

Abstract
Most evidence indicates that, as for family C G protein–coupled receptors (GPCRs), family A GPCRs form homo- and heteromers. Homodimers seem to be a predominant species, with potential dynamic formation of higher-order oligomers, particularly tetramers. Although monomeric GPCRs can activate G proteins, the pentameric structure constituted by one GPCR homodimer and one heterotrimeric G protein may provide a main functional unit, and oligomeric entities can be viewed as multiples of dimers. It still needs to be resolved if GPCR heteromers are preferentially heterodimers or if they are mostly constituted by heteromers of homodimers. Allosteric mechanisms determine a multiplicity of possible unique pharmacological properties of GPCR homomers and heteromers. Some general mechanisms seem to apply, particularly at the level of ligand-binding properties. In the frame of the dimer-cooperativity model, the two-state dimer model provides the most practical method to analyze ligand–GPCR interactions when considering receptor homomers. In addition to ligand-binding properties, unique properties for each GPCR oligomer emerge in relation to different intrinsic efficacy of ligands for different signaling pathways (functional selectivity). This gives a rationale for the use of GPCR oligomers, and particularly heteromers, as novel targets for drug development. Herein, we review the functional and pharmacological properties of GPCR oligomers and provide some guidelines for the application of discrete direct screening and high-throughput screening approaches to the discovery of receptor-heteromer selective compounds.
I. Morphologic Aspects of G Protein–Coupled Receptor Oligomerization
A. The Search for the Predominant Oligomeric G Protein–Coupled Receptor Species
Although G protein–coupled receptors (GPCRs) were initially thought to be, and function exclusively as, monomeric entities, evidence accumulated over the past two decades indicates that they can form homomers and heteromers in intact cells (Bouvier, 2001; Milligan and Bouvier, 2005; Pin et al., 2007; Ferré et al., 2009). It is now well accepted that family C GPCRs (e.g., metabotropic glutamate, calcium-sensing receptors, GABAB, and sweet and umami taste receptors) form constitutive homo- or heteromers (Kniazeff et al., 2011). Such observations raised a long debated question about whether family A (rhodopsin-like) GPCR dimers were also constitutive and required for G protein activation. A clear demonstration that this is not the case came from studies in which monomeric entities were trapped into nanodiscs. In such experiments, it was demonstrated that β2-adrenergic, rhodopsin, and μ-opioid receptors function effectively as monomers (Bayburt et al., 2007; Whorton et al., 2007, 2008; Kuszak et al., 2009). Also, monomeric rhodopsin in solution activated its G protein transducin at the diffusion limit (Ernst et al., 2007). Further support for the functionality of monomeric GPCR units came when isolated monomeric seven transmembrane (7TM) domains of family C GPCRs were shown to be capable of fully activating G protein when directly activated by synthetic small molecules (El Moustaine et al., 2012). Finally, a 1:1 stoichiometry has also been found to be sufficient for rhodopsin–β-arrestin binding (Tsukamoto et al., 2010; Bayburt et al., 2011). But those observations did not exclude that class A GPCR oligomers can spontaneously form in living cells, as it is well demonstrated with family C GPCRs, and raised the question of their functional significance. In fact, the three GPCRs shown to be functional as strict monomers were shown also to exist as dimers or higher-order oligomers in living cells (see below). One of the research groups that demonstrated the functionality of GPCR monomers in nanodiscs found evidence for the existence of stable β2 receptor oligomers, mostly tetramers, after reconstitution into phospholipid vesicles (Fung et al., 2009). Also, rhodopsin–β-arrestin binding stoichiometry in isolated rod outer segment membranes was found to depend on the percentage of activated receptors, increasing from 1:1 to 2:1 (Sommer et al., 2012).
An explosion of data supporting the existence of homo- and heteromers of GPCRs in intact cells (stored in the GPCR Oligomerization Knowledge Base; http://www.gpcr-okb.org; Khelashvili et al., 2010) came with the widespread use of biophysical techniques, such as resonance energy transfer [bioluminescence and fluorescence resonance energy transfer (BRET and FRET, respectively)], fluorescence complementation, or combination of these techniques (Milligan and Bouvier, 2005; Gandía et al., 2008; Pin et al., 2007, 2009; Bacart et al., 2008; Carriba et al., 2008; Guo et al., 2008; Cabello et al., 2009; Ferré et al., 2009; Urizar et al., 2011). These techniques, however, have largely fallen short in answering questions about the size of the oligomer complexes and their possible dynamic nature. Initial evidence for GPCR oligomerization in a physiologically relevant system came from atomic force microscopy experiments in native disk membranes from mice, which showed rhodopsin to be arranged in paracrystalline arrays of dimers (Fotiadis et al., 2003). These studies raised severe criticism (Chabre and le Maire, 2005), and more recent studies using single-molecule techniques have begun to address the details of the spatial and temporal organization of GPCR complexes in living cells by directly observing the state and behavior of individual proteins in the cell. Single-molecule total internal reflectance fluorescence microscopy (TIR-FM) was first used to track the position of individual molecules of muscarinic acetylcholine M1 and N-formyl peptide receptors using fluorescently labeled ligands (Hern et al., 2010; Kasai et al., 2011). Those ligands had such slow off-rates that they may be considered essentially irreversibly bound. Both studies suggested a transient (second-scale) formation and dissociation of dimers, with 30–40% proportion of dimers at any given time. These conclusions require that the fluorescent ligands bind with similar affinity to both protomers in the dimer (see below for discussion of ligand cooperativity within receptor dimers/oligomers; see Section II.B), and it is possible that the fluorescent ligand might stabilize the receptor in a nonrepresentative conformational state. Despite these concerns, similar studies have reached equivalent conclusions on the state of the muscarinic acetylcholine M2 receptor in cardiac muscle (Nenasheva et al., 2013). More recently, TIR-FM was used together with SNAP-tag technology to directly label cell-surface GPCRs with organic fluorophores to dynamically monitor individual β1- and β2-adrenoceptors as well as GABAB receptors on the surface of living, transiently transfected cells (Calebiro et al., 2013). This study showed that all three receptors form dimers and higher-order oligomers (also with estimations of dynamic second-scale receptor–receptor interactions) in a proportion dependent on the subtype of receptor and on the receptor density. At low densities, monomeric species were predominant for the β1-adrenoceptor, whereas β2-adrenoceptors displayed a higher proportion of dimers. A step-wise appearance of first dimers and successively trimers, tetramers, and upwards to higher-order complexes appeared with progressive increase in receptor density. At densities comparable to receptor expression in native tissue, dimers and higher-order oligomers were the predominant species for both receptors, and agonists did not modify the oligomerization status (Calebiro et al., 2013). As expected (Maurel et al., 2008; Comps-Agrar et al., 2011), GABAB receptors showed mostly dimeric and tetrameric species at lower densities, but higher-order species also appeared with increased expression levels (Calebiro et al., 2013).
Fluorescence correlation spectroscopy (FCS) is another indirect but useful technique to determine the oligomer status of protein clusters. It measures the molecular brightness of a fluorescence-tagged protein, which provides an estimate of the number of fluorescent molecules by recording fluctuations in fluorescence intensity arising from individual fluorescent molecules (Chen et al., 2003). FCS has been used to study μ-opioid receptor homo- and heteromerization with δ-opioid receptors, and number and brightness measurements suggested that μ-opioid receptors exist primarily as dimers that oligomerize with δ-opioid receptors into tetramers (Golebiewska et al., 2011). The recent FCS with a particle counting histogram approach by Herrick-Davis et al. (2013) also provides support for homodimers being the predominant, and perhaps only, species for several GPCRs, including α1B-adrenoceptor, β2-adrenoceptor, serotonin 5-HT2A and 5-HT2c, muscarinic acetylcholine M1 and M2, and dopamine D1 receptors. As with the results obtained with TIR-FM, the homodimer configuration was unaltered by agonist treatment (Calebiro et al., 2013; Herrick-Davis et al., 2013). Unlike the fast dynamic behavior of GPCR monomers/dimers shown in TIR-FM experiments (Hern et al., 2010; Kasai et al., 2011; Calebiro et al., 2013), however, the dimers were stable over a 10-fold range of receptor expression levels (Herrick-Davis et al., 2013). This is also in line with other findings that suggest much more stable interactions, such as higher-order dopamine D2 receptor oligomers, over a high range of receptor expression (Guo et al., 2008) and stable β2-adrenoceptor tetramers in phospholipid vesicles (Fung et al., 2009). Stability, at least over a substantial time period, of GPCR dimers/oligomers is also inferred from experiments that indicate these complexes are generated at an early stage of biosynthesis. In fact, it has been suggested that early-stage dimerization may be required for effective folding and maturation of the receptor (Salahpour et al., 2004; Bulenger et al., 2005; Milligan, 2010, 2013). The same reasoning has been used based on evidence for cointernalization, with the capacity of a selective ligand of one of the receptors in a GPCR heteromer to cointernalize the two different receptors constituting the heteromer (Hillion et al., 2002; Milligan, 2010, 2013; Ward et al., 2011; Tadagaki et al., 2012). Negative results should also be acknowledged, such as the recent study by Gavalas et al. (2013), which measured the recruitment of subsets of histidine-tagged GPCR protomers into artificial microdomains (containing immobilized metal affinity chromatography or streptavidin beads) on the surface of living cells and determines the simultaneous corecruitment of untagged protomers. The study only showed evidence for corecruitment of metabotropic glutamate receptor protomers, but not β2-adrenergic or μ-opioid protomers, into such artificial microdomains. Apart from differences between different receptors or when considering receptor homomers or heteromers, it might be that a number of the approaches used to study oligomerization are unable to resolve fluctuations in receptor interactions occurring on a second or subsecond scale. On the other hand, mammalian transfected cells might be lacking elements that increase the stability of the interfaces that determine the receptor–receptor interactions, which might also be different depending on the cellular compartment studied.
If different interfaces are involved in receptor oligomerization, we could expect dimers to be predominant species, determined by the most stable interaction, followed by tetramers (dimers of dimers), determined by a weaker interaction. Recently calculated estimates of the relative stability of different, putative dimeric interfaces of different GPCR subtypes using extended biased molecular dynamics simulations in explicit lipid-water environments argue in favor of a variable strength of association depending on the specific residue composition or shape of the interface, despite an overall transiency in receptor–receptor interactions (Johnston et al., 2012). Notably, simulations of β1- and β2-adrenoceptors suggested a model of oligomerization in which more stable homodimers involving TM1 diffuse through the membrane and transiently interact with other protomers/dimers via other TM helices (e.g., TM4). In agreement with these predictions are the results obtained in a recent study with the muscarinic acetylcholine M3 receptor, using quantitative FRET spectrometry techniques with controlled expression of the energy donor-tagged species (Patowary et al., 2013). Mathematical analysis of the FRET efficiencies obtained from spectral unmixing was compatible with the M3 receptor existing as stable dimeric complexes, a large fraction of which interacted dynamically to form tetramers that were specifically within a rhombic organization rather than a square or linear configuration (Patowary et al., 2013).
B. The Search for Oligomer Interfaces
A very intriguing set of inferences in the field of GPCR oligomerization has been based on recently obtained high-resolution crystallographic structures, including those of the chemokine CXCR4, the μ-opioid and κ-opioid receptors, the β1-adrenoceptor, and the smoothened receptor (Wu et al., 2010, 2012; Manglik et al., 2012; Huang et al., 2013; Wang et al., 2013). Each of these crystallized as parallel dimers and/or tetramers. Although some of the observed interfaces may reflect artifacts of crystal packing and the conditions employed for crystallization, a tentative first suggestion from these studies is the existence of different dimer interfaces for different GPCR homodimers. Nevertheless, some TM domains have been observed more often than others. TM5 and TM6 residues constituted the main interfaces for chemokine CXCR4 and μ-opioid receptor crystallized dimers, although there were marked differences between them, with only a few contacts between specific residues for chemokine CXCR4 and extensive contacts throughout the length of these TM helices in μ-opioid receptor dimers (Wu et al., 2010; Manglik et al., 2012). Involvement of TM6 was also suggested early on for β2-adrenoceptor dimers (Hebert et al., 1996) and for the leukotriene receptor BLT1 (Baneres and Parello, 2003) by the use of interfering synthetic peptides with the same sequence as TM6. Also, by using cysteine cross-linking techniques, TM5 was suggested to be involved in homodimerization of dopamine D2, muscarinic M3, and serotonin 5-HT2C receptors (Guo et al., 2005; Mancia et al., 2008; Hu et al., 2012).
Apart from the TM5-TM6 interface, crystallized chemokine CXCR4 dimers also showed contacts at the intracellular ends of TM3 and TM4 (Wu et al., 2010), and μ-opioid dimers also showed a second, less prominent symmetric interface, involving TM1, TM2, and, also, helix 8 (H8; the helix adjacent to TM7 running along the internal membrane surface) (Manglik et al., 2012). A TM1–TM2–H8 interface (with slightly different contact residues) was also found in crystals of κ-opioid receptor dimers (Wu et al., 2012), rhodopsin (Ruprecht et al., 2004; Salom et al., 2006), opsin (ligand-free rhodopsin; Park et al., 2008), and β1-adrenoceptor (Huang et al., 2013). An additional interface involving TM4 and TM5 was also obtained for the β1-adrenoceptor and smoothened receptor (Wang et al., 2013). Notably, the two crystallographic interfaces of the β1-adrenoceptor (TM1–TM2–H8 and TM4–TM5) were suggested to be physiologically relevant with cysteine-cross-linking experiments (Huang et al., 2013). A model of the potential rhodopsin dimer was initially built by docking the rhodopsin crystal structure into the unit cell constraints determined by atomic force microscopy. The model included a primary interface between protomers involving TM4 and TM5, which implied asymmetric binding of one heterotrimeric G protein to each protomer (Liang et al., 2003; Fotiadis et al., 2004). The model also suggested a secondary interface holding rows of dimers together, which was later shown to involve TM1, TM2, and H8 (Salom et al., 2006). The possible involvement of a TM1–TM2–H8 interface in rhodopsin quaternary organization was recently supported by cross-linking experiments of endogenous cysteines of rhodopsin in disk membranes (Knepp et al., 2012).
It is important to remember that crystal structures are not only the result of specific crystallization conditions, but they correspond to static, ligand-specific conformational states of receptors stripped of their natural lipid environment. For GPCRs, the majority of crystal structures that are currently available refer to antagonist-bound (inactive) structures. The inferred dimeric interfaces may, therefore, depend on those specific conformational states. As suggested by Huang et al. (2013), the comparison of the differences in the interfaces observed from the crystallized structures of the antagonist-bound μ-opioid and chemokine CXCR4 receptors and the ligand-free β1-adrenoceptor might suggest that the TM5 interface can partner in the interaction with TM4 or TM6, depending on the conformation of the receptor. As mentioned by Manglik et al. (2012), the TM5-TM6 interface inferred by the crystal structure of μ-opioid dimers could preclude either protomer from properly coupling to G protein, because the agonist-induced receptor–G protein interaction depends on rearrangements of TM5 and TM6 within the seven-helical domain bundle. Therefore, it is important to assess if, by stabilizing different receptor conformations, different ligands also promote different dimeric interfaces. By cross-linking substituted cysteines, Guo et al. (2005) suggested TM4 to be a main interface in the dopamine D2 receptor oligomer. Crosslinking of a different set of cysteines in TM4 was slowed by inverse agonists and accelerated by agonists. In fact, cross-linking of the latter set of cysteines locked the receptor in an active state, strongly suggesting that a conformational change at the TM4 dimer interface is part of dopamine D2 receptor activation (Guo et al., 2005). Mancia et al. (2008), also using the cysteine cross-linking approach, found two interfaces in serotonin 5-HT2C receptor dimers involving TM4–TM5 and TM1, with only TM4–TM5 being selectively sensitive to receptor activation. In summary, although a pattern of similar interfaces of GPCR homomers seems to be emerging, different interfaces can be found in different oligomers and even in different conformations of the same oligomer. Although agonists did not modify the dynamics of β2-adrenoceptor oligomerization in TIR-FM experiments (Calebiro et al., 2013) or the stability of β2-adrenoceptor oligomers in FCS–photon-counting histogram experiments (Herrick-Davis et al., 2013) or in FRET experiments in receptors into reconstituted phospholipid vesicles, an inverse agonist did modify the stability of β2-adrenoceptor oligomers in this latter preparation (Fung et al., 2009). Thus, selected ligands (which can stabilize specific conformations of the receptor) may still modify GPCR oligomeric interfaces and, therefore, the dynamics of receptor oligomerization.
C. Determinants of G Protein–Coupled Receptor Heteromerization
When attempting to understand the predominant oligomeric species within receptor heteromers, an additional issue is whether the complexes exist as heterodimers or as hetero-oligomers generated from individual homodimers. The lack of detection of trimers in situations in which interconversion between dimers and tetramers of the muscarinic M3 receptor was observed (Patowary et al., 2013) is consistent with a dimer + dimer model and with the organization of class C GABAB dimers and tetramers (Comps-Agrar et al., 2012). Some indirect biochemical data also support this model for class A receptors. These include cooperative binding of certain ligands to adenosine A2A and A1 receptor heteromers (see below and Orru et al., 2011a). Furthermore, oligomerization of more than two different GPCRs has been suggested from studies with sequential BRET-FRET, BRET plus bimolecular fluorescence complementation (Carriba et al., 2008; Cabello et al., 2009; Navarro et al., 2010). Tentatively, these apparent heterotrimers could represent heteromultimers of homomers, as suggested for adenosine A2A–dopamine D2–cannabinoid CB1 receptor heteromers (Navarro et al., 2010).
Apart from interactions between TM domains, several studies have provided evidence for disulfide bridges between extracellular domains of class C GPCR homomers (Kniazeff et al., 2011) and for a key role of electrostatic interactions between intracellular receptor domains in receptor heteromerization (Woods and Ferré, 2005). These electrostatic interactions depend on the very asymmetric and disordered structure of intracellular domains and have been suggested to be involved in several receptor heteromers (Ciruela et al., 2004; Woods and Ferré, 2005; Navarro et al., 2010; O’Dowd 2012, 2013). Several general features of the regions involved in these interactions have been outlined: one region, in one of the receptors, contains a series of adjacent arginine residues, and the other region, in the other receptor, contains acidic residues, with several adjacent residues or at least one phosphorylated serine. Once established, the polyarginine–phosphate electrostatic interaction possesses a strong stability. Thus, these bonds can withstand fragmentation by mass spectrometric collision-induced dissociation at energies similar to those that fragment covalent bonds, and they demonstrate an extremely low dissociation constant by surface plasmon resonance (Woods and Ferré, 2005). If electrostatic interactions between intracellular domains are more predominant in receptor heteromers (not reported yet in receptor homomers) and are stronger than those provided by TM interfaces, we could speculate that some receptor heteromers could be more stable than receptor homomers. An example of a functionally relevant electrostatic interaction is the one involved in adenosine A2A–dopamine D2 receptor heteromerization, with the arginine-rich domain localized in the N-terminal portion of the long third intracellular loop of the dopamine D2 receptor and the acidic domain in the distal part of the long C terminus of the adenosine A2A receptor (Ciruela et al., 2004; Navarro et al., 2010). Mutation- or peptide-mediated disruption of the A2A–D2 receptor electrostatic interaction produces a profound destabilization of the quaternary structure of the heteromer (Navarro et al., 2010) with disappearance of significantly relevant A2A–D2 receptor interactions in brain tissue, such as the adenosine A2A receptor–mediated inhibition of dopamine D2 receptor–induced depression of striatal neuronal firing (Azdad et al., 2009).
In summary, most evidence indicates that, as for family C GPCRs, which can be found as strict homodimers, heterodimers (Doumazane et al., 2011), as well as stable heteromers (Comps-Agrar et al., 2012), family A GPCRs form homo- and heteromers in heterologous systems. Homodimers seem to be a predominant species with potential dynamic formation of higher-order oligomers, particularly tetramers. It still needs to be resolved if class A GPCR heteromers are preferentially heterodimers or if they are mostly constituted by heteromers of homodimers. In the next sections, we will review the evidence that GPCR homo- and heteromers constitute functional and pharmacologic units.
II. Functional Aspects of G Protein–Coupled Receptor Oligomerization
A. Principles of Allosterism and Properties of Allosteric Modulators
Allosterism can be defined as the process by which the interaction of a chemical or protein at one location on a protein or macromolecular complex (the allosteric site) influences the binding or function of the same or another chemical or protein at a topographically distinct site (Christopoulos and Kenakin, 2002). This definition provides a framework to understand the biochemical properties of GPCR homomers and heteromers. As suggested by Kenakin and Miller (2010), it is useful to define allosterism in terms of three interacting species: the “modulator,” a ligand or protein that binds to the “conduit” (usually a protein; the GPCR protomer or oligomer in this review), which transmits the thermodynamic allosteric energy to the “guest,” the target of the allosteric modulation. With GPCRs, we can then consider three different kinds of allosterism depending on the location of the target of the allosteric modulation. If the target of the allosteric modulation is another ligand cobinding with the allosteric modulator, this is referred to as classic allostery. If the target of the allosteric modulation resides in the cytosol, it may be called cytosolic allosterism. Finally, if the target of the allosteric modulation interacts with the conduit of the allosteric modulation along the plane of the membrane, this is referred to as lateral allosterism, with a main example being allosterism in receptor oligomers (Kenakin and Miller, 2010).
An example of classic allosterism would be the case of a ligand that modulates allosterically the effect of an orthosteric agonist. An orthosteric agonist has two main independent properties: an affinity for the receptor and an intrinsic efficacy, which determines the power of the agonist to induce a functional response. The allosteric modulator can have different and independent effects on these properties of distinct orthosteric agonists (Kenakin and Miller, 2010; Smith and Milligan, 2010). Therefore, there may be two general effects of allosteric modulators on orthosteric ligands: on their affinity and on intrinsic efficacy. Analysis of functional responses will determine the contribution of both allosteric effects. Another property of allosteric modulators is saturability. A negative allosteric modulator that selectively modifies the affinity of an orthosteric agonist will displace the functional dose-response curve to higher concentrations, but only up to a certain extent. By contrast, for a competitive orthosteric antagonist, such a "right shift" would continue ad infinitum. This ceiling effect of classic allosteric modulators can have important therapeutic implications by reducing overdose effects, compared with orthosteric ligands (Kenakin and Miller, 2010; Smith and Milligan, 2010).
In cytosolic allosterism (as defined by Kenakin and Miller, 2010), modulated proteins are cytosolic signaling proteins, such as G proteins, GPCR kinases (GRKs), and β-arrestins. Importantly, this type of allosterism can lead to functional selectivity, i.e., the ability of a ligand to selectively promote a specific cellular signaling event (Reiter et al., 2012). The rationale is that different ligands can stabilize different conformations of the receptor. If different cytosolic proteins that mediate different signaling interact with different residues or phosphorylated residues of the GPCR, it may be expected that some conformations can favor or impair the binding of a particular signaling protein or that they can induce a different conformation of the cytosolic protein, leading to biased agonism or biased antagonism (Reiter et al., 2012). This can also have important therapeutic implications, i.e., when a particular signaling pathway or end point is associated with a therapeutic response, whereas another is associated with nonwanted or side effects. Agonist binding to GPCRs and G protein activation are rapidly followed by several coordinated events common to most GPCRs. These include recruitment of GRKs that phosphorylate the receptor at multiple intracellular sites, followed by the recruitment of β-arrestins, which trigger receptor endocytosis. But, in addition to canonical G protein–mediated signaling, GPCRs can also bind to other cytosolic adaptors, including β-arrestins, which elicit G protein–independent signaling through activation of mitogen-activated protein kinase (MAPK) and Akt. Most known endogenous and exogenous ligands can signal through both signaling mechanisms. There are already, however, several examples of biased ligands that preferentially signal through β-arrestins over G proteins. In some cases, this may be a therapeutically beneficial effect, in others an unwanted one. Carvedilol, for instance, has been reported to be a β-arrestin–biased ligand acting at β2-adrenoceptors (but also at β1-adrenoceptors), which may contribute to its clinical value in heart failure beyond its β-blocker property (Reiter et al., 2012). The work by Lefkowitz and coworkers indicates that a mechanism behind functional selectivity can be a ligand-dependent “phosphorylation barcoding” (concept developed by Tobin et al., 2008). Thus, the β-arrestin–biased agonism of carvedilol appears to depend on its ability to impair GRK2- and promote GRK6-mediated phosphorylation (Nobles et al., 2011). Another mechanism is the differential structural conformations that lead to a preferential binding for specific G proteins. For instance, different agonists of the cannabinoid CB1 receptor differentially regulate the binding of the three homologous Gi proteins to the receptor. Whereas one ligand (desacetyllevonantradol) behaved as an agonist for Gi1 and Gi2 and an inverse agonist for Gi3, another ligand [(R)-methanandamide] behaved in exactly the opposite manner, as an inverse agonist for Gi1 and Gi2 and an agonist for Gi3 (Mukhopadhyay and Howlett, 2005).
B. Allosterism in Receptor Oligomers: Modulation of Ligand Affinity
Evidence for the existence of GPCR homomers (and heteromers) could already be deduced some time ago from analysis of radioligand-binding experiments, with the evidence of complex binding, such as upward concave nonlinear Scatchard plots in saturation experiments and as biphasic curves in agonist–antagonist competitive-inhibition experiments. Such complex curves were usually explained by two nonoverlapping models (reviewed in Casadó et al., 2007). First is the monomer–G protein model, which considers receptors as monomers with two independent populations not in equilibrium. One population would be bound and the other would not be bound to G proteins, with the receptors coupled to G proteins having higher affinity for the agonist. Receptors with high affinity could then be converted into low affinity with the addition of GTP, because it would uncouple the G protein from the receptor (De Lean et al., 1980). To explain the complex radioligand binding curves, this first mechanism would assume a preexisting proportion of both populations of receptors and, therefore, a limited pool of G proteins. The second model, the dimer-cooperativity model, considers oligomerization, or at least GPCR dimers. In this case, allosteric communication through the two protomers allows negative cooperativity, meaning that the binding of a ligand to the first protomer decreases the affinity of the ligand for the second protomer. Cooperativity (positive or negative) is a particular type of allosteric modulation in receptor oligomers, where the protomers of a homodimer are the conduit of the allosteric modulation and the same ligand is the allosteric modulator (binding to the first protomer) and the modulated target (binding to the second protomer). This mechanism does not assume a limited pool of G proteins, which is always present and acts as an additional allosteric modulator that increases the affinity of the agonist, and provides the conformation of the dimer that allows negative cooperativity of a ligand through the protomers.
In apparent support for the first mechanism, in the study by Whorton et al. (2007) with reconstitution of monomers of β2-adrenoceptors and G proteins in high-lipoprotein nanoparticles, antagonist/agonist competitive inhibition curves showed biphasic curves, and the addition of a GTP analog converted the low affinity population into high affinity. The study also showed, however, that the proportion of receptors with high affinity state increases up to 100% by increasing the G protein pool, indicating that in situ, in membrane preparations of brain tissue, the detection of two populations of receptors would indicate a limited pool of G proteins. In this case, the monomeric model with two G protein–dependent populations of receptors could explain the upward concave Scatchard plots and the biphasic antagonist/agonist competition curves (Casadó et al., 2007). The upward concave Scatchard curves would be the result of the addition of two curves that describe the two independent populations, with high and low affinities for the agonist. The biphasic competition curves would be the result of two different affinities of the agonist, which would identify both populations, displacing the antagonist that would have the same affinity for both populations. However, some early studies indicated that antagonist binding could also be modulated by guanine nucleotides, which, according to the monomer–G protein model, would suggest that antagonist binding would also be dependent on G protein coupling (Burgisser et al., 1982; De Lean et al., 1982; Klotz et al., 1990). Dissociation kinetic experiments can resolve the enigma. Investigation of the dissociation kinetics of a tracer ligand in the absence and presence of a second ligand represents a sensitive method to detect cooperative interactions between two topographically distinct binding sites. Thus, ligands that compete for the same site on a monomeric receptor should not influence one another's dissociation kinetics. In contrast, allosteric modulation between two simultaneously bound and interacting sites, either within a receptor monomer or across a receptor dimer or oligomer, should alter ligand dissociation (May et al., 2007). This analysis has been used to demonstrate homomerization of several GPCRs (Urizar et al., 2005; Albizu et al., 2006, 2010; Springael et al., 2006; May et al., 2011). Therefore, at present, there is no doubt that negative cooperativity is an allosteric property of some ligands that bind to GPCR homomers.
It could be argued that the dimer-cooperativity model is an artifact of membrane preparations, where there would not be a limited pool of G proteins for the specific receptor under study compared with the in situ situation. Negative cooperativity has not only been observed in membrane preparations from different tissues, including brain, and from artificial systems, such as transfected cells, but by measuring dissociation kinetics with fluorescent GPCR ligands, negative cooperativity can also be demonstrated in living cells (May et al., 2011), indicating that it is not an artifact of membrane preparation and that it can be of functional and pharmacological significance. Finally, a significant study that strongly supports the dimer-cooperativity model not only in living cells, but also in native tissues, involved homogeneous time-resolved FRET with fluorescent ligands (Albizu et al., 2010). Homogeneous time-resolved FRET is based on an energy transfer between a lanthanide (europium or terbium) and a compatible fluorophore (lanthanide’s long-lasting fluorescence allows measurement of its fluorescence after a laser pulse at time points when background fluorescence has disappeared, greatly increasing signal-to-noise ratio) (Gandia et al., 2008). Having ligands for oxytocin receptors separately fused to donor and acceptor molecules demonstrated the existence of dimers, both in transfected cells and in native tissue (mammary glands; Albizu et al., 2010). Furthermore, the differential FRET obtained with labeled agonists and antagonists, with significantly less FRET obtained with agonists, indicated a stoichiometry of one agonist molecule per dimer consistent with negative cooperativity, and this was only observed with agonists and not antagonists (Albizu et al., 2010)
Results from radioligand binding experiments, therefore, can indicate the existence of GPCR oligomerization. Furthermore, apart from negative cooperativity, the monomeric–G protein model fails to explain other complex radioligand-binding data, such as downward concave Scatchard plots, as reported with the mixed vasopressin-oxytocin receptor antagonist OTA (d(CH2)5[Tyr(Me)2-Thr4-Orn8-Tyr(NH2)9]-oxytocin) (Albizu et al., 2006, 2010). A dimer-cooperativity model could explain these findings by the existence of positive cooperativity (Albizu et al., 2006, 2010). Other results of radioligand-binding experiments that cannot be explained by the monomer–G protein model are biphasic antagonist/antagonist competition curves—for instance, those recently described for the adenosine A2A receptor antagonists ZM-241385 [4-(2-[7-amino-2-(2-furyl)[1,2,4]triazolo[2,3-a][1,3,5]triazin-5-ylamino]ethyl)phenol] and SCH-442416 [2-(2-furanyl)-7-[3-(4-methoxyphenyl)propyl]-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine] as radioligand and competing compounds, respectively (Orru et al., 2011a). These complex competitive-inhibition curves could be explained by means of the dimer-cooperativity model. In this case, we have two possibilities: first, allosteric interactions with the same ligand, i.e., negative cooperativity, where the binding of one ligand, SCH-442416, to the first protomer modifies the affinity of the binding of the same ligand to the second protomer; or second, allosteric interactions between different ligands in the receptor homomer, where the binding of one ligand, i.e., ZM-241385, to the first protomer changes the affinity of the other ligand, SCH-442416, to the second protomer. This implies different types of allosterism, and their differentiation could have important therapeutic implications (see below and Fig. 1). Negative cooperativity has also been reported for some serotonin 5-HT2A receptor antagonists, like the atypical antipsychotics clozapine and risperidone, but not the typical antipsychotic haloperidol (Brea et al., 2009). Importantly, there was a correlation between the binding behavior of the different antipsychotics and their properties as antagonists of a serotonin 5-HT2A receptor-mediated signaling. Thus, negative cooperativity could explain a biphasic inhibition of serotonin-stimulated arachidonic acid release (Brea et al., 2009).
Fig. 1.

Analysis of radioligand-binding experiments considering GPCRs as dimers: the two-state dimer model. For saturation experiments, KD1 and KD2 are the macroscopic equilibrium dissociation constants, which define the dissociation equilibria involved in the binding of a ligand to the receptor dimer. DC represents the dimer cooperativity index. DC = 0 implies no cooperativity, whereas positive and negatives values imply positive and negative cooperativity, respectively. For competition experiments, KDB1 and KDB2 correspond to the macroscopic equilibrium dissociation constants for the binding of the competing ligand to the first and second receptor in the dimer. KDAB is a value of the association and dissociation of the competing ligand on a dimer semioccupied by the radioligand. Reciprocally, KDBA is a macroscopic equilibrium dissociation constant of the radioligand binding to a receptor dimer semioccupied by the competing ligand. DAB and DBA represent the corresponding dimer radioligand/competitor modulation indexes. DAB or DBA = 0 implies no modulation, whereas positive and negatives values imply positive and negative modulation, respectively. DCB defines a dimer cooperativity index for the competing ligand. DCB = 0 implies no cooperativity, whereas positive and negative values imply positive and negative cooperativity.
What would be the functional significance of negative (or positive) cooperativity of a receptor homomer? In a framework of symmetric signaling properties of both protomers in a receptor homodimer, we would expect two different levels of ligand-mediated signaling that would depend on the concentration of the ligand. Negative cooperativity could provide a mechanism that protects the biologic system against acute elevations of the endogenous ligand (Agnati et al., 2005). Positive cooperativity, on the other hand, could provide an amplificatory mechanism, although to our knowledge, no clear examples of positive cooperativity of endogenous ligands have been reported (but see below about symmetry).
Regarding allosterism in receptor oligomers, and according to Kenakin and Miller (2010), two different scenarios have to be considered. First, the receptor dimer becomes the new conduit of the allosteric modulation, the allosteric modulator binds to one of the protomers and the modulated target binds to the other protomer. When the allosteric modulator and the target of the allosteric modulation are the same ligand and the protomers are identical (GPCR homomers), the result of this allosterism is positive or negative cooperativity. When the allosteric modulator and the target of the allosteric modulation are different and the protomers identical, the result is allosteric interactions between different ligands in the receptor homomer. The same can apply, with different ligands and different protomers, in receptor heteromers. As an example, adenosine A2A receptor ligands modulate the affinity of dopamine D2 receptor ligands in the A2A–D2 receptor heteromer. This is a well known receptor heteromer localized in one of the two main neuronal populations in the striatum and suggested to be a target for the treatment of Parkinson’s disease (Ferré et al., 2008; Azdad et al., 2009). In the case of the δ-μ-opioid receptor heteromer, it was shown that binding and signaling by morphine or μ receptor agonists were potentiated by δ-opioid receptor antagonists, and reciprocally, binding and signaling by δ-opioid receptor agonists were potentiated by μ receptor selective antagonists (Gomes et al., 2004, 2011). Studies carried out with the δ-opioid–cannabinoid CB1 receptor heteromer have also revealed allosteric modulations of cannabinoid CB1 receptor ligands on δ-opioid receptor ligand binding properties (Bushlin et al., 2012; Rozenfeld et al., 2012). Both in recombinant systems expressing both receptors and endogenous tissues, binding and consequently signaling by δ opioid receptor could be potentiated by a low, nonsignaling dose of cannabinoid CB1 receptor agonist or a selective antagonist (Bushlin et al., 2012). These unique properties, taken with the fact that the δ-opioid–CB1 receptor heteromers are upregulated during neuropathic pain (Bushlin et al., 2012), make this receptor heteromer an attractive target for the treatment of this disease.
In the second scenario of allosterism in receptor oligomers (Kenakin and Miller, 2010), one of the protomers becomes the allosteric modulator, which is sometimes referred in the literature as ligand-independent allosteric modulation in the receptor heteromer. As we will see later, dopamine D2 receptor selectively modifies the binding of SCH-442416 to the adenosine A2A receptor in the A2A–D2 receptor heteromer (Orru et al., 2011a). The melatonin MT1-GPR50 receptor heteromer constitutes a particular example of this allosteric modulation, where the presence of the orphan GPR50 receptor has a negative allosteric effect on melatonin binding to the MT1 receptor (Levoye et al., 2006a).
C. Allosterism in Receptor Oligomers: The Two-State Dimer Model
In view of the existence of clear experimental evidence supporting the dimer-cooperativity model, there is a need to develop new models of analysis of radioligand-binding experiments that consider GPCRs as oligomers or, at least, GPCR dimers. From all the models that consider receptors as monomers, the most commonly used is the two-independent-site model, which can explain the upward concave Scatchard plots and the biphasic competition curves. This is assuming the existence of two independent interconvertible populations of receptors with two different affinities for the agonists. This model, however, has serious drawbacks. When trying to resolve complex radioligand-binding data, when using the two-independent-site model, the values of the equilibrium dissociation constants and number of receptors obtained vary significantly depending on the concentration of the radioligand (Casadó et al., 2009a), indicating a lack of robustness.
For the analysis of radioligand-binding experiments, several models that consider receptors as dimers/oligomers have been developed (Durroux, 2005; Casadó et al., 2007; Rovira et al., 2009; Giraldo, 2013). These models, however, involve quite complex initial mechanistic equations, with a high number of constants, which make them quite unpractical for the analysis of radioligand binding experiments. Nevertheless, the further development of empirical equations from one of these models, the two-state dimer model, makes it particularly insightful and easy to use (Franco et al., 2005, 2006; Casadó et al., 2007, 2009a,b). This is not entirely obvious when looking at the initial equations that include all the microscopic equilibrium isomerization and dissociation constants: seven for the binding of one ligand for saturation experiments, and 11 for two ligands for competition experiments (described in detail in Casadó et al., 2007). Hence empirical equations were derived that disclose a lower number of constants, macroscopic equilibrium dissociation constants, and this allows for a practical analysis of radioligand-binding data when considering the receptors as dimers (analyzed in Casadó et al., 2007)
Equation for saturation experiments:
 |
A is the concentration of radioligand and RT is the total amount of receptor dimers (the traditional Bmax from the two-independent-site model would be twice this value). KD1 and KD2 are the macroscopic equilibrium dissociation constants, which define the dissociation equilibria involved in the binding of a ligand to the receptor dimer as a whole (not to be confused with the intrinsic equilibrium dissociation constants) (Fig. 1). KD1 represents the equilibrium between free ligand, empty dimer, and semi-occupied dimer (the first ligand occupying a dimer). KD2 represents the equilibrium between free ligand and the occupied and semioccupied dimer (the second ligand occupying a dimer in which one binding site is already occupied by the first ligand). Assuming symmetry, the rate at which the ligand binds to the dimer is proportional to the number of unoccupied receptors in the dimer, therefore twice for the empty compared with the semi-occupied dimer. The rate at which the ligand dissociates from the dimer is proportional to the number of occupied receptors in the dimer, therefore twice in the occupied compared with the semi-occupied dimer. This implies, and it is easy to demonstrate, that when the intrinsic affinity of both receptors in the dimer is the same, i.e., when there is no cooperativity, KD2 = 4KD1. The two-state dimer model also introduces a dimer cooperativity index, DC, which is defined as log (4KD1/KD2). DC = 0 implies no cooperativity, whereas positive and negatives values imply positive and negative cooperativity, respectively (Fig. 1).
Equation for competitive-inhibition experiments:
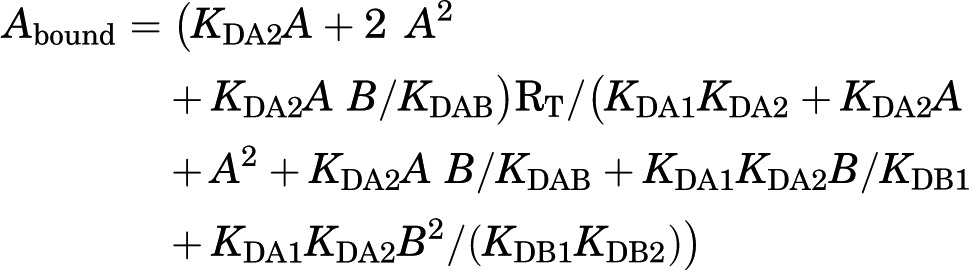 |
Although the equation might look complex, there are just a few constants to be determined. A is again the concentration of radioligand (fixed in competition experiments). B is the concentration of the competing compound. RT is again the total amount of receptor dimers. KDA1 and KDA2 are the macroscopic equilibrium dissociation constants for the binding of A, the radioligand, to the first and second receptor in the dimer, equivalent to KD1 and KD2 described above for saturation experiments. These constant values would have been calculated from previous saturation experiments with the radioligand and, therefore, they are already known. KDB1 and KDB2 correspond to the macroscopic equilibrium dissociation constants for the binding of B, the competing compound, to the first and second receptor in the dimer (Fig. 1). And there is one more constant, KDAB, which can be considered as a hybrid equilibrium dissociation constant, a value of the association and dissociation of B on a dimer semi-occupied by A (Fig. 1). KDAB is instrumental to explain a biphasic curve in competitive-inhibition experiments by means of a noncooperative binding. It measures an allosteric interaction between the two different ligands in the dimer, by which the binding of the radioligand to the first protomer decreases or increases the affinity of the competitor to the second protomer. It can easily be demonstrated that when the binding of the radioligand to one protomer in the dimer does not modify the binding of the competing ligand to the other empty protomer, KDAB = 2KDB1 (Figs. 1 and 2). Reciprocally, a KDBA value, which is the equilibrium dissociation constant of A binding to a receptor dimer semi-occupied by B, can be deduced (Figs. 1 and 2). The relation between these parameters is:
The two-state dimer model also introduces two dimer radioligand/competitor modulation indexes, DAB and DBA, which are defined as log (2KDB1/KDAB) and log (2KDA1/KDBA), respectively. DAB or DBA = 0 implies no modulation, whereas positive and negatives values imply positive and negative modulation, respectively (Figs. 1 and 2). Furthermore, we can also define a dimer cooperativity index for the competing ligand, DCB, as log (4KDB1/KDB2). DCB = 0 implies no cooperativity, whereas positive and negative values imply positive and negative cooperativity, respectively (Figs. 1 and 2). Finally, the two-state dimer model allows the calculation of the concentration of the competitor providing half saturation = (KDB1KDB2)1/2, independent of the biphasic or monophasic nature of the competition curve or of the radioligand concentration (Casadó et al., 2009a).
Fig. 2.

Application of the two-state dimer model. Two different competing ligands, the adenosine A2A receptor agonist CGS 21680 (A) and the A2A receptor antagonist SCH-442416 (B and C), are used to displace the A2A receptor antagonist [3H]ZM-241385 from membrane preparations of sheep striatum (A) or mammalian cells stably transfected with adenosine A2A and A1 receptors (B) or A2A and dopamine D2 receptors (C). (A) CGS 21680 displaces the binding of [3H]ZM-241385 in a biphasic manner; although not obvious by just looking at the graph, the analysis with a monomeric model gives a statistically significant better fit for two than for one binding site; analysis with the two-state dimer model indicates that the agonist does not show negative cooperativity (DCB = 0); in fact, previous studies with saturation experiments with [3H]CGS 21680 usually show noncurvilinear Scatchard plots (Jarvis et al., 1989; Borea et al., 1995); the analysis nevertheless indicates that the biphasic displacement can be explained by an allosteric modulation between ligands, by which the binding of [3H]ZM-241385 facilitates the binding of CGS 21680 to the A2A receptor dimer (DAB = 0.5). (B) Typical antagonist/antagonist competition, with SCH-442416 displacing in a monophasic manner [3H]ZM-241385 (DCB = 0; DAB = 0). On the other hand, in C, SCH-442416 displaces [3H]ZM-241385 in an obvious biphasic manner; the analysis with the two-state dimer model indicates that in this case, with coexpression of D2 receptors, SCH-442416 binding to A2A receptors displays a strong negative cooperativity (DCB = −2.30; DAB = 0). Results are modified from Casadó et al. (2009a) and Orru et al. (2011), where details of the methods, including radioligand concentrations, can be found. CGS 21680, 4-[2-[[6-amino-9-(N-ethyl-β-D-ribofuranuronamidosyl)-9H-purin-2-yl]amino]ethyl]benzenepropanoic acid.
Considering the evidence of receptor oligomerization and the validity of the dimer-cooperativity model, the two-state dimer model provides a better approach than the monomer–G protein models for fitting data that eventually give more accurate and physiologic relevant parameters. In terms of accuracy, the two-state dimer model is significantly more robust than the two-independent-site model. Thus, with the two-state dimer model the same parameters (RT, equilibrium dissociation constants) are obtained irrespective of the concentration of radioligand (Casadó et al., 2009a). In terms of physiologic relevance, the two-state dimer model does not only give equilibrium dissociation constants for high- and low-affinity binding to receptor dimers, but provides indexes of ligand cooperativity and of allosteric modulation between different ligands simultaneously binding to the dimer. For instance, the analysis with the two-state dimer model of the complex competition data obtained with the ligand SCH-442416 displacing [3H]ZM-241385 indicated the existence of a strong negative cooperativity of the binding of SCH-442416 to the adenosine A2A receptor, without evidence for allosteric interactions between both ligands (Orru et al., 2011a) (Fig. 2, B and C). This behavior of SCH-442416 was not observed with other adenosine A2A receptor ligands and was only observed in cells transfected with dopamine D2 receptors, which therefore act as allosteric modulators of adenosine A2A receptor dimers (Orru et al., 2011a). Thus, the observation of dopamine D2 receptor-mediated negative binding cooperativity of SCH-442416 suggests that the minimal A2A–D2 receptor heteromer unit is made of heteromers of homodimers (at least heteromers with A2A receptor dimers). This has important pharmacological implications that will be discussed below. The robust and significant fit of the equations provided by the two-state dimer model agrees with the above-reviewed evidences supporting dimers as predominant GPCR species and functional units. It is also of importance to underscore that the two-state dimer model should provide a good approximation, even in the context of a significant proportion of high-order oligomers, because we would probably be dealing with dimers or other multiples of dimers (see above).
Radioligand competition curves are usually analyzed for drug screening to determine the affinity constants for putative agonists and antagonist. If the target receptor is assumed to be primarily in its monomeric form, monomeric models should be used. However, if homomers have already been described for the target receptor, the two-state dimer model should be the most appropriate choice. In the absence of previous assumptions and in the absence of obvious biphasic competition curves, monomeric models could be used to provide initial estimates of drug affinities, because they imply a lesser number of constants to handle. When not obvious as biphasic, monomeric models can also be used to establish if the competition curves fit significantly better for one or two binding sites. In the latter case we should proceed with the analysis with the two-state model (see Fig. 2A). Nevertheless, when looking for the most appropriate model, the best approach would be implementing both a monomer-based model and a two-state dimer model and comparing results (preferably using a statistical test; see Casadó et al., 2009a). It is important to realize that choosing either model should not imply discarding the possibility of the existence of mixtures of monomers and dimers/oligomers.
D. Allosterism in Receptor Oligomers: Allosteric Modulation of Intrinsic Efficacy and Functional Asymmetry
The two-state dimer model assumes that both protomers have the same initial ligand binding properties and, therefore, the same probability to bind the first ligand molecule and the same ability of the first ligand–protomer complex to allosterically modulate the binding properties of the second protomer. That is, there should be an initial symmetry of both protomers in a receptor dimer at the binding level. Are there symmetric signaling properties of both protomers in a receptor dimer? This requires examining functional responses and to address questions about the minimal functional GPCR-G protein unit. Furthermore, above we only considered allosteric modulation of the affinity of ligands, whereas allosteric modulation of the intrinsic efficacy of the ligands should also be considered. When dealing with dimers, the most accepted model, at present, is that two protomers can only accommodate one heterotrimeric G protein complex (reviewed in Maurice et al., 2011). Extensive biochemical evidence indicates the involvement of at least two distinct G protein regions in interactions with the receptor. The primary docking site is in the C terminus of the Gα subunit, which penetrates into the crevice created in the intracellular surface of the receptor by the movement of cytoplasmic regions of TM5 and TM6 upon binding of the agonist (Oldham and Hamm, 2008; Rasmussen et al., 2011). Another putative site is in the C terminus of the Gγ subunit. These regions are 55 Å apart in the Gαβγ heterotrimer, a distance greater than the width of the monomeric GPCR (approximately 45 Å), indicating that for both contacts to take place simultaneously, one heterotrimeric G protein must contact two receptor protomers (Oldham and Hamm, 2008). Rasmussen et al. (2011) recently resolved the crystal structure of the active state ternary complex composed of agonist-occupied monomeric β2-adrenoceptor and nucleotide-free Gs heterotrimer. One of the most interesting findings was the lack of direct interactions between the β2-adrenoceptor and Gβγ. Given that the heterotrimer is required for efficient coupling to a GPCR, these results are consistent with the existence of β2-adrenoceptor dimers in cell membranes with one protomer interacting with Gα and the second promoter interacting with the Gβγ subunit (Rasmussen et al., 2011). Although a sequential interaction of simultaneously incompatible contacts could still be possible (Herrmann et al., 2004), this sequential fit mechanism would still be compatible with the existence of GPCR homodimers and with the pentameric structure consisting of one GPCR homodimer and one heterotrimeric G protein as a minimal functional unit (Baneres and Parello, 2003; Han et al., 2009; Pellissier et al., 2011). Even if we consider tetramers as the predominant oligomeric species, the most common minimal receptor–G protein stoichiometry would still probably be 2:1, and oligomeric entities can be viewed as multiples of dimers, as, for instance, suggested by the crystallographic structure of μ-opioid receptor and β1-adrenoceptor (Manglik et al., 2012; Huang et al., 2013).
The apparent asymmetric pentameric structure of GPCRs (homodimer plus heterotrimeric G protein) might seem incompatible with the two-state dimer model and the dimer-cooperativity model, because they assume there is an initial symmetry between both protomers regarding ligand binding properties (the same probability to bind the first ligand molecule) and that the G protein is always interacting with the receptor (Casadó et al., 2007). In this respect, two opposing models have been presented to explain the encounter between G proteins and the activated receptor. In the "collision coupling" model, the interactions occur as a result of the free lateral diffusion, and G proteins only interact with the agonist-bound receptor. In the "precoupling" model, G proteins are already interacting with receptors before agonist binding and the ligand modifies this interaction by creating the conformational change in the receptor (crevice) that allows the α subunit to “tightly” bind the receptor and induce G protein activation (reviewed in Oldham and Hamm, 2008). About the symmetry, the two-state dimer model only assumes an initial symmetry (at least in terms of ligand-binding probability) and fits very well with the precoupling model of receptor–G protein interaction. The two-state dimer model accepts that the ligand binding to the first protomer determines an asymmetric function of the pentameric functional unit. According to the model, this ligand-induced asymmetry determines the allosteric modulations at the binding level, such as the reduced affinity of the ligand for the second protomer (negative cooperativity).
What is the functional response that a ligand can produce when binding to one or both protomers in a GPCR homodimer? Is the binding of an agonist to one of the protomers enough to elicit a full functional response? Does the asymmetric constraint of G protein coupling to the two protomers determine an asymmetric functional response? Most experimental data agree with the model that proposes that ligand occupancy to the first protomer is enough to produce a significant G protein activation and functional response. Occupancy of the second protomer will then usually potentiate (Kniazeff et al., 2004; Pellissier et al., 2011), but could also reduce (Han et al., 2009) or not alter significantly (Hlavackova et al., 2005), the functional response (irrespective of the allosteric modulations at the binding level).
Therefore, when the ligand binds to the second protomer in a homodimer, it will often act as an allosteric modulator of the intrinsic efficacy of the ligand when binding to the first protomer.
We should also consider G protein–independent signaling, such as β-arrestin–dependent signaling. Similar to G proteins, GPCR oligomerization raises the question of whether GRK and β-arrestin binding to GPCRs occur in an asymmetric manner. The possible asymmetric nature of the receptor–β-arrestin interaction is still a matter of debate. As with G protein binding and activation, studies with artificially reconstituted monomeric GPCRs show that receptor dimers are not required for their functional interaction with GRK and β-arrestins (Tsukamoto et al., 2010; Bayburt et al., 2011). These results, however, do not exclude the possibility that GRK or β-arrestins bind to GPCR dimers (Modzelewska et al., 2006; Sommer et al., 2012). GPCR oligomerization potentially provides a larger platform to accommodate the different GPCR interacting proteins necessary for receptor function (reviewed in Maurice et al., 2011). This is demonstrated in the complex of melatonin MT1 receptor dimers, Gi protein and RGS20 (a protein that regulates the speed of G protein signal transduction), which both bind directly to helix 8 of the receptor (Maurice et al., 2010). In this complex, protomer 1 binds the ligand and the G protein and protomer 2 binds RGS20. In this case, RGS20, by slowing down the decay time of G protein inactivation, thus participates in prolonged signal transduction (Maurice et al., 2010).
Several examples of allosteric modulation of intrinsic efficacy have been reported for GPCR heteromers. As for ligand affinity, there are cases of allosteric modulation of the intrinsic efficacy of a ligand of one of the protomers by the other protomer or by a ligand that binds to the other protomer. As an example of the latter, in the α2A-adrenoceptor–μ-opioid receptor heteromer, morphine binding to the μ-opioid receptor inhibits α2A-adrenoceptor signaling (Jordan et al., 2003). This has been related to a morphine-induced conformational change of the α2A-adrenoceptor, as detected by dynamic intramolecular FRET techniques (Vilardaga et al., 2008). These studies indicated that activation of the μ-opioid receptor component in the α2A-adrenoceptor–μ-opioid receptor heteromer modulates α2A-adrenoceptor receptor signaling by a direct conformational change that propagates from the μ to the α2A-adrenoceptor within 0.4 seconds, slightly faster than the rate of G protein activation, implicating a direct allosteric modulation through the receptor heteromer (Vilardaga et al., 2008).
As an example of an allosteric modulation of intrinsic efficacy of a ligand of one of the protomers by the other protomer in the GPCR heteromer, in the metabotropic glutamate mGlu2-serotonin 5-HT2A receptor heteromer, glutamate produces a stronger mGlu2-mediated signaling and serotonin produces a weaker serotonin 5-HT2A–mediated signaling compared with when each receptor is expressed alone (Fribourg et al., 2011). In the same receptor heteromer, the serotonin 5-HT2A receptor antagonist clozapine also induced a positive allosteric modulation of the intrinsic efficacy of glutamate (Fribourg et al., 2011). These results could have implications for the understanding of the clinical efficacy of this compound in schizophrenia and establish the mGlu2–5-HT2A receptor heteromer as a promising target for the treatment of this disease.
Considering, therefore, receptor heteromers in the frame of allosteric modulation of GPCRs, there are examples of allosteric modulation by specific ligands of some receptors of the affinity and intrinsic efficacy of ligands for other receptors (see above). There are also significant examples of functional selectivity, where one of the protomers of the heteromer acts as an allosteric modulator that “forces” the other receptor protomer to signal predominantly through a distinct signaling pathway. An example is that of the δ-μ-opioid receptor heteromer (Rozenfeld and Devi, 2010). Under normal physiologic conditions, the μ-opioid receptor is found mostly in the homomeric state, and morphine stimulation induces G protein–mediated signaling, which is involved in its analgesic effects, and low β-arrestin–mediated signaling, which promotes unwanted effects, such as tolerance to the analgesic effects (Raehal et al., 2011). In contrast, upon chronic treatment with morphine, the abundance of δ-μ-opioid receptor heteromers increases (Gupta et al., 2010), and morphine stimulation induces β-arrestin–mediated signaling (Rozenfeld and Devi, 2007), which is thought to contribute to the tolerance to the analgesic effect of morphine upon its chronic administration (Raehal et al., 2011). Notably, occupancy of the δ-opioid receptor protomer with a selective δ-opioid receptor antagonist (Rozenfeld and Devi, 2007) or with a bivalent ligand in the δ-μ-opioid receptor heteromer (see below and Daniels et al., 2005) allows for the restoration of G protein–biased agonism of morphine and, therefore, its analgesic effects (Rozenfeld and Devi, 2010).
Other dramatic examples of functional selectivity in receptor heteromers are changes in G protein coupling, such as with the angiotensin AT1–cannabinoid CB1 receptor heteromer (Rozenfeld et al., 2011). Although, in the normal liver angiotensin AT1 receptor signals via Gq, under conditions of alcohol-induced liver fibrosis cannabinoid, CB1 receptor is upregulated and heteromerizes with angiotensin AT1 receptor and this, in turn, leads to Gi-mediated signaling by angiotensin AT1 receptor that can be blocked by cannabinoid CB1 receptor antagonist, suggesting that AT1–CB1 receptor heteromer represents a disease-specific and potentially tissue-specific therapeutic target. Another example of change in G protein coupling is the dopamine D1–histamine H3 receptor heteromer, where dopamine D1 receptor agonists activates Gi instead of Gs proteins (inhibition of cAMP formation) (Ferrada et al., 2009). Another important allosteric property of the D1–H3 heteromer is the ability of H3 receptor agonists to inhibit dopamine D1 receptor–mediated G protein and β-arrestin signaling, which allows histamine to provide a brake on dopamine D1 receptor–mediated effects, including cell death (Moreno et al., 2014). This heteromer also binds the two-transmembrane sigma σ1 receptors, which are well known targets for cocaine. Binding of cocaine to the D1–H3–σ1 complex disrupts the allosteric properties of the D1–H3 heteromer allows dopamine D1 receptor to couple to Gs and to eliminate the histamine H3 receptor–mediated signaling brake, which promotes cell death (Moreno et al., 2014). Therefore D1-H3 heteromers may provide a new therapeutic target for the D1 receptor–mediated neurotoxic effects of cocaine.
Another recent example of functional selectivity in receptor heteromers is the dopamine D2–ghrelin GHS1a receptor heteromer (Kern et al., 2012). These receptors are colocalized in the brain, in the hypothalamus and brain stem (Guan et al., 1997). Interestingly, ghrelin receptors seem to be orphan receptors in certain localizations, because the peptide ghrelin, which is produced in the stomach, can only reach the arcuate nucleus but not other hypothalamic nuclei and other brain regions. In the hypothalamus, heteromerization with ghrelin GHS1a receptor modifies dopamine D2 receptor signaling, resulting in Gβγ-dependent mobilization of Ca2+ (Kern et al., 2012). The anorexigenic effects of dopamine D2 receptor agonists seem to be mediated by the hypothalamic D2–GHS1a receptor heteromer, which therefore might become an important therapeutic target for eating disorders.
In summary, the allosteric analysis of GPCR homomers and heteromers provides overwhelming additional evidence for the results of in vitro experiments and experiments in heterologous systems, supporting the existence of functionally and pharmacologically relevant GPCR oligomers. The evidence points to the pentameric structure constituted by one GPCR homodimer and one heterotrimeric G protein as a main minimal functional unit and oligomeric entities can be viewed as multiples of dimers. Allosteric mechanisms determine a multiplicity of possible unique pharmacological properties of receptor homomers and heteromers. Some general mechanisms seem to apply, particularly at the level of ligand-binding properties. When considering receptor homomers, the two-state dimer model provides the most practical method to analyze ligand–GPCR interactions. If possible, similar practical models would need to be developed to study ligand–GPCR interactions for receptor heteromers and for dynamically changing oligomers. In addition to ligand-binding properties, unique properties for each GPCR oligomer seem to emerge in relation to different intrinsic efficacy of different ligands (intrinsic efficacy) for different signaling pathways (functional selectivity). This provides a rationale for the use of GPCR oligomers, and particularly heteromers, as novel targets for drug development.
III. G Protein–Coupled Receptor Oligomers as Novel Targets for Drug Development
A. Localization of Receptor Oligomers in Native Tissues
Before considering the possible role of GPCR oligomers in vivo, and particularly receptor heteromers, as targets for drug development, we must first readdress questions about the demonstration of receptor homo- and heteromers in native tissues, because most of the studies discussed above have been performed in heterologous expression systems, particularly in transfected mammalian cells. Next, we need to address the questions about localization in situ and significance of receptor homomers and heteromers in vivo (Levoye et al., 2006b; Pin et al., 2007; Ferré et al., 2007, 2009). Efforts toward addressing these important questions have been hampered by the lack of appropriate tools and techniques that would allow their clear demonstration in native tissues. Biochemical techniques such as coimmunoprecipitation cannot provide unambiguous evidence for direct protein–protein interactions, and biophysical techniques, such as BRET and FRET, are difficult to implement in tissues. In contrast to heterologous systems, where wild-type or mutated receptors fused with tags or fluorescent proteins can be easily expressed, the expression level of receptors in native tissues is difficult to modify and their sequences cannot be modified unless using knock-in strategies. Nevertheless, quantitative FRET in situ has been used to explore the presence of dopamine D1–D2 receptor heteromers in brain tissue (Hasbi et al., 2011).
An alternative approach to label and detect native GPCR oligomers has been the use of antibodies or fluorescent ligands. However, they need to be highly specific and to provide a reliable signal, and the levels of GPCR oligomers should be abundant or highly regulated. Recent efforts to identify tools that fit some of these criteria have begun to yield promising results. Using labeled ligands that could demonstrate proximity of endogenous receptors, the study by Albizu et al. (2010) provided quite a clear demonstration of the presence of oxytocin receptor homomers in the mammary gland. They showed differential FRET with labeled agonists and antagonists in tissue as they had seen with transfected cells, implying the existence of oxytocin receptor homomers. This is therefore an indirect but clear demonstration of the existence of receptor homomers in native tissues.
Efforts to generate heteromer-selective antibodies using a subtractive immunization strategy have begun to yield useful monoclonal antibodies with heteromer selectivity, because they recognize the epitope only in wild-type animals but not in animals lacking either protomers of the heteromer (Gomes et al., 2013a,c). These heteromer-selective antibodies have helped not only studies examining heteromer levels and regulation in endogenous tissue but also studies investigating the extent of heteromer-specific signaling (Gomes et al., 2013a,c). In the case of δ-μ-opioid receptor heteromers, the antibodies were helpful in demonstrating morphine-induced increases in heteromer abundance, as well as characterizing heteromer-mediated signaling (Gupta et al., 2010). The AT1–CB1 receptor heteromer selective antibody was useful in demonstrating upregulation of the heteromer in hepatic stellate cells after alcohol-induced fibrosis (Rozenfeld et al., 2011). The δ-opioid–CB1 receptor heteromer selective antibody was useful in demonstrating increases in δ-opioid–CB1 receptor heteromers in the brain after peripherally elicited neuropathic pain (Bushlin et al., 2012). Finally, the δ-κ-opioid receptor heteromer selective antibody was critical in demonstrating a role for this heteromer in sensory pain transmission, because the antibody was able to augment the effect of δ receptor agonist-mediated antinociception (Berg et al., 2012). Together these studies demonstrate that GPCR heteromer antibodies, if found to be highly selective, are critical and important tools in studies of GPCR heteromers in native tissue.
The proximity ligation assay (PLA) is an antibody-based method that also provides an approximation to the demonstration of receptor heteromers in tissues. It has been used for instance for the demonstration of close proximity of adenosine A2A and dopamine D2 receptors in brain slices (Trifilieff et al., 2011). In PLA, primary antibodies labeling the two putative protomers of the receptor heteromer are labeled with two different species-specific secondary antibodies conjugated to complementary oligonucleotides. When the antibodies are in close proximity, the complementary DNA strands can be ligated, amplified, and visualized with a fluorescent probe. The maximal distance between the secondary antibodies in this assay is ∼16 nm, only slightly larger than that for resonance energy transfer between fluorophores (∼10 nm) (Soderberg et al., 2006). However, PLA does not demonstrate GPCR heteromerization, but allows a validation in vivo of the molecular proximity of two GPCRs, giving the frame for heteromerization to take place in vivo.
Another indirect but easier and straightforward demonstration depends on the identification of a biochemical property (“biochemical fingerprint”) of the receptor oligomer (Ferré et al., 2009). To ascertain a “biochemical fingerprint” of the receptor heteromer, the putative biochemical property should be disrupted with molecular or chemical tools that significantly destabilize the quaternary structure of the heteromer (such as introducing mutations of key determinant residues at the oligomerization interfaces or using competing peptides that harbor the same sequence as that of the receptor heteromer interface). The recently reported dopamine D2–D4 receptor heteromer serves as a good example. A human polymorphism of the dopamine D4 receptor, D4.7 (with seven repeats of a proline-reach 16-amino acid sequence in the third intracellular loop) differs from the most common variant, D4.4 (with four repeats) in that it does not heteromerize with dopamine D2 receptor and it does not show a pharmacological D2–D4 receptor interaction (Gonzalez et al., 2012). This interaction consists of a dopamine D2 receptor agonist-mediated potentiation of MAPK activation induced by a dopamine D4 receptor agonist (Gonzalez et al., 2012). Thus, a synergistic MAPK activation could be used as a biochemical fingerprint of the heteromer, which could only be observed in mice expressing wild-type dopamine D4 receptor (which also heteromerized with mouse wild-type dopamine D2 receptor), but not in knock-in transgenic mice expressing the dopamine D4.7 human variant (Gonzalez et al., 2012). The use of transgenic animals is therefore a useful indirect method to study receptor heteromer function. An additional example is the recent study about the role of MT1–MT2 receptor heteromers on the effect of melatonin on rod photoreceptor light sensitivity, which was abolished in melatonin receptor MT1−/− and MT2−/− mice (Baba et al., 2013). The involvement of MT1–MT2 receptor heteromers in this effect was further underscored by the lack of effect of melatonin in transgenic mice overexpressing a nonfunctional melatonin MT2 receptor mutant that competes with the formation of functional MT1–MT2 heteromers in photoreceptor cells (Baba et al., 2013). These experiments are of therapeutic interest for the improvement of photoreceptor functioning in several clinical conditions and applications in the pathogenesis of age-related macular degeneration and glaucoma.
Therefore, use of several complementary approaches is needed to address the identification of GPCR oligomers in native tissues. It is essential that these approaches are critically evaluated and validated using artificial systems, such as mammalian transfected cells, especially when looking for specific biochemical properties of a GPCR oligomer. It is not enough just reproducing some findings in native tissues previously found in the artificial systems. A unique biochemical finding in mammalian transfected cells that express receptor heteromers (which can be demonstrated with properly applied biophysical techniques) could well be related to interactions between the two receptors that do not depend on oligomerization. As mentioned above, we need to obtain and implement tools that specifically disrupt GPCR oligomerization or the quaternary structure of the GPCR oligomer. When possible, those tools should also be applied in situ. For instance, by selectively interfering with the oligomer interfaces, particular mutations or the in vitro application or in vivo delivery of peptides altering the quaternary structure of a GPCR oligomer (as first demonstrated with biophysical techniques in artificial systems) should selectively alter the detection of the putative biochemical property of the GPCR oligomer.
B. Do Receptor Heteromers Constitute Possible Targets for Drug Development?
GPCRs are targeted by many therapeutic drugs aiming at restoring or ameliorating a variety of health disorders and remain a primary focus of biomedical research and programs of pharmacological drug discovery. However, despite the proven success of GPCRs as drug targets, clinically useful ligands do not exist for the majority of GPCRs. A main contributing factor is the lack of selectivity of orthosteric ligands across members of a GPCR subfamily (or that certain GPCRs do not have roles in major disease phenotypes), with the obvious selectivity problem, which leads to unwanted side effects. An alternative approach already found successful for ligand-gated ion channels, such as benzodiazepines, positive allosteric modulators of GABAA receptors, is the development of GPCR allosteric modulators (ligands that modulate the affinity or intrinsic efficacy of orthosteric agonists of the same GPCR unit) (Conn et al., 2009). Allosteric modulators can potentially overcome the selectivity problem. The orthosteric binding sites across members of a GPCR subfamily for a particular endogenous ligand, such as noradrenaline or dopamine, are highly conserved, but GPCRs offer numerous possible selective allosteric binding sites across different receptor subtypes. Furthermore, allosteric modulators possess pharmacologically appealing properties, such as saturability. In recent years, significant progress has been made in the discovery of various GPCR allosteric modulators, although not without difficulties, such as generating a structure–activity relationship in an allosteric compound series, or likelihood of lack of cross-species activity for validation/efficacy studies (Conn et al., 2009).
Receptor heteromers, with their allosteric properties, give rise to a new kind of pharmacological target. The ability of one of the protomers to act as an allosteric modulator of the second protomer in the receptor heteromer gives the possibility of finding selective ligands for the protomer acting as conduit of the allosteric modulation. A proof of concept was provided by the dramatically decreased binding affinity (strong negative cooperativity) of the adenosine A2A receptor antagonist SCH-442416 in cells cotransfected with dopamine D2 receptors compared with cells only expressing adenosine A2A receptors or also expressing adenosine A1 (Orru et al., 2011a) (Fig. 2, B and C). Several other antagonists, such as MSX-3, also showed a less dramatic yet significantly decreased affinity for adenosine A2A receptors when cotransfected with dopamine D2 receptors (Orru et al., 2011a). These results are important from a practical pharmacological point of view, because different striatal subpopulations of adenosine A2A receptors modulate different striatal functions (Orru et al., 2011a,b), and these subpopulations are constituted by different adenosine A2A receptor heteromers. A2A–D2 receptor heteromers are selectively localized postsynaptically in GABAergic striatopallidal neurons, where they exert a key role in the modulation of the activity of these neurons (see above and Azdad et al., 2009). On the other hand, A1–A2A receptor heteromers are selectively localized presynaptically in the striatal glutamatergic terminals that make synaptic contact with GABAergic striatonigral neurons (Quiroz et al., 2009). Through these heteromers, postsynaptic blockade of adenosine A2A receptors produces locomotor activation, and presynaptic blockade of adenosine A2A receptors produces inhibition of corticostriatal neurotransmission (Ciruela et al., 2006; Quiroz et al., 2009; Orru et al., 2011a). The postsynaptic effect has implications for Parkinson’s disease and, based on the existence of A2A–D2 receptor interactions, adenosine A2A receptor antagonists are being used for the treatment of Parkinson’s disease (see below and Armentero et al., 2011). On the other hand, presynaptic effects can have implications for drug addiction, but a concomitant postsynaptic adenosine A2A receptor blockade will determine opposing effects. In this regard, a low dose of the selective adenosine A2A receptor antagonist MSX-3 (shown to be preferentially presynaptic in rodents; Orru et al., 2011a) decreased tetrahydrocannabinol and anandamide self-administration in squirrel monkeys, whereas a slightly higher dose had potentiating effects (Justinová et al., 2011). The appearance of potentiating rather than suppressing effects on cannabinoid reinforcement at the slightly higher dose of MSX-3 would likely preclude the use of such a compound as a pharmacologic treatment of cannabis abuse. As expected, recent results showed a significant antagonistic effect of SCH-442416 over a much wider range of doses than with MSX-3 (Justinová et al., submitted manuscript), strongly supporting the use of this or other similar compounds for the treatment of drug abuse.
When the receptor heteromer is the conduit for the allosteric modulation, the allosteric modulator acts very much the same way as a classic allosteric modulator. The difference is that the binding of the allosteric modulator takes place in the orthosteric site of one of the receptors in the heteromer and the modulated ligand binds to the orthosteric site of the other receptor. For instance, adenosine A2A receptor antagonists increase the affinity of the dopamine D2 receptor for dopamine, which as mentioned earlier is being used as a strategy for the treatment of Parkinson’s disease. In fact, evidence has already been provided for an increase in the therapeutic index of l-dopa with adenosine A2A receptor antagonists (Armentero et al., 2011). Furthermore, adenosine A2A receptor antagonists can potentially be useful as monotherapy in early stages of Parkinson’s disease where some symptoms seem to be related to the loss of a tonic dopamine D2 receptor–mediated inhibition of adenosine A2A receptor signaling (Ferré et al., 2008; Armentero et al., 2011). Schizophrenia is another pathologic condition where the A2A–D2 receptor heteromer could be used as a therapeutic target (Ferré, 1997). Thus, different from dopamine D2 receptor antagonists, adenosine A2A receptor agonists behave as atypical antipsychotics (antipsychotics with low extrapyramidal side effects) in animal models (Rimondini et al., 1997). This profile could be related to the allosteric and therefore saturable effect on dopamine D2 receptor–mediated signaling. So far, the cardiovascular effects of adenosine A2A receptor agonists preclude their use as antipsychotics, but ligands with selectivity for the A2A–D2 receptor heteromer would be devoid of these peripheral side effects because of the lack of peripheral A2A–D2 receptor interactions (Schindler et al., 2004). In the A2A–D2 receptor heteromer as target for Parkinson’s disease and schizophrenia, we deal with an allosteric modulation of the affinity of dopamine D2 receptor ligands, although changes in intrinsic efficacy might also take place. As mentioned above, another candidate target for antipsychotic treatment is the mGlu2–5HT2A receptor heteromer, where the atypical antipsychotic clozapine, in addition to blocking serotonin 5-HT2A, induces an allosteric modulation of the intrinsic efficacy of glutamate (Fribourg et al., 2011). Functional selectivity is also being addressed in the search for antipsychotics with low liability for side effects.
A particular value of receptor heteromers as targets for drug development is that they can be involved in the pathophysiology of the disorder being targeted. In this regard, the receptor heteromer would be disease specific (Ferré et al., 2010; Gomes et al., 2013a). The dopamine D1–D3 receptor heteromer seems to be involved in the very common and troublesome secondary effect of l-dopa treatment in Parkinson’s disease, l-dopa–induced dyskinesia (reviewed in Ferré et al., 2010). The dopamine D1 receptor in the D1-D3 receptor heteromer has significantly more affinity for dopamine or other agonists than when not forming heteromers with dopamine D3 receptors (Fiorentini et al., 2008; Marcellino et al., 2008), which results in a significant increase in the potency of dopamine D1 receptor agonists to signal through the G protein and potentiate cAMP accumulation (Fiorentini et al., 2008). This is a very significant finding, because it is well known that, under normal conditions, striatal dopamine D1 receptors have a much lower affinity for dopamine than dopamine D2 or D3 receptors (Missale et al., 1998). There is a substantial amount of evidence that indicates that dopamine D1 and D3 receptors are colocalized in GABAergic striatonigral neurons (which constitutes the direct striatal efferent pathway), where they play a key role in the development of l-dopa–induced dyskinesia in Parkinson’s disease. The working hypothesis is that dopamine denervation followed by chronic l-dopa treatment leads to an increased dopamine D1 receptor signaling promoted by an upregulation of dopamine D3 receptors and by an increase in the expression of D1–D3 receptor heteromers, which produces an imbalance in basal ganglia processing due to an increased activation of the direct pathway (reviewed in Ferré et al., 2010). Significantly, treatment with dopamine D3 receptor partial agonists or antagonists attenuates l-dopa–induced dyskinesia in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)–treated monkeys (Bezard et al., 2003; Visanji et al., 2009). Finally, an increase in dopamine D3 receptor density has been observed in human cocaine fatalities (Staley and Mash, 1996), suggesting that a concomitant increase in D1–D3 receptor heteromers may also occur. The D1–D3 receptor heteromer therefore emerges as a target for the treatment of l-dopa–induced dyskinesia and cocaine addiction.
As mentioned earlier, the availability of the heteromer-selective antibodies has helped explore disease-specific modulation of their abundance and hence define them as novel therapeutic targets (Gomes et al., 2013c). A potential role for δ-μ-opioid receptor heteromers in morphine tolerance was implied by studies that demonstrated an increase in the abundance of δ-μ-opioid receptor heteromers in the brain and in dorsal root ganglion cells in response to a paradigm of morphine treatment that leads to development of tolerance (Gupta et al., 2010). AT1–CB1 receptor heteromers are upregulated in alcohol-induced liver fibrosis, and this leads to a shift in signaling from a Gi to Gq-coupled pathway implicating the CB1R–AT1R heteromer as a potential target for the treatment of liver fibrosis (Rozenfeld et al., 2011). Finally, the δ-opioid–CB1 receptor heteromer was found to be upregulated during neuropathic pain and the heteromer exhibited unique allosteric modulation, making this an attractive target for the development of therapeutic for the treatment of pain (Bushlin et al., 2012).
C. Approaches for the Identification of Receptor-Heteromer Selective Compounds
Despite their pharmacologic potential, there are very few studies reporting the identification of receptor-heteromer selective compounds. A receptor-heteromer selective compound would not only include ligands with significantly different affinity or intrinsic efficacy for one or both receptors in a receptor heteromer (compared with the same biochemical properties of the ligands for the same receptors when not forming heteromers), it would also include compounds that would exert significant allosteric modulations in the receptor heteromer. Two general approaches were recently used for the discovery of receptor-heteromer selective compounds: discrete direct screening (DDS) and high-throughput screening (HTS). DDS is used when a series of ligands targeting either of the receptors in the heteromer is available. In this case, relatively smaller number of ligands would allow the use of labor-intensive techniques to demonstrate differences in affinity (for agonists or antagonists) or intrinsic efficacy (agonists). The most important step for this strategy to be successful is the availability of appropriate cell lines. The study by Orru et al. (2011a) provides a successful example of DDS of adenosine A2A receptor antagonists in different comparable cell lines expressing only adenosine A2A receptors or A1–A2A or A2A–D2 receptor heteromers. As mentioned above, radioligand-binding experiments and analysis of the data with the two-state dimer model demonstrated a very specific behavior, a negative cooperativity of the adenosine A2A receptor antagonist SCH-442416 in cells expressing A2A–D2 receptor heteromers (Fig. 2, B and C). This biochemical result predicted a preferential striatal presynaptic profile of SCH-442416, which totally correlated with in vivo experiments in rodents and monkeys. Notably, preclinical data suggest that this compound could be useful in the treatment of drug addiction (see Section III.B). SCH-442416 can now be used as a lead compound for further chemical optimization leading to novel compounds exhibiting even lower affinity for the A2A–D2 receptor heteromer (striatal postsynaptic adenosine A2A receptors), whereas keeping a significant affinity for A1–A2A receptor heteromers (striatal presynaptic adenosine A2A receptors). The same DDS approach could be used to find adenosine A2A receptor antagonists useful for Parkinson’s disease that would have preferential affinity for the A2A–D2 versus the A1–A2A receptor heteromer or that would have stronger allosteric modulatory effects of the affinity or intrinsic efficacy of dopamine D2 receptor agonists. Other receptor-heteromer selective compounds obtained by the DDS approach include the putative D1–D2 receptor heteromer compound SKF-83959 [6-chloro-7,8-dihydroxy-3-methyl-1-(3-methylphenyl)-2,3,4,5-tetrahydro-1H-3-benzazepine] (Rashid et al., 2007; but see Chun et al., 2013) and clozapine, which not only acts as a potent serotonin 5-HT2A receptor antagonist but also increases the intrinsic efficacy of glutamate in the mGlu2–5-HT2A receptor heteromer (Fribourg et al., 2011).
DDS using bivalent ligands constitutes another approach for targeting receptor-heteromers (recently reviewed in Hiller et al., 2013). A bivalent ligand consists of two pharmacophoric entities linked by an appropriate spacer. In principle, it should be possible to target GPCR heteromers by selection of adequate, potent, and receptor-selective pharmacophores. The proof of principle came from the work of Daniels et al. (2005), who reported the effects of bivalent ligands incorporating the pharmacophores of the μ-opioid receptor agonist oxymorphone and the δ-opioid receptor antagonist naltrindole tethered by spacers of different lengths to selectively label δ-μ-opioid receptor heteromers. Only ligands with appropriate spacer length produced no tolerance to their analgesic effects (Daniels et al., 2005). Absence of tolerance was therefore most likely due to the appropriate tethering of the μ-opioid receptor agonist and δ-opioid receptor antagonist and again supported the involvement of δ-μ-opioid receptor heteromer in the tolerance to the analgesic effects of opioids. The same research group recently published results that suggest that some bivalent ligands can very selectively interact with the putative μ-opioid–glutamate metabotropic mGlu5 receptor heteromer, with potent antinociceptive effects (Akgun et al., 2013). Although clinical application of these big molecules might not be feasible for pharmacokinetic reasons, the same as any receptor-heteromer selective compound, bivalent ligands can provide important tools for the identification and study of the functional significance of receptor heteromers.
HTS is the most current initial strategy in the process of drug development. Availability of a simple, quick, and robust assay is critical for enabling HTS (reviewed in Franco et al., 2010). A frequently used assay for initial screens is the second-messenger cell-based assay using promiscuous G protein that is able to elicit a calcium response. Another common strategy is based on β-arrestin translocation that occurs as a result of receptor activation-dependent recruitment of β-arrestins. This latter approach was recently used for the identification of δ-μ-opioid receptor heteromer selective compounds, because a previous study suggested that this heteromer preferentially recruits β-arrestin (Rozenfeld and Devi, 2007). Screening of a small-molecule library was performed with the β-arrestin recruitment assay in cell lines expressing δ-μ-opioid receptor heteromers; the hits were subject to a secondary screening using cells expressing only δ-opioid or μ-opioid receptors (Gomes et al., 2013b). The compound named CYM51010 [ethyl 1-[4-(acetylamino)benzyl]-4-(2-phenylethyl)piperidine-4-carboxylate] was identified as a ligand that preferentially activates δ-μ-opioid receptor heteromer (Gomes et al., 2013b). The compound showed strong intrinsic efficacy not only for β-arrestin but also for G protein–mediated signaling [guanosine 5′-3-O-(thio)triphosphate binding]. The δ-μ-opioid receptor-heteromer selective antibody was able to block not only the CYM51010-mediated signaling in heterologous cells, but also intrathecal analgesia in a rodent model of thermal pain (Gomes et al., 2013b). The significant intrinsic efficacy of the compound for δ-μ-opioid receptor–induced β-arrestin and G protein–mediated signaling (compared with morphine) makes CYM51010 an attractive candidate for the development of a new analgesic with lower tolerance than morphine. In fact, using an in vivo assay of analgesia (tail-flick assay), CYM51010 was found to exert similar antinociceptive effects than morphine but with less tolerance (Gomes et al., 2013b). Compounds such as these that selectively target the heteromers are likely to serve as valuable tools to explore the physiologic role of heteromers in biologic systems.
In summary, GPCR heteromers offer very promising new targets for drug development. First, they tend to exhibit unique localization (for instance a type-specific neuron in the brain). Second, their distinct allosteric properties provide a framework for saturability, ligand selectivity, and functional selectivity. These are all properties that are now eagerly pursued in possibly new marketable compounds. Very few studies have addressed the search for receptor-heteromer selective compounds, which is quite surprising given the continuous discovery of functionally significant receptor heteromers (for recent review, see Gomes et al., 2013a). Compounds or reagents (monovalent antibodies or membrane permeant peptides) specifically targeting the heteromer interface are likely not only to serve as much needed tools to explore the physiologic significance of GPCR heteromers, but also serve as "leads" for development of highly selective drugs for the treatment of a variety of disorders. We provide some guidelines for the application of DDS and HTS approaches to the discovery of receptor-heteromer selective compounds. This should definitively serve as a key element in GPCR drug discovery programs.
Acknowledgments
This article was written as a result of a round-table meeting of the coauthors organized by Esteve Foundation in S’Agaro (Girona, Spain) in May 2013.
Abbreviations
- A
concentration of radioligand
- B
concentration of the nonradioactive competing compound in radioligand binding competition experiments
- BRET
bioluminescence resonance energy transfer
- CYM51010
ethyl 1-[4-(acetylamino)benzyl]-4-(2-phenylethyl)piperidine-4-carboxylate
- CGS 21680
4-[2-[[6-amino-9-(N-ethyl-β-D-ribofuranuronamidosyl)-9H-purin-2-yl]amino]ethyl]benzenepropanoic acid
- DAB
dimer radioligand/competitor modulation index in radioligand competition experiments
- DC
dimer cooperativity index in radioligand binding saturation experiments
- DCB
dimer cooperativity index for the competing ligand in radioligand binding competition experiments
- DDS
discrete direct screening
- FCS
fluorescence correlation spectroscopy
- FRET
fluorescence resonance energy transfer
- GPCR
G protein–coupled receptor
- GRK
GPCR kinase
- H8
helix 8
- HTS
high-throughput screening
- KD1 and KD2
macroscopic equilibrium dissociation constants for the binding of A to the first and second receptor in the receptor homodimer in radioligand binding saturation experiments
- KDA1 and KDA2
macroscopic equilibrium dissociation constants for the binding of A to the first and second receptor in the receptor homodimer in radioligand binding competition experiments
- KDAB
equilibrium dissociation constant of B on a receptor homodimer semi-occupied by A in radioligand binding competition experiments
- KDB1 and KDB2
macroscopic equilibrium dissociation constants for the binding of B to the first and second receptor in the receptor homodimer in radioligand binding competition experiments
- KDBA
equilibrium dissociation constant of A binding to a receptor homodimer semi-occupied by B in radioligand binding competition experiments
- MAPK
mitogen-activated protein kinase
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MSX-3
3,7-dihydro-8-[(1E)-2-(3-methoxyphenyl)ethenyl]-7-methyl-3-[3-(phosphonooxy)propyl-1-(2-propynyl)-1H-purine-2,6-dione
- OTA
d(CH2)5[Tyr(Me)2-Thr4-Orn8-Tyr(NH2)9]-oxytocin
- PLA
proximity ligation assay
- RGS20
protein that regulates the speed of G protein signal transduction
- RT
total amount of receptor dimers
- SCH-442416
2-(2-furanyl)-7-[3-(4-methoxyphenyl)propyl]-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine
- SKF-83959
6-chloro-7,8-dihydroxy-3-methyl-1-(3-methylphenyl)-2,3,4,5-tetrahydro-1H-3-benzazepine
- TIR-FM
total internal reflection fluorescence microscopy
- TM
transmembrane domain
- ZM-241385
4-(2-[7-amino-2-(2-furyl)[1,2,4]triazolo[2,3-a][1,3,5]triazin-5-ylamino]ethyl)phenol
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Ferré, Casadó, Devi, Filizola, Jockers, Lohse, Milligan, Pin, Guitart.
Footnotes
This work was supported by the Intramural Research Program of the National Institutes of Health [National Institute on Drug Abuse] (to S.F. and X.G.); the National Institutes of Health National Institute on Drug Abuse [Grants DA026434 and DA034049 (to M.F.) and Grants DA008863 and DA019521 (to L.A.D.)]; “Ministerio de Ciencia y Tecnología” [Grant SAF2011-23813], “Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas” [Grant PI2011/02-7], and “Fondation Jerome Lejeune” [FJL-01/01/2013] (to V.C.); “Agence Nationale de la Recherche” [Grants ANR RPIB 2012 “MED-HET-REC-2”, ANR-11-IDEX-0005-01, and ANR-11-LABX-0071 (to R.J.) and ANR-12-BSV2-0015 and ARN-09-BIOT-018 (to J.-P.P.)]; “Fondation Recherche Médicale” [Grant FRM DEQ20130326503], “Institut National de la Santé et de la Recherche Médicale,” and “Centre National de la Recherche Scientifique” (to R.J.); European Research Council Advanced Grant “Topas” (to M.J.L.); and Medical Research Council [Grant G0900050] (to G.M.).
References
- Agnati LF, Fuxe K, Ferré S. (2005) How receptor mosaics decode transmitter signals. Possible relevance of cooperativity. Trends Biochem Sci 30:188–193 [DOI] [PubMed] [Google Scholar]
- Akgün E, Javed MI, Lunzer MM, Smeester BA, Beitz AJ, Portoghese PS. (2013) Ligands that interact with putative MOR-mGluR5 heteromer in mice with inflammatory pain produce potent antinociception. Proc Natl Acad Sci USA 110:11595–11599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albizu L, Balestre MN, Breton C, Pin JP, Manning M, Mouillac B, Barberis C, Durroux T. (2006) Probing the existence of G protein-coupled receptor dimers by positive and negative ligand-dependent cooperative binding. Mol Pharmacol 70:1783–1791 [DOI] [PubMed] [Google Scholar]
- Albizu L, Cottet M, Kralikova M, Stoev S, Seyer R, Brabet I, Roux T, Bazin H, Bourrier E, Lamarque L, et al. (2010) Time-resolved FRET between GPCR ligands reveals oligomers in native tissues. Nat Chem Biol 6:587–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armentero MT, Pinna A, Ferré S, Lanciego JL, Müller CE, Franco R. (2011) Past, present and future of A(2A) adenosine receptor antagonists in the therapy of Parkinson’s disease. Pharmacol Ther 132:280–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azdad K, Gall D, Woods AS, Ledent C, Ferré S, Schiffmann SN. (2009) Dopamine D2 and adenosine A2A receptors regulate NMDA-mediated excitation in accumbens neurons through A2A-D2 receptor heteromerization. Neuropsychopharmacology 34:972–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba K, Benleulmi-Chaachoua A, Journé AS, Kamal M, Guillaume JL, Dussaud S, Gbahou F, Yettou K, Liu C, Contreras-Alcantara S, et al. (2013) Heteromeric MT1/MT2 melatonin receptors modulate photoreceptor function. Sci Signal 6:ra89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacart J, Corbel C, Jockers R, Bach S, Couturier C. (2008) The BRET technology and its application to screening assays. Biotechnol J 3:311–324 [DOI] [PubMed] [Google Scholar]
- Banères JL, Parello J. (2003) Structure-based analysis of GPCR function: evidence for a novel pentameric assembly between the dimeric leukotriene B4 receptor BLT1 and the G-protein. J Mol Biol 329:815–829 [DOI] [PubMed] [Google Scholar]
- Bayburt TH, Leitz AJ, Xie G, Oprian DD, Sligar SG. (2007) Transducin activation by nanoscale lipid bilayers containing one and two rhodopsins. J Biol Chem 282:14875–14881 [DOI] [PubMed] [Google Scholar]
- Bayburt TH, Vishnivetskiy SA, McLean MA, Morizumi T, Huang CC, Tesmer JJ, Ernst OP, Sligar SG, Gurevich VV. (2011) Monomeric rhodopsin is sufficient for normal rhodopsin kinase (GRK1) phosphorylation and arrestin-1 binding. J Biol Chem 286:1420–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg KA, Rowan MP, Gupta A, Sanchez TA, Silva M, Gomes I, McGuire BA, Portoghese PS, Hargreaves KM, Devi LA, et al. (2012) Allosteric interactions between δ and κ opioid receptors in peripheral sensory neurons. Mol Pharmacol 81:264–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bézard E, Ferry S, Mach U, Stark H, Leriche L, Boraud T, Gross C, Sokoloff P. (2003) Attenuation of levodopa-induced dyskinesia by normalizing dopamine D3 receptor function. Nat Med 9:762–767 [DOI] [PubMed] [Google Scholar]
- Borea PA, Dalpiaz A, Varani K, Guerra L, Gilli G. (1995) Binding thermodynamics of adenosine A2a receptor ligands. Biochem Pharmacol 49:461–469 [DOI] [PubMed] [Google Scholar]
- Bouvier M. (2001) Oligomerization of G-protein-coupled transmitter receptors. Nat Rev Neurosci 2:274–286 [DOI] [PubMed] [Google Scholar]
- Brea J, Castro M, Giraldo J, López-Giménez JF, Padín JF, Quintián F, Cadavid MI, Vilaró MT, Mengod G, Berg KA, et al. (2009) Evidence for distinct antagonist-revealed functional states of 5-hydroxytryptamine(2A) receptor homodimers. Mol Pharmacol 75:1380–1391 [DOI] [PubMed] [Google Scholar]
- Bulenger S, Marullo S, Bouvier M. (2005) Emerging role of homo- and heterodimerization in G-protein-coupled receptor biosynthesis and maturation. Trends Pharmacol Sci 26:131–137 [DOI] [PubMed] [Google Scholar]
- Bürgisser E, De Lean A, Lefkowitz RJ. (1982) Reciprocal modulation of agonist and antagonist binding to muscarinic cholinergic receptor by guanine nucleotide. Proc Natl Acad Sci USA 79:1732–1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushlin I, Gupta A, Stockton SD, Jr, Miller LK, Devi LA. (2012) Dimerization with cannabinoid receptors allosterically modulates delta opioid receptor activity during neuropathic pain. PLoS ONE 7:e49789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabello N, Gandía J, Bertarelli DC, Watanabe M, Lluís C, Franco R, Ferré S, Luján R, Ciruela F. (2009) Metabotropic glutamate type 5, dopamine D2 and adenosine A2a receptors form higher-order oligomers in living cells. J Neurochem 109:1497–1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calebiro D, Rieken F, Wagner J, Sungkaworn T, Zabel U, Borzi A, Cocucci E, Zürn A, Lohse MJ. (2013) Single-molecule analysis of fluorescently labeled G-protein-coupled receptors reveals complexes with distinct dynamics and organization. Proc Natl Acad Sci USA 110:743–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carriba P, Navarro G, Ciruela F, Ferré S, Casadó V, Agnati L, Cortés A, Mallol J, Fuxe K, Canela EI, et al. (2008) Detection of heteromerization of more than two proteins by sequential BRET-FRET. Nat Methods 5:727–733 [DOI] [PubMed] [Google Scholar]
- Casadó V, Cortés A, Ciruela F, Mallol J, Ferré S, Lluis C, Canela EI, Franco R. (2007) Old and new ways to calculate the affinity of agonists and antagonists interacting with G-protein-coupled monomeric and dimeric receptors: the receptor-dimer cooperativity index. Pharmacol Ther 116:343–354 [DOI] [PubMed] [Google Scholar]
- Casadó V, Cortés A, Mallol J, Pérez-Capote K, Ferré S, Lluis C, Franco R, Canela EI. (2009b) GPCR homomers and heteromers: a better choice as targets for drug development than GPCR monomers? Pharmacol Ther 124:248–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadó V, Ferrada C, Bonaventura J, Gracia E, Mallol J, Canela EI, Lluís C, Cortés A, Franco R. (2009a) Useful pharmacological parameters for G-protein-coupled receptor homodimers obtained from competition experiments. Agonist-antagonist binding modulation. Biochem Pharmacol 78:1456–1463 [DOI] [PubMed] [Google Scholar]
- Chabre M, le Maire M. (2005) Monomeric G-protein-coupled receptor as a functional unit. Biochemistry 44:9395–9403 [DOI] [PubMed] [Google Scholar]
- Christopoulos A, Kenakin T. (2002) G protein-coupled receptor allosterism and complexing. Pharmacol Rev 54:323–374 [DOI] [PubMed] [Google Scholar]
- Chun LS, Free RB, Doyle TB, Huang XP, Rankin ML, Sibley DR. (2013) D1-D2 dopamine receptor synergy promotes calcium signaling via multiple mechanisms. Mol Pharmacol 84:190–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciruela F, Burgueño J, Casadó V, Canals M, Marcellino D, Goldberg SR, Bader M, Fuxe K, Agnati LF, Lluis C, et al. (2004) Combining mass spectrometry and pull-down techniques for the study of receptor heteromerization. Direct epitope-epitope electrostatic interactions between adenosine A2A and dopamine D2 receptors. Anal Chem 76:5354–5363 [DOI] [PubMed] [Google Scholar]
- Ciruela F, Casadó V, Rodrigues RJ, Luján R, Burgueño J, Canals M, Borycz J, Rebola N, Goldberg SR, Mallol J, et al. (2006) Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J Neurosci 26:2080–2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Christopoulos A, Lindsley CW. (2009) Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov 8:41–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wei LN, Müller JD. (2003) Probing protein oligomerization in living cells with fluorescence fluctuation spectroscopy. Proc Natl Acad Sci USA 100:15492–15497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comps-Agrar L, Kniazeff J, Brock C, Trinquet E, Pin JP. (2012) Stability of GABAB receptor oligomers revealed by dual TR-FRET and drug-induced cell surface targeting. FASEB J 26:3430–3439 [DOI] [PubMed] [Google Scholar]
- Comps-Agrar L, Kniazeff J, Nørskov-Lauritsen L, Maurel D, Gassmann M, Gregor N, Prézeau L, Bettler B, Durroux T, Trinquet E, et al. (2011) The oligomeric state sets GABA(B) receptor signalling efficacy. EMBO J 30:2336–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels DJ, Lenard NR, Etienne CL, Law PY, Roerig SC, Portoghese PS. (2005) Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proc Natl Acad Sci USA 102:19208–19213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lean A, Kilpatrick BF, Caron MG. (1982) Dopamine receptor of the porcine anterior pituitary gland. Evidence for two affinity states discriminated by both agonists and antagonists. Mol Pharmacol 22:290–297 [PubMed] [Google Scholar]
- De Lean A, Stadel JM, Lefkowitz RJ. (1980) A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J Biol Chem 255:7108–7117 [PubMed] [Google Scholar]
- Doumazane E, Scholler P, Zwier JM, Trinquet E, Rondard P, Pin JP. (2011) A new approach to analyze cell surface protein complexes reveals specific heterodimeric metabotropic glutamate receptors. FASEB J 25:66–77 [DOI] [PubMed] [Google Scholar]
- Durroux T. (2005) Principles: a model for the allosteric interactions between ligand binding sites within a dimeric GPCR. Trends Pharmacol Sci 26:376–384 [DOI] [PubMed] [Google Scholar]
- El Moustaine D, Granier S, Doumazane E, Scholler P, Rahmeh R, Bron P, Mouillac B, Banères JL, Rondard P, Pin JP. (2012) Distinct roles of metabotropic glutamate receptor dimerization in agonist activation and G-protein coupling. Proc Natl Acad Sci USA 109:16342–16347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst OP, Gramse V, Kolbe M, Hofmann KP, Heck M. (2007) Monomeric G protein-coupled receptor rhodopsin in solution activates its G protein transducin at the diffusion limit. Proc Natl Acad Sci USA 104:10859–10864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrada C, Moreno E, Casadó V, Bongers G, Cortés A, Mallol J, Canela EI, Leurs R, Ferré S, Lluís C, et al. (2009) Marked changes in signal transduction upon heteromerization of dopamine D1 and histamine H3 receptors. Br J Pharmacol 157:64–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S. (1997) Adenosine-dopamine interactions in the ventral striatum. Implications for the treatment of schizophrenia. Psychopharmacology (Berl) 133:107–120 [DOI] [PubMed] [Google Scholar]
- Ferré S, Baler R, Bouvier M, Caron MG, Devi LA, Durroux T, Fuxe K, George SR, Javitch JA, Lohse MJ, et al. (2009) Building a new conceptual framework for receptor heteromers. Nat Chem Biol 5:131–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Ciruela F, Woods AS, Lluis C, Franco R. (2007) Functional relevance of neurotransmitter receptor heteromers in the central nervous system. Trends Neurosci 30:440–446 [DOI] [PubMed] [Google Scholar]
- Ferré S, Lluis C, Lanciego JL, Franco R. (2010) Prime time for G-protein-coupled receptor heteromers as therapeutic targets for CNS disorders: the dopamine D₁-D₃ receptor heteromer. CNS Neurol Disord Drug Targets 9:596–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Quiroz C, Woods AS, Cunha R, Popoli P, Ciruela F, Lluis C, Franco R, Azdad K, Schiffmann SN. (2008) An update on adenosine A2A-dopamine D2 receptor interactions: implications for the function of G protein-coupled receptors. Curr Pharm Des 14:1468–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorentini C, Busi C, Gorruso E, Gotti C, Spano P, Missale C. (2008) Reciprocal regulation of dopamine D1 and D3 receptor function and trafficking by heterodimerization. Mol Pharmacol 74:59–69 [DOI] [PubMed] [Google Scholar]
- Fotiadis D, Liang Y, Filipek S, Saperstein DA, Engel A, Palczewski K. (2003) Atomic-force microscopy: Rhodopsin dimers in native disc membranes. Nature 421:127–128 [DOI] [PubMed] [Google Scholar]
- Fotiadis D, Liang Y, Filipek S, Saperstein DA, Engel A, Palczewski K. (2004) The G protein-coupled receptor rhodopsin in the native membrane. FEBS Lett 564:281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R, Canela EI, Casadó V, Ferré S. (2010) Platforms for the identification of GPCR targets, and of orthosteric and allosteric modulators. Expert Opin Drug Discov 5:391–403 [DOI] [PubMed] [Google Scholar]
- Franco R, Casadó V, Mallol J, Ferrada C, Ferré S, Fuxe K, Cortés A, Ciruela F, Lluis C, Canela EI. (2006) The two-state dimer receptor model: a general model for receptor dimers. Mol Pharmacol 69:1905–1912 [DOI] [PubMed] [Google Scholar]
- Franco R, Casadó V, Mallol J, Ferré S, Fuxe K, Cortés A, Ciruela F, Lluis C, Canela EI. (2005) Dimer-based model for heptaspanning membrane receptors. Trends Biochem Sci 30:360–366 [DOI] [PubMed] [Google Scholar]
- Fribourg M, Moreno JL, Holloway T, Provasi D, Baki L, Mahajan R, Park G, Adney SK, Hatcher C, Eltit JM, et al. (2011) Decoding the signaling of a GPCR heteromeric complex reveals a unifying mechanism of action of antipsychotic drugs. Cell 147:1011–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung JJ, Deupi X, Pardo L, Yao XJ, Velez-Ruiz GA, Devree BT, Sunahara RK, Kobilka BK. (2009) Ligand-regulated oligomerization of beta(2)-adrenoceptors in a model lipid bilayer. EMBO J 28:3315–3328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavalas A, Lan TH, Liu Q, Corrêa IR, Jr, Javitch JA, Lambert NA. (2013) Segregation of family A G protein-coupled receptor protomers in the plasma membrane. Mol Pharmacol 84:346–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandía J, Lluís C, Ferré S, Franco R, Ciruela F. (2008) Light resonance energy transfer-based methods in the study of G protein-coupled receptor oligomerization. Bioessays 30:82–89 [DOI] [PubMed] [Google Scholar]
- Giraldo J. (2013) Modeling cooperativity effects in dimeric G protein-coupled receptors. Prog Mol Biol Transl Sci 115:349–373 [DOI] [PubMed] [Google Scholar]
- Golebiewska U, Johnston JM, Devi L, Filizola M, Scarlata S. (2011) Differential response to morphine of the oligomeric state of μ-opioid in the presence of δ-opioid receptors. Biochemistry 50:2829–2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Fujita W, Chandrakala MV, Devi LA. (2013a) Disease-specific heteromerization of G-protein-coupled receptors that target drugs of abuse. Prog Mol Biol Transl Sci 117:207–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Fujita W, Gupta A, Saldanha SA, Negri A, Pinello CE, Roberts E, Filizola M, Hodder P, Devi LA. (2013b) Identification of a μ-δ opioid receptor heteromer-biased agonist with antinociceptive activity. Proc Natl Acad Sci USA 110:12072–12077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Gupta A, Devi LA. (2013c) G-protein-coupled heteromers: regulation in disease. Methods Enzymol 521:219–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Gupta A, Filipovska J, Szeto HH, Pintar JE, Devi LA. (2004) A role for heterodimerization of mu and delta opiate receptors in enhancing morphine analgesia. Proc Natl Acad Sci USA 101:5135–5139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Ijzerman AP, Ye K, Maillet EL, Devi LA. (2011) G protein-coupled receptor heteromerization: a role in allosteric modulation of ligand binding. Mol Pharmacol 79:1044–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González S, Rangel-Barajas C, Peper M, Lorenzo R, Moreno E, Ciruela F, Borycz J, Ortiz J, Lluís C, Franco R, et al. (2012) Dopamine D4 receptor, but not the ADHD-associated D4.7 variant, forms functional heteromers with the dopamine D2S receptor in the brain. Mol Psychiatry 17:650–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan XM, Yu H, Palyha OC, McKee KK, Feighner SD, Sirinathsinghji DJ, Smith RG, Van der Ploeg LH, Howard AD. (1997) Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Brain Res Mol Brain Res 48:23–29 [DOI] [PubMed] [Google Scholar]
- Guo W, Shi L, Filizola M, Weinstein H, Javitch JA. (2005) Crosstalk in G protein-coupled receptors: changes at the transmembrane homodimer interface determine activation. Proc Natl Acad Sci USA 102:17495–17500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Urizar E, Kralikova M, Mobarec JC, Shi L, Filizola M, Javitch JA. (2008) Dopamine D2 receptors form higher order oligomers at physiological expression levels. EMBO J 27:2293–2304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Mulder J, Gomes I, Rozenfeld R, Bushlin I, Ong E, Lim M, Maillet E, Junek M, Cahill CM, et al. (2010) Increased abundance of opioid receptor heteromers after chronic morphine administration. Sci Signal 3:ra54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Moreira IS, Urizar E, Weinstein H, Javitch JA. (2009) Allosteric communication between protomers of dopamine class A GPCR dimers modulates activation. Nat Chem Biol 5:688–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasbi A, O’Dowd BF, George SR. (2011) Dopamine D1-D2 receptor heteromer signaling pathway in the brain: emerging physiological relevance. Mol Brain 4:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert TE, Moffett S, Morello JP, Loisel TP, Bichet DG, Barret C, Bouvier MA. (1996) A peptide derived from a beta2-adrenergic receptor transmembrane domain inhibits both receptor dimerization and activation. J Biol Chem 271:16384–16392 [DOI] [PubMed] [Google Scholar]
- Hern JA, Baig AH, Mashanov GI, Birdsall B, Corrie JE, Lazareno S, Molloy JE, Birdsall NJ. (2010) Formation and dissociation of M1 muscarinic receptor dimers seen by total internal reflection fluorescence imaging of single molecules. Proc Natl Acad Sci USA 107:2693–2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann R, Heck M, Henklein P, Henklein P, Kleuss C, Hofmann KP, Ernst OP. (2004) Sequence of interactions in receptor-G protein coupling. J Biol Chem 279:24283–24290 [DOI] [PubMed] [Google Scholar]
- Herrick-Davis K, Grinde E, Cowan A, Mazurkiewicz JE. (2013) Fluorescence correlation spectroscopy analysis of serotonin, adrenergic, muscarinic, and dopamine receptor dimerization: the oligomer number puzzle. Mol Pharmacol 84:630–642 10.1124/mol.113.087072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller C, Kühhorn J, Gmeiner P.(2013) Class A G-protein-coupled receptor (GPCR) dimers and bivalent ligands. J Med Chem 56:6542–6559 [DOI] [PubMed] [Google Scholar]
- Hillion J, Canals M, Torvinen M, Casadó V, Scott R, Terasmaa A, Hansson A, Watson S, Olah ME, Mallol J, et al. (2002) Coaggregation, cointernalization, and codesensitization of adenosine A2A receptors and dopamine D2 receptors. J Biol Chem 277:18091–18097 [DOI] [PubMed] [Google Scholar]
- Hlavackova V, Goudet C, Kniazeff J, Zikova A, Maurel D, Vol C, Trojanova J, Prézeau L, Pin JP, Blahos J. (2005) Evidence for a single heptahelical domain being turned on upon activation of a dimeric GPCR. EMBO J 24:499–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Thor D, Zhou Y, Liu T, Wang Y, McMillin SM, Mistry R, Challiss RA, Costanzi S, Wess J. (2012) Structural aspects of M₃ muscarinic acetylcholine receptor dimer formation and activation. FASEB J 26:604–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Chen S, Zhang JJ, Huang XY. (2013) Crystal structure of oligomeric β1-adrenergic G protein-coupled receptors in ligand-free basal state. Nat Struct Mol Biol 20:419–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis MF, Schulz R, Hutchison AJ, Do UH, Sills MA, Williams M. (1989) [3H]CGS 21680, a selective A2 adenosine receptor agonist directly labels A2 receptors in rat brain. J Pharmacol Exp Ther 251:888–893 [PubMed] [Google Scholar]
- Johnston JM, Wang H, Provasi D, Filizola M. (2012) Assessing the relative stability of dimer interfaces in g protein-coupled receptors. PLOS Comput Biol 8:e1002649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan BA, Gomes I, Rios C, Filipovska J, Devi LA. (2003) Functional interactions between mu opioid and alpha 2A-adrenergic receptors. Mol Pharmacol 64:1317–1324 [DOI] [PubMed] [Google Scholar]
- Justinová Z, Ferré S, Redhi GH, Mascia P, Stroik J, Quarta D, Yasar S, Müller CE, Franco R, Goldberg SR. (2011) Reinforcing and neurochemical effects of cannabinoid CB1 receptor agonists, but not cocaine, are altered by an adenosine A2A receptor antagonist. Addict Biol 16:405–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai RS, Suzuki KG, Prossnitz ER, Koyama-Honda I, Nakada C, Fujiwara TK, Kusumi A. (2011) Full characterization of GPCR monomer-dimer dynamic equilibrium by single molecule imaging. J Cell Biol 192:463–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T, Miller LJ. (2010) Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev 62:265–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern A, Albarran-Zeckler R, Walsh HE, Smith RG. (2012) Apo-ghrelin receptor forms heteromers with DRD2 in hypothalamic neurons and is essential for anorexigenic effects of DRD2 agonism. Neuron 73:317–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khelashvili G, Dorff K, Shan J, Camacho-Artacho M, Skrabanek L, Vroling B, Bouvier M, Devi LA, George SR, Javitch JA, et al. (2010) GPCR-OKB: the G Protein-Coupled Receptor Oligomer Knowledge Base. Bioinformatics 26:1804–1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotz KN, Keil R, Zimmer FJ, Schwabe U. (1990) Guanine nucleotide effects on 8-cyclopentyl-1,3-[3H]dipropylxanthine binding to membrane-bound and solubilized A1 adenosine receptors of rat brain. J Neurochem 54:1988–1994 [DOI] [PubMed] [Google Scholar]
- Knepp AM, Periole X, Marrink SJ, Sakmar TP, Huber T. (2012) Rhodopsin forms a dimer with cytoplasmic helix 8 contacts in native membranes. Biochemistry 51:1819–1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kniazeff J, Bessis AS, Maurel D, Ansanay H, Prézeau L, Pin JP. (2004) Closed state of both binding domains of homodimeric mGlu receptors is required for full activity. Nat Struct Mol Biol 11:706–713 [DOI] [PubMed] [Google Scholar]
- Kniazeff J, Prézeau L, Rondard P, Pin JP, Goudet C. (2011) Dimers and beyond: The functional puzzles of class C GPCRs. Pharmacol Ther 130:9–25 [DOI] [PubMed] [Google Scholar]
- Kuszak AJ, Pitchiaya S, Anand JP, Mosberg HI, Walter NG, Sunahara RK. (2009) Purification and functional reconstitution of monomeric mu-opioid receptors: allosteric modulation of agonist binding by Gi2. J Biol Chem 284:26732–26741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levoye A, Dam J, Ayoub MA, Guillaume JL, Couturier C, Delagrange P, Jockers R. (2006a) The orphan GPR50 receptor specifically inhibits MT1 melatonin receptor function through heterodimerization. EMBO J 25:3012–3023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levoye A, Jockers R, Ayoub MA, Delagrange P, Savaskan E, Guillaume JL. (2006b) Are G protein-coupled receptor heterodimers of physiological relevance?—Focus on melatonin receptors. Chronobiol Int 23:419–426 [DOI] [PubMed] [Google Scholar]
- Liang Y, Fotiadis D, Filipek S, Saperstein DA, Palczewski K, Engel A. (2003) Organization of the G protein-coupled receptors rhodopsin and opsin in native membranes. J Biol Chem 278:21655–21662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancia F, Assur Z, Herman AG, Siegel R, Hendrickson WA. (2008) Ligand sensitivity in dimeric associations of the serotonin 5HT2c receptor. EMBO Rep 9:363–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. (2012) Crystal structure of the µ-opioid receptor bound to a morphinan antagonist. Nature 485:321–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcellino D, Ferré S, Casadó V, Cortés A, Le Foll B, Mazzola C, Drago F, Saur O, Stark H, Soriano A, et al. (2008) Identification of dopamine D1-D3 receptor heteromers. Indications for a role of synergistic D1-D3 receptor interactions in the striatum. J Biol Chem 283:26016–26025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurel D, Comps-Agrar L, Brock C, Rives ML, Bourrier E, Ayoub MA, Bazin H, Tinel N, Durroux T, Prézeau L, et al. (2008) Cell-surface protein-protein interaction analysis with time-resolved FRET and snap-tag technologies: application to GPCR oligomerization. Nat Methods 5:561–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice P, Daulat AM, Turecek R, Ivankova-Susankova K, Zamponi F, Kamal M, Clement N, Guillaume JL, Bettler B, Galès C, et al. (2010) Molecular organization and dynamics of the melatonin MT₁ receptor/RGS20/G(i) protein complex reveal asymmetry of receptor dimers for RGS and G(i) coupling. EMBO J 29:3646–3659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice P, Kamal M, Jockers R. (2011) Asymmetry of GPCR oligomers supports their functional relevance. Trends Pharmacol Sci 32:514–520 [DOI] [PubMed] [Google Scholar]
- May LT, Leach K, Sexton PM, Christopoulos A. (2007) Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol 47:1–51 [DOI] [PubMed] [Google Scholar]
- May LT, Bridge LJ, Stoddart LA, Briddon SJ, Hill SJ. (2011) Allosteric interactions across native adenosine-A3 receptor homodimers: quantification using single-cell ligand-binding kinetics. FASEB J 25:3465–3476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G. (2010) The role of dimerisation in the cellular trafficking of G-protein-coupled receptors. Curr Opin Pharmacol 10:23–29 [DOI] [PubMed] [Google Scholar]
- Milligan G. (2013) The prevalence, maintenance, and relevance of G protein-coupled receptor oligomerization. Mol Pharmacol 84:158-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G, Bouvier M. (2005) Methods to monitor the quaternary structure of G protein-coupled receptors. FEBS J 272:2914–2925 [DOI] [PubMed] [Google Scholar]
- Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. (1998) Dopamine receptors: from structure to function. Physiol Rev 78:189–225 [DOI] [PubMed] [Google Scholar]
- Moreno E, Moreno-Delgado D, Navarro G, Hoffmann HM, Fuentes S, Rosell-Vilar S, Gasperini P, Rodriguez-Ruiz M, Medrano M, Mallol J, et al. (2014) Cocaine disrupts histamine H3 receptor modulation of dopamine D1 receptor signaling via σ1 receptor: a key target for cocaine toxicity. J Neurosci (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modzelewska A, Filipek S, Palczewski K, Park PS. (2006) Arrestin interaction with rhodopsin: conceptual models. Cell Biochem Biophys 46:1–15 [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, Howlett AC. (2005). Chemically distinct ligands promote differential CB1 cannabinoid receptor-Gi protein interactions. Mol Pharmacol 67:2016–2024 [DOI] [PubMed] [Google Scholar]
- Navarro G, Ferré S, Cordomi A, Moreno E, Mallol J, Casadó V, Cortés A, Hoffmann H, Ortiz J, Canela EI, et al. (2010) Interactions between intracellular domains as key determinants of the quaternary structure and function of receptor heteromers. J Biol Chem 285:27346–27359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nenasheva TA, Neary M, Mashanov GI, Birdsall NJ, Breckenridge RA, Molloy JE. (2013) Abundance, distribution, mobility and oligomeric state of M₂ muscarinic acetylcholine receptors in live cardiac muscle. J Mol Cell Cardiol 57:129–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobles KN, Xiao K, Ahn S, Shukla AK, Lam CM, Rajagopal S, Strachan RT, Huang TY, Bressler EA, Hara MR, et al. (2011) Distinct phosphorylation sites on the β(2)-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci Signal 4:ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Dowd BF, Ji X, Nguyen T, George SR. (2012) Two amino acids in each of D1 and D2 dopamine receptor cytoplasmic regions are involved in D1-D2 heteromer formation. Biochem Biophys Res Commun 417:23–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Dowd BF, Nguyen T, Ji X, George SR. (2013) D5 dopamine receptor carboxyl tail involved in D5-D2 heteromer formation. Biochem Biophys Res Commun 431:586–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham WM, Hamm HE. (2008) Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol 9:60–71 [DOI] [PubMed] [Google Scholar]
- Orru M, Bakešová J, Brugarolas M, Quiroz C, Beaumont V, Goldberg SR, Lluís C, Cortés A, Franco R, Casadó V, et al. (2011a) Striatal pre- and postsynaptic profile of adenosine A(2A) receptor antagonists. PLoS ONE 6:e16088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrú M, Quiroz C, Guitart X, Ferré S. (2011b) Pharmacological evidence for different populations of postsynaptic adenosine A2A receptors in the rat striatum. Neuropharmacology 61:967–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. (2008) Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature 454:183–187 [DOI] [PubMed] [Google Scholar]
- Patowary S, Alvarez-Curto E, Xu TR, Holz JD, Oliver JA, Milligan G, Raicu V. (2013) The muscarinic M3 acetylcholine receptor exists as two differently sized complexes at the plasma membrane. Biochem J 452:303–312 [DOI] [PubMed] [Google Scholar]
- Pellissier LP, Barthet G, Gaven F, Cassier E, Trinquet E, Pin JP, Marin P, Dumuis A, Bockaert J, Banères JL, et al. (2011) G protein activation by serotonin type 4 receptor dimers: evidence that turning on two protomers is more efficient. J Biol Chem 286:9985–9997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pin JP, Comps-Agrar L, Maurel D, Monnier C, Rives ML, Trinquet E, Kniazeff J, Rondard P, Prézeau L. (2009) G-protein-coupled receptor oligomers: two or more for what? Lessons from mGlu and GABAB receptors. J Physiol 587:5337–5344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pin JP, Neubig R, Bouvier M, Devi L, Filizola M, Javitch JA, Lohse MJ, Milligan G, Palczewski K, Parmentier M, et al. (2007) International Union of Basic and Clinical Pharmacology. LXVII. Recommendations for the recognition and nomenclature of G protein-coupled receptor heteromultimers. Pharmacol Rev 59:5–13 [DOI] [PubMed] [Google Scholar]
- Quiroz C, Luján R, Uchigashima M, Simoes AP, Lerner TN, Borycz J, Kachroo A, Canas PM, Orru M, Schwarzschild MA, et al. (2009) Key modulatory role of presynaptic adenosine A2A receptors in cortical neurotransmission to the striatal direct pathway. ScientificWorldJournal 9:1321–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raehal KM, Schmid CL, Groer CE, Bohn LM. (2011) Functional selectivity at the μ-opioid receptor: implications for understanding opioid analgesia and tolerance. Pharmacol Rev 63:1001–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid AJ, O’Dowd BF, Verma V, George SR. (2007) Neuronal Gq/11-coupled dopamine receptors: an uncharted role for dopamine. Trends Pharmacol Sci 28:551–555 [DOI] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477:549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. (2012) Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu Rev Pharmacol Toxicol 52:179–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimondini R, Ferré S, Ogren SO, Fuxe K. (1997) Adenosine A2A agonists: a potential new type of atypical antipsychotic. Neuropsychopharmacology 17:82–91 [DOI] [PubMed] [Google Scholar]
- Rovira X, Vivó M, Serra J, Roche D, Strange PG, Giraldo J. (2009) Modelling the interdependence between the stoichiometry of receptor oligomerization and ligand binding for a coexisting dimer/tetramer receptor system. Br J Pharmacol 156:28–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenfeld R, Bushlin I, Gomes I, Tzavaras N, Gupta A, Neves S, Battini L, Gusella GL, Lachmann A, Ma’ayan A, et al. (2012) Receptor heteromerization expands the repertoire of cannabinoid signaling in rodent neurons. PLoS ONE 7:e29239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenfeld R, Devi LA. (2007) Receptor heterodimerization leads to a switch in signaling: beta-arrestin2-mediated ERK activation by mu-delta opioid receptor heterodimers. FASEB J 21:2455–2465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenfeld R, Devi LA. (2010) Receptor heteromerization and drug discovery. Trends Pharmacol Sci 31:124–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenfeld R, Gupta A, Gagnidze K, Lim MP, Gomes I, Lee-Ramos D, Nieto N, Devi LA. (2011) AT1R-CB₁R heteromerization reveals a new mechanism for the pathogenic properties of angiotensin II. EMBO J 30:2350–2363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruprecht JJ, Mielke T, Vogel R, Villa C, Schertler GF. (2004) Electron crystallography reveals the structure of metarhodopsin I. EMBO J 23:3609–3620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salahpour A, Angers S, Mercier JF, Lagacé M, Marullo S, Bouvier M. (2004) Homodimerization of the beta2-adrenergic receptor as a prerequisite for cell surface targeting. J Biol Chem 279:33390–33397 [DOI] [PubMed] [Google Scholar]
- Salom D, Lodowski DT, Stenkamp RE, Le Trong I, Golczak M, Jastrzebska B, Harris T, Ballesteros JA, Palczewski K. (2006) Crystal structure of a photoactivated deprotonated intermediate of rhodopsin. Proc Natl Acad Sci USA 103:16123–16128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler CW, Karcz-Kubicha M, Thorndike EB, Müller CE, Tella SR, Goldberg SR, Ferré S. (2004) Lack of adenosine A1 and dopamine D2 receptor-mediated modulation of the cardiovascular effects of the adenosine A2A receptor agonist CGS 21680. Eur J Pharmacol 484:269–275 [DOI] [PubMed] [Google Scholar]
- Smith NJ, Milligan G. (2010) Allostery at G protein-coupled receptor homo- and heteromers: uncharted pharmacological landscapes. Pharmacol Rev 62:701–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderberg O, Gullberg M, Jarvius M, Ridderstråle K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, et al. (2006) Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 3:995–1000 [DOI] [PubMed] [Google Scholar]
- Sommer ME, Hofmann KP, Heck M. (2012) Distinct loops in arrestin differentially regulate ligand binding within the GPCR opsin. Nat Commun 3:995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springael JY, Le Minh PN, Urizar E, Costagliola S, Vassart G, Parmentier M. (2006) Allosteric modulation of binding properties between units of chemokine receptor homo- and hetero-oligomers. Mol Pharmacol 69:1652–1661 [DOI] [PubMed] [Google Scholar]
- Staley JK, Mash DC. (1996) Adaptive increase in D3 dopamine receptors in the brain reward circuits of human cocaine fatalities. J Neurosci 16:6100–6106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadagaki K, Tudor D, Gbahou F, Tschische P, Waldhoer M, Bomsel M, Jockers R, Kamal M. (2012) Human cytomegalovirus-encoded UL33 and UL78 heteromerize with host CCR5 and CXCR4 impairing their HIV coreceptor activity. Blood 119:4908–4918 [DOI] [PubMed] [Google Scholar]
- Tobin AB, Butcher AJ, Kong KC. (2008) Location, location, location...site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol Sci 29:413–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifilieff P, Rives ML, Urizar E, Piskorowski RA, Vishwasrao HD, Castrillon J, Schmauss C, Slättman M, Gullberg M, Javitch JA. (2011) Detection of antigen interactions ex vivo by proximity ligation assay: endogenous dopamine D2-adenosine A2A receptor complexes in the striatum. Biotechniques 51:111–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto H, Sinha A, DeWitt M, Farrens DL. (2010) Monomeric rhodopsin is the minimal functional unit required for arrestin binding. J Mol Biol 399:501–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urizar E, Montanelli L, Loy T, Bonomi M, Swillens S, Gales C, Bouvier M, Smits G, Vassart G, Costagliola S. (2005) Glycoprotein hormone receptors: link between receptor homodimerization and negative cooperativity. EMBO J 24:1954–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urizar E, Yano H, Kolster R, Galés C, Lambert N, Javitch JA. (2011) CODA-RET reveals functional selectivity as a result of GPCR heteromerization. Nat Chem Biol 7:624–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilardaga JP, Nikolaev VO, Lorenz K, Ferrandon S, Zhuang Z, Lohse MJ. (2008) Conformational cross-talk between alpha2A-adrenergic and mu-opioid receptors controls cell signaling. Nat Chem Biol 4:126–131 [DOI] [PubMed] [Google Scholar]
- Visanji NP, Fox SH, Johnston T, Reyes G, Millan MJ, Brotchie JM. (2009) Dopamine D3 receptor stimulation underlies the development of L-DOPA-induced dyskinesia in animal models of Parkinson’s disease. Neurobiol Dis 35:184–192 [DOI] [PubMed] [Google Scholar]
- Wang C, Wu H, Katritch V, Han GW, Huang XP, Liu W, Siu FY, Roth BL, Cherezov V, Stevens RC. (2013) Structure of the human smoothened receptor bound to an antitumour agent. Nature 497:338–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward RJ, Pediani JD, Milligan G. (2011) Heteromultimerization of cannabinoid CB(1) receptor and orexin OX(1) receptor generates a unique complex in which both protomers are regulated by orexin A. J Biol Chem 286:37414–37428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton MR, Bokoch MP, Rasmussen SG, Huang B, Zare RN, Kobilka B, Sunahara RK. (2007) A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci USA 104:7682–7687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton MR, Jastrzebska B, Park PS, Fotiadis D, Engel A, Palczewski K, Sunahara RK. (2008) Efficient coupling of transducin to monomeric rhodopsin in a phospholipid bilayer. J Biol Chem 283:4387–4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods AS, Ferré S. (2005) Amazing stability of the arginine-phosphate electrostatic interaction. J Proteome Res 4:1397–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, et al. (2010) Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330:1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang XP, Carroll FI, et al. (2012) Structure of the human κ-opioid receptor in complex with JDTic. Nature 485:327–332 [DOI] [PMC free article] [PubMed] [Google Scholar]