described [18]. The respective lipoprotein fraction lipid concentrations were calculated after integration of the individual chromatograms. ApoAI and apoE content was determined in the lipoprotein fractions by western blot analysis using an anti-mouse apoA-I (Rockland, Gilbertsville, PA, USA) and an anti-mouse apoE (Calbiochem, Merk, Darmstadt, Germany) as primary antibody. Lipids were extracted from ∼100 mg liver sample using 6 mL chloroform-methanol (2∶1 v/v). The organ was removed from the tubes containing the lipid extract, which was then dried down under N2 and re-dissolved in a measured volume of 2∶1 chloroform/methanol. Diluted H2SO4 was added to the extract, which was then vortexed and centrifuged to split the phases. The aqueous upper phase was aspirated and discarded, and an aliquot of the bottom phase was removed and dried down. 1% Triton X-100 in chloroform was then added, and the solvent was evaporated [19]. Lipids were quantified on Tecan GENios plate reader equipped with Magellan Software (Tecan Group Ltd, Switzerland) using the respective enzymatic kit: Cholesterol/HP, Triglyceride/GB (Roche Diagnostics, Indianapolis, USA), and Free Cholesterol C (Wako Chemicals USA Inc.). The amount of esterified cholesterol was calculated by subtracting the unesterified cholesterol from the total cholesterol, and this difference was multiplied by 1.67 to convert it to CE mass. Liver lipid levels were normalized for the hepatic protein content measured according to Lowry method in the tissues digested with NaOH (1 mol/L). To obtain a relative index of hepatic bile acid synthesis, the concentration of 7α-hydroxy-4-cholesten-3-one (C4) was assayed in the liver lipid extract by isotope dilution-mass spectrometry as previously reported, and the ratio of C4 to total cholesterol was calculated [20], [21].
Cell culture and materials
Cell culture media, trypsin-EDTA, and gentamicin were purchased from Gibco/Invitrogen (Paisley, Scotland). Fetal bovine serum (FBS), serum albumin, the ACAT inhibitor Sandoz 58-035, and 8-(4-Chlorophenylthio)adenosine 3′,5′-cyclic monophosphate sodium salt (cpt-cAMP) were from Sigma-Aldrich (Stockholm, Sweden). [1,2-3H(N)]-cholesterol was from PerkinElmer (Uppland Väsby, Sweden). Tissue culture flasks, plates, and tubes were from Thermo Fisher Scientific/Nunc (Roskilde Site, Denmark) or Falcon (Lincoln, NY, USA). Human serum, prepared from blood of healthy donors, was provided by the Dept. of Clinical Immunology and Transfusion Medicine, Karolinska University Hospital, Huddinge, Sweden (http://www.karolinska.se/Karolinska-Universitetslaboratoriet/Kliniker/Immunologi-transfusionsmedicin/). The blood was donated for general research purposes. The donors gave informed consent that the blood could be used for preparation of serum for research purposes, but were not explicitly asked about consent to publish data. All samples were anonymized, i.e. not possible to trace back to the blood donor. Ethical permission was not warranted for requiring blood components per se, as this did not pose any extra harm to the donors (regular blood donation was performed). Lipid poor apoA-I isolated was isolated and purified from human serum as previously described [22]. J774.A1 murine macrophages were purchased from American type culture cell (LGC standards AB), and maintained in RPMI with L-Glutamine plus 10% FBS and gentamicin in 5% CO2. Fu5AH rat hepatoma cells [23]–[26] were a kind gift of Prof. Franco Bernini (Dept. of Pharmacy, University of Parma, Italy). Fu5AH were cultured in High glucose DMEM plus 10% FBS and gentamicin. Ultima Gold™ was from PerkinElmer (Uppland Väsby, Sweden).
Quantification of serum cholesterol efflux capacity (CEC)
Mouse sera from the 4-weeks and 0-weeks experiment were tested as cholesterol acceptors in different cell models to evaluate the CEC. J774 murine macrophages incubated with cpt-cAMP were used as a model of total cholesterol efflux, since all the efflux pathways are active [27], [28]. J774 cultured under basal conditions were used to evaluate the aqueous diffusion [26], [27]. The ABCA1-mediated cholesterol efflux was the difference between the cholesterol efflux measured by J774 incubated with cpt-cAMP and the cholesterol efflux measured by J774 cultured in basal condition [26], [27]. SR-BI mediated cholesterol efflux was measured using Fu5AH rat hepatoma cells, a stable highly SR-BI-expressing cell line [29]. In brief, cells were plated into 24-well plates in medium containing 10% FBS. Monolayers were washed with PBS and incubated for 24 h in medium containing 1% FBS, [1,2-3H(N)]-cholesterol and ACAT inhibitor (2 μCi/mL). Cells were then incubated for 18 h with medium plus 0.2% BSA and ACAT inhibitor (2 μCi/mL), with 0.3 mmol/L cpt-cAMP when appropriate. Cells were then harvested with NaOH (1 M) and counted by liquid scintillation. These cells provided baseline (time 0) values for total [1,2-3H(N)]-cholesterol content. Cell monolayers were then incubated with 1% (v/v) mouse serum in medium for 4 h. Cell media were centrifuged to remove floating cells, and radioactivity in the supernatant was determined by liquid scintillation counting. Cholesterol efflux was calculated as: (cpm in medium at 4 h/cpm at time 0) x 100. For each experiment aimed to measure the serum CEC by ABCA1, we monitored ABCA1 up-regulation in J774 cells as increased efflux to human apoA-I (20 μg/mL) from cells treated with cpt-cAMP compared to untreated cells. Pooled human serum from 20 healthy donors was tested in every experiment as an acceptor (2% v/v) in order to monitor and correct for the inter-assay variability in cholesterol efflux.
2D gel electrophoresis
Serum HDL subclasses were separated by 2D electrophoresis, in which agarose gel electrophoresis was followed by non-denaturing polyacrylamide gradient gel electrophoresis and subsequent immunoblotting [23]. In the first dimension, serum (5 μL) was run on a 0.5% agarose gel; agarose gel strips containing the separated lipoproteins were then transferred to a 3-20% polyacrylamide gradient gel. Separation in the second dimension was performed at 30 mA for 4 h. Fractionated HDLs were then electroblotted onto a nitrocellulose membrane and detected with an anti-mouse apoA-I (Rockland, Gilbertsville, PA, USA) or an anti-mouse apoE (Calbiochem, Merck, Darmstadt, Germany) antibody and visualized by enhanced chemiluminescence (GE Healthcare Biosciences, Uppsala, Sweden). Densitometric analysis was performed with a GS-690 Imaging Densitometer and the Multi-Analyst software (Bio-Rad Laboratories, Hercules, CA, USA). Serum content of preβ-HDL was expressed as percentage of total apoA-I.
RNA extraction, cDNA synthesis, and qPCR analysis of mRNA expression levels
RNA from liver tissue was isolated using RNeasy Mini Kit (QIAGEN GmbH, Hilden, Germany) according to the manufacturer's protocol. The concentration and quality of the purified total RNA were determined spectrophotometrically at OD260 nm and by the OD260/280 ratio, respectively. Synthesis of single-stranded cDNA was carried out on 0.5 μg RNA using iScript cDNA synthesis kit (Bio-Rad Laboratories, Inc., Hercules, CA) following a standard protocol. PCR primers were designed using Primer Express Software version 2.0, a program especially provided for primer design using ABI qPCR machines. qPCR assay on the basis of SYBR Green I technology was performed with ABI 7500 fast qPCR system (Applied Biosystems, Foster City, CA). For each pair of primers, a dissociation curve analysis was conducted to validate the specificity of the PCR amplification. Primers were used at a concentration of 100 mmol/L in qPCR analyses and the sequences are listed in Table S1. We calculated relative changes employing the comparative method using Tfiib as the reference gene and controls as calibrators as indicated in the figures.
Statistical analysis
Statistics was calculated using Statistica software (Stat Soft Inc., USA). As indicated in the text or in each figure, differences between the treatment groups were determined by the Mann Whitney test. Multi-way analysis of variance (ANOVA) test followed by post-hoc comparisons of group means according to the least significant difference (LSD) method was used when comparing the effect of treatment in the different mouse genotypes. Outlier rejection was performed prior to the analysis. Mean, standard error of the mean (SEM), and p-values were used for descriptive purposes. A p-value <0.05 was considered statistically significant.
Results
In order to control for the efficacy of ASO6 treatment, we examined Soat2 expression in the two organs where ACAT2 is exclusively expressed [3], the intestine and liver. In agreement with previous studies [13], ASO6 administration led to a specific down-regulation of ACAT2 in the liver (Figure 1 A) without modifying the expression in proximal or distal intestine (Figure 1 B, C). When the feeding with the western type of diet was started 4 weeks after the ASO injection (4-weeks), ASO6 reduced hepatic Soat2 mRNA by about 80% in both WT and DOKO mice. The reduction of Soat2 was coupled with more than 80% reduction in the liver microsomal activity of ACAT2 in both genotypes (ASO6 vs ASOctrl: 0.08±0.01 vs 1.21±0.14 ng/mg/min in WT, p<0.005; and 0.15±0.03 vs 0.86±0.06 ng/mg/min, p<0.01; in DOKO respectively). Also when the diet challenge was started concomitantly with the treatment (0-weeks), a similar reduction of liver microsomal activity of ACAT2 was observed in both WT (ASO6 vs ASOctrl: 0.03±0.004 vs 0.55±0.10 ng/mg/min; p<0.005) and DOKO (ASO6 vs ASOctrl: 0.14±0.02 vs 0.68±0.06; p<0.005) mice. As seen in the 4-weeks experiment, the reduction in ACAT2 activity followed a significant reduction in hepatic Soat2 mRNA expression in both genotypes (Figure 1A).
Figure 1. Effect of hepatic ACAT2 down regulation on Soat2 expression, and on the liver lipid content.

Soat2 mRNA was quantified by real-time RT-PCR in samples from liver (A), proximal (B) and distal intestine (C). Data were standardized for Tfiib mRNA expression, and normalized to WT Ctrl in each experiment. Cholesteryl ester (CE; D), unesterified cholesterol (UC; E), and triglyceride (TG; F) mass was measured in liver lipid extracts by enzymatic assays as described in the methods section. Cholesteryl esters were calculated subtracting the mass of unesterified cholesterol to total cholesterol, and adjusted for the mass of the moiety of fatty acid in CE. Liver lipid levels were normalized for the hepatic protein content. 7α-hydroxy-4-cholesten-3-one (C4; G) mass was measured in the liver lipid extracts by LC-MS/MS, and normalized for the hepatic total cholesterol (TC) mass. Error bars represent the median. Mann Whitney test, * p<0.05, ** p< 0.01, *** p<0.001.
Liver lipid content in ASO6 treated mice
We evaluated the lipid content in the liver samples from all the experiments performed. As ASO6 treatment reduced hepatic ACAT2 activity more then 80% in both the 4-weeks and 0-weeks experiment, hepatic CE mass was diminished. As shown in Figure 1D, ASO6 treatment in the 4-weeks experiment reduced hepatic CE levels by 91% in WT mice, whereas only a 30% reduction was observed in DOKO mice receiving ASO6. A similar effect was observed in the 0-weeks experiment where ASO6 lowered hepatic CE levels up to 91% in WT and up to 50% in DOKO mice. Hepatic ACAT2 knock down by ASO6 did not result in accumulation of UC in the liver as previously observed in a different mouse genotype [13]. In the 4-weeks experiment hepatic UC was not affected by ASO treatment in either WT or DOKO mice, whereas in the 0-weeks experiment ASO6 led to a 21% reduction of hepatic UC only in WT mice (Figure 1E). ACAT2 knockdown has been reported to alter triglyceride (TG) metabolism in mice, thus lowering hepatic TG levels [30]. We quantified the hepatic TG content in both the 4-weeks and 0-weeks experiments (Figure 1F). In WT mice ASO6 lowered hepatic TG, although significantly only in the 0-weeks experiment. Liver TG mass was not affected by ASO6 in DOKO mice, which showed decreased hepatic TG levels compared to their wild type counterparts independently of the treatment (p<0.001 and p<0.01 in the 4-weeks and 0-weeks experiments respectively; Factorial ANOVA). In order to assess whether hepatic ACAT2 knock down induces BA synthesis, and to confirm that LXR DOKO mice have a reduced synthesis, we also quantified hepatic C4. As shown in Figure 1G, in both experimental conditions ASO6 led to an increase of liver C4 content in both WT and DOKO mice. As expected C4 levels in DOKO mice were significantly lower that in WT mice independently of the treatment (4-weeks and 0-weeks: p<0.001, Factorial ANOVA).
ACAT2 activity and ABCA1 expression in the liver
Since we did not observe any accumulation of UC following hepatic ACAT2 down-regulation, we hypothesized that UC could be channelled into nascent HDL through the ABCA1 transporter, whose expression was analysed at both mRNA, and protein levels in liver membranes. In the 4-weeks experiment, ASO6 treatment of WT induced a 133% increase in hepatic ABCA1 protein, whereas no effects were observed at the mRNA level (Figure 2A). In DOKO mice (Figure 2B) ASO6 led to a 171% increase of the ABCA1 protein compared to ASOctrl. Abca1 mRNA was slightly reduced by ACAT2 down-regulation in this mouse genotype. Also in the 0-weeks experiment, hepatic ACAT2 depletion by ASO6 led to about a doubling of the ABCA1 protein expression in liver membranes of both WT and DOKO mice (Figure 2 C and D. However, under these experimental conditions the Abca1 mRNA levels increased in WT mice (Figure 2C).
Figure 2. Hepatic ACAT2 down regulation increases the ABCA1 protein independently of the mRNA expression.
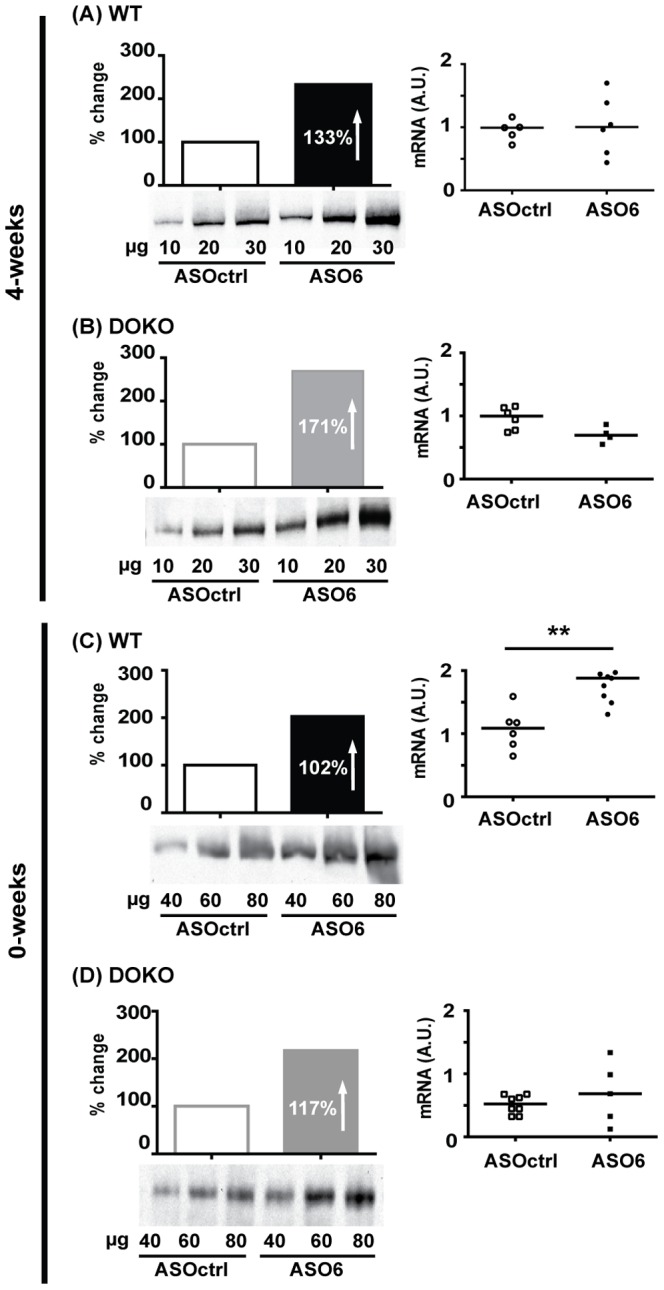
Liver membrane proteins were pooled group-wise, loaded, and separated on Tris-Acetate Gels. After transfer onto nitrocellulose membrane, samples were incubated with anti-mouse ABCA1 antibody. ABCA1 band (≈ 250 kD) was detected by chemiluminescence, and signals were plotted by μg-loaded protein. The slope of the curves was calculated by method of least square, and the slope of the ASOctrl group was set equal to 100%. Hepatic Abca1 mRNA was quantified by real-time RT-PCR. Data were standardized for Tfiib mRNA expression, and normalized to WT Ctrl in each experiment. In the mRNA data error bars represent the median. Mann Whitney test, ** p< 0.01.
Serum lipid lipoprotein profile
The analysis of the serum lipid profile revealed that in WT mice ASO6 did not affect the total levels of TC or UC (Table 1). Instead it led to a redistribution of cholesterol within the different lipoprotein fractions (Figure 3 A, B, E, F). As expected the reduction of ACAT2 expression in the liver reduced TC in the VLDL fractions (4-weeks: 0.25±0.02 vs 0.56±0.09 mmol/L, p<0.005; 0-weeks: 0.15±0.03 vs 0.66±0.09 mmol/L, p<0.005). Interestingly in the TC serum lipoprotein profile, ASO6 treatment led to the appearance of a new peak between LDL and HDL particles in both experiments (Figure 3A and E). This effect was even more pronounced when the distribution of UC in serum lipoproteins was analysed (Figure 3B, F), and it was also present when phospholipids (PL) were analysed (Figure 4). In order to further characterize this new peak, pooled serum from WT mice from the 0-weeks experiment was separated by size exclusion chromatography (SEC), and lipoprotein fractions were collected. Analysis of apoA-I and apoE content by Western blot revealed an increased content of both these apolipoproteins in the fractions (nr 30-39) corresponding to the lipoprotein peak formed by ASO6 treatment (Figure 4). In DOKO mice ASO6 treatment did not change the plasma levels of TC, but led to an increase in serum UC (Table 1). This effect was more pronounced in the 0-weeks experiment. In the 4-weeks experiment DOKO mice receiving ASO6 showed higher levels of TC in the HDL lipoprotein fractions compared to ASOctrl treated animals (Figure 3E, F). In both experiments ASO6 treatment strongly increased the HDL-UC in DOKO mice (Table 1 and Figure 3D, H). In both WT and DOKO mice from the 4-weeks experiment down-regulation of ACAT2 increased serum TG levels in the apoB-containing lipoprotein fraction (1.16±0.06 vs 0.74±0.09 mmol/L in WT, p<0.01; 0.29±0.05 vs 0.09±0.01 mmol/L in DOKO, p<0.01). In the 0-weeks experiment, this effect was only observed in DOKO mice (0.72±0.08 vs 0.52±0.05 mmol/L in WT; 0.35±0.03 vs 0.08±0.01 mmol/L in DOKO, p<0.005).
Table 1. Lipid quantification in serum and HDL lipoproteins.
| 4-weeks | 0-weeks | |||||||
| mmol/L | WT ASOctrl | WT ASO6 | DOKO ASOctrl | DOKO ASO6 | WT ASOctrl | WT ASO6 | DOKO ASOctrl | DOKO ASO6 |
| TC | 5.30±0.33 | 5.54±0.21 | 6.10±0.30 | 5.30±0.27 | 4.87±0.36 | 4.26±0.41 | 7.67±0.24 | 6.33±0.59 |
| HDL-TC | 4.23±0.26 | 4.62±0.17 | 2.15±0.05 | 2.87±0.28 | 3.78±0.34 | 3.68±0.33 | 2.65±0.12 | 2.49±0.39 |
| UC | 1.45±0.11 | 1.64±0.17 | 1.45±0.07 | 2.01±0.29 | 1.29±0.11 | 1.39±0.14 | 2.55±0.14 | 4.66±0.88* |
| HDL-UC | 1.13±0.12 | 1.17±0.14 | 0.41±0.01 | 1.04±0.10† | 0.91±0.08 | 1.04±0.10 | 0.72±0.06 | 1.94±0.27† |
| PL | 5.87±0.61 | 7.07±0.29 | 4.45±0.23 | 6.41±0.60* | 5.65±0.26 | 5.33±0.27 | 5.11±0.21 | 5.44±0.57 |
| HDL-PL | 5.03±0.61 | 6.45±0.32 | 2.46±0.17 | 4.60±0.17† | 4.82±0.22 | 4.61±0.22 | 2.50±0.37 | 3.54±0.39 |
| TG | 1.09±0.13 | 1.29±0.06 | 0.22±0.02 | 0.43±0.05* | 0.58±0.04 | 0.78±0.07 | 0.13±0.01 | 0.48±0.11‡ |
| HDL-TG | 0.35±0.09 | 0.13±0.03 | 0.12±0.03 | 0.14±0.01 | 0.06±0.01 | 0.06±0.01 | 0.06±0.01 | 0.05±0.02 |
Serum lipoproteins were separated by size exclusion chromatography, and the concentration of total (TC) and unesterified (UC) cholesterol, phospholipids (PL), and triglycerides (TG) was determined by a system for on-line detection. Data are expressed as average ± SEM (n = 6–8). Mann Whitney test, *p<0.05, † p<0.01, ‡ p<0.005.
Figure 3. Effect of hepatic ACAT2 down regulation on serum lipoprotein profile.

Serum lipoproteins were separated by size exclusion chromatography, and the total (left panels) and unesterified cholesterol (right panels) content was determined by a system for on-line detection. Black solid lines are the average chromatogram for the ASOctrl treated group, and grey solid lines are the average chromatogram for the ASO6 treated group (n = 6–8). Lipoprotein profiles of WT mice are shown in panels A, B, E, and F. Lipoprotein profiles of LXR DOKO mice are represented in panels C, D, H and G.
Figure 4. Hepatic ACAT2 down regulation led to formation of apoE-rich HDL.

Serum samples from WT mice treated with ASO6 or ASOctrl were pooled (n = 3–6) and ultracentrifugated. Lipoproteins with a density < 1.21 g/L were then separated by SEC, collecting the lipoprotein fractions every 3 min. As described in the method section, apolipoprotein (apo) AI and E content (A) were determined in the different fractions by western blot analysis. Phospholipids (PL) were quantified enzymatically.
Effect of ACAT2 disruption on serum cholesterol efflux capacity (CEC)
Since changes in serum lipid profiles were observed in both WT and DOKO mice treated with ASO6, we investigated whether the effect on HDL fractions would affect the capacity of serum to accept cholesterol from macrophages. CEC is strictly related to the composition of the lipoproteins present in the serum, and more importantly to the capacity of the different HDL subclasses to act as a lipid acceptor [26]. Thus, serum samples from all the experiments were tested using cAMP-treated J774 macrophages as cholesterol donors [27]. As shown in Figure 5, serum from both WT and DOKO mice treated with ASO6 was a more efficient cholesterol acceptor than serum from ASOctrl treated mice, but this was seen only in the 4-weeks experiment. No differences in serum CEC were observed comparing serum from ASO6 treated WT and DOKO mice with their respective control from the 0-weeks experiment.
Figure 5. Impact of hepatic ACAT2 down regulation on serum CEC.
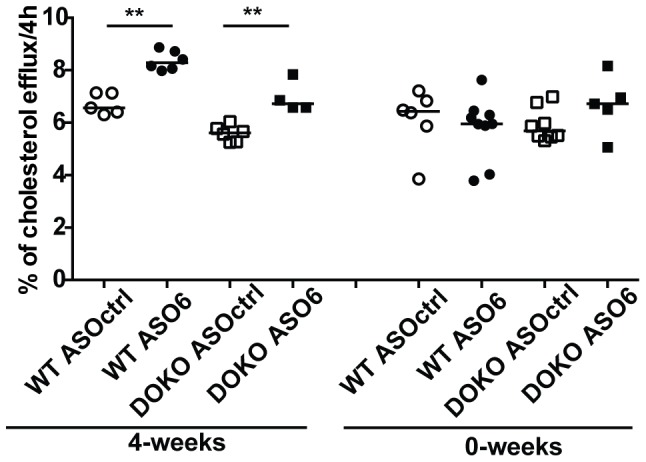
J774 murine macrophages were radiolabeled with [3H]-cholesterol for 24 h, equilibrated in a BSA-containing medium ± cpt-cAMP 0.3 mmol/L for 18 h. Efflux was measured after 4 h of cell incubation with medium containing 1% serum from WT (black bars) and LXR DOKO (grey bars) mice treated with ASOctrl (empty bars) or ASO6 (filled bars). Efflux is expressed as cpm in medium/cpm T0 × 100. Error bars represent the median. Mann Whitney test, ** p<0.01.
Effect of ASO6 on HDL subclasses
The presence of specific HDL subclasses determines HDL functionality and serum CEC. Therefore, we investigated whether ASO6 could affect the distribution of the HDL particle, providing us with a possible explanation for the effects we observed on the lipid lipoprotein profile and serum CEC. HDL were separated by 2D gel electrophoresis, and immunoblotted against apoA-I or apoE. As shown in Figure 6 (left panels), in WT mice from the 4-weeks experiment ASO6 treatment did not affect the pre-β-HDL, but led to the appearance of larger α-particles. Conversely in DOKO mice, hepatic ACAT2 down-regulation induced the appearance of smaller α-HDL, and significantly increased the pre-β-HDL content. For both mouse genotypes from the 0-weeks experiment, no differences were observed in HDL subclasses between ASO6 and ASOctrl treatment (Figure 6 right panels).
Figure 6. Hepatic ACAT2 knock-down modifies HDL subclasses.

HDL subclasses were separated by non-denaturing two-dimensional gel electrophoresis and transferred onto a nitrocellulose membrane, on which lipoproteins were detected with an anti-mouse apoA-I or apoE antibody; a representative animal in each group is shown. Preβ-HDL content was calculated as percentage of total apoA-I signal from 2D-electrophoresis developed against apoA-I. Error bars represent the median. Mann Whitney test, * p<0.05.
In both experiments ASO6 increased the amount of apoE-containing HDL, without modifying the particle size (Figure 6 bottom panels). In the 4-weeks experiment we observed an average of 26% increase in apoE-containing HDL when WT ASO6 treated mice were compared to randomly selected ASOctrl treated animals. For DOKO mice the average increase was 128%. In the 0-weeks experiment the average increase in apoE-containing HDL of ASO6 treated WT mice was of 122%, while in DOKO mice was about 100%. In both genotypes from the 4-weeks and 0-weeks experiment, apoE-containing HDL from ASO6 treated animals showed a progressive shift of migration towards preβ position.
Changes in HDL subclasses and cholesterol efflux mechanisms
In the 4-weeks experiment, hepatic ACAT2 down-regulation led to changes in apoA-I-containing particle distribution and resulted in increased capacity of serum to promote cholesterol efflux from macrophages. Since cholesterol efflux occurs by several mechanisms characterized by a preferential acceptor [31], serum samples from the 4-weeks experiment were tested for their cholesterol acceptor capacity using different cell lines selectively expressing specific lipid transporters. Reflecting the presence of larger mature HDL secondary to hepatic ACAT2 knockdown, serum from ASO6 treated WT mice showed increased capacity to promote cholesterol efflux by aqueous diffusion and by SR-BI transporter when compared to serum from ASOctrl treated WT (Figure S1 A and B). Serum from ASO6 treated DOKO mice was more efficient than serum from DOKO mice treated by ASOctrl, in promoting cholesterol efflux through the ABCA1 transporters (Figure S1 C). Indeed, in DOKO mice ASO6 treatment resulted in increased serum levels of pre-β-HDL (Figure 4), which have been demonstrated to be a preferential acceptor for the ABCA1-mediated efflux [32]. Efflux via aqueous diffusion (AD) was also increased to serum from DOKO mice when hepatic ACAT2 was reduced.
Effect of hepatic ACAT2 down-regulation on genes involved in lipid metabolism
The reduced hepatic ACAT2 activity by ASO treatment ought to affect the pool of UC that can signal to and activate the LXR system. Surprisingly, no consistent LXR activation was observed in any of the experiments, as assessed by the hepatic gene profile of LXR target genes (Table 2). Cyp7a1 and Abcg5; were not affected by the ASO6 treatment, whereas Abcg8 and Scd1 were respectively increased only in WT and DOKO from the 0-weeks experiment. A decreased hepatic ACAT2 activity appeared to result in a down-regulation of the genes under control of the SREBP2 system, but only in WT mice from the 4-weeks experiment (Table 2). Indeed ASO6 treatment resulted in reduced mRNA expression of Ldlr, Pcsk9, Hmgcr and Hmgcs (see Table 2 and Figure S2). Some of these effects were also seen in DOKO mice (Table 2). However, hepatic ACAT2 down-regulation did not reduce the expression of SREBP2 target genes when the dietary challenge was started together with ASO6 treatment (0-weeks; Table 2). As shown in Figure S2, LDLr protein expression was analysed in liver membrane pools. In the 4-weeks experiment hepatic ACAT2 down-regulation did not affect LDLr protein in WT (Figure S2 A), whereas it reduced the receptor by 98% in DOKO mice (Figure S2 C). A similar reduction was also observed in DOKO animals from the 0-weeks experiment (Figure S2 G).
Table 2. Hepatic expression of genes involved in lipid metabolism.
| 4-weeks | 0-weeks | |||||||
| Gene (a.u.) | WT ASOctrl | WT ASO6 | DOKO ASOctrl | DOKO ASO6 | WT ASOctrl | WT ASO6 | DOKO ASOctrl | DOKO ASO6 |
| Srebp2 | 0.92±0.04 | 0.63±0.13 | 1.25±0.17 | 0.93±0.05 | 0.99±0.14 | 1.17±0.41 | 0.30±0.04 | 0.30±0.01 |
| Hmgcr | 1.15±0.12 | 0.524±0.10* | 0.84±0.06 | 0.57±0.05* | 2.77±1.03 | 1.88±0.33 | 0.17±0.02 | 0.28±0.10 |
| Hmgcs | 1.25±0.23 | 0.43±0.16* | 0.90±0.09 | 0.55±0.11* | 0.40±0.13 | 0.32±0.08 | 0.10±0.01 | 0.18±0.06* |
| Pcsk9 | 1.51±0.39 | 0.18±0.07† | 0.23±0.08 | 0.08±0.02 | 1.47±0.35 | 1.67±0.48 | 0.19±0.05 | 0.30±0.14 |
| Lcat | 0.75±0.09 | 0.81±0.15 | 0.96±0.10 | 0.65±0.11 | 0.78±0.07 | 0.81±0.04 | 0.23±0.03 | 0.25±0.0 |
| Lipc | 1.32±0.21 | 1.50±0.29 | 0.81±0.09 | 0.67±0.24 | 0.71±0.21 | 0.68±0.11 | 0.23±0.0 | 0.15±0.07 |
| Cyp7a1 | 2.30±1.17 | 4.23±1.10 | 1.29±0.25 | 0.80±0.16 | 1.74±0.50 | 1.98±0.37 | 0.24±0.08 | 0.37±0.16 |
| Abcg5 | 1.37±0.22 | 1.18±0.30 | 0.24±0.03 | 0.16±0.02 | 2.78±1.03 | 1.88±0.33 | 0.17±0.02 | 0.28±0.10 |
| Abcg8 | 1.54±0.26 | 1.74±0.60 | 0.34±0.03 | 0.24±0.04 | 1.54±0.23 | 2.63±0.41* | 0.65±0.10 | 0.45±0.12 |
| Scd1 | 2.86±0.77 | 1.69±0.52 | 0.02±0.01 | 0.02±0.00 | 1.43±0.27 | 1.14±0.17 | 0.004±0.00 | 0.01±0.01* |
| Fasn | 1.50±0.33 | 0.15±0.05‡ | 0.04±0.01 | 0.03±0.01 | 1.45±0.56 | 1.47±0.31 | 0.05±0.00 | 0.05±0.02 |
| Acc1 | 1.50±0.33 | 0.42±0.10‡ | 0.38±0.02 | 0.26±0.03 | 1.43±0.27 | 1.13±0.17 | 0.004±0.00 | 0.01±0.01* |
| Srebp1c | 0.60±0.15 | 0.34±0.17 | 0.001±0.0 | 0.00±0.00 | 0.61±0.14 | 1.21±0.23 | 0.00±0.00 | 0.00±0.00 |
| Tgh1 | 1.84±0.30 | 1.64±0.17 | 0.50±0.04 | 0.72±0.0† | 1.29±0.18 | 1.47±0.36 | 0.26±0.04 | 0.37±0.15 |
| Tgh2 | 1.55±0.16 | 1.44±0.04 | 0.17±0.03 | 0.07±0.01 | 1.26±0.20 | 0.84±0.21 | 0.05±0.01 | 0.13±0.07 |
| Ctsd | 3.94±1.88 | 0.68±0.17 | 5.78±1.45 | 2.87±0.47 | 0.83±0.14 | 0.91±0.20 | 1.51±0.24 | 0.77±0.24 |
| Zdhhc8 | 1.13±0.23 | 1.28±0.24 | 3.02±0.96 | 2.40±0.53 | 0.59±0.09 | 0.74±0.12 | 0.49±0.06 | 0.47±0.10 |
| Rab8a | 0.84±0.07 | 1.06±0.24 | 2.26±0.41 | 1.59±0.39 | 0.66±0.10 | 0.94±0.20 | 0.28±0.02 | 0.43±0.14 |
mRNA was quantified by real-time RT-PCR. Data were standardized for Tfiib mRNA expression, and normalized to the expression of each gene in WT Ctrl in each single experiment. mRNA data are expressed as average ± SEM (n = 6–8). Mann Whitney test, *p<0.05, † p<0.01, ‡ p<0.005.
In spite of the minor effects seen for TG content in the liver of WT animals (see above), in the 4-weeks experiment ASO6 treatment reduced the mRNA of genes involved in fatty acid synthesis, which was likely mediated by a SREBP1-c down-regulation (i.e. Fasn and Acc1; Table 2). However, in the 0-weeks experiment, we did not observe this effect. In these experimental conditions, ASO6 increased Scd1 and Acc1 mRNA levels in the liver of DOKO mice (Table 2). Triacylglycerol hydrolase 1 and 2 mRNA expression was significantly reduced in LXR DOKO mice compared to WT independently of the treatment (4-weeks: Tgh1, p<0.01 and Tgh2, p<0.001; 0-weeks: Tgh1, p<0.001 and Tgh2, p<0.001; Factorial ANOVA). ASO6 did affect the expression of these genes in WT mice (Table 2). Only in the 4-weeks experiment and in DOKO mice did ACAT2 down-regulation lead to slightly increased Tgh1 mRNA levels (Table 2). We also evaluated the hepatic expression of the HDL receptor SR-BI, both at mRNA and protein levels. As shown in Figure S2 (right panels), hepatic ACAT2 down-regulation led to a reduction of SR-BI protein expression in the liver membranes, without affecting the mRNA. In the 4-weeks experiment ASO6 reduced SR-BI by 28% and 6% in WT and DOKO respectively (Figure S2 B and D). In the 0-weeks experiment hepatic ACAT2 down-regulation led to a reduction in SR-BI protein expression by 8% in WT and 36% in DOKO mice (Figure S2 F and H).
Discussion
Our main aim with this study was to uncover a possible link between hepatic ACAT2 activity and HDL metabolism in mice. We hypothesized that the excess of UC in the hepatocytes due to a lower activity of ACAT2 may be also shunted into nascent HDL by ABCA1. In order to better unravel this mechanism we decided to include LXR α/β double knockout mice in our experiments. In these animals UC should be more available to secretion in to nascent HDL because of lower bile acid synthesis paralleled by a maintained ABCA1 expression in the liver.
We have previously reported that the hepatic ABCA1 protein was up-regulated when the expression of hepatic ACAT2 was reduced, independently of the levels of Abca1 mRNA expression [33]; an effect also seen in apoB100only-LDLr−/− mice [13]. In mice where ACAT2 was selectively knocked out in the liver, hepatic Abca1 mRNA was increased [14]. However, the protein expression of ABCA1 was not reported in these studies. Here we clearly demonstrate that down-regulation of hepatic ACAT2 activity results in a strong stimulation of ABCA1 protein expression in liver membranes, and that this process is definitively independent from LXRs. The up-regulation of the ABCA1 protein expression results from an as yet unidentified post-transcriptional process, since it is clearly independent of Abca1 mRNA expression. Intracellular UC may play a role since it has been shown that UC loading induces ABCA1 expression in several cell models [34]. Wang et al showed that hepatocytes from mice depleted of NPC1 accumulate FC and have an up-regulation of ABCA1 protein. Cathespin D, a lysomal proteinase, has been proposed as a possible player in this upregulation [35]. Furthermore Rab8 over-expression was found to increase ABCA1 protein in human primary macrophages, without affecting the mRNA levels [36]. Finally, palmitoylation of ABCA1 by the palmitoyl transferase DHHC8 is essential for its localization at the plasma membrane and contributes to its efflux function [37]. In the present work, we quantified the mRNA expression of these genes (Table 2), and found that ACAT2 downregulation did not affect their expression. However, we cannot completely exclude their participation in the ABCA1 up-regulation, since only the mRNA expression was evaluated.
The liver is the major organ for HDL synthesis (by lipidation of apoA-I through ABCA1) as shown by the study of liver-specific ABCA1 knockout mice [38]. Whether ACAT2 disruption in mice would have an impact on HDL metabolism was unclear, since previous studies have shown contrasting results on the lipid content in HDL [4], [6], [11]–[14]. In apoE knockout [4] and LDLr knockout [6] mice an increase in HDL cholesterol was observed when ACAT2 was knocked out. However, no changes in HDL lipid composition were observed in different ACAT2−/− mouse genotypes [12], [13], or in mice where ACAT2 was selectively knocked out in the liver or intestine [14]. In the present work we showed that after ASO6 treatment decreased hepatic ACAT2, HDL lipid composition changed only in DOKO mice (increased levels of HDL-UC). Hepatic Lcat mRNA was not affected by ASO6 treatment suggesting that the HDL-UC increase is not mediated by a reduction in the levels of this enzyme.
HDL lipoproteins are not just simple lipid carriers, but are a complex and heterogeneous class of particles, differing in physical and chemical properties, protein and lipid composition, metabolism and functions [39]. Treatment of WT mice with ASO6 revealed the appearance of a new peak in the serum lipoprotein profile, which elutes between the typical peak for LDL and HDL particles. We demonstrated that these newly generated lipoproteins are large HDL particles enriched in apoA-I and apoE. Thus, reduction of hepatic ACAT2 activity does not change plasma lipid levels, but rather leads to a redistribution of lipids within the different lipoprotein particles. The newly generated particle appeared independently of the dietary conditions, as was the increase in hepatic ABCA1 expression. This effect could not be observed from the lipid profile of DOKO mice. The big peak generated by the increase in HDL-UC could have possibly masked the peak of the new generated particles for this mouse genotype. Interestingly, large apoE-rich HDL particles seem to be associated with increased hepatic ABCA1 protein expression. This phenomenon has been observed in mice treated with the LXR agonist T090137 [40], in mice overexpressing hepatic ABCA1 [41], and in transgenic mice expressing human NPC1L1in the liver [42]. Lipid poor apoE can directly interact with ABCA1 and be lapidated [43]. This process generates particles with a preβ-migration, and a diameter ranging between 9 and 15 nm [43]. By 2D-gel electrophoresis analysis, we found that ASO6 treatment increased the amount of apoE-containing HDL in both WT and LXR DOKO mice, and that the apoE-containing HDL formed show a progressive shift of migration towards preβ position and an estimated size between 12 and 15 nm. This effect was independent of the dietary challenges.
We also investigated if the changes in HDL metabolism secondary to hepatic ACAT2 knock down resulted in improved HDL functionality. Thus, we measured the capacity of serum to promote cholesterol efflux. This parameter and the different mechanisms involved in cholesterol efflux have been shown to be strongly dependent on the different HDL sub-classes present in the circulation [26], [44]. Indeed, we observed that only when the apoA-I containing particle distribution was modified by ASO6 treatment, serum cholesterol efflux capacity was increased. Importantly, we showed that changes in the efflux pathways involved reflected the changes in preβ- and α-HDL subclasses. These effects were observed only when the western type diet was started prior to the ASO6 treatment, but not when diet and treatment were started concomitantly. In our study we found that serum CEC is positively correlated with both serum and HDL PL content (Figure S3). When the diet was started concomitantly with the treatment ASO6 did not alter the serum PL content, conversely to what observed when the diet was started 4 weeks before the ASO6 treatment. This effect may partly explain the lack of increased serum CEC we observed when the diet was started concomitantly with the ASO6 treatment. It has been shown previously that cellular cholesterol efflux is strongly dependent on the phospholipid content of the extracellular acceptor [32], [45], [46].
In the hepatocytes cholesterol can be esterified by ACAT2 and packed into VLDL, or fluxed into nascent HDL or converted into bile acids. In the present study we also demonstrated that hepatic ACAT2 knock down led to an increased bile acid synthesis, as shown by the higher levels of 7α-hydroxy-4-cholesten-3-one in the liver of the ASO6 treated mice.
Another important finding in this study is the unexpected lack of LXR activation in response to the down-regulation of hepatic ACAT2 activity. No evident changes in mRNA abundance of the classical LXR target genes were observed upon ASO6 treatment. Conversely, decreased hepatic cholesterol esterification in mice led to a down-regulation of the SREBP2 transcriptional pathway since the abundance of Ldlr, Pcsk9, Hmgc reductase and synthase mRNA were decreased. However, this was only evident when the liver was preloaded with dietary cholesterol before down-regulation of ACAT2 activity, since no effects on SREBP2 target genes were observed when ASO6 treatment was started concomitantly with dietary cholesterol challenge. In this latter experimental condition treatment of WT mice with ASO6, the levels of hepatic UC were reduced, and this difference may explain some of the discrepancies observed between the 4-weeks and 0-weeks experiment.
In LXR DOKO compared to WT mice, ACAT2 down-regulation was less effective in reducing hepatic CE in both experiments. For the first time, we have shown here that Tgh1 and Tgh2 expression is extremely low in LXRα/β double knockout mice. Since carboxylesterases can also catalyse the hydrolysis of CE, the reduced expression of Tgh1 and Tgh2 observed in LXR DOKO may explain the elevated CE content in the livers of LXR DOKO mice, as well as the failure to mobilize it by hydrolysis upon reduction of cholesterol esterification by ASO6 treatment.
A limitation of our study can be represented by the fact that we only investigated liver lipid storage/synthesis, hepatic conversion of cholesterol into bile acids and HDL formation process. Two other important pathways of the hepatic lipid metabolism (i.e. VLDL production and biliary cholesterol secretion) have not been evaluated even if they are known to play a role in HDL metabolism. However the effect of ACAT2 deletion on these pathways has been previously studied. It has been shown that the total apoB accumulation rate in liver perfusate from ACAT2 KO mice was not different from that of their respective littermates [47], and ASO6 treatment led to a reduction of biliary cholesterol secretion in apoB100only-LDLr−/− mice [13].
It is known that ASOs can present non-target effect. In the present work we did not test the effect of ASO6 treatment in ACAT2 knock out (KO) mice to rule out that possibility. Nevertheless, we ran a third experiment (data not shown) feeding ACAT2 KO animals and their respective littermates for 6 weeks with the same western type of diet used in the ASOs experiments. ACAT2 KO showed a similar phenotype to that we observed by ASO6 treatment. Compared to their littermates, ACAT2 KO mice showed double the expression of ABCA1 protein in the hepatic liver membranes together with the appearance of the new HDL lipoprotein particle peaks in the lipid profile.
In conclusion, the use of anti-sense oligonucleotide targeted to hepatic ACAT2 revealed a new pathway by which the liver may contribute to HDL metabolism in mice. ACAT2 seems to be an important hepatocyte player that influences intracellular cholesterol fluxes either into VLDL lipoproteins or into HDL particles, the latter via the up-regulated ABCA1 transporter. The relevance of these findings for the human condition does not seem to be futile. A negative correlation between the hepatic ACAT2 activity and the plasma levels of HDL cholesterol and apoA-I was described in normolipidemic non-obese Chinese patients [48], and a functional variant of the SOAT2 gene was identified as an independent genetic determinant of plasma HDL cholesterol levels in the LCAS and TexGen cohorts [49]. Thus SOAT2 has been listed among those genes that influence HDL plasma level in humans [50]. Also, it is worth to remember that treatment with ASO6 generates a condition in mice similar to that in humans, where a low hepatic activity of ACAT2 [51] is present together with elevated intestinal ACAT2 activity [52]. Mice and non-human primates have high hepatic ACAT2 activity [3]. However, the lack of plasma CETP activity [53] or the high rate of hydrolysis of triglyceride rich lipoproteins positively influences the genesis of HDL respectively in these two species. In humans, low hepatic ACAT2 activity may be needed to keep up the lipidation of apoAI. Considering the role of HDL for innate immunity [54], [55], a low hepatic ACAT2 activity may have been a strong evolutionary advantage.
Supporting Information
Efflux mechanisms involved in serum cholesterol efflux capacity (CEC). Cells were radiolabeled with [3H]-cholesterol for 24 h, equilibrated in a BSA-containing medium for 18 h and exposed for 4 h to 1% serum from WT (left panels) and LXR DOKO (right panels) mice treated with ASOctrl or ASO6. SR-BI-mediated efflux (A) was assessed in Fu5AH rat hepatoma cells; aqueous diffusion-mediated efflux (B) was assessed in J774 macrophages; ABCA1-mediated efflux (C) was assessed in J774 macrophages: ABCA1 contribution was calculated as the difference between the efflux determined in J774 cells treated with cpt-cAMP 0.3 mM or grown under basal condition. Mean and SEM are shown. Mann Whitney test, *p<0.05; **p<0.01.
(TIF)
Effect of hepatic ACAT2 down regulation on LDLr and SR-B expression in the liver. Liver membrane proteins were pooled group-wise, loaded, and separated on Tris-Acetate Gels. After transfer onto nitrocellulose membrane, samples were incubated with anti-mouse LDLr or SR-BI antibody. LDLr band (≈ 140 kD), and SR-BI bands (≈ 75 kD free and ≈50 kD glycosylated form) were detected by chemiluminescence, and signals were plotted by μg-loaded protein. The slope of the curves was calculated by method of least square, and the slope of the ASOctrl group was set equal to 100%. Hepatic Ldlr and Srb1 mRNA were quantified by real-time RT-PCR. Data were standardized for Tfiib mRNA expression, and normalized to WT Ctrl in each experiment. mRNA data are expressed as average ± SEM (n = 6–8). Mann Whitney test, * p< 0.05.
(TIF)
Correlation between serum cholesterol efflux capacity, serum and HDL phospholipids.
(TIF)
Sequences of the mouse primers.
(DOCX)
Acknowledgments
We thank Prof. Franco Bernini (Department of Pharmacy, University of Parma, Italy) for kindly donating us the Fu5AH rat hepatoma cells, Prof. Hiroshi Tomoda, Department of Microbial Chemistry, Graduate School of Pharmaceutical Sciences, Kitasato University, 5-9-1 Shirokane, Minato-ku, Tokyo 108-8641, Japan for kindly donating us the pyripyropene A, Isis Pharmaceuticals, Inc. (Carlsbad, CA, USA) for providing us with the antisense oligonucleotides, and Dr Maura Heverin (Division of Clinical Chemistry, Department of Laboratory Medicine, Karolinska Institutet, Sweden) for proofreading the text.
Funding Statement
The studies described in this paper were supported by the Swedish Heart-Lung Foundation, the Swedish Research Council, the Karolinska Institutet, the Swedish Medical Association, the Stockholm County Council, and Novo Nordisk Research Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Fazio S, Linton M (2006) Failure of ACAT inhibition to retard atherosclerosis. N Engl J Med 354: 1307–1309. [DOI] [PubMed] [Google Scholar]
- 2. Pramfalk C, Eriksson M, Parini P (2012) Cholesteryl esters and ACAT. Eur J Lipid Sci Tech 114: 624–633. [Google Scholar]
- 3. Rudel LL, Lee RG, Parini P (2005) ACAT2 is a target for treatment of coronary heart disease associated with hypercholesterolemia. Arterioscler Thromb Vasc Biol 25: 1112–1118. [DOI] [PubMed] [Google Scholar]
- 4. Willner EL, Tow B, Buhman KK, Wilson M, Sanan DA, et al. (2003) Deficiency of acyl CoA:cholesterol acyltransferase 2 prevents atherosclerosis in apolipoprotein E-deficient mice. Proc Natl Acad Sci U S A 100: 1262–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bell TA 3rd, Kelley K, Wilson MD, Sawyer JK, Rudel LL (2007) Dietary fat-induced alterations in atherosclerosis are abolished by ACAT2-deficiency in ApoB100 only, LDLr-/- mice. Arterioscler Thromb Vasc Biol 27: 1396–1402. [DOI] [PubMed] [Google Scholar]
- 6. Lee RG, Kelley KL, Sawyer JK, Farese RV Jr, Parks JS, et al. (2004) Plasma cholesteryl esters provided by lecithin:cholesterol acyltransferase and acyl-coenzyme a:cholesterol acyltransferase 2 have opposite atherosclerotic potential. Circ Res 95: 998–1004. [DOI] [PubMed] [Google Scholar]
- 7. Ma J, Folsom AR, Lewis L, Eckfeldt JH (1997) Relation of plasma phospholipid and cholesterol ester fatty acid composition to carotid artery intima-media thickness: the Atherosclerosis Risk in Communities (ARIC) Study. The Am J Clin Nutr 65: 551–559. [DOI] [PubMed] [Google Scholar]
- 8. Warensjo E, Sundstrom J, Vessby B, Cederholm T, Riserus U (2008) Markers of dietary fat quality and fatty acid desaturation as predictors of total and cardiovascular mortality: a population-based prospective study. Am J Clin Nutr 88: 203–209. [DOI] [PubMed] [Google Scholar]
- 9. Miller CD, Thomas MJ, Hiestand B, Samuel MP, Wilson MD, et al. (2012) Cholesteryl esters associated with acyl-CoA:cholesterol acyltransferase predict coronary artery disease in patients with symptoms of acute coronary syndrome. Acad Emerg Med 19: 673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bell TA 3rd, Brown JM, Graham MJ, Lemonidis KM, Crooke RM, et al. (2006) Liver-specific inhibition of acyl-coenzyme a:cholesterol acyltransferase 2 with antisense oligonucleotides limits atherosclerosis development in apolipoprotein B100-only low-density lipoprotein receptor-/- mice. Arterioscler Thromb Vasc Biol 26: 1814–1820. [DOI] [PubMed] [Google Scholar]
- 11. Buhman KK, Accad M, Novak S, Choi RS, Wong JS, et al. (2000) Resistance to diet-induced hypercholesterolemia and gallstone formation in ACAT2-deficient mice. Nat Med 6: 1341–1347. [DOI] [PubMed] [Google Scholar]
- 12. Temel RE, Lee RG, Kelley KL, Davis MA, Shah R, et al. (2005) Intestinal cholesterol absorption is substantially reduced in mice deficient in both ABCA1 and ACAT2. J Lipid Res 46: 2423–2431. [DOI] [PubMed] [Google Scholar]
- 13. Brown JM, Bell TA 3rd, Alger HM, Sawyer JK, Smith TL, et al. (2008) Targeted depletion of hepatic ACAT2-driven cholesterol esterification reveals a non-biliary route for fecal neutral sterol loss. J Biol Chem 283: 10522–10534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang J, Kelley KL, Marshall SM, Davis MA, Wilson MD, et al. (2012) Tissue-specific knockouts of ACAT2 reveal that intestinal depletion is sufficient to prevent diet-induced cholesterol accumulation in the liver and blood. J Lipid Res 53: 1144–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Alberti S, Schuster G, Parini P, Feltkamp D, Diczfalusy U, et al. (2001) Hepatic cholesterol metabolism and resistance to dietary cholesterol in LXRbeta-deficient mice. J Clin Invest 107: 565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carr TP, Parks JS, Rudel LL (1992) Hepatic ACAT activity in African green monkeys is highly correlated to plasma LDL cholesteryl ester enrichment and coronary artery atherosclerosis. Arterioscler Thromb 12: 1274–1283. [DOI] [PubMed] [Google Scholar]
- 17. Lada AT, Davis M, Kent C, Chapman J, Tomoda H, et al. (2004) Identification of ACAT1- and ACAT2-specific inhibitors using a novel, cell-based fluorescence assay: individual ACAT uniqueness. J Lipid Res 45: 378–386. [DOI] [PubMed] [Google Scholar]
- 18. Parini P, Johansson L, Bröijersén A, Angelin B, Rudling M (2006) Lipoprotein profiles in plasma and interstitial fluid analyzed with an automated gel-filtration system. Eur J Clin Invest 36: 98–104. [DOI] [PubMed] [Google Scholar]
- 19. Carr TP, Andresen CJ, Rudel LL (1993) Enzymatic determination of triglyceride, free cholesterol, and total cholesterol in tissue lipid extracts. Clin Biochem 26: 39–42. [DOI] [PubMed] [Google Scholar]
- 20. Lovgren-Sandblom A, Heverin M, Larsson H, Lundstrom E, Wahren J, et al. (2007) Novel LC-MS/MS method for assay of 7alpha-hydroxy-4-cholesten-3-one in human plasma. Evidence for a significant extrahepatic metabolism. J Chromatogr B Analyt Technol Biomed Life Sci 856: 15–19. [DOI] [PubMed] [Google Scholar]
- 21. Slatis K, Gafvels M, Kannisto K, Ovchinnikova O, Paulsson-Berne G, et al. (2010) Abolished synthesis of cholic acid reduces atherosclerotic development in apolipoprotein E knockout mice. J Lipid Res 51: 3289–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Duong PT, Collins HL, Nickel M, Lund-Katz S, Rothblat GH, et al. (2006) Characterization of nascent HDL particles and microparticles formed by ABCA1-mediated efflux of cellular lipids to apoA-I. J Lipid Res 47: 832–843. [DOI] [PubMed] [Google Scholar]
- 23. Favari E, Lee M, Calabresi L, Franceschini G, Zimetti F, et al. (2004) Depletion of pre-beta-high density lipoprotein by human chymase impairs ATP-binding cassette transporter A1- but not scavenger receptor class B type I-mediated lipid efflux to high density lipoprotein. J Biol Chem 279: 9930–9936. [DOI] [PubMed] [Google Scholar]
- 24. Rothblat GH (1974) Cholesteryl ester metabolism in tissue culture cells. I. Accumulation in Fu5AH rat hepatoma cells. Lipids 9: 526–535. [DOI] [PubMed] [Google Scholar]
- 25. Rothblat GH, Arbogast L, Kritchevsky D, Naftulin M (1976) Cholesteryl ester metabolism in tissue culture cells: II. Source of accumulated esterified cholesterol in Fu5AH rat hepatoma cells. Lipids 11: 97–108. [DOI] [PubMed] [Google Scholar]
- 26. Zanotti I, Poti F, Pedrelli M, Favari E, Moleri E, et al. (2008) The LXR agonist T0901317 promotes the reverse cholesterol transport from macrophages by increasing plasma efflux potential. J Lipid Res 49: 954–960. [DOI] [PubMed] [Google Scholar]
- 27. de la Llera-Moya M, Drazul-Schrader D, Asztalos BF, Cuchel M, Rader DJ, et al. (2010) The ability to promote efflux via ABCA1 determines the capacity of serum specimens with similar high-density lipoprotein cholesterol to remove cholesterol from macrophages. Arterioscler Thromb Vasc Biol 30: 796–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, et al. (2011) Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med 364: 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. de la Llera-Moya M, Rothblat GH, Connelly MA, Kellner-Weibel G, Sakr SW, et al. (1999) Scavenger receptor BI (SR-BI) mediates free cholesterol flux independently of HDL tethering to the cell surface. J Lipid Res 40: 575–580. [PubMed] [Google Scholar]
- 30. Alger HM, Brown JM, Sawyer JK, Kelley KL, Shah R, et al. (2010) Inhibition of acyl-coenzyme A:cholesterol acyltransferase 2 (ACAT2) prevents dietary cholesterol-associated steatosis by enhancing hepatic triglyceride mobilization. J Biol Chem 285: 14267–14274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yancey PG, Bortnick AE, Kellner-Weibel G, de la Llera-Moya M, Phillips MC, et al. (2003) Importance of different pathways of cellular cholesterol efflux. Arterioscler Thromb Vasc Biol 23: 712–719. [DOI] [PubMed] [Google Scholar]
- 32. Favari E, Calabresi L, Adorni MP, Jessup W, Simonelli S, et al. (2009) Small discoidal pre-beta1 HDL particles are efficient acceptors of cell cholesterol via ABCA1 and ABCG1. Biochemistry 48: 11067–11074. [DOI] [PubMed] [Google Scholar]
- 33. Pedrelli M, Davoodpour P, Degirolamo C, Gomaraschi M, Larsson L, et al. (2010) HDL Metabolism and Hepatic Acyl Coenzyme A: Cholesterol Acyltransferase (ACAT) 2 Inhibition in Mice. Arterioscler Thromb Vasc Biol 30: E183–E321. [Google Scholar]
- 34. Oram JF (2003) HDL apolipoproteins and ABCA1: partners in the removal of excess cellular cholesterol. Arterioscler Thromb Vasc Biol 23: 720–727. [DOI] [PubMed] [Google Scholar]
- 35. Wang MD, Franklin V, Sundaram M, Kiss RS, Ho K, et al. (2007) Differential regulation of ATP binding cassette protein A1 expression and ApoA-I lipidation by Niemann-Pick type C1 in murine hepatocytes and macrophages. J Biol Chem 282: 22525–22533. [DOI] [PubMed] [Google Scholar]
- 36. Linder MD, Mayranpaa MI, Peranen J, Pietila TE, Pietiainen VM, et al. (2009) Rab8 regulates ABCA1 cell surface expression and facilitates cholesterol efflux in primary human macrophages. Arterioscler Thromb Vasc Biol 29: 883–888. [DOI] [PubMed] [Google Scholar]
- 37. Singaraja RR, Kang MH, Vaid K, Sanders SS, Vilas GL, et al. (2009) Palmitoylation of ATP-binding cassette transporter A1 is essential for its trafficking and function. Circ Res 105: 138–147. [DOI] [PubMed] [Google Scholar]
- 38. Timmins JM, Lee JY, Boudyguina E, Kluckman KD, Brunham LR, et al. (2005) Targeted inactivation of hepatic Abca1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of apoA-I. J Clin Invest 115: 1333–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Camont L, Chapman MJ, Kontush A (2011) Biological activities of HDL subpopulations and their relevance to cardiovascular disease. Trends Mol Med 17: 594–603. [DOI] [PubMed] [Google Scholar]
- 40. Tang W, Ma Y, Jia L, Ioannou YA, Davies JP, et al. (2008) Niemann-Pick C1-like 1 is required for an LXR agonist to raise plasma HDL cholesterol in mice. Arterioscler Thromb Vasc Biol 28: 448–454. [DOI] [PubMed] [Google Scholar]
- 41. Vaisman BL, Lambert G, Amar M, Joyce C, Ito T, et al. (2001) ABCA1 overexpression leads to hyperalphalipoproteinemia and increased biliary cholesterol excretion in transgenic mice. J Clin Invest 108: 303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Temel RE, Tang W, Ma Y, Rudel LL, Willingham MC, et al. (2007) Hepatic Niemann-Pick C1-like 1 regulates biliary cholesterol concentration and is a target of ezetimibe. J Clin Invest 117: 1968–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Krimbou L, Denis M, Haidar B, Carrier M, Marcil M, et al. (2004) Molecular interactions between apoE and ABCA1: impact on apoE lipidation. J Lipid Res 45: 839–848. [DOI] [PubMed] [Google Scholar]
- 44. Zanotti I, Pedrelli M, Poti F, Stomeo G, Gomaraschi M, et al. (2011) Macrophage, but not systemic, apolipoprotein E is necessary for macrophage reverse cholesterol transport in vivo. Arterioscler Thromb Vasc Biol 31: 74–80. [DOI] [PubMed] [Google Scholar]
- 45. Yancey PG, de la Llera-Moya M, Swarnakar S, Monzo P, Klein SM, et al. (2000) High density lipoprotein phospholipid composition is a major determinant of the bi-directional flux and net movement of cellular free cholesterol mediated by scavenger receptor BI. J Biol Chem 275: 36596–36604. [DOI] [PubMed] [Google Scholar]
- 46. Camont L, Lhomme M, Rached F, Le Goff W, Negre-Salvayre A, et al. (2013) Small, dense high-density lipoprotein-3 particles are enriched in negatively charged phospholipids: relevance to cellular cholesterol efflux, antioxidative, antithrombotic, anti-inflammatory, and antiapoptotic functionalities. Arterioscler Thromb Vasc Biol 33: 2715–2723. [DOI] [PubMed] [Google Scholar]
- 47. Lee RG, Shah R, Sawyer JK, Hamilton RL, Parks JS, et al. (2005) ACAT2 contributes cholesteryl esters to newly secreted VLDL, whereas LCAT adds cholesteryl ester to LDL in mice. J Lipid Res 46: 1205–1212. [DOI] [PubMed] [Google Scholar]
- 48. Parini P, Jiang ZY, Einarsson C, Eggertsen G, Zhang SD, et al. (2009) ACAT2 and human hepatic cholesterol metabolism: identification of important gender-related differences in normolipidemic, non-obese Chinese patients. Atherosclerosis 207: 266–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen SN, Cilingiroglu M, Todd J, Lombardi R, Willerson JT, et al. (2009) Candidate genetic analysis of plasma high-density lipoprotein-cholesterol and severity of coronary atherosclerosis. BMC Med Genet 10: 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Qiu S, Luo S, Evgrafov O, Li R, Schroth GP, et al. (2012) Single-neuron RNA-Seq: technical feasibility and reproducibility. Front Genet 3: 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Parini P, Davis M, Lada AT, Erickson SK, Wright TL, et al. (2004) ACAT2 is localized to hepatocytes and is the major cholesterol-esterifying enzyme in human liver. Circulation 110: 2017–2023. [DOI] [PubMed] [Google Scholar]
- 52. Jiang ZY, Jiang CY, Wang L, Wang JC, Zhang SD, et al. (2009) Increased NPC1L1 and ACAT2 expression in the jejunal mucosa from Chinese gallstone patients. Biochem Biophys Res Commun 379: 49–54. [DOI] [PubMed] [Google Scholar]
- 53. Jiao S, Cole TG, Kitchens RT, Pfleger B, Schonfeld G (1990) Genetic heterogeneity of lipoproteins in inbred strains of mice: analysis by gel-permeation chromatography. Metabolism 39: 155–160. [DOI] [PubMed] [Google Scholar]
- 54. Levine DM, Parker TS, Donnelly TM, Walsh A, Rubin AL (1993) In vivo protection against endotoxin by plasma high density lipoprotein. Proceedings of the National Academy of Sciences of the United States of America 90: 12040–12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Feingold KR, Grunfeld C (2011) The role of HDL in innate immunity. J Lipid Res 52: 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Efflux mechanisms involved in serum cholesterol efflux capacity (CEC). Cells were radiolabeled with [3H]-cholesterol for 24 h, equilibrated in a BSA-containing medium for 18 h and exposed for 4 h to 1% serum from WT (left panels) and LXR DOKO (right panels) mice treated with ASOctrl or ASO6. SR-BI-mediated efflux (A) was assessed in Fu5AH rat hepatoma cells; aqueous diffusion-mediated efflux (B) was assessed in J774 macrophages; ABCA1-mediated efflux (C) was assessed in J774 macrophages: ABCA1 contribution was calculated as the difference between the efflux determined in J774 cells treated with cpt-cAMP 0.3 mM or grown under basal condition. Mean and SEM are shown. Mann Whitney test, *p<0.05; **p<0.01.
(TIF)
Effect of hepatic ACAT2 down regulation on LDLr and SR-B expression in the liver. Liver membrane proteins were pooled group-wise, loaded, and separated on Tris-Acetate Gels. After transfer onto nitrocellulose membrane, samples were incubated with anti-mouse LDLr or SR-BI antibody. LDLr band (≈ 140 kD), and SR-BI bands (≈ 75 kD free and ≈50 kD glycosylated form) were detected by chemiluminescence, and signals were plotted by μg-loaded protein. The slope of the curves was calculated by method of least square, and the slope of the ASOctrl group was set equal to 100%. Hepatic Ldlr and Srb1 mRNA were quantified by real-time RT-PCR. Data were standardized for Tfiib mRNA expression, and normalized to WT Ctrl in each experiment. mRNA data are expressed as average ± SEM (n = 6–8). Mann Whitney test, * p< 0.05.
(TIF)
Correlation between serum cholesterol efflux capacity, serum and HDL phospholipids.
(TIF)
Sequences of the mouse primers.
(DOCX)