

Abstract
Pharmacological treatment of any maternal illness during pregnancy warrants consideration of the consequences of the illness and/or medication for both the mother and unborn child. In the case of major depressive disorder, which affects up to 10–20% of pregnant women, the deleterious effects of untreated depression on the offspring can be profound and long lasting. Progress has been made in our understanding of the mechanism(s) of action of antidepressants, fetal exposure to these medications, and serotonin’s role in development. New technologies and careful study designs have enabled the accurate sampling of maternal serum, breast milk, umbilical cord serum, and infant serum psychotropic medication concentrations to characterize the magnitude of placental transfer and exposure through human breast milk. Despite this progress, the extant clinical literature is largely composed of case series, population-based patient registry data that are reliant on nonobjective means and retrospective recall to determine both medication and maternal depression exposure, and limited inclusion of suitable control groups for maternal depression. Conclusions drawn from such studies often fail to incorporate embryology/neurotransmitter ontogeny, appropriate gestational windows, or a critical discussion of statistically versus clinically significant. Similarly, preclinical studies have predominantly relied on dosing models, leading to exposures that may not be clinically relevant. The elucidation of a defined teratological effect or mechanism, if any, has yet to be conclusively demonstrated. The extant literature indicates that, in many cases, the benefits of antidepressant use during pregnancy for a depressed pregnant woman may outweigh potential risks.
I. Introduction
A. Historical Perspective
Modern psychopharmacology began in the early 1950s with the introduction of chlorpromazine, followed by other phenothiazines, and by the end of the decade, the introduction of the tricyclic antidepressants (TCA) that gave clinicians new and effective tools to treat mental health disorders. Regulatory agencies such as the U.S. Food and Drug Administration (FDA) were establishing standards for reproductive safety categories (Table 1) for the use of drugs during pregnancy, underscoring the need to consider acute morphologic effects as well as potential long-term adverse effects of perinatal medication exposure.
TABLE 1.
U.S. FDA Use-in-Pregnancy Ratings
| Category | Interpretation |
|---|---|
| A | Controlled studies show no risk: Adequate, well-controlled studies in pregnant women have failed to demonstrate risk to the fetus. |
| B | No evidence of risk in humans: Either animal findings show risk, but human findings do not; or, if no adequate human studies have been done, animal findings are negative. |
| C | Risk cannot be ruled out: Human studies are lacking, and animal studies are either positive for fetal risk or lacking as well. However, potential benefits may justify the potential risk. |
| D | Positive evidence of risk: Investigational or postmarketing data show risk to the fetus. Nevertheless, potential benefits may outweigh risks. |
| X | Contraindicated in pregnancy: Studies in animals or humans, or investigational or postmarketing reports, have shown fetal risk that clearly outweighs any possible benefit to the patient. |
Source: Food and Drug Administration (2011).
Tricyclic antidepressants (TCA) were first introduced in 1959 (imipramine), with the active desmethyl metabolite of imipramine, desipramine, introduced in 1964 (Drugs Co, 1965). The timing of the introduction of TCAs and potential effects of TCA exposure in pregnancy coincided with the timing of the thalidomide tragedy, and there were early reports of potential limb anomalies associated with amitriptyline. In animal studies, it was observed that imipramine may have produced fetal abnormalities in rabbits (Robson and Sullivan, 1963). Dr. William McBride, who first reported deformities due to thalidomide, began examining mothers and their infants and reported increased limb deformities associated with iminodibenzyl hydrochloride (associated with TCA use) during pregnancy (McBride, 1972). The article was reported by several press agencies including The Times of London (March 4, 1972), spurring a reaction from physicians reporting the safety of these medications during pregnancy in their patients (Crombie et al., 1972; Kuenssberg and Knox, 1972; Levy, 1972; Sim, 1972). Notably, at the same time, the Lithium Registry of Babies (Schou et al., 1973) reported a higher rate of a specific cardiac defect, Ebstein’s Anomaly associated with lithium. This is still taught as dogma to young clinicians despite additional studies and critical reviews (Cohen et al., 1994) showing a more modest risk. These experiences contributed to increased reluctance to treat mood disorders in pregnancy as well as a decreased tendency to less aggressively treat nonthreatening conditions such as nausea. In both cases (vide infra), recent evidence suggests that untreated psychiatric disorders or nausea are associated with risk.
In 1987, the first selective serotonin reuptake inhibitor (SSRI), fluoxetine, was introduced in the United States. In the subsequent 26 years, the SSRIs have essentially replaced the TCAs in North America, much of Europe, and modern Asia as first- and second-line treatments for depression. Their widespread use for a variety of mood and anxiety disorders has been accompanied by significant utilization in the perinatal period. A review of health care data bases indicated that >6% of women were prescribed an antidepressant at some point during pregnancy (Andrade et al., 2008). The reproductive safety of the SSRIs has undergone considerable scrutiny utilizing a variety of methodologies, and unfortunately, only the studies demonstrating potential risk are highly publicized relative to studies reporting their apparent safety or lack of observed untoward effects.
B. Role of Serotonin in Development
“Teratogenic agents act in specific ways (mechanisms) on developing cells and tissues to initiate sequences of abnormal developmental events (pathogenesis).”—James G. Wilson, Environment and Birth Defects, 1973
The serotonin transporter is a sodium-serotonin symporter that transports serotonin from the extracellular space into the cell (Talvenheimo et al., 1979; Hoffman et al., 1991; Masson et al., 1999). After vesicular release of serotonin into the synaptic cleft, serotonin binds to pre- and postsynaptic serotonin receptors to mediate serotonin innervation (Pineyro and Blier, 1999). The most commonly used pharmacological treatment of depression currently relies on selective-serotonin reuptake inhibitors that act by binding to the serotonin transporter and preventing uptake of serotonin into the presynaptic neuron (Stafford et al., 2001). Increased serotonin in the synaptic cleft and activation of serotonergic receptors or local paracrine actions on neuronal migration represent the most probable mechanism(s) of action to induce developmental effects on the fetus.
Serotonin has diverse ontogeneological functions in utero to guide development. These functions should be considered in the context of laboratory and clinical evidence as a mechanistic approach to investigate the effects of in utero exposure to antidepressants. Serotonin increases neurite outgrowth ex vivo in mouse embryo thalamic neurons (Lotto et al., 1999). It is likely that the mechanism of this neurite extension is via serotonin-mediated stimulation of S100β release from astrocytes through 5-HT1A agonism (Whitaker-Azmitia, 2001). Serotonin also plays an important role in axonal guidance, because disruption of serotonin availability in the forebrain can lead to abnormal thalamocortical axon trajectories (Bonnin et al., 2007, 2011). Additionally, a switch from a placental source of serotonin to an endogenous fetal source of serotonin occurs in the second trimester of mice (Bonnin et al., 2011). Therefore, disruption of serotonin signaling during these critical times of development may form the basis of any underlying long-term effects on the fetus.
Another possible mechanism of antidepressant-mediated effects in utero is through direct effects on the uterus and uterine blood flow. The 5-HT2B receptor has been examined in human uterine smooth muscle cells and agonists increase phosphoinositide hydrolysis, which may lead to smooth muscle cell contraction (Kelly and Sharif, 2006). Serotonin produces relaxation of the porcine oviduct (Inoue et al., 2003) and inhibits myometrial contractility (Kitazawa et al., 1998), a finding somewhat at odds with the data above. These effects are antagonized by mianserin, a 5-HT receptor antagonist. Vedernikov et al. (2000) isolated uterine rings of Sprague-Dawley rats on gestational (G) days 14 and 21 and used these rings for isometric tension recording and direct stimulation of the uterine rings with serotonergic compounds. These studies and others would indicate that the serotonergic system has some role in uterine musculature. Not surprisingly, no effect was observed for direct application of fluoxetine, imipramine, nortriptyline; however, 5-HT itself also had no effect on uterine contractile activity on isolated uterine rings from rats. Ex vivo experimentation on human placentas revealed that a high concentration of doxepin, which possess both transporter antagonism as well as potent receptor antagonism at multiple targets (e.g., adrenergic, muscarinic, and histaminergic), decreases nonneuronal acetylcholine release. Although the clinical relevancy of the concentrations used are not clear, this has been speculated to be a possible mechanism for some clinical findings reporting low birth weight due to prenatal antidepressant exposure, because acetylcholine may control vascularization and alter energy availability to the fetus (Wessler et al., 2007). However, similar studies have not been performed using other antidepressants. In a prospective study of uterine and umbilical artery blood flow at 20 weeks gestation, there were no effects of maternal depression and SSRIs on indices of uterine or umbilical artery blood flow. However, bupropion had a very modest effect on decreasing uterine blood flow (Monk et al., 2012).
Serotonin also plays a role in the cardiovascular system. Serotonin receptors 5-HT1A, 5-HT3, and 5-HT7 have been shown to play a physiologic role in parasympathetic regulation of the cardiovascular system (Ramage and Villalón, 2008). Therefore, it has been posited that interference of this system in utero may disrupt normal development of the cardiovascular system. Sloot et al. (2009) treated whole-rat embryos in culture with 12 monoamine transporter inhibitors. The group found both cardiac defects and major malformations when treating these embryos. However, the lowest concentration of drug used was 0.3 µg/ml, which is approximately five to six times human exposure (Sloot et al., 2009). This article was disputed as not indicative of teratogenicity, because the concentrations used are much higher than clinically relevant serum concentrations, and it is unclear whether this is a direct effect of the drug or via SERT antagonism (Brent, 2010; Scialli and Iannucci, 2010).
James Wilson (1973) has identified six principles of teratology, and if a xenobiotic is thought to be teratogenic, it should attempt to satisfy these criteria. One of these principles posits that a xenobiotic must act by a defined cellular mechanism to initiate abnormal developmental effects (Wilson, 1973). Current and future studies investigating in utero exposure of antidepressants must consider that if there is a teratogenic mechanism, presumably via increased synaptic 5-HT concentrations, it has not been thoroughly investigated or replicated.
II. Animal Exposure Studies
There are several requisite components in understanding the potential relevance of preclinical data for import into the clinical decision including: 1) comparison of developmental time points; 2) clinical relevance of exposure; and 3) outcomes of interest across species.
A. Translation of Development between Animals and Humans
Translating equivalent gestational time points of neurodevelopment across mammalian species is problematic because of a human fetus's rapid neural development in gestation compared with other animals (Gottlieb et al., 1977; Clancy et al., 2001). For comparison of rats and humans, different morphologic characteristics can be compared but there is no numeric relationship that could easily compare rat development and human development (e.g., 30 human years ≈ 1 rat year), and to do so would oversimplify the field of embryology. However, brain growth velocity peaks at birth for humans, whereas for rats, growth rate peaks on postnatal day (PND) 8, with further spurts of growth postnatally. In contrast, multiple markers of neural development in the hippocampus suggest that prenatal and postnatal development have remarkably similar temporal patterns in rats and humans (Avishai-Eliner et al., 2002). Because no ontogeneological data are available on the development of the prenatal serotonin system in humans, for the purpose of this review, it is reasonable to consider the first few postnatal weeks in rodents to be similar to the very end of human gestation (reviewed in (Watson et al., 2006). However, the in utero environment is not present during the first postnatal week of a rodent’s life, and therefore, exposure studies are complicated by difficulties in maintaining clinically relevant exposure of the offspring to medications following birth. Therefore, this review will focus on in utero animal exposure as a translatable approach to present findings from animal studies using these compounds (Tables 2 and 3).
TABLE 2.
Summary of endpoints after prenatal tricyclic/tetracyclic antidepressant exposure in animals
| Drug | Species | Dose (mg/kg per day) | Route | Exposure | Maternal Serum Monitored for Drug Conc. | 5-HT | NE | DA | Behavior | Cardio | Health | Endpoint | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CLO, IPD, MNS | Rat | 10 | SC | G6–G21 | No | + | + | + | ø | P25 | De Ceballos et al., 1985b | ||
| AMI | Rat | 10 | SC | G2–G21 | No | + | ø | P60 | Henderson et al., 1993 | ||||
| CLO, DSP, NOM | Rat | 10 | OG | G6–G21 | No | + | P25 | Montero et al., 1990 | |||||
| AMI | Rat | 10 | SC | G2–G21 | No | ø | ø | ø | P30, 60, 180 | Henderson et al., 1991 | |||
| CLO, IPD, TNP | Rat | 10 | SC | G6–G21 | No | ø | P25 | Montero et al., 1991 | |||||
| IMI | Rat | 15 | OG | G8–G20 | No | + | + | + | + | P14, 30 | Jason et al., 1981 | ||
| IMI | Rat | 0, 5, 10 | SC | G8–G20 | No | + | ø | + | ø | P1, 7 | Ali et al., 1986 | ||
| IMI | Rat | Unknown | DW | G0–P21 | No | ø | ø | P290 | Tonge, 1972, 1973 | ||||
| IMI | Rat | 0, 5, 10 | SC | G8–G20 | No | ø | + | ø | P4, 7 | Harmon et al., 1986 | |||
| CLO, NOM, IPR, MNS | Rat | 10 | SC | G6–G21 | No | + | + | P25 | De Ceballos et al., 1985a | ||||
| DSP | Rat | 10 | SC | G8–G20 | No | ø | ø | ø | P20 | Stewart et al., 1998 | |||
| IMI | Rat | 5 | OG | G14–G19 | No | + | + | P21 | Coyle, 1975 | ||||
| CLO | Rat | 3, 10, 30 | IP | G8–G21 | No | + | + | P35 | File and Tucker, 1983 | ||||
| DSP, MNS, VX | Rat | 1.25/5/10 | SC | G8–G20 | No | + | + | P23, 60 | Cuomo et al., 1984 | ||||
| AMI | Rat | 10 | SC | G2–G21 | No | + | + | P30 | Henderson et al., 1990 | ||||
| CLO | Rat | 3 | DW | G14–P2 | No | + | ø | P50, 110 | Rodríguez et al., 1983 | ||||
| IMI | Rat | 5 | OG | G21–P25 | No | + | P25, 80 | Coyle and Singer, 1975a | |||||
| CLO | Rat | 7.5, 15 | IP | G8–G21 | No | + | P32, P42 | File and Tucker, 1984 | |||||
| IMI | Rat | 5 | OG | G14–G19 | No | ø | + | P60 | Coyle and Singer, 1975b | ||||
| DOX, IMI | Rat | 30 | IP | Varied | No | + | + | P35–70 | Simpkins et al., 1985 | ||||
| AMI | Hamster | 75 | IP | G8 | No | + | G15 | Beyer et al., 1984 | |||||
| IMI | Rat | 5, 15, 30 | SC | G21–G21 | No | + | P1 | Singer and Coyle, 1973 | |||||
| IMI | Rat | 10 | IP | G9–G11 | No | + | G21 | Swerts et al., 2010 | |||||
| IMI | Rat | 5 | SC | G1–21 | No | +, ø | P56, 90 | Fujii, 1997; Fujii and Ohtaki, 1985 | |||||
| IMI | Primate | 20–244 | OG | Varied | No | ø | NA | Hendrickx, 1975 |
+, Positive association; ø, null association; AMI, amitriptyline; CLO, clomipramine; DSP, deprenyl; DOX, doxepin; DSP, desipramine; DW, drinking water; G, gestation day; IMI, imipramine; IP, intraperitoneal; IPD, iprindole; MNS, mianserin; NOM, nomifensine; OG, oral gavage; P, postnatal day; SC, subcutaneous; TNP, tianeptine; VX, viloxazine.
TABLE 3.
Summary of endpoints after prenatal SSRI exposure in animals
| Drug | Species | Dose | Administration | Exposure | Maternal Serum Monitored for Drug Conc. | 5-HT | DA | Behavior | Cardio | Health | Endpoint | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mg/kg | ||||||||||||
| FLX, FVA | Mouse | 0–4.2 | IP | G8–G18 | Placental transfer | + | + | + | + | P20, P90 | Noorlander et al., 2008 | |
| ESC, FLX, PRX, SRT, VEN | Rat | 3–80 | OM | G12–G21 | Yes | + | ø | ø | G21–P7, P70–P100 | Capello et al., 2011 | ||
| FLX | Rat | 10 | OM | G14–P7 | No | + | + | + | P30–P120 | Forcelli and Heinrichs, 2008 | ||
| CIT | Rat | 0–20 | SC | G11–P21 | No | + | + | P22–P25 | Simpson et al., 2011 | |||
| FLX | Rat | 2.5 | OG | G6–G21 | No | + | P25 | Montero et al., 1990 | ||||
| FLX | Rat | 10 | SC | G13–G20 | No | + | P26, P70 | Cabrera-Vera et al., 1997 | ||||
| FLX | Rat | 10 | SC | G13–G20 | No | + | P26 | Cabrera-Vera and Battaglia 1998 | ||||
| ZIM | Rat | 5 | SC | G10–G20 | No | ø | +, ø | P20–P90 | Grimm and Frieder, 1987 | |||
| FLX | Rat | 12.5 | OG | G8–G20 | No | ø | ø | ø | P20 | Stewart et al., 1998 | ||
| FLX | Rat | 8, 12 | DW | G6–G20 | No | + | + | P18–P56 | Bairy et al., 2007 | |||
| FLX | Mouse | 5, 10 | SC | G13–G20 | No | + | + | P1–P120 | Cagiano et al., 2008 | |||
| FLX | Guinea pig | 7 | OM | G1–G54 | No | + | ø | P63 | Vartazarmian et al., 2005 | |||
| FLX | Mouse | 7.5 | OG | G0–P21 | No | + | ø | P40, P70 | Lisboa et al., 2007 | |||
| FLX | Mouse | 7.5 | OG | G0–P20 | No | + | P90 | Gouvêa et al. 2008 | ||||
| FLX | Rat | 10 | IP | G13–G21 | No | + | P56 | Singh et al., 1998 | ||||
| FLX | Sheep | 0.099 | IV | G120–G128 | Yes | + | G128 | Morrison et al., 2001 | ||||
| FLX | Mouse | 7.5 | OG | G0–P21 | No | +, ø | + | P40 | Favaro et al., 2008 | |||
| PRX | Mouse | 30 | Chow | G14–G16.5 | Yes | +, ø | P13–P90 | Coleman et al., 1999 | ||||
| FLX | Sheep | 0.099 | IV | G120–-G128 | Yes | ø | ø | G128 | Morrison et al., 2005 | |||
| PRX | Mouse | 30 | Chow | G14–P0 | No | ø | P34–P105 | Christensen et al., 2000 | ||||
| FLX | Rat | 10 | OG | G11–G21 | No | + | + | G21–P3 | Fornaro et al., 2007 | |||
| FLX, VEN | Rat | 8–80 | OG | G15–G20 | No | + | P1 | da-Silva et al.1999 | ||||
| FLX | Sheep | 0.099 | IV | G120–G128 | Yes | + | G128 | Morrison et al., 2004 | ||||
| FLX | Mouse | 10 | DW | G0–G21 | No | + | G14, G21 | Bauer et al., 2010 | ||||
| FLX | Rat | 10 | IP | G0–BF | No | + | P3–P30 | Periera et al., 2007 | ||||
| PRX | Rat | 10 | DW | G14–G21 | No | + | P0 | Van den Hove et al., 2008 | ||||
| FLX | Rat | 10 | IP | G9–G11 | No | ø | G21 | Swerts et al., 2010 | ||||
| FLX | Rat/rabbit | 0–15 | OG | G6–G15 | No | ø | G20/G28 | Byrd and Markham, 1994 |
+, Positive association; ø, null association; BF, breast feeding; CIT, citalopram; DW, drinking water; ESC, escitalopram; FLX, fluoxetine; FVA, fluvoxamine; G, gestation day; IV, intravenous; IP, intraperitoneal; OG, oral gavage; OM, subcutaneous osmotic minipump; P, postnatal day; SC, subcutaneous; SRT, sertraline; VEN, venlafaxine; ZIM, zimelidine.
B. Exposure Studies
Studies in laboratory animals are vital to investigate the possible effects of prenatal antidepressant exposure. However, translatable exposure concentrations between animals and humans can be difficult attributable to differences in pharmacokinetics between species.
C. Tricyclic/Tetracyclic Antidepressant Animal Studies
The majority of TCA prenatal exposure studies in animals have relied upon single, daily administration of the compound or dissolving the compound in drinking water. To extrapolate animal data and apply it to the human population, exposure studies should rely on a clinically relevant dosing model that approximates human exposure. Pharmacokinetics dictates that there is a bolus effect after a single injection that may lead to suprapharmacological exposures and possible acute toxicity. Most xenobiotics are more quickly metabolized in laboratory animals compared with humans and dosing paradigms must attempt to maintain exposure throughout the dosing interval. Animal exposure studies investigating non–SSRI drugs have largely focused on imipramine, which is metabolized into desipramine. Desipramine and imipramine are norepinephrine transporter antagonists, although imipramine potently inhibits the serotonin transporter as well (Owens et al., 1997). Devane and Simpkins (1985) used a bolus injection of imipramine in pregnant rats to show imipramine and desipramine at higher concentrations in the fetal brain than the maternal serum, indicating, unsurprisingly, that these medications pass readily through the placenta and expose the offspring to a significant dose (DeVane and Simpkins, 1985). Because of the lipophilic nature of most psychotropic drugs, concentrations in brain tissue are frequently much higher than in serum or cerebrospinal fluid, but it is only drug in the brain extracellular fluid compartment that has access to its target and not that intercalated nonspecifically in lipid membranes, both of which are measured by studies utilizing “brain” concentration data. Nevertheless, studies measuring brain concentrations signify brain exposure. When administered intraperitoneally to a pregnant rat, a 30 mg/kg dose of imipramine on G18–19 resulted in ∼0.5 µg/ml in the dam plasma, ∼20 µg/ml in the maternal brain as well as a persistent (>5 µg/ml) concentration of its active metabolite desipramine for 18 hours (DeVane et al., 1984). A 10 mg/kg dose of imipramine or desipramine administered intramuscularly during the third trimester of pregnancy resulted in significant placental transfer with significant concentration in the fetal serum (2 µg/ml in fetal plasma, 1 µg/ml in maternal plasma for imipramine) (Douglas and Hume, 1967; Hume and Douglas, 1968). These studies indicate significant placental transfer in rodents, similar to clinical data.
D. Selective Serotonin Reuptake Inhibitor Antidepressant Animal Studies
The majority of SSRI animal studies have used a model of daily injections subcutaneously or intraperitoneally to investigate the effects on the pups. As noted above, daily injections in rodents are problematic because of bolus effects that may lead to transient, possibly toxic, serum concentrations of the compound. In contrast, human exposure leads to lower Cmax and steadier serum concentrations of the compound compared with rodents (Fig. 1). Similar to TCA studies, some SSRI antidepressants have active metabolites with relatively long half-lives; this is particularly true for fluoxetine and the active metabolite norfluoxetine (half-life ∼24 hours in rats), which shares nearly identical pharmacology with fluoxetine (Owens et al., 1997). Recently, animal exposure studies using SSRIs have begun to devise more steady exposure models and measure serum concentrations of compounds to quantify the level of exposure (Bourke et al., 2013a).
Fig. 1.
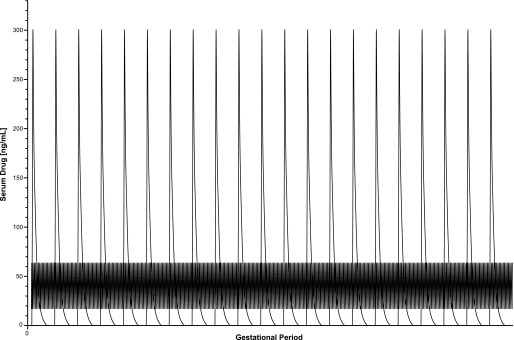
Comparison of human and rat pharmacokinetics of the antidepressant escitalopram during pregnancy. Based on the published half-life of escitalopram and observed peak and trough serum concentrations observed in humans and rats. Theoretical serum sampling of escitalopram after single injections (10 mg/kg free base) during pregnancy in rats would yield a peak bolus concentration of approximately 300 ng/ml followed by rapid clearance. The clinically observed range of escitalopram in human women with no attention paid to time of sampling reveals peak serum concentrations of ∼65 ng/ml and trough serum concentrations of ∼17 ng/ml.
Studies quantifying exposure in animals are, unfortunately, uncommon. Continuous infusion of fluoxetine in sheep via maternal femoral vein catheter yielded concentrations in maternal and fetal serum of ∼150 and ∼60 ng/ml, respectively; no measurements of norfluoxetine were obtained (Morrison et al., 2004). Similar results were found in mice: placental transfer of fluoxetine was 69%, whereas human placental transfer is 73% (Noorlander et al., 2008). Placental transfer comparing rodents and humans is also reported for fluvoxamine (30% in mice, 35% in humans). Comparing maternal serum concentrations and amniotic fluid concentrations, our group observed variable placental transfer depending on the antidepressant used (Capello et al., 2011).
Only recently has the degree of central nervous system (CNS) exposure been quantified in response to these data on placental transfer (Fig. 2). Although we observed variable placental transfer, fetal CNS exposure is similar to that of the pregnant dam (Capello et al., 2011). Direct measurement of monoamine transporter occupancy in human infants would be limited to postabortion tissue, and radioligand studies (e.g., PET) are not feasible in the intact fetus or neonate. Whether whole-blood serotonin concentrations are a viable proxy is unknown. Our group has quantified SERT occupancy in rat pups after continuous prenatal exposure to SRIs. Osmotic minipumps were used to administer steady serum concentrations similar to human exposure, and this resulted in approximately 80–95% occupancy of the serotonin transporter in embryonic day 21 pups (the end of gestation). This is essentially the same SERT occupancy in embryonic day 21 pups and their dams. Postnatally, SERT occupancy rapidly dropped after parturition as predicted from the pharmacokinetics of the drugs in rodents. Exposure via breast milk resulted in measurable but generally low SERT occupancy (Capello et al., 2011). This was demonstrated independently for escitalopram, fluoxetine, paroxetine, sertraline, and venlafaxine (Fig. 2). These data indicate that CNS exposure in utero to SRIs is similar between dams and their fetuses. In contrast, the postnatal only exposure via lactation did not result in SERT occupancy at the level observed therapeutically in humans. The presence of SERT occupancy in the pups exposed only via lactation is measurable even if serum drug concentrations are below the limit of detection. These data would suggest CNS exposure in humans occurs and that the undetectable levels for most antidepressants in human nursing infants may still be associated with some degree of CNS exposure. The magnitude of this exposure and its physiologic relevance are not known but are much less than that targeted for therapeutic activity in adults.
Fig. 2.

Representative autoradiographs of in utero exposure to SRIs. Representative autoradiographs of escitalopram (ESCIT), fluoxetine (fluoxetine), paroxetine (PAROX), sertraline (SRT), and venlafaxine (VEN), including representative pregnant dams exposed to VEN during pregnancy. Each treatment group had its own vehicle run in the same assay, but one series is shown for reference. Total binding representative images are displayed for comparison. Dense patches of total binding in the somatosensory cortex are consistent with previous studies investigating SERT binding during the early postnatal period (D'Amato et al., 1987; Kelly et al., 2002). PNC, postnatal clearance. Adapted from Capello et al. (2011).
E. Growth, Developmental, Gross Anatomical, and Physiological Outcomes
Gross anatomic differences due to prenatal NRI exposure have been investigated in animal models. Changes in birth weight or growth attributed to prenatal NRI exposure have been reported in the clinical literature. Animal studies show conflicting results; some report no changes in pup weight or litter size (Rodríguez Echandía and Broitman, 1983; De Ceballos et al., 1985b; Harmon et al., 1986; Stewart et al., 1998), an increase in fetal and neonatal weights (Cuomo et al., 1984; Swerts et al., 2010), or reduced birth weight in pups and reduced litter size (Singer and Coyle, 1973; Jason et al., 1981; Simpkins et al., 1985; Henderson and McMillen, 1990). Congenital malformations have also been investigated in prenatal exposure animal studies, showing a hint of encephalocele in Golden hamsters prenatally exposed to a single suprapharmacological (70.3 mg/kg) dose of amitriptyline (Beyer et al., 1984). Another study investigating teratological outcomes observed no differences due to prenatal exposure to imipramine in Bonnet or Rhesus primates at a dose of 244 mg/kg per day (Hendrickx, 1975). An important aspect of the thalidomide tragedy was the choice of the test species; rats and mice were not susceptible to the teratogenic effects, whereas rabbits and nonhuman primates were susceptible (Delahunt and Lassen, 1964; Fratta et al., 1965; Schumacher et al., 1968). Prenatal NRI exposure studies investigated several test species without showing any reproducible teratogenicity.
The noradrenergic system participates in thermoregulation and, therefore, has been examined in the context of prenatal NRI exposure (Mills et al., 2004). Adult male rat offspring prenatally exposed to imipramine displayed a baseline hyperthermic (difference of 1.31°C from controls) body temperature (Fujii and Ohtaki, 1985). Additionally, prenatal exposure resulted in a hyperthermic reaction to chlorpromazine (control rats were hypothermic in response to chlorpromazine), and this effect was observed on PND57 and persisted through PND90 (Fujii and Ohtaki, 1985; Fujii, 1997). Female control and females prenatally exposed to imipramine had a hypothermic response similar to male controls, indicating a sex-specific difference in thermoregulation in response to prenatal exposure.
Cardiovascular function is strongly regulated by norepinephrine (Singewald and Philippu, 1996), and some have hypothesized that disturbances in catecholamine homeostasis due to prenatal antidepressant exposure may alter cardiovascular function in the offspring. Prenatal exposure to 10 mg/kg imipramine from G8 to G20 has been shown to reduce heart weight in neonatal rats (Harmon et al., 1986). Another study examining prenatal doxepin or imipramine exposure found no differences measured in systolic blood pressure, but early prenatal exposure to doxepin increased offspring heart rate measured between PND35 and 70. Testing of aortal tissue in vitro revealed that third trimester exposure to doxepin or imipramine increased isoproterenol-induced relaxation of aortic tissue, but direct measurement of β-adrenergic receptor function or binding density was not undertaken (Simpkins et al., 1985). These studies demonstrate a role of prenatal antidepressant (TCA) exposure in the development of the cardiovascular system, whereby changes in the prenatal environment may cause long-term cardiovascular alterations; the physiologic significance is unclear.
Morrison and colleagues (2001) used Dorset Suffolk sheep and continuous infusion of fluoxetine to examine sleep endpoints. During the last trimester, a bolus injection of fluoxetine was administered followed by 8 days of continuous intravenous infusion. The measured plasma concentration of fluoxetine in the ewe was 106 ng/ml, whereas the fetal fluoxetine concentration was 58 ng/ml, showing substantial fetal transfer as expected. Fluoxetine infusion disrupted fetal sleep measured by low-voltage and high-voltage electrocortical activity (Morrison et al., 2001). A follow-up study showed no alterations in fetal plasma melatonin or prolactin during the infusion. No differences were observed in fetal behavioral state, fetal arterial pressure, heart rate, breathing, or circadian rhythm (Morrison et al., 2005).
The clinical literature has, for the most part, focused on gross indices of development such as weight, growth, or fetal mortality. Several animal studies examined these properties in the context of prenatal SSRI exposure with conflicting results. Impaired weight gain, low birth weight, or small litter size after prenatal SSRI exposure has been reported in several studies (da-Silva et al., 1999; Bairy et al., 2007; Pereira et al., 2007; Cagiano et al., 2008; Favaro et al., 2008; Forcelli and Heinrichs, 2008; Noorlander et al., 2008; Van den Hove et al., 2008; Bauer et al., 2010), whereas other studies observe no differences in weight gain or birth weight (Byrd and Markham, 1994; Stewart et al., 1998; Vartazarmian et al., 2005; Lisboa et al., 2007). Congenital malformations are reported in the clinical literature but have only been reported in the animal literature by extracting and exposing mouse embryos in vitro to 20 times the clinically observed serum concentration of sertraline for 48 hours (Shuey et al., 1992). Current studies do not support a role of clinically relevant exposure to SSRIs as a cause of congenital malformations or overt, long-term developmental consequences.
These data are consistent with the human literature suggesting alterations in sleep architecture, although the effects appear to be time limited and not associated with persistent alterations. The pup outcome data, like the human data, are also discordant. However, low birth weight in several studies is consistent with several of the clinical reports. The congenital malformation data are difficult to interpret given the relatively massive exposure that would be more consistent with daily overdose in a clinical population. Notably, the reported defects are not consistent with the purported cardiovascular defects in the human literature (see below and reviewed later).
F. Cardiovascular Outcomes
Chambers et al. (2006) reported that some children exposed prenatally to SSRIs after 20 weeks gestation displayed persistent pulmonary hypertension. Only two animal studies have tried to mechanistically determine the effects of prenatal antidepressant exposure on cardiovascular and pulmonary endpoints. Mice prenatally exposed to fluoxetine exhibited pulmonary hypertension as measured by increased right ventricle: left ventricle + septum ratio or pulmonary arterial medial thickness. Functionally, pulmonary arterial smooth muscle response to serotonin was significantly reduced. The lung concentration of serotonin was unaltered by prenatal exposure, but placental serotonin content was significantly reduced; the role of placental serotonin on cardiovascular development is unclear. Fetal mortality was increased in the first 3 days of life following fluoxetine exposure (0% in controls compared with 15% in fluoxetine-exposed pups). Newborn arterial oxygen saturation was lower in fluoxetine-exposed pups, but this normalized by the 3rd day of life. These findings were noted to be strongly indicative of pulmonary hypertension (Fornaro et al., 2007). Noorlander et al. (2008) also reported that mice prenatally exposed to fluoxetine manifested decreased left ventricle wall thickness, a measure of dilated cardiomyopathy, on PND20 and 90. These findings provide some concordant data regarding persistent pulmonary hypertension reported in the clinical population.
The issue of persistent pulmonary hypertension of the newborn (PPHN) is of serious concern and, thankfully, a relatively rare clinical condition affecting 1:1000. The limited preclinical data do suggest that more subtle changes in the cardiovascular/pulmonary systems (e.g., nonteratogenic) may be a consequence of prenatal SSRI exposure as noted in a recent review (Occhiogrosso et al., 2012).
G. In Utero Exposure and Effects on Monoaminergic Function: Molecular and Biochemical Outcomes
Initial radioligand binding and catecholamine detection studies of prenatal antidepressant exposure investigated catecholamine function in the context of receptor binding, affinity, and catecholamine concentrations. Several studies have shown reduced [3H]imipramine (used as a marker for the SERT), β-adrenergic, and dopamine receptor (D2) binding after prenatal NRI exposure (Jason et al., 1981; De Ceballos et al., 1985b; Ali et al., 1986; Montero et al., 1990), but other studies observed no changes in D1 or D2 receptor binding (Henderson et al., 1991; Stewart et al., 1998), adrenergic receptor binding (Harmon et al., 1986; Henderson et al., 1991), or 5-HT2 receptor binding (Henderson et al., 1991). These measures in offspring were conducted during the late postnatal period, up to PND25, and therefore may not be long-term alterations persisting into adulthood but may still be important for normal development. Dopamine affinity for dopamine receptors was reported to be increased by prenatal NRI exposure (De Ceballos et al., 1985a); however, a mechanistic explanation on how this (a change in affinity) is physically accomplished is unclear. Norepinephrine and dopamine concentrations in several brain areas in adolescent and adult male rats and norepinephrine turnover were shown to be unaffected by prenatal exposure to imipramine (Tonge, 1972, 1973; Ali et al., 1986), although another study reported reduced hypothalamic dopamine concentrations on PND30 (Jason et al., 1981). NRIs typically have weak activity for the serotonin transporter. 5-HT1B receptors have been shown to be unaffected by prenatal clomipramine exposure (Montero et al., 1991) but 5-HT and its metabolite, 5-hydroxyindoleacetic acid, were reduced in the striatum on PND60 following prenatal amitriptyline exposure (Henderson and McMillen, 1993); amitriptyline has SERT and NET antagonism properties, and its metabolite, nortriptyline, is a potent NET antagonist. These studies indicate some effects of prenatal NRIs on central adrenergic function, but the physiologic significance is unclear.
Two studies investigating the serotonergic system after prenatal fluoxetine exposure have been conducted by Battaglia and colleagues (Cabrera-Vera and Battaglia, 1998). Prenatal exposure via maternal fluoxetine injection resulted in an increased density of [3H]citalopram-labeled SERT sites in the CA2 (+47%) and CA3 (+38%) areas of the hippocampus, as well as the basolateral (+32%) and medial (+44%) amygdaloid nuclei in prepubescent progeny. In the diencephalon, the lateral hypothalamus displayed an increased SERT density (+21%) in prepubescent progeny. In contrast, the density of 5-HT transporters was significantly decreased in the dorsomedial nucleus of the hypothalamus (−21%) and in the substantia nigra (−19%) in prepubescent progeny. At PND90, there were no significant differences between control and fluoxetine animals (Cabrera-Vera and Battaglia, 1998). This model also resulted in 28% reduction in 5-HT content in the frontal cortex of prepubescent offspring and the midbrain of adult progeny as well as attenuated p-chloroamphetamine-induced reduction in midbrain 5-HT content (Cabrera-Vera et al., 1997). The physiologic significance of these changes is unknown.
After prenatal fluoxetine exposure, rats exhibited a lower Bmax of [3H]imipramine binding sites (predominantly SERT) in the cerebral cortex up to PND25 compared with controls, indicative of a reduction in SERT expression and opposite of that reported by Cabrera-Vera and Battaglia (1998) and Montero et al. (1990). Pregnant rats exposed to a steady concentration of 10 mg/kg per day of fluoxetine from osmotic minipumps resulted in decreased cell count in the nucleus accumbens and decreased SERT immunoreactivity in the raphe on PND120 (Forcelli and Heinrichs, 2008). The serum concentrations of fluoxetine and norfluoxetine were not determined in this study, but our experience suggests that this may be a clinically relevant exposure. A similar study examining prenatal fluvoxamine exposure reported decreased SERT immunoreactivity in the raphe on PND20 and 90 (Noorlander et al., 2008). Only one study has investigated nonserotonergic receptors and reported no changes in D1 or D2 receptor binding in the striatum on PND20 after prenatal fluoxetine exposure (Stewart et al., 1998). Other non–CNS studies of monoamine systems have shown that vas deferens tissue from PND30 rats exhibited decreased affinity for 5-HT, norepinephrine, and phenylephrine using a bioassay, but as noted earlier, changes in affinity are difficult to reconcile mechanistically (Pereira et al., 2007). Prenatal fluoxetine exposure seems to selectively affect the serotonergic system but not other monoaminergic systems. In the absence of other data, this is likely true for all of the SSRIs as they generally share a very similar pharmacology.
The known serotonergic regulation of the HPA axis (Owens et al., 1991a,b) led Morrison and colleagues (2004) to investigate pituitary and adrenal output in the context of prenatal fluoxetine exposure. During continuous infusion of fluoxetine to pregnant sheep, on certain days, fluoxetine decreased plasma ACTH concentrations compared with preinfusion day but not compared with control in the pregnant sheep. In the fetus, basal plasma ACTH concentrations were increased on G127 compared with preinfusion day. Cortisol was unaffected in the ewes, but on G127 and 128, fetal cortisol in fluoxetine-exposed animals was significantly elevated compared with controls (Morrison et al., 2004). Further studies of the HPA axis may help elucidate the function of the developing serotonin system on the fetal HPA axis.
Serotonin’s role in early development has been explored using prenatal citalopram exposure. Simpson and colleagues (2011) examined prenatal and postnatal exposures to citalopram and its possible effects on cortical network functioning in rats. A comparison of gestational citalopram exposure and postnatal (PND8–21) exposure showed disruptive behavioral effects of postnatal exposure only. Additionally, abnormal callosal connectivity was observed in postnatally exposed animals (Simpson et al., 2011). This study describes an exquisitely regulated developmental system with serotonin at the center. However, because the effects were mostly ascribed to the postnatal exposure period and two daily 10 mg/kg bolus injections were used for citalopram exposure, direct translation to prenatal antidepressant exposure is uncertain as is the overall clinical relevance.
Our group recently conducted studies on the long-term effects of prenatal antidepressant and/or stress exposure in rats. After steady exposure to escitalopram throughout pregnancy and/or chronic variable stress exposure, the offspring were analyzed in adulthood. Adult male rats had no significant changes in gene expression using a microarray and real-time PCR in the hippocampus, hypothalamus, or amygdala due to prenatal stress and/or escitalopram exposure. Behavior was also unaffected by prenatal treatments. After stimulation of the HPA axis with an acute air puff startle, plasma ACTH and cortisol concentrations were not physiologically affected by prenatal stress and/or escitalopram exposure (Bourke et al., 2013b).
These data, similar to the human data, indicate that antidepressant exposure in pregnancy can influence some measures of monoaminergic function and, albeit inconsistent between the preclinical and clinical data, some degree of effects on measures of neuroendocrine function.
H. Behavioral Outcomes
Disrupted behavior in laboratory animals may indicate a phenotypic difference due to prenatal antidepressant exposure. Several groups have investigated exploratory behaviors and social interaction after prenatal NRI exposure. The open-field test for rodents is used to measure exploratory behavior in a novel environment (possible inquisitive/anxiety-like behavior), and several groups have reported that prenatal NRI exposure decreased rearing behavior in adolescence (Coyle, 1975; File and Tucker, 1983; Drago et al., 1985). Prenatal NRI exposure also decreased exploration in adolescence and adulthood in male rats (Rodríguez Echandía and Broitman, 1983). Additionally, prenatal and prenatal + lactational NRI exposure to 3 mg/kg chloroimipramine delivered via tap water produced similar effects while lactation alone did not, indicating that exposure via lactation as predicted from earlier studies of quantifying exposure may not produce any behavioral effects (Rodríguez Echandía and Broitman, 1983). Measures of social interaction were shown to be increased in adolescence due to prenatal NRI exposure (File and Tucker, 1983, 1984) in male and female rats. However, other groups report the opposite: prenatal NRI exposure decreased social interaction in adolescence and adulthood in male rats (Coyle and Singer, 1975b; Rodríguez Echandía and Broitman, 1983).
The antidepressant neonatal syndrome was first reported in 1973 (Webster, 1973) with imipramine, and more recently, it has garnered considerable attention (Moses-Kolko et al., 2005). Clinically, the description has consistently included jitteriness and twitching. Although we do not believe that this is the classic “serotonin syndrome,” it may be indicative of alterations in the 5-HT system. Animal studies have recapitulated the classic “serotonin syndrome” through postnatal coadministration of clorgyline, a monoamine oxidase inhibitor, followed by administration of 5-hydroxytryptophan, the immediate precursor of serotonin. De Ceballos et al. (1985b) explored this in rats as a measure of the involvement of the developing serotonin system. Prenatal chloroimipramine, a potent SRI with some NRI activity, prevented drug-induced serotonin syndrome-like behaviors such as head twitches and a resting tremor. However, the 5-HT2 antagonists, iprindole or mianserin, both potentiated animal measures of a serotonin syndrome. Nomifensine, an NRI/dopamine reuptake inhibitor, had no effect on the behavioral characteristics of the serotonin syndrome. Although the conflicting effects seen in this study preclude a definitive interpretation, there does seem to be a serotonergic influence on postnatal jitteriness observed in laboratory animal infants prenatally exposed to antidepressants.
Measures of learning, the startle response, and hyperactivity have also been investigated in the context of prenatal NRI exposure. Only one study has examined prenatal NRI exposure in swim tests used to assess cognitive function on PND60 and PND125 but did not find any association between prenatal imipramine exposure and the cognitive outcomes in a Henderson-type maze (Coyle and Singer, 1975a). The acoustic startle response, which measures the startle reflex after a loud acoustic tone, has been shown to be mediated in part by the noradrenergic system (Olson et al., 2011). Prenatal NRI exposure has been shown to decrease the acoustic startle response in PND18 male rats (Ali et al., 1986) and adolescent (PND42–47) male rats (File and Tucker, 1984), although a dose-response relationship was not observed in either study. Haloperidol-induced catalepsy was unaltered by prenatal amitriptyline exposure in adolescent or adult males (Henderson and McMillen, 1993). Locomotion has been shown to be increased with prenatal NRI but not prenatal SRI exposure in male and female adolescent and adult rats (Cuomo et al., 1984) and adolescent male rats (Henderson and McMillen, 1990). However, when challenged with amphetamine, imipramine-exposed rats did not display any changes in locomotion compared with unexposed rats and completed negative geotaxis faster than control groups (Ali et al., 1986). Prenatal desipramine also did not produce any differences in quinpirole-induced stereotypy or locomotion (Stewart et al., 1998). Behavioral studies investigating prenatal NRI exposure are inconsistent, and although some groups have found behavioral differences, several endpoints have a null result or other studies conflict with positive associations with prenatal NRI exposure. Moreover, mechanistic explanations for many of the reported findings are lacking.
Studies investigating anxiety-like or depressive-like behavior have been performed in animals to elucidate any effects due to prenatal SRI exposure. Mice exposed prenatally to fluoxetine exhibited increases in anxiety-like behavior in adolescence and adulthood in the elevated plus maze, open-field test, and novelty suppressed feeding test (Noorlander et al., 2008). Others have reported increased immobility in the forced swim test for females in adolescence and adulthood (Lisboa et al., 2007). However, others report no differences in anxiety- or depressive-like behaviors (Coleman et al., 1999; Hsiao et al., 2005; Lisboa et al., 2007; Favaro et al., 2008). Ultrasonic vocalizations during separation or behavioral testing were increased in pups prenatally exposed to fluoxetine or paroxetine (Coleman et al., 1999; Cagiano et al., 2008). Prenatal exposure to the atypical antidepressant bupropion produced a decrease in rearing and ambulatory activity in the open-field test in adult male mice, although the doses used have not been commented upon as clinically relevant (Hsiao et al., 2005; Su et al., 2007). Alterations in locomotion may be a potential confound in behavioral tests, and false-positives in anxiety-like behavior, for example, may be attributed to altered locomotion. Studies have shown that prenatal fluoxetine exposure increases activity and improves motor coordination in the rotorod test (Bairy et al., 2007). A similar finding was reported with prenatal bupropion exposure (Su et al., 2007). In contrast, Coleman et al. (1999) and Stewart et al. (1998) observed no changes in baseline locomotion or quinpirole-induced stereotypy attributable to prenatal SSRI exposure.
Behavioral testing of learning and memory has been conducted in animals prenatally exposed to SSRIs. The Morris water maze and passive avoidance tests have been used to assess learning and memory in rodents. Prenatal exposure to fluoxetine resulted in an increase in learning in male and female adolescent rats (Bairy et al., 2007) but others have reported no differences due to prenatal SSRI exposure in the same tests (Grimm and Frieder, 1987; Christensen et al., 2000; Cagiano et al., 2008). Therefore, in these limited studies that do not attempt to use clinically relevant dosing, there does not seem to be a consistent role of prenatal SSRI exposure in the development of learning and memory skills.
Indices of social interaction have been investigated in animal studies of prenatal SSRI exposure. Prenatal fluoxetine exposure produced impaired sexual motivation in adult mice measured by no difference in preference for social contact with a male versus a female (Gouvêa et al., 2008). Aggressive behavior and foot shock-induced aggressive behavior have been shown to increase following prenatal SSRI exposure (Singh et al., 1998; Coleman et al., 1999). Several studies report no changes in social interaction or sexual behavior (Lisboa et al., 2007; Cagiano et al., 2008).
Several other indices of behavior have been investigated in the context of prenatal SSRI exposure. The acoustic startle response, a test of the fear-induced startle response (Koch, 1999), and prepulse inhibition, a test of sensorimotor gating, typically used to test animal models of schizophrenia and/or antipsychotic drug efficacy (Geyer et al., 2002), were examined by Vartazarmian et al. (2005) in guinea pigs prenatally exposed to fluoxetine. Prepulse inhibition was increased in both males and females after prenatal fluoxetine exposure, but the acoustic startle response was unchanged (Vartazarmian et al., 2005). Pain sensitivity has also been investigated after prenatal SSRI exposure, with one study showing an increase in pain threshold assessed by the hot plate test in adult guinea pigs after prenatal fluoxetine exposure via osmotic minipumps (7 mg/kg per day) (Vartazarmian et al., 2005); however, another study showed no changes in pain sensitivity in mice prenatally exposed to fluoxetine (7.5 mg/kg per day via oral gavage) (Lisboa et al., 2007). Forcelli and Heinrichs (2008) examined drug-seeking behavior after prenatal fluoxetine exposure using a steady exposure model via osmotic minipump implantation on G14 in the dam. Prenatal fluoxetine increased cocaine-induced place conditioning at PND60. Prenatal fluoxetine additionally increased the nose-poke response rate during the extinction phase of cocaine self-administration behavior on PND90, indicating an increase in the conditioned reinforcing effects of cocaine (Forcelli and Heinrichs, 2008). Others observed no changes in cocaine-induced place conditioning attributable to prenatal SSRI exposure (Hsiao et al., 2005). As described above, many behavioral studies examining the effects of prenatal SSRI exposure are often contradictory and a reproducible endpoint has not been identified as indicative of behavioral teratogenicity.
Concise ontogeneological data on human serotonergic function in the CNS is lacking, and definitive mapping of in utero exposure and postnatal exposures onto human fetal/infant serotonergic development is not possible at present (Avishai-Eliner et al., 2002). Importantly, the distribution of serotonergic neurons in rats is similar on G19 to adult groupings (Lidov and Molliver, 1982). Gingrich and colleagues investigated the use of serotonin reuptake inhibitors during postnatal development and find significant, long-term behavioral alterations in mice (Ansorge et al., 2004, 2008). Importantly, these studies employ a dosing regimen (i.e., daily administration) during the postnatal period (PND4–21) that may not adequately recapitulate human in utero exposure. To their credit, the dosing regimens used by Gingrich and colleagues postnatally lead to clinically relevant levels of SERT transporter occupancy, and drug exposure is maintained throughout their studies. However, the use of postnatal dosing may not be fully relevant to humans because, after birth, exposure in humans via lactation is low and infants are unlikely to receive any type of therapeutic dose of antidepressants for medical reasons for the first few years of life. Additionally, daily dosing does result in transient peaks that can be suprapharmacological, possibly toxic, and may be partially responsible for their observations.
III. Human Exposure Studies
Postmarketing surveillance and retrospective human studies examining the effects on the fetus and/or offspring of prenatal antidepressant exposure are the most common methods to assess possible adverse effects of perinatal exposure. Similarly, case reports and case series provide initial avenues for re-examination of existing data sets and future investigations. These data should contribute to delineation of potential mechanisms that can subsequently be tested in preclinical and clinical investigations with appropriate controls.
A. Defining the Clinical Problem
Pregnancy expands a woman's health considerations beyond herself to include her unborn child. In the case of a woman diagnosed with major depressive disorder, which has been reported to affect up to 10–20% of pregnant women (Gavin et al., 2005), the deleterious effects of untreated depression on the offspring can be profound and long lasting. Although the literature is replete with studies suggesting adverse effects, it is noteworthy that the definition of maternal depression has varied significantly across the investigations. Maternal depression and stress are often considered synonymous, making it difficult to delineate the impact, if any, of depressive symptoms from a clinical diagnosis of major depression. Depression during pregnancy has been associated with poor maternal health behaviors, including tobacco use, alcohol and drug use, and poor compliance with prenatal care (Zuckerman et al., 1989). Maternal depressive symptoms during pregnancy are associated with a higher risk of low birth weight and preterm delivery (Steer et al., 1992; Orr and Miller, 1995; Halbreich, 2005; Dunkel Schetter and Tanner, 2012). Behaviorally, neonates (8–72 hours postnatal) of depressed mothers are more inconsolable and cry excessively as assessed by the Neurologic and Adaptive Capacity Scale (Zuckerman et al., 1990). Children also exhibit increased internalizing behavior such as emotional reactivity, depression, anxiety, irritability, and withdrawal (Misri et al., 2006; Tronick and Reck, 2009). In a detailed prospective investigation conducted at 6 months postpartum, salivary cortisol reactivity was elevated in infants born to mothers with a history of major depression during pregnancy (Brennan et al., 2008; Field, 2011). Other groups have also demonstrated HPA axis abnormalities associated with maternal depression (Field, 2011). Additionally, mothers with a history of an affective disorder have a 2.5- to 5-fold increased risk of giving birth to a child with attention deficit hyperactivity disorder (Figueroa, 2010). Pregnant women who experienced a significant psychosocial stress exposure during pregnancy are reported to have an increased risk of giving birth to children later diagnosed with schizophrenia or shortened leukocyte telomere length (Khashan et al., 2008; Entringer et al., 2011). Stress during pregnancy has also been associated with an increased risk of developing an autism spectrum disorder in the offspring (Kinney et al., 2008). Human studies have shown that maternal depression, anxiety, and stress during pregnancy are associated with a greater risk of poor maternal health behaviors, obstetrical complications, offspring with HPA axis abnormalities, and neonatal and later behavioral aberrations. These data underscore the potential risks of untreated maternal symptoms during pregnancy, although the severity of symptoms warranting intervention remains obscure.
Preclinical studies typically use stress paradigms to model the potential impact of maternal depression during the perinatal period (see Newport et al., 2001 for review). Rodent studies utilizing prenatal stress exposures to the pregnant dam show disruption of stress responsivity in the offspring when examined at postnatal day 70 (Mueller and Bale, 2008). Chronic stress during pregnancy alters expression of key regulators and mediators of the stress pathway in the offspring, including decreased protein expression of the mineralocorticoid and glucocorticoid receptor in the hippocampus and prolonged increases in serum corticosterone concentrations in response to a stressor suggestive of impaired negative feedback (Maccari and Morley-Fletcher, 2007). Disruptions due to prenatal stress have been shown to persist via epigenetic miRNA mechanisms to offspring, and these may be passed on to subsequent generations (Darnaudery and Maccari, 2008; Dunn et al., 2011; Morgan and Bale, 2011).
Defining comparable stressor paradigms in preclinical models that appropriately reflect the clinical scenario of maternal depression is difficult at best. The key intersection might be summarized as “significant perinatal stress is associated with potentially quantifiable long lasting changes that may influence the developmental trajectory of the offspring.” Consistent with this area of concern is the American Congress of Obstetrics and Gynecology (ACOG) practice bulletin for psychotropic medications in pregnancy citing evidence that untreated maternal depression may pose significant risks (American College of Obstetricians and Gynecologists, 2007; ACOG Committee on Practice Bulletins–Obstetrics, 2008). Very recently, adult offspring (18 years of age) were at a significantly greater risk of having a diagnosis of depression if their mothers had depression during the pregnancy for that child (Pearson et al., 2013).
The use of antidepressants in the perinatal period appears to be increasing. For example, in the Netherlands, based on patient health records, 2% of all pregnant women were prescribed antidepressants (SSRIs or TCAs) during the 1st trimester of pregnancy (Ververs et al., 2006). In the United States of America, the number of women filling antidepressant prescriptions during pregnancy is as high as 8.7% (Cooper et al., 2007). The National Birth Defects Prevention Study in the United States has also observed that antidepressant use during pregnancy has increased 300% from 1998 to 2005 (Alwan et al., 2011). Given the increased use of antidepressants during pregnancy (Fig. 3), there is a great need for more rigorous studies that control for the potential confounds of other exposures and maternal symptoms to delineate potential adverse effects, if any, on the child. At present, a lack of clear, long-term consequences that coincide in preclinical and clinical studies should be acknowledged before a treatment decision is made and further argues for additional studies. Care providers need to be aware of the baseline percentage of untoward outcomes that naturally occur rather than attribute any untoward effects to prenatal medication exposure (Kaye and Weinstein, 2005).
Fig. 3.

Use of antidepressants during pregnancy: 1996–2005. The line with diamonds indicates any antidepressant use, the line with squares indicates SSRI use, the line with triangles indicates tricyclic antidepressant use, the line with crosses indicates tetracyclic antidepressant use, the line with asterisks indicates monoamine oxidase inhibitor use, and the line with circles indicates other miscellaneous antidepressant use. Less than 0.1% of pregnant women were exposed to tetracyclic antidepressants and MAO inhibitors. The pregnancy period is considered to be the period from 1 to 270 days before delivery, with three 90-day trimesters: first trimester incorporates the period from 181 to 270 days before delivery; second trimester incorporates the period from 91 to 180 days before delivery; third trimester incorporates the period from 1 to 90 days before delivery. Data for 2005 are not available for 1 of the 7 sites included in the analyses. Antidepressant use in the seven health plans for the period 1996–2000 were calculated using data from an earlier CERT study by Andrade et al. (2008) that evaluated medication use during pregnancy using similar methods (definitions and measures) as the present study. Adapted from Andrade et al. (2008).
Although increased usage during pregnancy is occurring, discontinuation of antidepressant treatment during pregnancy is also increasing, and antidepressant use decreases over each trimester (Ververs et al., 2006; Alwan et al., 2011). Although many medications may be used transiently, affective disorders belong to a class of complex diseases that typically are best controlled by continuous treatment (pharmacotherapy or proven psychotherapy techniques) (Kupfer et al., 2012). Pregnancy itself is a major determinant of antidepressant treatment discontinuation, and confirmation of pregnancy is associated with a 1.8- to 3.5-fold increase in antidepressant discontinuation, leading to a steady decline in antidepressant use over the course of pregnancy (Ramos et al., 2007; Bennett et al., 2010; Petersen et al., 2011).
The efficacy of the newer antidepressant medications in the treatment of major depression in nonpregnant populations has undergone increasing scrutiny relative to the treatment effects compared with placebo over the past decade (Thase, 2008). This raises important issues with respect to the potential use of antidepressants during pregnancy: are they efficacious in the treatment of depression during pregnancy? There are no randomized clinical trials of antidepressants during pregnancy despite agreement that such trials are warranted and satisfy ethical critique (Coverdale et al., 2008; Howland, 2013). As such, the only insights into the expected benefits of antidepressant treatment, defined as a reduction in depressive symptoms, are obtained from the studies of depression recurrence across pregnancy. A high relapse rate was observed in a prospective study in women who discontinued antidepressants (Fig. 4) and suggests efficacy in reducing risk for recurrence (Cohen et al., 2006). However, this was not confirmed in a community-derived sample (Yonkers et al., 2011). Unfortunately, neither study commented on whether reintroduction of antidepressant medication ameliorated symptoms. It is noteworthy, that in the Cohen study, the recurrence rate in women continuing antidepressants during pregnancy exceeded 25% over a 40-week period. This recurrence rate is higher than typically reported in nonpregnant samples. This lack of sustained efficacy may be attributable to changes in plasma concentrations of antidepressants across pregnancy (Sit et al., 2008). It is also plausible that pregnancy represents a unique medical condition in which antidepressants are less effective over time as reported in patients with diabetes. In the absence of randomized clinical trials, the efficacy of antidepressants in the short-term treatment of depressive episodes during pregnancy is unknown, although there is evidence supporting a decreased risk for recurrence in clinical populations.
Fig. 4.

Kaplan-Meier curves illustrating the time to relapse after discontinuation of antidepressants during pregnancy. Adapted from Cohen et al. (2006).
The limited studies and apparent contradictory results raise questions with respect to the historical premise that pregnancy protects the mother from the psychiatric disorders (Zajicek, 1981; Kendell et al., 1987). Proper counseling by providing balanced information is vital to making an informed decision about continuation and/or initiation of antidepressant treatment through pregnancy. The FDA safety classifications for pregnancy are shown in Table 1. The majority of antidepressants are currently category C; although there are animal studies indicating an adverse effect on the fetus, there are no adequate and well-controlled human studies, and the drug can be used during pregnancy only if the potential benefit justifies the potential risk to the fetus (Fig. 5). Paroxetine and the TCAs are rated category D; there are adequate and well-controlled human studies indicating a risk to the fetus, but the use of the drug in the pregnant woman may be acceptable despite the risk (Food and Drug Administration, 2011). Overall, the FDA recommends antidepressant use in pregnancy based on a risk-benefit decision on a case-by-case basis.
Fig. 5.

Percentage of pregnant women taking prescription drugs arranged by the Food and Drug Administration-labeling categories between 1996 and 2000 in the United States. Category A represents drugs that have well-controlled and adequate human and animal studies that show a low risk to the fetus. Category B drugs are classified as having animal studies that do not show a major risk to the fetus and the absence of well-controlled and adequate human studies. In Category C, a drug during pregnancy has an adverse effect in animal studies but there is an absence of well-controlled and adequate human studies. Category D drugs have a clear adverse effect in human and animal studies but the benefit to the pregnant woman may outweigh the risk. Category X drugs have a clear adverse effect in human and animal studies, and the benefit to the pregnant woman does not outweigh the risk to the fetus (Food and Drug Administration, 2011). Data adapted from (Andrade et al., 2004).
Investigators in the MotheRisk study in Toronto have commented on the absence of evidence-based information in the assessment of treatment decisions (Einarson, 2009). “Positive” studies that have an outcome or purported association of prenatal antidepressant exposure with some kind of malformation and/or adverse event are quickly reported by news outlets and disseminated to the lay public. Even more problematic in informing clinical decision making is the selective reporting of antidepressant effects and an apparent de-emphasis on the impact of maternal depression as was seen in the study by Oberlander and colleagues (2006). More recently, Domar and colleagues (2013) provided a selected review and noted the potential benefits of psychotherapy and exercise in nonpregnant groups as alternative treatment strategies. Although this article was quoted on several news media, they failed to note the >20% of women in their reproductive years do not have health insurance and that 48% of pregnancies in the United States are covered by Medicaid (Marcus et al., 2013). These are important considerations with respect to the availability and affordability of nonpharmacological treatment options. “Negative” studies that fail to identify any adverse effects garner far less media attention. This dichotomy in informing the lay public as well as bringing research data to the attention of clinicians contributes to the clinical conundrum and may lead to clinical decisions lacking evidence-based information (Einarson and Koren, 2007). Patients routinely overestimate the risk to the fetus when not presented with proper medical information (Einarson et al., 2005). The potential impact of reporting bias is illustrated in a study by Bonari and colleagues (2005) (see Table 4). For example, gastrointestinal medications, antibiotics, and antidepressants may have similar overall risks to the fetus, and in most cases, the antidepressant literature is more extensive. However, 87% of women believe that for antidepressants, the risk to the fetus is higher than the actual risk (Table 4) (Bonari et al., 2005).
TABLE 4.
Impact of MotheRisk counseling on perception of teratogenic risk
| Perception of Risk (Precounseling) * | Perception of Risk (Postcounseling) | P Value |
|---|---|---|
| 87% of depressed women rated risk of antidepressants as greater than 1–3% | 12% of depressed women rated risk of antidepressants as greater than 1–3% | <0.001 |
| 56% of women with gastric problems rated risk of medications as greater than 1–3% | 4% of women with gastric problems rated risk of medications as greater than 1–3% | <0.001 |
| 22% of women with infections rated the risk of medications greater than 1–3% | 2% of women with infections rated the risk of medications greater than 1–3% | <0.001 |
Reproduced with permission from (Bonari et al., 2005).
Actual baseline rate for major malformations in the general population is 1–3%
B. Quantification of Fetal and Nursing Infant Antidepressant Exposure
The extent of fetal psychotropic drug exposure is governed by the pharmacokinetic properties of the maternal, placental, and fetal compartment. The primary routes of fetal exposure are through umbilical circulation and amniotic fluid. All psychotropics studied in vivo to date cross the placenta and are found in amniotic fluid (Hostetter et al., 2000; Loughhead et al., 2006a,b). Yet there are significant differences in placental passage rates. Bidirectional placental transfer is primarily mediated by simple diffusion, and the determinants of the transplacental diffusion rate include molecular weight, lipid solubility, degree of ionization, and plasma protein binding affinity (Moore et al., 1966; Pacifici and Nottoli, 1995; Audus, 1999). In addition, placental P-glycoprotein (multidrug resistance protein 1), which actively transports substrates from the fetal to the maternal circulation, is likely a partial determinant of fetal psychotropic exposure. Consequently, agents with greater affinity for P-glycoprotein may be associated with lower fetal-to-maternal medication ratios, as demonstrated in an investigation of antipsychotic placental passage (Newport et al., 2007). Further clarification of the factors governing placental passage may ultimately contribute to the development of psychotropic agents with minimal rates of placental transfer (Wang et al., 2007).
Ex vivo studies of isolated human placentas show that tricyclic antidepressants and selective serotonin reuptake inhibitors pass across the placental barrier. Steady-state placental perfusion studies of amitriptyline, citalopram, or fluoxetine and their metabolites can reach up to 10% (Heikkinen et al., 2001, 2002b). In contrast, in vivo studies of umbilical cord medication distribution show much higher transfer of antidepressants as predicted for lipophilic substances.
Collection of paired maternal/umbilical cord blood at delivery as an index of placental passage has produced some interesting results. The TCAs nortriptyline, clomipramine, and their metabolites pass readily into umbilical cord serum in in vivo studies (Loughhead et al., 2006b). SSRI exposure results in higher umbilical cord transfer rates compared with TCAs. SSRI cord serum: maternal serum ratios are between 0.52 and 1.1 depending on the compound, illustrating significant placental transfer. Metabolites, which in some cases (e.g., norfluoxetine, desmethylvenlafaxine) are also active at inhibiting the serotonin transporter, readily transfer across the placental barrier (Rampono et al., 2004, 2009; Kim et al., 2006). Maternal/cord serum ratios have been rated as follows sertraline < paroxetine < fluoxetine < citalopram (Hendrick et al., 2003b). In a study of prenatal antidepressant exposure and pharmacogenetics, human infant drug concentrations were virtually undetectable and no relationship was associated with the drug-metabolizing enzymes CYP2D6 or CYP2C19 genotypes (Berle et al., 2004). Antidepressants are also present in human amniotic fluid (Loughhead et al., 2006a), providing additional routes of exposure.
Pregnancy can affect the metabolism and apparent clearance of medications. The clearance of several SSRIs has been shown to increase over the course of pregnancy (Sit et al., 2008), and our group demonstrated an increase in depressive scores for some women requiring dose adjustment during pregnancy (Hostetter et al., 2000). Similarly, postpartum, mothers taking citalopram, escitalopram, or sertraline have also been shown to be in a refractory metabolic state (Sit et al., 2008).
The majority of professional and lay organizations support breast milk as the "ideal" form of nutrition for the neonate through the first year of life. Studies of antidepressant exposure in breast milk are extensive relative to other classes of nonpsychotropic medications. Postpartum TCA exposure through breastfeeding results in a low exposure to the infant compared with in utero exposure. TCAs and their metabolites pass readily into breast milk, but because of the low absolute amounts (typically nanograms per milliliter range, with an estimated daily consumption of 100 ml/kg body weight), the nursing infant is exposed to a relatively small dose (Brixen-Rasmussen et al., 1982; Stancer and Reed, 1986; Wisner and Perel, 1991; Breyer-Pfaff et al., 1995; Yoshida et al., 1997). In many studies of TCAs, infant serum concentrations for drug or metabolite are undetectable at the level of analytic sensitivity used in those studies. Wisner et al. (1997) performed a controlled study of mother-infant breastfeeding pairs who had not received antidepressants during pregnancy. Nortriptyline and its metabolites were measured in the serum of six mother-infant pairs given nortriptyline 24 hours after birth for 4 weeks. Although nortriptyline and its hydroxylated metabolites were measured in maternal serum, infant serum concentrations were low or nondetectable (<4 ng/ml) compared with the typical clinical range of 50–150 ng/ml used in adults (Wisner et al., 1997).
SSRIs appear to transfer into breast milk at a much higher rate than TCAs, but exposure to the infant is still minimal. The first step in lactational exposure is transfer from the maternal serum to the breast milk. Breast milk has a natural gradient of fat content, with increasing fat content as one goes from fore to hind milk, and this can affect drug concentrations within the milk available at a feeding (Stowe et al., 1997, 2000, 2003). Additionally, breast milk concentrations change in direct relationship to maternal serum concentrations over the course of the day. The presence of both a temporal and fore to hind milk gradients for antidepressants should be kept in mind when examining the methodology of various research reports. Newport et al. (2009) examined the excretion of venlafaxine into breast milk. The infant/maternal plasma ratio was 0.062:1 for venlafaxine and 0.58:1 for the active metabolite desvenlafaxine, indicating significant exposure through lactation, although the effect was primarily driven by an extremely high ratio in a particular mother-infant pair from a cohort of five pairs. Otherwise, the ratios for desvenlafaxine in four other mother-infant pairs were 0.006, 0.009, 0.24, and 0.6:1 (Newport et al., 2009). Examination of other antidepressants has shown that the maternal serum and milk concentration ratio was paroxetine (0.7) < sertraline (1.8) < citalopram (2.1) < venlafaxine (2.4). Notably, the increasing milk: plasma ratios roughly correlate with the degree of protein binding for individual medications. Our previous work utilizing octanol partition coefficients as an index of lipophilicity of antidepressants (Capello et al., 2011) suggests that venlafaxine should have the lowest transfer into breast milk if driven primarily by lipophilicity.
Following transfer into breast milk, the question arises of whether the infant is exposed to an appreciable dose. Several antidepressants are detectable in human breast milk but are low or undetectable in infant serum (Altshuler et al., 1995; Epperson et al., 1997; Mammen et al., 1997; Stowe et al., 1997; Wisner et al., 1998; Heikkinen et al., 2002a; Briggs et al., 2009). A single study of potential functional consequences of nursing infant exposure estimated infant platelet serotonin uptake, measured by whole-blood serotonin concentrations, were shown to be unaffected in mother-infant breastfeeding pairs (Epperson et al., 2001). Weissman et al. (2004) conducted a large scale meta-analysis of 57 studies that examined mother-infant breastfeeding pairs to determine the amount of antidepressant exposure via lactational transfer. Overall, nortriptyline, sertraline, and paroxetine usually had undetectable concentrations in infant serum. Typically, fluoxetine, citalopram, and venlafaxine-exposed infants have detectable serum concentrations, but these concentrations are typically less than 10% of those clinically observed in the mother (Weissman et al., 2004). The extant literature indicates that fetal exposure in utero is substantially more pronounced than exposure via breast milk.
Atypical antidepressants (e.g., bupropion, trazodone, and mirtazapine) distribute into breast milk at a similar ratio as TCAs and SSRIs. Mother-infant breastfeeding pairs exposed to bupropion had zero detectable drug in the serum of infants (Baab et al., 2002). A very small milk/plasma ratio was observed (0.142), indicating that the infant would be exposed to less than 1% of the maternal dose (Verbeeck et al., 1986). A single patient dosed with mirtazapine had detectable concentrations in milk, but no drug was detectable in the plasma of the infant (Klier et al., 2007). A similar study (Kristensen et al., 2007) administered a dosing regimen of mirtazapine that reached steady-state concentrations. Breast milk mirtazapine and desmethylmirtazapine concentrations were 1.5% and 0.4% of those observed in maternal serum (Kristensen et al., 2007). Although the number of studies investigating exposure to atypical antidepressants is small, there appears to be a similar level of exposure via breast milk among the different classes of antidepressants.
In summary, all antidepressants studied to date cross the human placenta and are found in amniotic fluid and human breast milk; therefore the baby is exposed. However, fetal/neonatal exposure and subsequent plasma concentration analyses may underestimate the extent of exposure in the developing brain. The human fetus/neonate has several physiologic attributes, including 1) high cardiac output; 2) increased blood-brain barrier permeability; 3) low plasma protein concentrations and plasma protein binding affinity; and 4) low hepatic enzyme activity (Morgan, 1997; Oesterheld, 1998; Bertossi et al., 1999) that may conceivably lead to relatively higher fetal central nervous system (CNS) psychotropic concentrations. To delineate the extent and potential consequences of CNS exposure during early development warrants an appreciation of the preclinical data.
C. Growth, Developmental, Gross Anatomical, and Physiological Outcomes
Birth weight and gestational age have been examined in relation to prenatal SRI (serotonin reuptake inhibitor) exposure, because low birth weight has been associated with impaired development (Vohr et al., 2000). Several studies have shown an association of prenatal SRI use (may be dependent on trimester of SRI use) with low birth weight, shortened gestational length, or small size for gestational age (Ericson et al., 1999; Costei et al., 2002; Källén, 2004; Zeskind and Stephens, 2004; Malm et al., 2005; Oberlander et al., 2006; Lennestål and Källén, 2007; Diav-Citrin et al., 2008; Lund et al., 2009; Toh et al., 2009; Reis and Källén, 2010). One of these studies reported a higher risk of shortened gestational length due to prenatal TCA exposure compared with prenatal SRI exposure (Källén, 2004). However, a conflicting study reported that shortened gestational length was associated with SRIs but not TCAs, although TCAs can share the same SERT antagonism properties (Simon et al., 2002). Several other studies reported no association of prenatal SRI use and adverse birth outcomes (Nulman et al., 1997; Kulin et al., 1998b; Cohen et al., 2000; Einarson et al., 2001; Suri et al., 2004; Lund et al., 2009). Only a few of these studies control for the mental health status of the mother in their analyses, and the outcomes differ between studies (Suri et al., 2004; Lund et al., 2009). A population-based study of 860,215 women in the Swedish medical birth registry observed an increased risk for preterm birth, defined as less than 37 weeks, correlating with antidepressant prescription redemption. Preterm birth risk was increased for selective-norepinephrine reuptake inhibitors (SNRIs) versus SSRIs and late versus early exposure (Lennestål and Källén, 2007). Until quality meta-analyses are performed examining gestational length, it is not possible to conclude with any certainty the effect of prenatal antidepressant exposure on gestational length. It is noteworthy that maternal depressive symptoms have been consistently associated with similar outcomes. It is not known whether women with a history of major depression who remain asymptomatic and do not use antidepressants in pregnancy have the same outcomes.
Sporadic reports linking other symptoms and prenatal antidepressant exposure have been reported but have not been replicated. These include an association with jaundice and jitteriness (Diav-Citrin et al., 2008) and infantile hypertrophic pyloric stenosis (Bakker et al., 2010). Infant jaundice has been reported in certain cases of prenatal SRI exposure (Costei et al., 2002), although another study found no effect (Källén, 2004). As birth registry information becomes more expansive and readily available, studies using this endpoint and others, may become better statistically powered.
Several studies investigating prenatal exposure to SRIs have used Appearance pulse grimace activity respiration (Apgar) scores as outcomes, because the longstanding Apgar test is typically applied to every neonate (Casey et al., 2001). Studies have reported decreased Apgar scores due to prenatal SRI exposure (Casper et al., 2003), prenatal SRI and NRI exposure (Lennestål and Källén, 2007; Reis and Källén, 2010), and some studies found this association remains when controlling for maternal psychiatric history (Lund et al., 2009). Prenatal TCA exposure has been shown to increase risk of low Apgar scores compared with prenatal SRI exposure (Källén, 2004). Two of these studies reported decreased Apgar scores due to prenatal SRI exposure when controlling for maternal mood (Casper et al., 2003; Lund et al., 2009), but another study did not show an association when controlling for maternal mood (Suri et al., 2004). As noted above, quality meta-analyses of these datasets are sorely needed, although methodological differences may hinder conclusions.
The neonatal intensive care unit in hospitals admits infants that are typically premature or congenitally ill. Admission into the neonatal intensive care unit is therefore an easily identifiable endpoint that has been examined in the context of prenatal antidepressant exposure. One study by Casper and colleagues (2003) found that longer exposure to prenatal SSRIs correlated with increased neonatal intensive care unit admission. Timing of exposure appeared to be an important variable because third trimester exposure increased the risk of neonatal intensive care unit admission compared with first trimester exposure (Malm et al., 2005). This association of SSRI exposure and neonatal intensive care unit admission is still correlated even when controlling for maternal psychiatric condition (Sivojelezova et al., 2005). As noted above, a conflicting study reported that when controlling for maternal mood, prenatal SRI exposure did not increase the risk for neonatal intensive care unit admission (Suri et al., 2004). Recently, a study of birth registries in Nordic countries between 1996 and 2007 found no significant association between SSRI use in pregnancy and risk for stillbirth or postnatal mortality (Stephansson et al., 2013).
D. Associations with Respiratory Distress and Pulmonary Hypertension
Serotonin plays an important modulatory role in respiratory control, and much of the extant literature indicates that the serotonin system produces an inhibitory effect on respiration, although the meaning of “inhibitory” is not entirely clear (Mueller et al., 1982). Case reports have called attention to respiratory problems in infants prenatally exposed to TCAs, characterized by respiratory acidosis, cyanosis, and/or tachypnea (Ostergaard and Pedersen, 1982; Schimmell et al., 1991; Bloem et al., 1999; Frey et al., 1999). Oberlander's group used birth registry data from 120,000 patients and found an association of respiratory distress in newborns with prenatal SRI exposure even when controlling for severity of mental illness (Oberlander et al., 2006). Others have reported that an association with respiratory distress is dependent upon the antidepressant type: risk of respiratory distress was higher for prenatal TCA exposure compared with prenatal SRI exposure in two studies (Källén, 2004; Lennestål and Källén, 2007), but prenatal exposure to SRIs still has an increased risk for respiratory distress (Costei et al., 2002; Diav-Citrin et al., 2008). Although another study confirmed that SRI-exposed infants exhibited mild respiratory distress, outcomes were normal at 2 and 8 months of age (Oberlander et al., 2004). Serotonin plays a significant role in vasoconstriction and has been hypothesized to play a role in pulmonary arterial hypertension (Maclean and Dempsie, 2010). The role of the serotonin system in hypertension pathology has led to the focus on this endpoint in infants prenatally exposed to antidepressants. The Chambers study (Chambers et al., 2006) has been frequently cited to illustrate the danger of prenatal antidepressant exposure. In this retrospective study, authors observed an increased risk for persistent pulmonary hypertension only during late pregnancy prenatal SSRI exposure (odds ratio 6.0) (Chambers et al., 2006). A second study found a more modest (odds ratio 2.0) association of persistent pulmonary hypertension of the newborn (PPHN) with SRIs and NRIs (Reis and Källén, 2010). More recently, a study of birth registries in Nordic countries between 1996 and 2007 found an association of PPHN and SSRIs (adjusted odds ratio 2.1), a similar effect to the Reis and Källén study (Kieler et al., 2012). Although these studies may elucidate an important area to examine in the future, mental state and other potential exposures were not controlled for and may alter this association. The issue of antidepressants and the risk for PPHN was recently reviewed in detail (Occhiogrosso et al., 2012).
E. Associations with Congenital Malformations
The best established endpoints of teratology are congenital malformations that may be indicative of a significantly disrupted fetal environment. The extant literature indicates that congenital malformations are not associated with prenatal exposure to TCAs or SRIs (McElhatton et al., 1996; Ericson et al., 1999; Djulus et al., 2006; Lennestål and Källén, 2007), SRIs alone (Pastuszak et al., 1993; Goldstein et al., 1997; Kulin et al., 1998b; Simon et al., 2002; Hendrick et al., 2003a; Yaris et al., 2004; Malm et al., 2005; Sivojelezova et al., 2005; Källén and Otterblad Olausson, 2007; Pedersen et al., 2009), or atypical antidepressants (Einarson et al., 2003; Yaris et al., 2004; Chun-Fai-Chan et al., 2005). A meta-analysis of first trimester exposure to several classes of antidepressant confirmed these observations (Einarson and Einarson, 2005). Additionally, a meta-analysis found an increased risk of major malformations with fluoxetine and paroxetine exposure specifically in the first trimester (Myles et al., 2013). Although studies continue to examine this endpoint, the data do not point to a significant association of prenatal antidepressant exposure and a specific pattern of congenital malformations.
GlaxoSmithKline conducted their own study of first trimester paroxetine exposure compared with other antidepressants. An increased risk (adjusted odds ratio = 1.89) of congenital malformations was associated with paroxetine exposure (Cole et al., 2007). This observational study was conducted from patient medical records of 791 mothers in the absence of controls for potential confounding variables such as maternal mental state and other exposures. Venlafaxine, a dual SRI/NRI antidepressant, was compared in the MotheRisk study to determine any associated risk compared with other SRIs or nonteratogenic drugs. No increased risk of malformations or any other health outcomes was observed (Einarson et al., 2001). The Toronto MotheRisk Study also observed no association with congenital malformations and overall prenatal antidepressant exposure (Einarson et al., 2009). A meta-analysis has been conducted of first trimester fluoxetine exposure. Using the Chambers study and three others, the authors found no association of malformations with fluoxetine use. Importantly, based on the power calculations within this study, 26 controlled studies would be needed to reverse this finding of no association between antidepressants and congenital malformations (Addis and Koren, 2000).
Cardiovascular defects, a subset of congenital malformations, have also been investigated in the context of prenatal antidepressant exposure due to serotoninergic regulation of cardiovascular function. Birth registry studies have shown an increased risk of cardiovascular defects (ventral/atrial septum defects) in infants prenatally exposed to TCAs. This effect was predominately attributed to prenatal exposure to clomipramine (Källén and Otterblad Olausson, 2003). A follow up study showed that prenatal exposure to paroxetine or clomipramine, but not other SRIs or TCAs, was associated with an increased risk of ventricular/atrial septum defects using birth registries but not controlling for mental state (Källén and Otterblad Olausson, 2006, 2007). Septal heart defects typically resolve during early life, but may require surgery in some cases. A follow up study found that this observed effect was primarily due to clomipramine exposure (Reis and Källén, 2010). GlaxoSmithKline's own study of first trimester paroxetine exposure compared with other antidepressants showed an increased risk of cardiovascular malformations, mostly composed of ventricular septal defects, associated with paroxetine exposure (Cole et al., 2007; Louik et al., 2007) but another study found the opposite effect; first trimester paroxetine exposure resulted in fewer cardiovascular malformations than unexposed infants (Einarson et al., 2008). A study of birth registries in Finland found an increased risk for septal defects for both paroxetine and fluoxetine exposure, but not SSRIs as a whole (Malm et al., 2011). Prenatal exposure to the atypical antidepressant bupropion showed an increased risk for left outflow tract heart defects (Alwan et al., 2010). A study of Danish birth registries found an association of SSRI prescription redemption and septal heart defects (Pedersen et al., 2009). A meta-analysis of seven studies in the United States and Europe report an overall increased risk for cardiovascular malformations (Bar-Oz et al., 2007). Functional studies using echocardiograms have shown a twofold increased risk for mild nonsyndromic heart defects due to prenatal exposure to SRIs (Merlob et al., 2009).
There are several mitigating factors that should be considered in the context of the evidence of potential associations of congenital malformations and prenatal antidepressant exposure. Although a Finnish study of congenital malformations found craniofacial malformations (cleft palate/hydrocephalus) due to an imipramine/chloropyramine combination, it is interesting to note that the incidence was not above the national average of 1.25% (Idänpään-Heikkilä and Saxén, 1973). A study of the Danish population examining prescription redemption showed an increased risk of congenital malformations and SRI prescription redemption; however, there was a significant number of congenital malformations in women who discontinued SSRI use before pregnancy (4.5% compared with 4.9% in early and 6.8% in mid/late), indicating mental state may be involved (Wogelius et al., 2006).
F. Monoamine and Hormone Studies
Examination of 5-HT concentrations in infants reveals some short-term effects of in utero antidepressant exposure. SSRI exposure in utero yields a cord blood 5-HT concentration of 25% compared with controls and represents the inhibition of the platelet SERT and the platelet’s capacity to take up and store serotonin. Platelet serotonin represents >99% of whole-blood serotonin. Whole-blood 5-HT in newborns is correlated with their mothers’ (r = 0.42, P = 0.02). This was a short-term effect because infants older than 4 weeks had 5-HT concentrations similar to adult concentrations as predicted based upon the pharmacology and clearance of the drug (Anderson et al., 2004). This effect is isolated to the prenatal period of exposure when fetal exposure is considerable compared with SSRI exposure postpartum in mother-infant breastfeeding pairs that results in no changes in concentrations of 5-HT in newborns (Epperson et al., 2003).
Although the field is still in its infancy, biochemical monitoring in maternal and fetal biologic fluids after antidepressant treatment may provide an alternative to blood concentrations of SSRIs. S100β protein concentrations, a regulator of neurite outgrowth modulated by 5-HT1A receptor activation, are decreased in the cord serum after prenatal exposure to SSRIs (Pawluski et al., 2009).
Studies investigating the effects of prenatal antidepressant exposure on hormone systems are currently limited. Davidson and colleagues (2009) investigated 21 infants exposed to SSRIs throughout pregnancy compared with 20 unexposed controls. SSRI exposure in utero decreased cord blood cortisol concentrations, whereas thyroid stimulating hormone was increased compared with unexposed controls. Placental insulin-like growth factor-1 receptor expression, which participates in fetal growth and has significant cross-talk with the HPA axis, was higher in SSRI-exposed groups. The Finnegan score, as a measure of gross CNS and respiratory function, positively correlated with placental insulin-like growth factor-1 receptor expression and cord cortisol concentrations. Finnegan scores also correlated with dehydroepiandrosterone and its metabolite, dehydroepiandrosterone sulfate, which play an important role in regulating serotonin, dopamine, glutamate, and GABA neurotransmitter systems through modulation of receptor activation (Pérez-Neri et al., 2008). These correlations were only observed in SSRI-exposed groups and not healthy control groups (Davidson et al., 2009). Mental health was not controlled for in the mothers, so it is not possible to determine whether the observed effects were due to maternal depressive symptoms or SSRI exposure. Another study examined the cortisol response of second and third trimester SSRI-exposed infants. Although mothers had higher anxiety and depressive-like symptoms, at 3 months of age SSRI-exposed infants had lower evening basal salivary cortisol (∼0.9 ng/ml) compared with controls (∼2.2 ng/ml) (Oberlander et al., 2008b). These studies indicate a potential role of prenatal SSRI exposure in the regulation of fetal/infant adrenal output and/or responsivity.
G. Genome Association Studies
The advent of shotgun sequencing demonstrated by Venter and colleagues (2001) led to a rapid rise in genetic studies investigating the susceptibility to disease. The genome era has significantly affected the field of psychiatry and a large number of genome-wide association studies have been performed, although important outcomes have been less than originally hoped. Only a few studies have investigated the prenatal environment in the context of genomics. One study investigated 20 children (age 2–6 years old) exposed to citalopram or fluoxetine during pregnancy or via lactation in patients (i.e., mothers) diagnosed with major depressive disorder or panic disorder. This study found significant associations with genes coding for the degradation of monoamines such as monoamine oxidase (Maoa) and catechol-O-methyltransferase (Comt). High-activity Maoa alleles correlated with high cord blood concentrations of the norepinephrine metabolite dihydroxyphenylglycol and serotonergic symptoms in newborns (see Hegerl et al., 1998 for description of these symptoms). The high-activity Comt alleles correlated with high prolactin. Serotonin receptor or transporter gene polymorphism genotypes did not correlate with cord monoamine concentrations or serotonergic symptoms (Hilli et al., 2009). Although this study was part of a larger study with a control group (Laine et al., 2003), no control group was used so it is unclear whether in utero SSRI exposure interacted with genetics to produce these outcomes. However, a mechanistic explanation of how SSRI exposure could lead to genomic changes in allelic status in these studies is not at all clear or even possible.
Oberlander and colleagues in Canada performed extensive work associating maternal conditions, genetics, and endpoints in the child. These studies are in the minority that control for maternal mood or diagnosis of an affective disorder in relation to prenatal antidepressant exposure. Oberlander’s group investigated the behavior of the infant as it relates to prenatal antidepressant exposure but also genotyped the offspring to examine the serotonin transporter 5′-promoter region. The serotonin-transporter-linked polymorphic region (5HTTLPR) in the 5′-promoter region had originally been proposed to modulate the expression of the serotonin transporter. A 43-base pair insertion/deletion polymorphism in this region was previously reported to affect serotonin transporter expression and treatment response to antidepressants (Heils et al., 1996; Kim et al., 2000; Pollock et al., 2000). The short variant (ss) in this region has been reported to reduce expression of the serotonin transporter in vitro, and the short variant or heterozygous genotypes (ss, ls) correlate with anxiety-related traits (Lesch et al., 1996). However, association of the 5HTTLPR genotype and anxiety-like behavior has not been replicated in subsequent studies or through a meta-analysis (Sen et al., 2004). Oberlander's group examined infants prenatally exposed to antidepressant and correlated infant behavior with the 5HTTLPR variants. In a model controlling for maternal depressed mood, higher internalizing behaviors in the infant were only associated with maternal mood and not SSRI exposure. Externalizing behaviors classified as attention and aggression (Oberlander et al., 2010) in the infant were associated with anxious or depressed mothers and interacted with anxiety and the ll 5HTTLPR variant. Child scores for traits of anxiety and depression, as measured by the Child Behavior Checklist and reported by the mother, increased because of maternal anxiety and the ss 5HTTLPR variant. Internalizing behaviors were classified as anxious/depressed, emotionally reactive, somatic complaints, sleep problems, and withdrawn. Further studies replicating these findings and expanding upon the interaction of the 5HTTLPR and behavioral outcomes may be valuable to understanding the etiology of infant behavior and the relationship with the serotonin system and potential impact, if any, of prenatal medication exposure.
An additional study by Oberlander’s group examined respiratory measures at birth as they relate to maternal mood, prenatal antidepressant exposure, and the 5HTTLPR variants. This study examined women using the SRIs paroxetine, fluoxetine, sertraline, venlafaxine, and citalopram for an average of 220 days during pregnancy. When controlling for maternal mood, Apgar scores [Appearance Pulse Grimace Activity Respiration (Apgar, 1953)] were reduced in 5HTTLPR genotype ss. Exposed infants with heterozygous ls genotypes had lower birth weight. Disrupted respiration was observed in exposed ll genotypes, whereas ss neonates had an increased risk of neuromotor symptoms when exposed to SRIs in utero (Oberlander et al., 2008a). Although empirical data are absent, Oberlander and colleagues postulate that the combination of prenatal SRI exposure and presumably low expression of the serotonin transporter leads to an overabundance of serotonin in the presynaptic cleft as well as hypersensitivity of postsynaptic serotonin receptors and thus produces neuromotor irritability.
In a genomewide study of infant DNA methylation, Schroeder et al. (2012) suggest that there are no large effects of maternal psychiatric illness, depressive symptoms, or prenatal exposure to antidepressants on neonatal DNA methylation in 201 infant DNA samples collected at delivery from peripheral blood.
H. Behavioral Studies in Neonates and Children
The concept of behavioral teratogenicity denotes that prenatal exposure to xenobiotics may manifest as subtle changes in infant behavior that may point to novel neurotoxic effects or systems to explore. Although several animal studies (covered later in this review) explored behavioral teratogenicity in the context of prenatal antidepressant exposure, this is an area that warrants greater exploration in human studies of prenatal psychotropic exposure. Heel lances are typically performed in neonates to diagnose phenylketoneuria and are thus a convenient and efficient way to test the behavioral response to pain. Oberlander's group has assessed prenatal exposure to fluoxetine, paroxetine, or sertraline (n = 22) and SSRI + clonazepam (n = 16) compared with 23 unexposed controls and reported a decreased facial action to the pain of a heel lance in the recovery period. Facial action was assessed by the Neonatal Facial Coding System (Grunau and Craig, 1987; Grunau et al., 1990). Mean heart rate after heel lance displayed a blunted increase in the SRI-exposed offspring (Oberlander et al., 2002). The same group replicated this study, again finding a decreased facial action to the pain of a heel lance in the recovery period. Somewhat surprisingly, the combination of prenatal and postnatal (breast milk) exposure, but not prenatal exposure alone, to these medications resulted in greater parasympathetic control of heart rate variability in the recovery period after heel lance (control ∼175 beats per minute, prenatally exposed ∼145 beats per minute, prenatally and postnatally exposed ∼155 beats per minute) (Oberlander et al., 2005). The clinical significance of heart rate response to painful stimuli remains obscure.
Rating scales of infant development have also been used to assess the behavioral effects of prenatal SRI exposure. Utilizing the Brazelton Neonatal Behavioral Assessment Scale (Als et al., 1977) in a group of 64 infants, Suri and colleagues (2011) failed to find any effects of prenatal antidepressants and maternal depression at 6–8 weeks of age. In contrast, a second study using the Brazelton Neonatal Behavioral Assessment Scale reported that exposed neonates (determined by clinically established steady-state serum drug concentrations in the pregnant women) had lower habituation, social-interactive, motor, and autonomic behavior, although maternal mood was not controlled for (Rampono et al., 2009). Although neurologically normal as assessed by a pediatric neurologist, children examined at 12–40 months of age utilizing Bayley Scales of Infant Development showed that length of exposure, assessed by number of trimesters of SRI use, increased the risk for lower Psychomotor Developmental Index and Behavioral Rating Scale scores (Casper et al., 2003). The investigators also reported that prenatal exposure to SRIs (sertraline, fluoxetine, paroxetine, or fluvoxamine) resulted in lower psychomotor development at ages 6–40 months. This effect was still significant when controlling for maternal mood and comparing antidepressant-exposed children to children born to women with major depressive disorder (Casper et al., 2003). Sleep has also been considered to be modulated by prenatal SRI exposure, because a known side effect of current SSRI use is disruption of normal sleep patterns (Sharpley and Cowen, 1995; Trivedi et al., 1999). A study using median and higher doses of SRIs observed increased fetal motor activity in first and second trimesters compared with control or unmedicated, depressed mothers. Additionally, SRI exposure increased fetal movements in the 3rd trimester; purported to be indicative of disrupted non–rapid eye movement sleep. Covarying for mental state also resulted in an association with the observed results and potentially confounds the association with SRI exposure (Mulder et al., 2011). Examination of neonates prenatally exposed to SRIs showed disrupted rapid eye movement sleep, slightly disrupted autonomic homeostasis, and increased motor activity in 1- to 2-day-old neonates (Zeskind and Stephens, 2004). Some animal studies, especially in sheep, have observed a similar disruption in sleep patterns (vide infra). SRIs are known to produce some withdrawal symptoms upon discontinuation; it is not clear whether that is what is being observed in this study.
Only a few studies have examined long-term neurodevelopment in relation to prenatal SRI exposure. A separate study evaluating fluoxetine or TCA-exposed infants (ages 1–7 years old at follow up, n = 135 exposed infants) revealed no differences in perinatal complications, gestation age, cognition, IQ, or behavior compared with unexposed controls (Nulman et al., 1997). A series of studies utilizing a small cohort of infants from well-characterized women has produced some interesting results. Misri et al. (2006) evaluated both prenatal exposure to fluoxetine, paroxetine, or sertraline and maternal mood and observed no changes in internalizing behavior compared with controls for prenatal SRI exposure. Exposure was defined as enrollment in the Reproductive Mental Health Program at British Columbia Women's Hospital. Maternal anxiety and depressive symptoms did correlate with increased internalizing behavior in the offspring (Misri et al., 2006). Oberlander et al. (2007) conducted a 4-year follow up study of infants exposed prenatally to sertraline, paroxetine, or fluoxetine (N = 22) to examine internalizing and externalizing behaviors. Umbilical cord serum concentrations of antidepressants correlated with increased externalizing behaviors, but this effect was lost when controlling for maternal mood. Increased measures of aggressive behavior were correlated with maternal stress levels and poor neonatal adaptation. Controls were not exposed to prenatal antidepressants, and the maternal mood was neither depressed nor anxious (Oberlander et al., 2007). These studies illustrate an important role(s) of maternal mood in discriminating the effects of prenatal antidepressant exposure from prenatal stress/maternal affective disorders.
Increased awareness of childhood neurodevelopmental disorders in recent years has prompted further investigation in relation to prenatal antidepressant exposure. Using the MarketScan database, which uses insurance claims data, exposure to bupropion during the second trimester has been shown to be correlated with increased risk of attention deficit hyperactivity disorder in children, but SRI exposure as a whole was not correlated with increased risk. Psychiatric disorders exhibit differing degrees of heritability and, as might be predicted, psychiatric disorders in the mother were also positively correlated with attention-deficit hyperactivity disorder risk in the child (Figueroa, 2010). A population-based case control study reported an increased risk of developing childhood autism spectrum disorder after prenatal antidepressant exposure (Croen et al., 2011). Further studies of these behavioral endpoints will be valuable as more attention is given to this disorder.
I. Summary of Human Exposure Studies
Human studies over the last 20 years have steadily incorporated more rigorous designs that define exposure and have increasingly recognized the potential impact of maternal illnesses and other exposures. Problems with current human studies arise from issues regarding a proper control group of nonexposed, depressed, pregnant mothers (Tuccori et al., 2010). Because most of these studies are case control or cohort studies, there are a large number of confounding factors that may influence the endpoint such as tobacco use during pregnancy, alcohol and drug exposure, and consistent prenatal care. Likewise, the literature on the potential adverse effects of maternal depression in pregnancy may be equally confounded. This, in fact, may explain the overlap in purported outcomes such as preterm delivery, low birth weight, and neonatal complications. Two recent studies from our group underscore these limitations. Retrospective recall at 6 months postpartum versus prospective documentation indicates that women have poor recall of depressive symptoms in pregnancy and exposure to nonpsychotropic medications (Newport et al., 2008). In a second prospective study, we found a significant association of maternal depressive symptoms and anxiety in pregnancy with exposure to nonpsychiatric medications, increased tobacco use, and poor compliance with prenatal vitamins (Newport et al., 2012). Despite these limitations and other potential confounds, the reproductive outcome literature on antidepressants in pregnancy and breast feeding is very large compared with other classes of medications used during the perinatal period such as antiemetics and pain medications.
Numerous reviews cite the apparent safety of antidepressants during pregnancy (Koren et al., 1998; Kulin et al., 1998a; Davanzo et al., 2011). Others point out that the risk of administering these medications during pregnancy is still uncertain (Tuccori et al., 2009; Ellfolk and Malm, 2010). The issue sparks intense debate, and some point out that the risk is too great and psychotherapy should supplant antidepressants (Campagne, 2007), although this is contested by others who conduct large center studies investigating these issues (Campagne, 2008; Einarson and Eberhard-Gran, 2013; Einarson, 2009). Nevertheless, informed consent is advised in all circumstances, and antidepressant use should be considered on a case by case basis (Gupta et al., 1998). The clinical conundrum of balancing the risks and benefits of antidepressant use in the perinatal period is often distilled to decisions being made on a "case by case" basis in the majority of review articles cited above and others. Unfortunately, this is not always a meaningful guide to the clinician. In many cases, alternative nonpharmacological treatments may not be available or affordable. There is no current gold standard test for the level of maternal depression associated with adverse outcomes that would guide the need for intervention.
To complement the information in the clinical decision process in light of the inconclusive clinical literature, the incorporation of results from preclinical studies may be of benefit. For example, antidepressants share many common pharmacological properties and if the primary mechanism of action is responsible for the potential impact, more consistency between individual medications and broader classes (SSRI and TCA) would be expected. Scrutiny of the preclinical literature for lines of convergence and agreement may serve to substantiate concerns in light of contradictory data as well as direct future investigations.
III. Conclusions and Future Directions
The use of antidepressants as a primary treatment modality during pregnancy will continue to generate debate. Fortunately, the ongoing scrutiny has generated a relatively enormous literature compared with other classes of medication that are often used in the prenatal period. Unfortunately, the literature remains laden with methodological differences that preclude definitive conclusions and clinical recommendations. Tragically, the studies reporting adverse effects tend to garner greater coverage in the "lay" media and even notoriety in the scientific literature relative to those studies that have failed to identify adverse effects even when these "negative" studies have been rigorously designed. This has all too often resulted in the "reflexive" changes in clinical care (e.g., abruptly discontinuing antidepressants at knowledge of conception) that are not evidence based and may pose potential risks for both the mental health of the mother and overall outcome. For example, the rate of inadvertent conception is over 50%, hence, changing treatment at knowledge of conception would not reduce the potential liability for birth defects, if any, related to organogenesis. Similarly, there is not uniform availability or affordability of nonpharmacological interventions for depression even during the perinatal period—an issue beyond the scope of this review.
The standard recommendation of providing a risk-benefit assessment is not unique to antidepressant medications. The potential risks to the fetus versus benefits of intervention are default in the FDA category and extend to all classes of medications. For example, a risk-benefit assessment is advised before prescribing many drugs such as antihistamines or proton pump inhibitors to a pregnant woman (Gilbert et al., 2005; Majithia and Johnson, 2012). Antidepressant use during the perinatal period should be considered carefully, weighing all the evidence equally and impartially. Definitive conclusions from the available evidence are limited by the potential confounds and methodological limitations of the individual studies.
The perfect perinatal exposure study does not exist in the clinical or preclinical literature. The major limitations within the clinical literature are seldom incorporated in the discussion and/or conclusions of these studies and include, but are not limited to 1) assessment and verification of antidepressant use; 2) control for exposure to other substances; 3) control for maternal psychiatric illness using validated instruments of symptoms and/or diagnoses; and 4) defining appropriate control/comparison groups. Briefly, there is considerable evidence that patients are not 100% compliant with antidepressant medications, even in clinical trials where the patient knows that some assessment will be conducted. Noncompliance or poor compliance in patients receiving treatment of psychiatric disorders has been shown to be as high as 60% (Colom et al., 2000). For our conclusions, we have defined “reported antidepressant use” as those studies where the assessment was either prospective or retrospective patient reporting or medical record documentation. “Probable antidepressant use” are for those studies that used pharmacy or prescription database information. “Confirmed antidepressant use” is reserved for those studies that obtained objective laboratory confirmation of exposure, and this is by far the smallest number of studies. The same criteria could be applied to the use of concomitant substances such as nicotine, drugs of abuse, and other medications; however, such rigor would essentially negate all the clinical literature. It is well documented that nicotine use during pregnancy contributes significantly to long-term diseases and disorders in the offspring (Bruin et al., 2010) and 12.4–21.7% of pregnant women in the United States smoke during pregnancy (Goodwin et al., 2007). Investigators have shown that maternal reporting of smoking does not consistently reflect measures of urine cotinine in late pregnancy. Studies of the effects of antidepressants and maternal depression have often relied on measures (retrospective and cross-sectional) of depressive symptoms, and although there have been studies examining the validity of such measures across pregnancy with respect to meeting diagnostic criteria (Ji et al., 2011), it is unclear whether these symptoms influence the outcome relative to a lifetime diagnosis of major depression. Defining appropriate control groups is one of the greatest challenges. Controlled, randomized trials for antidepressants in pregnancy do not exist. Similarly, comparing antidepressant exposure to a group of women without a history of depression fails to account for the illness. We are aware of no data on obstetrical outcome in women with a history of depression that remain well and off medications during pregnancy.
Controlled studies that use pregnant women with a concurrent history of mental illness and antidepressant use compared with unmedicated pregnant women with a history of mental illness are necessary to determine possible ill effects (Fig. 6). The need for this important control group becomes evident when examining the effects of prenatal stress exposure. There are more studies of the effects of stress during pregnancy than studies examining the effects of prenatal antidepressant exposure that predominately indicate a detrimental effect of prenatal stress on behavior and aspects of stress physiology. Controlling for illness, symptoms, and maternal stress will be essential in future clinical studies if we hope to definitively develop clinical guidelines for antidepressant use in pregnancy.
Fig. 6.

Teratological relevance of animal studies versus clinical relevance of human studies. The majority of human studies investigating prenatal antidepressant exposure compare medicated and depressed pregnant women to unmedicated and nondepressed control groups to investigated clinical outcomes. The majority of animal studies investigating prenatal antidepressant exposure compare a medicated animal to an unmedicated animal to determine teratological consequences.
The current preclinical investigations of prenatal antidepressant exposure also have several limitations that warrant attention, including 1) dosing models; 2) species differences; and 3) comparable outcome variables. The majority of animal studies use short-term dosing paradigms to examine adverse effects in the fetus and/or offspring. The clinical relevance of these studies may be of limited clinical import if exposures do not mirror those seen in humans (Fig. 6). Species differences in the transporter affinity of individual antidepressants are minimal (Owens et al., 1997); however, animal drug clearance is typically much greater than that of humans, precipitating a need for a much higher dose in animals than humans to produce equivalent serum concentrations/transporter occupancy. When the logistics of conducting animal experiments and the actual physiochemical properties of the medications themselves are taken into account, many extant dosing strategies lead to a pronounced bolus effect in animals that may lead to transient toxic serum concentrations (Fig. 1). Human dosing regimens and pharmacokinetics lead to much shallower peak and trough serum concentrations of psychotropics. The advent of continuous drug delivery systems such as osmotic minipumps or dosing in chow or water can circumvent these concerns by delivery of a more consistent dose (i.e., exposure) of medication over a long period of time. These continuous delivery systems begin to achieve clinically relevant, documented levels of exposure to enhance the clinical import of preclinical studies.
The decision of a model species to use for testing teratogenicity is central to the investigation of the effects of prenatal antidepressant exposure. Current studies have used sheep, guinea pigs, rabbits, nonhuman primates, rats, and mice, but the majority of animal studies have relied on rats as the model species. Unfortunately, the majority of the endpoints examined in rats are not dose dependent or reproducible. In contrast, studies in mice have often shown toxicity. The difference(s) between rat and mouse studies may be important and may be related to species differences in maternal and other behaviors, at baseline or in response to stressful environments. Indeed, it is clear that even strain differences within species exist in animal behavior studies. Until definitive human data are obtained, it seems prudent to use multiple species in experimental studies
We sought to provide a synopsis of both the clinical and preclinical data with the hopes of identifying lines of convergence between the clinical and preclinical literature to assist in the clinical decision process. These are summarized in Tables 5 and 6. The conclusions across species for perinatal antidepressant exposure are limited, but include 1) confirmed antidepressant use in pregnancy results in significant fetal exposure across the placenta that has been confirmed preclinically; 2) umbilical cord nursing infant serum antidepressant concentrations do not accurately reflect the extent of CNS exposure and preclinical data would indicate significant transporter binding; 3) reported antidepressant use in pregnancy results in short-term changes in indices of monoamine systems and neuroendocrine systems that do not persist after discontinuing exposure—the preclinical literature confirms alterations albeit not consistent for the neuroendocrine effects; 4) reported antidepressant use in pregnancy is associated with decreased birth weight but it is not clear that this is a clinically significant effect (e.g., use of weight as a continuous variable is positive, but negative when defined categorically)—this is also reflected in the preclinical literature; 5) reported antidepressant use during pregnancy is associated with alterations in sleep architecture in the neonatal period in the limited number of both clinical and preclinical studies; 6) reported and confirmed antidepressant use during pregnancy is associated with increased motor activity and/or jitteriness in a subgroup of neonates—this has been confirmed in the preclinical literature, but it remains unclear if this is driven by short-term exposure, withdrawal, and/or potential toxicity; and 7) reported antidepressant use has been associated with an increased risk for persistent pulmonary hypertension of the newborn (PPHN), although subsequent studies in larger populations with improved methodology have demonstrated a steadily decreasing odds ratio compared with the initial retrospective report. The preclinical literature has suggested that there may be more subtle effects that may potentially contribute to this risk. The convergence of these data indicates that the majority of effects are associated with the short-term effects of antidepressant exposure and, with the exception of the potential risk for PPHN, there is no evidence supporting adverse consequences.
TABLE 5.
Summary of convergent data
| Preclinical | Clinical | Comments | |
|---|---|---|---|
| Exposure | Confirmed placental passage | Confirmed placental passage | In utero fetal exposure is likely similar to that in the mother. |
| Confirmed presence in breast milk | Confirmed presence in breast milk | Exposure is substantially less but measurable. Physiologic significance not known but probably minimal. | |
| Obstetrical outcome | Decreased birth weight | Decreased birth weight | The majority use weight as continuous variable and clinical significance is not clear. |
| Neonatal period | Increased motor activity/jitteriness | Increased motor activity/jitteriness | Limited to subgroup of the exposed offspring. |
| Altered sleep architecture | Altered sleep architecture | ||
| Persistent pulmonary hypertension | Increased risk | Increased risk |
TABLE 6.
Summary of nonconvergent data
| Preclinical | Clinical | Comments | |
|---|---|---|---|
| Birth defects | No substantial evidence | Sporadic case reports and statistical evidence of increased risk in some large scale studies. | Numerous potential confounds in clinical literature preclude conclusions in the presence of prolonged record of antidepressant use during pregnancy (>40 years). |
| Long-term outcome | No persistent adverse effects | Limited studies from neonatal to early childhood do not demonstrate significant risk. No studies of long-term outcomes in offspring into adolescence or adulthood. | Preclinical studies of postnatal exposure reveal long term behavioral alterations, however many findings are not replicable. Clinical relevancy of dosing regimens must be taken into account. |
| Monoamine/neuroendocrine | Altered monoamine | Altered salivary cortisol in children | Preclinical studies may have questionable dosing relevancy |
| Altered cortisol and insulin | |||
| Behavioral outcome | Some evidence for behavioral outcome | Some studies show behavioral differences in children | Preclinical studies of postnatal exposure reveal long term behavioral alterations |
| Studies often conflicting or not replicated |
In contrast, the clinical and preclinical literature on antidepressant use in pregnancy have failed to identify an increased risk for birth defects, a consistent pattern of organ dysgenesis, persistent aberrations in behavior and/or development in the neonatal period, and the limited long-term outcome data are devoid of consistent adverse effects.
In summary, the decision to use antidepressants during pregnancy either as an short-term intervention and/or preventative strategy for recurrent illness remains controversial and is hindered by the lack of evidence-based studies. The relative reproductive safety data compared with other classes of medication used to treat conditions of far less severity than depression are promising and support them as a viable treatment option. The importance of serotonin and other neurotransmitter systems in development underscore the need for more rigorous methodology to delineate the impact, if any, of confirmed antidepressant exposure in pregnancy. Future preclinical investigations with clinically relevant exposures and clinical studies with improved methodology provide the requisite foundation to develop scientifically derived guidelines for the treatment of these women.
Abbreviations
- Apgar
Appearance Pulse Grimace Activity Respiration
- CNS
central nervous system
- FDA
Food and Drug Administration
- G
gestational day
- HPA
hypothalamic-pituitary-adrenal
- 5-HT
5-hydroxytryptamine (serotonin)
- 5HTTLPR
serotonin-transporter-linked polymorphic
- NRI
norepinephrine reuptake inhibitor
- PND
postnatal day
- PPHN
persistent pulmonary hypertension of the newborn
- TCA
tricyclic antidepressant
- SERT
serotonin transporter
- SRI
serotonin reuptake inhibitor
- SSRI
selective-serotonin reuptake inhibitor
Authorship Contributions
Wrote and contributed to the writing of the manuscript: Bourke, Stowe, Owens.
Footnotes
This work was supported by the National Institutes of Health National Institute of Mental Health [Grant MH077928] (to C.H.B. and M.J.O.); and the National Institutes of Health National Institute of Environmental Health Sciences [Grant ES012870] (to C.H.B.). Financial disclosure (preceding 12 months or anticipated in next 12 months) are indicated as follows: C.H.B. has no conflicts of interest or financial disclosures; M.J.O. received research grants from the National Institutes of Health, Lundbeck A/S, Cyberonics, Dainippon Sumitomo Pharma, SK Life Sciences, Sunovion Pharmaceuticals; serves as a consultant to H. Lundbeck A/S, R.J. Reynolds, and the United States Department of Justice; and receives compensation for these services (the terms of this arrangement have been reviewed and approved by Emory University in accordance with its conflict of interest policies); and holds a patent [Owens MJ and Nemeroff CB (2004) Method of assessing antidepressant drug therapy via transport inhibition of monoamine neurotransmitters. U.S. Patent 7,148,027 B2, 12 Dec 2006.]; Z.N.S. received research support from the National Institutes of Health, GlaxoSmithKline, Pfizer, and Wyeth; served on speakers and/or advisory boards for Pfizer, Eli Lilly, Wyeth, BristolMyers-Squibb, and GlaxoSmithKline; and received honoraria from Eli Lilly, GlaxoSmithKline, Pfizer, and Wyeth.
Address correspondence to: Dr. Michael J. Owens, Laboratory of Neuropsychopharmacology, Department of Psychiatry and Behavioral Sciences, 101 Woodruff Circle, Suite 4000, Emory University, Atlanta, GA 30322. E-mail: mowens@emory.edu
References
- ACOG Committee on Practice Bulletins–Obstetrics (2008) ACOG Practice Bulletin: Clinical management guidelines for obstetrician-gynecologists number 92, April 2008 (replaces practice bulletin number 87, November 2007). Use of psychiatric medications during pregnancy and lactation. Obstet Gynecol 111:1001–1020 [DOI] [PubMed] [Google Scholar]
- Addis A, Koren G. (2000) Safety of fluoxetine during the first trimester of pregnancy: a meta-analytical review of epidemiological studies. Psychol Med 30:89–94 [DOI] [PubMed] [Google Scholar]
- Ali SF, Buelke-Sam J, Newport GD, Slikker W., Jr (1986) Early neurobehavioral and neurochemical alterations in rats prenatally exposed to imipramine. Neurotoxicology 7:365–380 [PubMed] [Google Scholar]
- Als H, Tronick E, Lester BM, Brazelton TB. (1977) The Brazelton Neonatal Behavioral Assessment Scale (BNBAS). J Abnorm Child Psychol 5:215–231 [DOI] [PubMed] [Google Scholar]
- Altshuler LL, Burt VK, McMullen M, Hendrick V. (1995) Breastfeeding and sertraline: a 24-hour analysis. J Clin Psychiatry 56:243–245 [PubMed] [Google Scholar]
- Alwan S, Reefhuis J, Botto LD, Rasmussen SA, Correa A, Friedman JM. (2010) Maternal use of bupropion and risk for congenital heart defects. Am J Obstet Gynecol 203:e1–6 [DOI] [PubMed] [Google Scholar]
- Alwan S, Reefhuis J, Rasmussen SA, Friedman JM, National Birth Defects Prevention Study (2011) Patterns of antidepressant medication use among pregnant women in a United States population. J Clin Pharmacol 51:264–270 [DOI] [PubMed] [Google Scholar]
- American College of Obstetricians and Gynecologists (2007) ACOG Practice Bulletin No. 87 November 2007: Use of psychiatric medications during pregnancy and lactation. Obstet Gynecol 110:1179–1198 [DOI] [PubMed] [Google Scholar]
- Anderson GM, Czarkowski K, Ravski N, Epperson CN. (2004) Platelet serotonin in newborns and infants: ontogeny, heritability, and effect of in utero exposure to selective serotonin reuptake inhibitors. Pediatr Res 56:418–422 [DOI] [PubMed] [Google Scholar]
- Andrade SE, Gurwitz JH, Davis RL, Chan KA, Finkelstein JA, Fortman K, McPhillips H, Raebel MA, Roblin D, Smith DH, et al. (2004) Prescription drug use in pregnancy. Am J Obstet Gynecol 191:398–407 [DOI] [PubMed] [Google Scholar]
- Andrade SE, Raebel MA, Brown J, Lane K, Livingston J, Boudreau D, Rolnick SJ, Roblin D, Smith DH, Willy ME, et al. (2008) Use of antidepressant medications during pregnancy: a multisite study. Am J Obstet Gynecol 198:e1–e5 [DOI] [PubMed] [Google Scholar]
- Ansorge MS, Morelli E, Gingrich JA. (2008) Inhibition of serotonin but not norepinephrine transport during development produces delayed, persistent perturbations of emotional behaviors in mice. J Neurosci 28:199–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansorge MS, Zhou M, Lira A, Hen R, Gingrich JA. (2004) Early-life blockade of the 5-HT transporter alters emotional behavior in adult mice. Science 306:879–881 [DOI] [PubMed] [Google Scholar]
- Apgar V. (1953) A proposal for a new method of evaluation of the newborn infant. Curr Res Anest Anal 32:260–267 [PubMed] [Google Scholar]
- Audus KL. (1999) Controlling drug delivery across the placenta. Eur J Pharm Sci 8:161–165 [DOI] [PubMed] [Google Scholar]
- Avishai-Eliner S, Brunson KL, Sandman CA, Baram TZ. (2002) Stressed-out, or in (utero)? Trends Neurosci 25:518–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baab SW, Peindl KS, Piontek CM, Wisner KL. (2002) Serum bupropion levels in 2 breastfeeding mother-infant pairs. J Clin Psychiatry 63:910–911 [DOI] [PubMed] [Google Scholar]
- Bairy KL, Madhyastha S, Ashok KP, Bairy I, Malini S. (2007) Developmental and behavioral consequences of prenatal fluoxetine. Pharmacology 79:1–11 [DOI] [PubMed] [Google Scholar]
- Bakker MK, De Walle HEK, Wilffert B, de Jong-Van den Berg LTW. (2010) Fluoxetine and infantile hypertrophic pylorus stenosis: a signal from a birth defects-drug exposure surveillance study. Pharmacoepidemiol Drug Saf 19:808–813 [DOI] [PubMed] [Google Scholar]
- Bar-Oz B, Einarson T, Einarson A, Boskovic R, O’Brien L, Malm H, Bérard A, Koren G. (2007) Paroxetine and congenital malformations: meta-Analysis and consideration of potential confounding factors. Clin Ther 29:918–926 [DOI] [PubMed] [Google Scholar]
- Bauer S, Monk C, Ansorge M, Gyamfi C, Myers M. (2010) Impact of antenatal selective serotonin reuptake inhibitor exposure on pregnancy outcomes in mice. Am J Obstet Gynecol 203:e1–e4 [DOI] [PubMed] [Google Scholar]
- Bennett IM, Marcus SC, Palmer SC, Coyne JC. (2010) Pregnancy-related discontinuation of antidepressants and depression care visits among Medicaid recipients. Psychiatr Serv 61:386–391 [DOI] [PubMed] [Google Scholar]
- Berle JØ, Steen VM, Aamo TO, Breilid H, Zahlsen K, Spigset O. (2004) Breastfeeding during maternal antidepressant treatment with serotonin reuptake inhibitors: infant exposure, clinical symptoms, and cytochrome p450 genotypes. J Clin Psychiatry 65:1228–1234 [DOI] [PubMed] [Google Scholar]
- Bertossi M, Virgintino D, Errede M, Roncali L. (1999) Immunohistochemical and ultrastructural characterization of cortical plate microvasculature in the human fetus telencephalon. Microvasc Res 58:49–61 [DOI] [PubMed] [Google Scholar]
- Beyer BK, Guram MS, Geber WF. (1984) Incidence and potentiation of external and internal fetal anomalies resulting from chlordiazepoxide and amitriptyline alone and in combination. Teratology 30:39–45 [DOI] [PubMed] [Google Scholar]
- Bloem BR, Lammers GJ, Roofthooft DW, De Beaufort AJ, Brouwer OF. (1999) Clomipramine withdrawal in newborns. Arch Dis Child Fetal Neonatal Ed 81:F77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonari L, Koren G, Einarson TR, Jasper JD, Taddio A, Einarson A. (2005) Use of antidepressants by pregnant women: evaluation of perception of risk, efficacy of evidence based counseling and determinants of decision making. Arch Women Ment Health 8:214–220 [DOI] [PubMed] [Google Scholar]
- Bonnin A, Goeden N, Chen K, Wilson ML, King J, Shih JC, Blakely RD, Deneris ES, Levitt P. (2011) A transient placental source of serotonin for the fetal forebrain. Nature 472:347–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnin A, Torii M, Wang L, Rakic P, Levitt P. (2007) Serotonin modulates the response of embryonic thalamocortical axons to netrin-1. Nat Neurosci 10:588–597 [DOI] [PubMed] [Google Scholar]
- Bourke CH, Capello CF, Rogers SM, Yu ML, Boss-Williams KA, Weiss JM, Stowe ZN, Owens MJ. (2013a) Prenatal exposure to escitalopram and/or stress in rats: a prenatal stress model of maternal depression and its treatment. Psychopharmacology (Berl) 228:231–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourke CH, Stowe ZN, Neigh GN, Olson DE, Owens MJ. (2013b) Prenatal exposure to escitalopram and/or stress in rats produces limited effects on endocrine, behavioral, or gene expression measures in adult male rats. Neurotoxicol Teratol 39:100–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan PA, Pargas R, Walker EF, Green P, Newport DJ, Stowe Z. (2008) Maternal depression and infant cortisol: influences of timing, comorbidity and treatment. J Child Psychol Psychiatry 49:1099–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brent RL. (2010) Reproductive toxicology. Reprod Toxicol 29:251–253, author reply 254–255 [DOI] [PubMed] [Google Scholar]
- Breyer-Pfaff U, Nill K, Entenmann KN, Gaertner HJ. (1995) Secretion of amitriptyline and metabolites into breast milk. Am J Psychiatry 152:812–813 [DOI] [PubMed] [Google Scholar]
- Briggs GG, Ambrose PJ, Ilett KF, Hackett LP, Nageotte MP, Padilla G. (2009) Use of duloxetine in pregnancy and lactation. Ann Pharmacother 43:1898–1902 [DOI] [PubMed] [Google Scholar]
- Brixen-Rasmussen L, Halgrener J, Jørgensen A. (1982) Amitriptyline and nortriptyline excretion in human breast milk. Psychopharmacology (Berl) 76:94–95 [DOI] [PubMed] [Google Scholar]
- Bruin JE, Gerstein HC, Holloway AC. (2010) Long-term consequences of fetal and neonatal nicotine exposure: a critical review. Toxicol Sci 116:364–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd RA, Markham JK. (1994) Developmental toxicology studies of fluoxetine hydrochloride administered orally to rats and rabbits. Fundam Appl Toxicol 22:511–518 [DOI] [PubMed] [Google Scholar]
- Cabrera-Vera TM, Battaglia G. (1998) Prenatal exposure to fluoxetine (Prozac) produces site-specific and age-dependent alterations in brain serotonin transporters in rat progeny: evidence from autoradiographic studies. J Pharmacol Exp Ther 286:1474–1481 [PubMed] [Google Scholar]
- Cabrera-Vera TM, Garcia F, Pinto W, Battaglia G. (1997) Effect of prenatal fluoxetine (Prozac) exposure on brain serotonin neurons in prepubescent and adult male rat offspring. J Pharmacol Exp Ther 280:138–145 [PubMed] [Google Scholar]
- Cagiano R, Flace P, Bera I, Maries L, Cioca G, Sabatini R, Benagiano V, Auteri P, Marzullo A, Vermesan D, et al. (2008) Neurofunctional effects in rats prenatally exposed to fluoxetine. Eur Rev Med Pharmacol Sci 12:137–148 [PubMed] [Google Scholar]
- Campagne DM. (2007) Fact: antidepressants and anxiolytics are not safe during pregnancy. Eur J Obstet Gynecol Reprod Biol 135:145–148 [DOI] [PubMed] [Google Scholar]
- Campagne DM. (2008) Author's response: Antidepressants and anxiolytics in pregnancy: The facts stand. Eur J Obstet Gynecol Reprod Biol 171:e2–e3 [DOI] [PubMed] [Google Scholar]
- Capello CF, Bourke CH, Ritchie JC, Stowe ZN, Newport DJ, Nemeroff A, Owens MJ. (2011) Serotonin transporter occupancy in rats exposed to serotonin reuptake inhibitors in utero or via breast milk. J Pharmacol Exp Ther 339:275–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey BM, McIntire DD, Leveno KJ. (2001) The continuing value of the Apgar score for the assessment of newborn infants. N Engl J Med 344:467–471 [DOI] [PubMed] [Google Scholar]
- Casper RC, Fleisher BE, Lee-Ancajas JC, Gilles A, Gaylor E, DeBattista A, Hoyme HE. (2003) Follow-up of children of depressed mothers exposed or not exposed to antidepressant drugs during pregnancy. J Pediatr 142:402–408 [DOI] [PubMed] [Google Scholar]
- Chambers CD, Hernandez-Diaz S, Van Marter LJ, Werler MM, Louik C, Jones KL, Mitchell AA. (2006) Selective serotonin-reuptake inhibitors and risk of persistent pulmonary hypertension of the newborn. N Engl J Med 354:579–587 [DOI] [PubMed] [Google Scholar]
- Christensen HD, Rayburn WF, Gonzalez CL. (2000) Chronic prenatal exposure to paroxetine (Paxil) and cognitive development of mice offspring. Neurotoxicol Teratol 22:733–739 [DOI] [PubMed] [Google Scholar]
- Chun-Fai-Chan B, Koren G, Fayez I, Kalra S, Voyer-Lavigne S, Boshier A, Shakir S, Einarson A. (2005) Pregnancy outcome of women exposed to bupropion during pregnancy: a prospective comparative study. Am J Obstet Gynecol 192:932–936 [DOI] [PubMed] [Google Scholar]
- Clancy B, Darlington RB, Finlay BL. (2001) Translating developmental time across mammalian species. Neuroscience 105:7–17 [DOI] [PubMed] [Google Scholar]
- Cohen LS, Altshuler LL, Harlow BL, Nonacs R, Newport DJ, Viguera AC, Suri R, Burt VK, Hendrick V, Reminick AM, et al. (2006) Relapse of major depression during pregnancy in women who maintain or discontinue antidepressant treatment. JAMA 295:499–507 [DOI] [PubMed] [Google Scholar]
- Cohen LS, Friedman JM, Jefferson JW, Johnson EM, Weiner ML. (1994) A reevaluation of risk of in utero exposure to lithium. JAMA 271:146–150 [PubMed] [Google Scholar]
- Cohen LS, Heller VL, Bailey JW, Grush L, Ablon JS, Bouffard SM. (2000) Birth outcomes following prenatal exposure to fluoxetine. Biol Psychiatry 48:996–1000 [DOI] [PubMed] [Google Scholar]
- Cole JA, Ephross SA, Cosmatos IS, Walker AM. (2007) Paroxetine in the first trimester and the prevalence of congenital malformations. Pharmacoepidemiol Drug Saf 16:1075–1085 [DOI] [PubMed] [Google Scholar]
- Coleman FH, Christensen HD, Gonzalez CL, Rayburn WF. (1999) Behavioral changes in developing mice after prenatal exposure to paroxetine (Paxil). Am J Obstet Gynecol 181:1166–1171 [DOI] [PubMed] [Google Scholar]
- Colom F, Vieta E, Martínez-Arán A, Reinares M, Benabarre A, Gastó C. (2000) Clinical factors associated with treatment noncompliance in euthymic bipolar patients. J Clin Psychiatry 61:549–555 [DOI] [PubMed] [Google Scholar]
- Cooper WO, Willy ME, Pont SJ, Ray WA. (2007) Increasing use of antidepressants in pregnancy. Am J Obstet Gynecol 196:e1–5 [DOI] [PubMed] [Google Scholar]
- Costei AM, Kozer E, Ho T, Ito S, Koren G. (2002) Perinatal outcome following third trimester exposure to paroxetine. Arch Pediatr Adolesc Med 156:1129–1132 [DOI] [PubMed] [Google Scholar]
- Coverdale JH, McCullough LB, Chervenak FA. (2008) The ethics of randomized placebo-controlled trials of antidepressants with pregnant women: a systematic review. Obstet Gynecol 112:1361–1368 [DOI] [PubMed] [Google Scholar]
- Coyle IR. (1975) Changes in developing behavior following prenatal administration of imipramine. Pharmacol Biochem Behav 3:799–807 [DOI] [PubMed] [Google Scholar]
- Coyle IR, Singer G. (1975a) The interaction of post-weaning housing conditions and prenatal drug effects on behaviour. Psychopharmacology (Berl) 41:237–244 [DOI] [PubMed] [Google Scholar]
- Coyle IR, Singer G. (1975b) The interactive effects of prenatal imipramine exposure and postnatal rearing conditions on behaviour and histology. Psychopharmacology (Berl) 44:253–256 [DOI] [PubMed] [Google Scholar]
- Croen LA, Grether JK, Yoshida CK, Odouli R, Hendrick V. (2011) Antidepressant use during pregnancy and childhood autism spectrum disorders. Arch Gen Psychiatry 68:1104–1112 [DOI] [PubMed] [Google Scholar]
- Crombie DL, Pinsent RJ, Fleming D. (1972) Imipramine in pregnancy. BMJ 1:745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuomo V, Cortese I, Cagiano R, Renna G, Racagni G. (1984) Behavioural changes in rats after prenatal administration of typical and atypical antidepressants. Arch Toxicol Suppl 7:504–507 [DOI] [PubMed] [Google Scholar]
- D’Amato RJ, Blue ME, Largent BL, Lynch DR, Ledbetter DJ, Molliver ME, Snyder SH. (1987) Ontogeny of the serotonergic projection to rat neocortex: transient expression of a dense innervation to primary sensory areas. Proc Natl Acad Sci USA 84:4322–4326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- da-Silva VA, Altenburg SP, Malheiros LR, Thomaz TG, Lindsey CJ. (1999) Postnatal development of rats exposed to fluoxetine or venlafaxine during the third week of pregnancy. Braz J Med Biol Res 32:93–98 [DOI] [PubMed] [Google Scholar]
- Darnaudéry M, Maccari S. (2008) Epigenetic programming of the stress response in male and female rats by prenatal restraint stress. Brain Res Brain Res Rev 57:571–585 [DOI] [PubMed] [Google Scholar]
- Davanzo R, Copertino M, De Cunto A, Minen F, Amaddeo A. (2011) Antidepressant drugs and breastfeeding: a review of the literature. Breastfeed Med 6:89–98 [DOI] [PubMed] [Google Scholar]
- Davidson S, Prokonov D, Taler M, Maayan R, Harell D, Gil-Ad I, Weizman A. (2009) Effect of exposure to selective serotonin reuptake inhibitors in utero on fetal growth: potential role for the IGF-I and HPA axes. Pediatr Res 65:236–241 [DOI] [PubMed] [Google Scholar]
- De Ceballos ML, Benedi A, De Felipe C, Del Rio J. (1985a) Prenatal exposure of rats to antidepressants enhances agonist affinity of brain dopamine receptors and dopamine-mediated behaviour. Eur J Pharmacol 116:257–262 [DOI] [PubMed] [Google Scholar]
- De Ceballos ML, Benedi A, Urdin C, Del Rio J. (1985b) Prenatal exposure of rats to antidepressant drugs down-regulates beta-adrenoceptors and 5-HT2 receptors in cerebral cortex. Lack of correlation between 5-HT2 receptors and serotonin-mediated behaviour. Neuropharmacology 24:947–952 [DOI] [PubMed] [Google Scholar]
- Delahunt CS, Lassen LJ. (1964) Thalidomide syndrome in monkeys. Science 146:1300–1305 [DOI] [PubMed] [Google Scholar]
- DeVane CL, Simpkins JW. (1985) Pharmacokinetics of imipramine and its major metabolites in pregnant rats and their fetuses following a single dose. Drug Metab Dispos 13:438–442 [PubMed] [Google Scholar]
- DeVane CL, Simpkins JW, Stout SA. (1984) Cerebral and blood pharmacokinetics of imipramine and its active metabolites in the pregnant rat. Psychopharmacology (Berl) 84:225–230 [DOI] [PubMed] [Google Scholar]
- Diav-Citrin O, Shechtman S, Weinbaum D, Wajnberg R, Avgil M, Di Gianantonio E, Clementi M, Weber-Schoendorfer C, Schaefer C, Ornoy A. (2008) Paroxetine and fluoxetine in pregnancy: a prospective, multicentre, controlled, observational study. Br J Clin Pharmacol 66:695–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djulus J, Koren G, Einarson TR, Wilton L, Shakir S, Diav-Citrin O, Kennedy D, Voyer Lavigne S, De Santis M, Einarson A. (2006) Exposure to mirtazapine during pregnancy: a prospective, comparative study of birth outcomes. J Clin Psychiatry 67:1280–1284 [DOI] [PubMed] [Google Scholar]
- Domar AD, Moragianni VA, Ryley DA, Urato AC. (2013) The risks of selective serotonin reuptake inhibitor use in infertile women: a review of the impact on fertility, pregnancy, neonatal health and beyond. Hum Reprod 28:160–171 [DOI] [PubMed] [Google Scholar]
- Douglas BH, Hume AS. (1967) Placental transfer of imipramine, a basic, lipid-soluble drug. Am J Obstet Gynecol 99:573–575 [DOI] [PubMed] [Google Scholar]
- Drago F, Continella G, Alloro MC, Scapagnini U. (1985) Behavioral effects of perinatal administration of antidepressant drugs in the rat. Neurobehav Toxicol Teratol 7:493–497 [PubMed] [Google Scholar]
- Drugs Co (1965) A new antidepressant: desipramine hydrochloride (Norpramin, Pertofrane). JAMA 194:82–83 [PubMed] [Google Scholar]
- Dunkel Schetter C, Tanner L. (2012) Anxiety, depression and stress in pregnancy: implications for mothers, children, research, and practice. Curr Opin Psychiatry 25:141–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn GA, Morgan CP, Bale TL. (2011) Sex-specificity in transgenerational epigenetic programming. Horm Behav 59:290–295 [DOI] [PubMed] [Google Scholar]
- Einarson A. (2009) Comment on Campagne “Fact: antidepressants and anxiolytics are not safe during pregnancy”. Eur J Obstet Gynecol Reprod Biol 142:163. [DOI] [PubMed] [Google Scholar]
- Einarson A, Bonari L, Voyer-Lavigne S, Addis A, Matsui D, Johnson Y, Koren G. (2003) A multicentre prospective controlled study to determine the safety of trazodone and nefazodone use during pregnancy. Can J Psychiatry 48:106–110 [DOI] [PubMed] [Google Scholar]
- Einarson A, Choi J, Einarson TR, Koren G. (2009) Incidence of major malformations in infants following antidepressant exposure in pregnancy: results of a large prospective cohort study. Can J Psychiatry 54:242–246 [DOI] [PubMed] [Google Scholar]
- Einarson A, Eberhard-Gran M. (2013) Comment on Campagne "Fact: antidepressants and anxiolytics are not safe during pregnancy”. Eur J Obstet Gynecol Reprod Biol 171:e2. [DOI] [PubMed] [Google Scholar]
- Einarson A, Fatoye B, Sarkar M, Lavigne SV, Brochu J, Chambers C, Mastroiacovo P, Addis A, Matsui D, Schuler L, et al. (2001) Pregnancy outcome following gestational exposure to venlafaxine: a multicenter prospective controlled study. Am J Psychiatry 158:1728–1730 [DOI] [PubMed] [Google Scholar]
- Einarson A, Koren G. (2007) Prescribing antidepressants to pregnant women: what is a family physician to do? Can Fam Physician 53:1412–1414, 1423–1415 [PMC free article] [PubMed] [Google Scholar]
- Einarson A, Pistelli A, DeSantis M, Malm H, Paulus WD, Panchaud A, Kennedy D, Einarson TR, Koren G. (2008) Evaluation of the risk of congenital cardiovascular defects associated with use of paroxetine during pregnancy. Am J Psychiatry 165:749–752 [DOI] [PubMed] [Google Scholar]
- Einarson A, Schachtschneider AK, Halil R, Bollano E, Koren G. (2005) SSRI’S and other antidepressant use during pregnancy and potential neonatal adverse effects: impact of a public health advisory and subsequent reports in the news media. BMC Pregnancy Childbirth 5:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einarson TR, Einarson A. (2005) Newer antidepressants in pregnancy and rates of major malformations: a meta-analysis of prospective comparative studies. Pharmacoepidemiol Drug Saf 14:823–827 [DOI] [PubMed] [Google Scholar]
- Ellfolk M, Malm H. (2010) Risks associated with in utero and lactation exposure to selective serotonin reuptake inhibitors (SSRIs). Reprod Toxicol 30:249–260 [DOI] [PubMed] [Google Scholar]
- Entringer S, Epel ES, Kumsta R, Lin J, Hellhammer DH, Blackburn EH, Wüst S, Wadhwa PD. (2011) Stress exposure in intrauterine life is associated with shorter telomere length in young adulthood. Proc Natl Acad Sci USA 108:E513–E518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epperson CN, Anderson GM, McDougle CJ. (1997) Sertraline and breast-feeding. N Engl J Med 336:1189–1190 [DOI] [PubMed] [Google Scholar]
- Epperson CN, Jatlow PI, Czarkowski K, Anderson GM. (2003) Maternal fluoxetine treatment in the postpartum period: effects on platelet serotonin and plasma drug levels in breastfeeding mother-infant pairs. Pediatrics 112:e425–e429 [DOI] [PubMed] [Google Scholar]
- Epperson N, Czarkowski KA, Ward-O’Brien D, Weiss E, Gueorguieva R, Jatlow P, Anderson GM. (2001) Maternal sertraline treatment and serotonin transport in breast-feeding mother-infant pairs. Am J Psychiatry 158:1631–1637 [DOI] [PubMed] [Google Scholar]
- Ericson A, Källén B, Wiholm B. (1999) Delivery outcome after the use of antidepressants in early pregnancy. Eur J Clin Pharmacol 55:503–508 [DOI] [PubMed] [Google Scholar]
- Favaro Pd, Costa LC, Moreira EG. (2008) Maternal fluoxetine treatment decreases behavioral response to dopaminergic drugs in female pups. Neurotoxicol Teratol 30:487–494 [DOI] [PubMed] [Google Scholar]
- Field T. (2011) Prenatal depression effects on early development: a review. Infant Behav Dev 34:1–14 [DOI] [PubMed] [Google Scholar]
- Figueroa R. (2010) Use of antidepressants during pregnancy and risk of attention-deficit/hyperactivity disorder in the offspring. J Dev Behav Pediatr 31:641–648 [DOI] [PubMed] [Google Scholar]
- File SE, Tucker JC. (1983) Prenatal treatment with clomipramine has an anxiolytic profile in the adolescent rat. Physiol Behav 31:57–61 [DOI] [PubMed] [Google Scholar]
- File SE, Tucker JC. (1984) Prenatal treatment with clomipramine: effects on the behaviour of male and female adolescent rats. Psychopharmacology (Berl) 82:221–224 [DOI] [PubMed] [Google Scholar]
- Food and Drug Administration (2011) 21CFR201.57: Title 21–Food and Drugs, in (Department of Health and Human Services ed). http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm?fr=201.57 [Google Scholar]
- Forcelli PA, Heinrichs SC. (2008) Teratogenic effects of maternal antidepressant exposure on neural substrates of drug-seeking behavior in offspring. Addict Biol 13:52–62 [DOI] [PubMed] [Google Scholar]
- Fornaro E, Li D, Pan J, Belik J. (2007) Prenatal exposure to fluoxetine induces fetal pulmonary hypertension in the rat. Am J Respir Crit Care Med 176:1035–1040 [DOI] [PubMed] [Google Scholar]
- Fratta ID, Sigg EB, Maiorana K. (1965) Teratogenic effects of thalidomide in rabbits, rats, hamsters, and mice. Toxicol Appl Pharmacol 7:268–286 [DOI] [PubMed] [Google Scholar]
- Frey OR, Scheidt P, von Brenndorff AI. (1999) Adverse effects in a newborn infant breast-fed by a mother treated with doxepin. Ann Pharmacother 33:690–693 [DOI] [PubMed] [Google Scholar]
- Fujii T. (1997) Transgenerational effects of maternal exposure to chemicals on the functional development of the brain in the offspring. Cancer Causes Control 8:524–528 [DOI] [PubMed] [Google Scholar]
- Fujii T, Ohtaki Y. (1985) Sex-related hyperthermic response to chlorpromazine in the offspring of rats treated with imipramine. Dev Pharmacol Ther 8:364–373 [DOI] [PubMed] [Google Scholar]
- Gavin NI, Gaynes BN, Lohr KN, Meltzer-Brody S, Gartlehner G, Swinson T. (2005) Perinatal depression: a systematic review of prevalence and incidence. Obstet Gynecol 106:1071–1083 [DOI] [PubMed] [Google Scholar]
- Geyer MA, McIlwain KL, Paylor R. (2002) Mouse genetic models for prepulse inhibition: an early review. Mol Psychiatry 7:1039–1053 [DOI] [PubMed] [Google Scholar]
- Gilbert C, Mazzotta P, Loebstein R, Koren G. (2005) Fetal safety of drugs used in the treatment of allergic rhinitis: a critical review. Drug Saf 28:707–719 [DOI] [PubMed] [Google Scholar]
- Goldstein DJ, Corbin LA, Sundell KL. (1997) Effects of first-trimester fluoxetine exposure on the newborn. Obstet Gynecol 89:713–718 [DOI] [PubMed] [Google Scholar]
- Goodwin RD, Keyes K, Simuro N. (2007) Mental disorders and nicotine dependence among pregnant women in the United States. Obstet Gynecol 109:875–883 [DOI] [PubMed] [Google Scholar]
- Gottlieb A, Keydar I, Epstein HT. (1977) Rodent brain growth stages: an analytical review. Biol Neonate 32:166–176 [DOI] [PubMed] [Google Scholar]
- Gouvêa TS, Morimoto HK, de Faria MJSS, Moreira EG, Gerardin DCC. (2008) Maternal exposure to the antidepressant fluoxetine impairs sexual motivation in adult male mice. Pharmacol Biochem Behav 90:416–419 [DOI] [PubMed] [Google Scholar]
- Grimm VE, Frieder B. (1987) Prenatal and early postnatal exposure to zimelidine: behavioral, neurochemical and histological findings in rats. Int J Neurosci 33:225–235 [DOI] [PubMed] [Google Scholar]
- Grunau RV, Craig KD. (1987) Pain expression in neonates: facial action and cry. Pain 28:395–410 [DOI] [PubMed] [Google Scholar]
- Grunau RV, Johnston CC, Craig KD. (1990) Neonatal facial and cry responses to invasive and non-invasive procedures. Pain 42:295–305 [DOI] [PubMed] [Google Scholar]
- Gupta S, Masand PS, Rangwani S. (1998) Selective serotonin reuptake inhibitors in pregnancy and lactation. Obstet Gynecol Surv 53:733–736 [DOI] [PubMed] [Google Scholar]
- Halbreich U. (2005) The association between pregnancy processes, preterm delivery, low birth weight, and postpartum depressions—the need for interdisciplinary integration. Am J Obstet Gynecol 193:1312–1322 [DOI] [PubMed] [Google Scholar]
- Harmon JR, Webb PJ, Kimmel GL, Delongchamp RR. (1986) Effect of prenatal imipramine exposure on development of the postnatal rat heart and brain. Teratog Carcinog Mutagen 6:173–184 [DOI] [PubMed] [Google Scholar]
- Hegerl U, Bottlender R, Gallinat J, Kuss HJ, Ackenheil M, Möller HJ. (1998) The serotonin syndrome scale: first results on validity. Eur Arch Psychiatry Clin Neurosci 248:96–103 [DOI] [PubMed] [Google Scholar]
- Heikkinen T, Ekblad U, Kero P, Ekblad S, Laine K. (2002a) Citalopram in pregnancy and lactation. Clin Pharmacol Ther 72:184–191 [DOI] [PubMed] [Google Scholar]
- Heikkinen T, Ekblad U, Laine K. (2001) Transplacental transfer of amitriptyline and nortriptyline in isolated perfused human placenta. Psychopharmacology (Berl) 153:450–454 [DOI] [PubMed] [Google Scholar]
- Heikkinen T, Ekblad U, Laine K. (2002b) Transplacental transfer of citalopram, fluoxetine and their primary demethylated metabolites in isolated perfused human placenta. BJOG 109:1003–1008 [DOI] [PubMed] [Google Scholar]
- Heils A, Teufel A, Petri S, Stöber G, Riederer P, Bengel D, Lesch KP. (1996) Allelic variation of human serotonin transporter gene expression. J Neurochem 66:2621–2624 [DOI] [PubMed] [Google Scholar]
- Henderson MG, McConnaughey MM, McMillen BA. (1991) Long-term consequences of prenatal exposure to cocaine or related drugs: effects on rat brain monoaminergic receptors. Brain Res Bull 26:941–945 [DOI] [PubMed] [Google Scholar]
- Henderson MG, McMillen BA. (1990) Effects of prenatal exposure to cocaine or related drugs on rat developmental and neurological indices. Brain Res Bull 24:207–212 [DOI] [PubMed] [Google Scholar]
- Henderson MG, McMillen BA. (1993) Changes in dopamine, serotonin and their metabolites in discrete brain areas of rat offspring after in utero exposure to cocaine or related drugs. Teratology 48:421–430 [DOI] [PubMed] [Google Scholar]
- Hendrick V, Smith LM, Suri R, Hwang S, Haynes D, Altshuler L. (2003a) Birth outcomes after prenatal exposure to antidepressant medication. Am J Obstet Gynecol 188:812–815 [DOI] [PubMed] [Google Scholar]
- Hendrick V, Stowe ZN, Altshuler LL, Hwang S, Lee E, Haynes D. (2003b) Placental passage of antidepressant medications. Am J Psychiatry 160:993–996 [DOI] [PubMed] [Google Scholar]
- Hendrickx AG. (1975) Teratologic evaluation of imipramine hydrochloride in bonnet (Macaca radiata) and rhesus monkeys (Macaca mulatta). Teratology 11:219–221 [DOI] [PubMed] [Google Scholar]
- Hilli J, Heikkinen T, Rontu R, Lehtimäki T, Kishida I, Aklillu E, Bertilsson L, Vahlberg T, Laine K. (2009) MAO-A and COMT genotypes as possible regulators of perinatal serotonergic symptoms after in utero exposure to SSRIs. Eur Neuropsychopharmacol 19:363–370 [DOI] [PubMed] [Google Scholar]
- Hoffman BJ, Mezey E, Brownstein MJ. (1991) Cloning of a serotonin transporter affected by antidepressants. Science 254:579–580 [DOI] [PubMed] [Google Scholar]
- Hostetter A, Stowe ZN, Strader JR, Jr, McLaughlin E, Llewellyn A. (2000) Dose of selective serotonin uptake inhibitors across pregnancy: clinical implications. Depress Anxiety 11:51–57 [PubMed] [Google Scholar]
- Howland RH. (2013) Antidepressant medication and pregnancy: time for randomized controlled trials. J Psychosoc Nurs Ment Health Serv 51:11–14 [DOI] [PubMed] [Google Scholar]
- Hsiao SY, Cherng CFG, Yang YK, Yeh TL, Yu L. (2005) Prenatal bupropion exposure enhances the cocaine reward and stress susceptibility in adult mice. Chin J Physiol 48:223–229 [PubMed] [Google Scholar]
- Hume AS, Douglas BH. (1968) Placental transfer of desmethylimipramine. Am J Obstet Gynecol 101:915–917 [DOI] [PubMed] [Google Scholar]
- Idänpään-Heikkilä J, Saxén L. (1973) Possible teratogenicity of imipramine-chloropyramine. Lancet 2:282–284 [DOI] [PubMed] [Google Scholar]
- Inoue M, Kitazawa T, Cao J, Taneike T. (2003) 5-HT7 receptor-mediated relaxation of the oviduct in nonpregnant proestrus pigs. Eur J Pharmacol 461:207–218 [DOI] [PubMed] [Google Scholar]
- Jason KM, Cooper TB, Friedman E. (1981) Prenatal exposure to imipramine alters early behavioral development and beta adrenergic receptors in rats. J Pharmacol Exp Ther 217:461–466 [PubMed] [Google Scholar]
- Ji S, Long Q, Newport DJ, Na H, Knight B, Zach EB, Morris NJ, Kutner M, Stowe ZN. (2011) Validity of depression rating scales during pregnancy and the postpartum period: impact of trimester and parity. J Psychiatr Res 45:213–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Källén B. (2004) Neonate characteristics after maternal use of antidepressants in late pregnancy. Arch Pediatr Adolesc Med 158:312–316 [DOI] [PubMed] [Google Scholar]
- Källén B, Otterblad Olausson P. (2006) Antidepressant drugs during pregnancy and infant congenital heart defect. Reprod Toxicol 21:221–222 [DOI] [PubMed] [Google Scholar]
- Källén BAJ, Otterblad Olausson P. (2003) Maternal drug use in early pregnancy and infant cardiovascular defect. Reprod Toxicol 17:255–261 [DOI] [PubMed] [Google Scholar]
- Källén BAJ, Otterblad Olausson P. (2007) Maternal use of selective serotonin re-uptake inhibitors in early pregnancy and infant congenital malformations. Birth Defects Res A Clin Mol Teratol 79:301–308 [DOI] [PubMed] [Google Scholar]
- Kaye BA, Weinstein J. (2005) Neonatal signs after in utero exposure to selective serotonin reuptake inhibitors. JAMA 294:2299–2300; author reply 2300–2291 [DOI] [PubMed] [Google Scholar]
- Kelly CR, Sharif NA. (2006) Pharmacological evidence for a functional serotonin-2B receptor in a human uterine smooth muscle cell line. J Pharmacol Exp Ther 317:1254–1261 [DOI] [PubMed] [Google Scholar]
- Kelly PAT, Ritchie IM, Quate L, McBean DE, Olverman HJ. (2002) Functional consequences of perinatal exposure to 3,4-methylenedioxymethamphetamine in rat brain. Br J Pharmacol 137:963–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendell RE, Chalmers JC, Platz C. (1987) Epidemiology of puerperal psychoses. Br J Psychiatry 150:662–673 [DOI] [PubMed] [Google Scholar]
- Khashan AS, Abel KM, McNamee R, Pedersen MG, Webb RT, Baker PN, Kenny LC, Mortensen PB. (2008) Higher risk of offspring schizophrenia following antenatal maternal exposure to severe adverse life events. Arch Gen Psychiatry 65:146–152 [DOI] [PubMed] [Google Scholar]
- Kieler H, Artama M, Engeland A, Ericsson O, Furu K, Gissler M, Nielsen RB, Nørgaard M, Stephansson O, Valdimarsdottir U, et al. (2012) Selective serotonin reuptake inhibitors during pregnancy and risk of persistent pulmonary hypertension in the newborn: population based cohort study from the five Nordic countries. BMJ 344:d8012. [DOI] [PubMed] [Google Scholar]
- Kim DK, Lim SW, Lee S, Sohn SE, Kim S, Hahn CG, Carroll BJ. (2000) Serotonin transporter gene polymorphism and antidepressant response. Neuroreport 11:215–219 [DOI] [PubMed] [Google Scholar]
- Kim J, Riggs KW, Misri S, Kent N, Oberlander TF, Grunau RE, Fitzgerald C, Rurak DW. (2006) Stereoselective disposition of fluoxetine and norfluoxetine during pregnancy and breast-feeding. Br J Clin Pharmacol 61:155–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney DK, Munir KM, Crowley DJ, Miller AM. (2008) Prenatal stress and risk for autism. Neurosci Biobehav Rev 32:1519–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa T, Kubo O, Satoh M, Taneike T. (1998) Involvement of 5-hydroxytryptamine7 receptors in inhibition of porcine myometrial contractility by 5-hydroxytryptamine. Br J Pharmacol 123:173–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klier CM, Mossaheb N, Lee A, Zernig G. (2007) Mirtazapine and breastfeeding: maternal and infant plasma levels. Am J Psychiatry 164:348–349 [DOI] [PubMed] [Google Scholar]
- Koch M. (1999) The neurobiology of startle. Prog Neurobiol 59:107–128 [DOI] [PubMed] [Google Scholar]
- Koren G, Pastuszak A, Ito S. (1998) Drugs in pregnancy. N Engl J Med 338:1128–1137 [DOI] [PubMed] [Google Scholar]
- Kristensen JH, Ilett KF, Rampono J, Kohan R, Hackett LP. (2007) Transfer of the antidepressant mirtazapine into breast milk. Br J Clin Pharmacol 63:322–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuenssberg EV, Knox JD. (1972) Imipramine in pregnancy. BMJ 2:292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulin NA, Pastuszak A, Koren G. (1998a) Are the new SSRIs safe for pregnant women? Can Fam Physician 44:2081–2083 [PMC free article] [PubMed] [Google Scholar]
- Kulin NA, Pastuszak A, Sage SR, Schick-Boschetto B, Spivey G, Feldkamp M, Ormond K, Matsui D, Stein-Schechman AK, Cook L, et al. (1998b) Pregnancy outcome following maternal use of the new selective serotonin reuptake inhibitors: a prospective controlled multicenter study. JAMA 279:609–610 [DOI] [PubMed] [Google Scholar]
- Kupfer DJ, Frank E, Phillips ML. (2012) Major depressive disorder: new clinical, neurobiological, and treatment perspectives. Lancet 379:1045–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine K, Heikkinen T, Ekblad U, Kero P. (2003) Effects of exposure to selective serotonin reuptake inhibitors during pregnancy on serotonergic symptoms in newborns and cord blood monoamine and prolactin concentrations. Arch Gen Psychiatry 60:720–726 [DOI] [PubMed] [Google Scholar]
- Lennestål R, Källén B. (2007) Delivery outcome in relation to maternal use of some recently introduced antidepressants. J Clin Psychopharmacol 27:607–613 [DOI] [PubMed] [Google Scholar]
- Lesch KP, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S, Benjamin J, Müller CR, Hamer DH, Murphy DL. (1996) Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science 274:1527–1531 [DOI] [PubMed] [Google Scholar]
- Levy L. (1972) Imipramine and fetal deformities. Can Med Assoc J 106:1057–1058 [PMC free article] [PubMed] [Google Scholar]
- Lidov HG, Molliver ME. (1982) Immunohistochemical study of the development of serotonergic neurons in the rat CNS. Brain Res Bull 9:559–604 [DOI] [PubMed] [Google Scholar]
- Lisboa SFS, Oliveira PE, Costa LC, Venâncio EJ, Moreira EG. (2007) Behavioral evaluation of male and female mice pups exposed to fluoxetine during pregnancy and lactation. Pharmacology 80:49–56 [DOI] [PubMed] [Google Scholar]
- Lotto B, Upton L, Price DJ, Gaspar P. (1999) Serotonin receptor activation enhances neurite outgrowth of thalamic neurones in rodents. Neurosci Lett 269:87–90 [DOI] [PubMed] [Google Scholar]
- Loughhead AM, Fisher AD, Newport DJ, Ritchie JC, Owens MJ, DeVane CL, Stowe ZN. (2006a) Antidepressants in amniotic fluid: another route of fetal exposure. Am J Psychiatry 163:145–147 [DOI] [PubMed] [Google Scholar]
- Loughhead AM, Stowe ZN, Newport DJ, Ritchie JC, DeVane CL, Owens MJ. (2006b) Placental passage of tricyclic antidepressants. Biol Psychiatry 59:287–290 [DOI] [PubMed] [Google Scholar]
- Louik C, Lin AE, Werler MM, Hernández-Díaz S, Mitchell AA. (2007) First-trimester use of selective serotonin-reuptake inhibitors and the risk of birth defects. N Engl J Med 356:2675–2683 [DOI] [PubMed] [Google Scholar]
- Lund N, Pedersen LH, Henriksen TB. (2009) Selective serotonin reuptake inhibitor exposure in utero and pregnancy outcomes. Arch Pediatr Adolesc Med 163:949–954 [DOI] [PubMed] [Google Scholar]
- Maccari S, Morley-Fletcher S. (2007) Effects of prenatal restraint stress on the hypothalamus-pituitary-adrenal axis and related behavioural and neurobiological alterations. Psychoneuroendocrinology 32 (Suppl 1):S10–S15 [DOI] [PubMed] [Google Scholar]
- Maclean MR, Dempsie Y. (2010) The serotonin hypothesis of pulmonary hypertension revisited. Adv Exp Med Biol 661:309–322 [DOI] [PubMed] [Google Scholar]
- Majithia R, Johnson DA. (2012) Are proton pump inhibitors safe during pregnancy and lactation? Evidence to date. Drugs 72:171–179 [DOI] [PubMed] [Google Scholar]
- Malm H, Artama M, Gissler M, Ritvanen A. (2011) Selective serotonin reuptake inhibitors and risk for major congenital anomalies. Obstet Gynecol 118:111–120 [DOI] [PubMed] [Google Scholar]
- Malm H, Klaukka T, Neuvonen PJ. (2005) Risks associated with selective serotonin reuptake inhibitors in pregnancy. Obstet Gynecol 106:1289–1296 [DOI] [PubMed] [Google Scholar]
- Mammen OK, Perel JM, Rudolph G, Foglia JP, Wheeler SB. (1997) Sertraline and norsertraline levels in three breastfed infants. J Clin Psychiatry 58:100–103 [DOI] [PubMed] [Google Scholar]
- Masson J, Sagné C, Hamon M, El Mestikawy S. (1999) Neurotransmitter transporters in the central nervous system. Pharmacol Rev 51:439–464 [PubMed] [Google Scholar]
- Markus AR, Andres E, West KD, Garro N, Pellegrini C. (2013) Medicaid covered births, 2008 through 2010, in the context of the implementation of health reform. Womens Health Issues 23:e273–e280 [DOI] [PubMed] [Google Scholar]
- McBride WG. (1972) Limb deformities associated with iminodibenzyl hydrochloride. Med J Aust 1:492. [PubMed] [Google Scholar]
- McElhatton PR, Garbis HM, Eléfant E, Vial T, Bellemin B, Mastroiacovo P, Arnon J, Rodríguez-Pinilla E, Schaefer C, Pexieder T, et al. (1996) The outcome of pregnancy in 689 women exposed to therapeutic doses of antidepressants. A collaborative study of the European Network of Teratology Information Services (ENTIS). Reprod Toxicol 10:285–294 [DOI] [PubMed] [Google Scholar]
- Merlob P, Birk E, Sirota L, Linder N, Berant M, Stahl B, Klinger G. (2009) Are selective serotonin reuptake inhibitors cardiac teratogens? Echocardiographic screening of newborns with persistent heart murmur. Birth Defects Res A Clin Mol Teratol 85:837–841 [DOI] [PubMed] [Google Scholar]
- Mills EM, Rusyniak DE, Sprague JE. (2004) The role of the sympathetic nervous system and uncoupling proteins in the thermogenesis induced by 3,4-methylenedioxymethamphetamine. J Mol Med (Berl) 82:787–799 [DOI] [PubMed] [Google Scholar]
- Misri S, Reebye P, Kendrick K, Carter D, Ryan D, Grunau RE, Oberlander TF. (2006) Internalizing behaviors in 4-year-old children exposed in utero to psychotropic medications. Am J Psychiatry 163:1026–1032 [DOI] [PubMed] [Google Scholar]
- Monk C, Newport DJ, Korotkin JH, Long Q, Knight B, Stowe ZN. (2012) Uterine blood flow in a psychiatric population: impact of maternal depression, anxiety, and psychotropic medication. Biol Psychiatry 72:483–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero D, de Ceballos ML, Del Rio J. (1990) Down-regulation of 3H-imipramine binding sites in rat cerebral cortex after prenatal exposure to antidepressants. Life Sci 46:1619–1626 [DOI] [PubMed] [Google Scholar]
- Montero D, De Felipe MC, Del Río J. (1991) Acute or chronic antidepressants do not modify [125I]cyanopindolol binding to 5-HT1B receptors in rat brain. Eur J Pharmacol 196:327–329 [DOI] [PubMed] [Google Scholar]
- Moore WM, Hellegers AE, Battaglia FC. (1966) In vitro permeability of different layers of the human placenta to carbohydrates and urea. Am J Obstet Gynecol 96:951–955 [DOI] [PubMed] [Google Scholar]
- Morgan CP, Bale TL. (2011) Early prenatal stress epigenetically programs dysmasculinization in second-generation offspring via the paternal lineage. J Neurosci 31:11748–11755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan DJ. (1997) Drug disposition in mother and foetus. Clin Exp Pharmacol Physiol 24:869–873 [DOI] [PubMed] [Google Scholar]
- Morrison JL, Chien C, Gruber N, Rurak D, Riggs W. (2001) Fetal behavioural state changes following maternal fluoxetine infusion in sheep. Brain Res Dev Brain Res 131:47–56 [DOI] [PubMed] [Google Scholar]
- Morrison JL, Riggs KW, Chien C, Gruber N, McMillen IC, Rurak DW. (2004) Chronic maternal fluoxetine infusion in pregnant sheep: effects on the maternal and fetal hypothalamic-pituitary-adrenal axes. Pediatr Res 56:40–46 [DOI] [PubMed] [Google Scholar]
- Morrison JL, Rurak DW, Chien C, Kennaway DJ, Gruber N, McMillen IC, Riggs KW. (2005) Maternal fluoxetine infusion does not alter fetal endocrine and biophysical circadian rhythms in pregnant sheep. J Soc Gynecol Investig 12:356–364 [DOI] [PubMed] [Google Scholar]
- Moses-Kolko EL, Bogen D, Perel J, Bregar A, Uhl K, Levin B, Wisner KL. (2005) Neonatal signs after late in utero exposure to serotonin reuptake inhibitors: literature review and implications for clinical applications. JAMA 293:2372–2383 [DOI] [PubMed] [Google Scholar]
- Mueller BR, Bale TL. (2008) Sex-specific programming of offspring emotionality after stress early in pregnancy. J Neurosci 28:9055–9065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller RA, Lundberg DB, Breese GR, Hedner J, Hedner T, Jonason J. (1982) The neuropharmacology of respiratory control. Pharmacol Rev 34:255–285 [PubMed] [Google Scholar]
- Mulder EJH, Ververs FF, de Heus R, Visser GHA. (2011) Selective serotonin reuptake inhibitors affect neurobehavioral development in the human fetus. Neuropsychopharmacology 36:1961–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myles N, Newall H, Ward H, Large M. (2013) Systematic meta-analysis of individual selective serotonin reuptake inhibitor medications and congenital malformations. Aust N Z J Psychiatry 47:1002–1012 [DOI] [PubMed] [Google Scholar]
- Newport DJ, Brennan PA, Green P, Ilardi D, Whitfield TH, Morris N, Knight BT, Stowe ZN. (2008) Maternal depression and medication exposure during pregnancy: comparison of maternal retrospective recall to prospective documentation. BJOG 115:681–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newport DJ, Calamaras MR, DeVane CL, Donovan J, Beach AJ, Winn S, Knight BT, Gibson BB, Viguera AC, Owens MJ, et al. (2007) Atypical antipsychotic administration during late pregnancy: placental passage and obstetrical outcomes. Am J Psychiatry 164:1214–1220 [DOI] [PubMed] [Google Scholar]
- Newport DJ, Ji S, Long Q, Knight BT, Zach EB, Smith EN, Morris NJ, Stowe ZN. (2012) Maternal depression and anxiety differentially impact fetal exposures during pregnancy. J Clin Psychiatry 73:247–251 [DOI] [PubMed] [Google Scholar]
- Newport DJ, Ritchie JC, Knight BT, Glover BA, Zach EB, Stowe ZN. (2009) Venlafaxine in human breast milk and nursing infant plasma: determination of exposure. J Clin Psychiatry 70:1304–1310 [DOI] [PubMed] [Google Scholar]
- Newport DJ, Wilcox MM, Stowe ZN. (2001) Antidepressants during pregnancy and lactation: defining exposure and treatment issues. Semin Perinatol 25:177–190 [DOI] [PubMed] [Google Scholar]
- Noorlander CW, Ververs FFT, Nikkels PGJ, van Echteld CJA, Visser GHA, Smidt MP. (2008) Modulation of serotonin transporter function during fetal development causes dilated heart cardiomyopathy and lifelong behavioral abnormalities. PLoS ONE 3:e2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nulman I, Rovet J, Stewart DE, Wolpin J, Gardner HA, Theis JG, Kulin N, Koren G. (1997) Neurodevelopment of children exposed in utero to antidepressant drugs. N Engl J Med 336:258–262 [DOI] [PubMed] [Google Scholar]
- Oberlander TF, Bonaguro RJ, Misri S, Papsdorf M, Ross CJD, Simpson EM. (2008a) Infant serotonin transporter (SLC6A4) promoter genotype is associated with adverse neonatal outcomes after prenatal exposure to serotonin reuptake inhibitor medications. Mol Psychiatry 13:65–73 [DOI] [PubMed] [Google Scholar]
- Oberlander TF, Eckstein Grunau R, Fitzgerald C, Ellwood A-L, Misri S, Rurak D, Riggs KW. (2002) Prolonged prenatal psychotropic medication exposure alters neonatal acute pain response. Pediatr Res 51:443–453 [DOI] [PubMed] [Google Scholar]
- Oberlander TF, Grunau R, Mayes L, Riggs W, Rurak D, Papsdorf M, Misri S, Weinberg J. (2008b) Hypothalamic-pituitary-adrenal (HPA) axis function in 3-month old infants with prenatal selective serotonin reuptake inhibitor (SSRI) antidepressant exposure. Early Hum Dev 84:689–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberlander TF, Grunau RE, Fitzgerald C, Papsdorf M, Rurak D, Riggs W. (2005) Pain reactivity in 2-month-old infants after prenatal and postnatal serotonin reuptake inhibitor medication exposure. Pediatrics 115:411–425 [DOI] [PubMed] [Google Scholar]
- Oberlander TF, Misri S, Fitzgerald CE, Kostaras X, Rurak D, Riggs W. (2004) Pharmacologic factors associated with transient neonatal symptoms following prenatal psychotropic medication exposure. J Clin Psychiatry 65:230–237 [DOI] [PubMed] [Google Scholar]
- Oberlander TF, Papsdorf M, Brain UM, Misri S, Ross C, Grunau RE. (2010) Prenatal effects of selective serotonin reuptake inhibitor antidepressants, serotonin transporter promoter genotype (SLC6A4), and maternal mood on child behavior at 3 years of age. Arch Pediatr Adolesc Med 164:444–451 [DOI] [PubMed] [Google Scholar]
- Oberlander TF, Reebye P, Misri S, Papsdorf M, Kim J, Grunau RE. (2007) Externalizing and attentional behaviors in children of depressed mothers treated with a selective serotonin reuptake inhibitor antidepressant during pregnancy. Arch Pediatr Adolesc Med 161:22–29 [DOI] [PubMed] [Google Scholar]
- Oberlander TF, Warburton W, Misri S, Aghajanian J, Hertzman C. (2006) Neonatal outcomes after prenatal exposure to selective serotonin reuptake inhibitor antidepressants and maternal depression using population-based linked health data. Arch Gen Psychiatry 63:898–906 [DOI] [PubMed] [Google Scholar]
- Occhiogrosso M, Omran SS, Altemus M. (2012) Persistent pulmonary hypertension of the newborn and selective serotonin reuptake inhibitors: lessons from clinical and translational studies. Am J Psychiatry 169:134–140 [DOI] [PubMed] [Google Scholar]
- Oesterheld JR. (1998) A review of developmental aspects of cytochrome P450. J Child Adolesc Psychopharmacol 8:161–174 [DOI] [PubMed] [Google Scholar]
- Olson VG, Rockett HR, Reh RK, Redila VA, Tran PM, Venkov HA, Defino MC, Hague C, Peskind ER, Szot P, et al. (2011) The role of norepinephrine in differential response to stress in an animal model of posttraumatic stress disorder. Biol Psychiatry 70:441–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr ST, Miller CA. (1995) Maternal depressive symptoms and the risk of poor pregnancy outcome. Review of the literature and preliminary findings. Epidemiol Rev 17:165–171 [DOI] [PubMed] [Google Scholar]
- Ostergaard GZ, Pedersen SE. (1982) Neonatal effects of maternal clomipramine treatment. Pediatrics 69:233–234 [PubMed] [Google Scholar]
- Owens MJ, Knight DL, Ritchie JC, Nemeroff CB. (1991a) The 5-hydroxytryptamine2 agonist, (+-)-1-(2,5-dimethoxy-4-bromophenyl)-2-aminopropane stimulates the hypothalamic-pituitary-adrenal (HPA) axis. I. Acute effects on HPA axis activity and corticotropin-releasing factor-containing neurons in the rat brain. J Pharmacol Exp Ther 256:787–794 [PubMed] [Google Scholar]
- Owens MJ, Knight DL, Ritchie JC, Nemeroff CB. (1991b) The 5-hydroxytryptamine2 agonist, (+-)-1-(2,5-dimethoxy-4-bromophenyl)-2-aminopropane stimulates the hypothalamic-pituitary-adrenal (HPA) axis. II. Biochemical and physiological evidence for the development of tolerance after chronic administration. J Pharmacol Exp Ther 256:795–800 [PubMed] [Google Scholar]
- Owens MJ, Morgan WN, Plott SJ, Nemeroff CB. (1997) Neurotransmitter receptor and transporter binding profile of antidepressants and their metabolites. J Pharmacol Exp Ther 283:1305–1322 [PubMed] [Google Scholar]
- Pacifici GM, Nottoli R. (1995) Placental transfer of drugs administered to the mother. Clin Pharmacokinet 28:235–269 [DOI] [PubMed] [Google Scholar]
- Pastuszak A, Schick-Boschetto B, Zuber C, Feldkamp M, Pinelli M, Sihn S, Donnenfeld A, McCormack M, Leen-Mitchell M, Woodland C, et al. (1993) Pregnancy outcome following first-trimester exposure to fluoxetine (Prozac). JAMA 269:2246–2248 [PubMed] [Google Scholar]
- Pawluski JL, Galea LAM, Brain U, Papsdorf M, Oberlander TF. (2009) Neonatal S100B protein levels after prenatal exposure to selective serotonin reuptake inhibitors. Pediatrics 124:e662–e670 [DOI] [PubMed] [Google Scholar]
- Pearson RM, Evans J, Kounali D, Lewis G, Heron J, Ramchandani PG, O'Connor TG, Stein A. (2013) Maternal depression during pregnancy and the postnatal period: risks and possible mechanisms for offspring depression at age 18 years. JAMA Psychiatry 70:1312–1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen LH, Henriksen TB, Vestergaard M, Olsen J, Bech BH. (2009) Selective serotonin reuptake inhibitors in pregnancy and congenital malformations: population based cohort study. BMJ 339:b3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira JD, Caricati-Neto A, Jurkiewicz A, Jurkiewicz NH. (2007) Decreased noradrenergic and serotonergic reactivity of vas deferens of newborn rats from mothers treated with the serotonin reuptake inhibitor fluoxetine during pregnancy and breast-feeding. Life Sci 81:1501–1508 [DOI] [PubMed] [Google Scholar]
- Pérez-Neri I, Montes S, Ojeda-López C, Ramírez-Bermúdez J, Ríos C. (2008) Modulation of neurotransmitter systems by dehydroepiandrosterone and dehydroepiandrosterone sulfate: mechanism of action and relevance to psychiatric disorders. Prog Neuropsychopharmacol Biol Psychiatry 32:1118–1130 [DOI] [PubMed] [Google Scholar]
- Petersen I, Gilbert RE, Evans SJW, Man S-L, Nazareth I. (2011) Pregnancy as a major determinant for discontinuation of antidepressants: an analysis of data from The Health Improvement Network. J Clin Psychiatry 72:979–985 [DOI] [PubMed] [Google Scholar]
- Piñeyro G, Blier P. (1999) Autoregulation of serotonin neurons: role in antidepressant drug action. Pharmacol Rev 51:533–591 [PubMed] [Google Scholar]
- Pollock BG, Ferrell RE, Mulsant BH, Mazumdar S, Miller M, Sweet RA, Davis S, Kirshner MA, Houck PR, Stack JA, et al. (2000) Allelic variation in the serotonin transporter promoter affects onset of paroxetine treatment response in late-life depression. Neuropsychopharmacology 23:587–590 [DOI] [PubMed] [Google Scholar]
- Ramage AG, Villalón CM. (2008) 5-hydroxytryptamine and cardiovascular regulation. Trends Pharmacol Sci 29:472–481 [DOI] [PubMed] [Google Scholar]
- Ramos E, Oraichi D, Rey E, Blais L, Bérard A. (2007) Prevalence and predictors of antidepressant use in a cohort of pregnant women. BJOG 114:1055–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampono J, Proud S, Hackett LP, Kristensen JH, Ilett KF. (2004) A pilot study of newer antidepressant concentrations in cord and maternal serum and possible effects in the neonate. Int J Neuropsychopharmacol 7:329–334 [DOI] [PubMed] [Google Scholar]
- Rampono J, Simmer K, Ilett KF, Hackett LP, Doherty DA, Elliot R, Kok CH, Coenen A, Forman T. (2009) Placental transfer of SSRI and SNRI antidepressants and effects on the neonate. Pharmacopsychiatry 42:95–100 [DOI] [PubMed] [Google Scholar]
- Reis M, Källén B. (2010) Delivery outcome after maternal use of antidepressant drugs in pregnancy: an update using Swedish data. Psychol Med 40:1723–1733 [DOI] [PubMed] [Google Scholar]
- Robson JM, Sullivan FM. (1963) The production of foetal abnormalities in rabbits by imipramine. Lancet 1:638–639 [DOI] [PubMed] [Google Scholar]
- Rodríguez Echandía EL, Broitman ST. (1983) Effect of prenatal and postnatal exposure to therapeutic doses of chlorimipramine on emotionality in the rat. Psychopharmacology (Berl) 79:236–241 [DOI] [PubMed] [Google Scholar]
- Schimmell MS, Katz EZ, Shaag Y, Pastuszak A, Koren G. (1991) Toxic neonatal effects following maternal clomipramine therapy. J Toxicol Clin Toxicol 29:479–484 [DOI] [PubMed] [Google Scholar]
- Schou M, Goldfield MD, Weinstein MR, Villeneuve A. (1973) Lithium and pregnancy. I. Report from the Register of Lithium Babies. BMJ 2:135–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder JW, Smith AK, Brennan PA, Conneely KN, Kilaru V, Knight BT, Newport DJ, Cubells JF, Stowe ZN. (2012) DNA methylation in neonates born to women receiving psychiatric care. Epigenetics 7:409–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher H, Blake DA, Gurian JM, Gillette JR. (1968) A comparison of the teratogenic activity of thalidomide in rabbits and rats. J Pharmacol Exp Ther 160:189–200 [PubMed] [Google Scholar]
- Scialli AR, Iannucci AR. (2010) Whole embryo culture and the identification of “teratogenicity”. Reprod Toxicol 29:247–248, author reply 249–250 [DOI] [PubMed] [Google Scholar]
- Sen S, Burmeister M, Ghosh D. (2004) Meta-analysis of the association between a serotonin transporter promoter polymorphism (5-HTTLPR) and anxiety-related personality traits. Am J Med Genet B Neuropsychiatr Genet 127B:85–89 [DOI] [PubMed] [Google Scholar]
- Sharpley AL, Cowen PJ. (1995) Effect of pharmacologic treatments on the sleep of depressed patients. Biol Psychiatry 37:85–98 [DOI] [PubMed] [Google Scholar]
- Shuey DL, Sadler TW, Lauder JM. (1992) Serotonin as a regulator of craniofacial morphogenesis: site specific malformations following exposure to serotonin uptake inhibitors. Teratology 46:367–378 [DOI] [PubMed] [Google Scholar]
- Sim M. (1972) Imipramine and pregnancy. BMJ 2:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon GE, Cunningham ML, Davis RL. (2002) Outcomes of prenatal antidepressant exposure. Am J Psychiatry 159:2055–2061 [DOI] [PubMed] [Google Scholar]
- Simpkins JW, Field FP, Torosian G, Soltis EE. (1985) Effects of prenatal exposure to tricyclic antidepressants on adrenergic responses in progeny. Dev Pharmacol Ther 8:17–33 [DOI] [PubMed] [Google Scholar]
- Simpson KL, Weaver KJ, de Villers-Sidani E, Lu JY-F, Cai Z, Pang Y, Rodriguez-Porcel F, Paul IA, Merzenich M, Lin RCS. (2011) Perinatal antidepressant exposure alters cortical network function in rodents. Proc Natl Acad Sci USA 108:18465–18470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer G, Coyle IR. (1973) The effect of imipramine administered before and during pregnancy on litter size in the rat. Psychopharmacology (Berl) 32:337–342 [DOI] [PubMed] [Google Scholar]
- Singewald N, Philippu A. (1996) Involvement of biogenic amines and amino acids in the central regulation of cardiovascular homeostasis. Trends Pharmacol Sci 17:356–363 [PubMed] [Google Scholar]
- Singh Y, Jaiswal AK, Singh M, Bhattacharya SK. (1998) Effect of prenatal diazepam, phenobarbital, haloperidol and fluoxetine exposure on foot shock induced aggression in rats. Indian J Exp Biol 36:1023–1024 [PubMed] [Google Scholar]
- Sit DK, Perel JM, Helsel JC, Wisner KL. (2008) Changes in antidepressant metabolism and dosing across pregnancy and early postpartum. J Clin Psychiatry 69:652–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivojelezova A, Shuhaiber S, Sarkissian L, Einarson A, Koren G. (2005) Citalopram use in pregnancy: prospective comparative evaluation of pregnancy and fetal outcome. Am J Obstet Gynecol 193:2004–2009 [DOI] [PubMed] [Google Scholar]
- Sloot WN, Bowden HC, Yih TD. (2009) In vitro and in vivo reproduction toxicology of 12 monoaminergic reuptake inhibitors: possible mechanisms of infrequent cardiovascular anomalies. Reprod Toxicol 28:270–282 [DOI] [PubMed] [Google Scholar]
- Stafford RS, MacDonald EA, Finkelstein SN. (2001) National Patterns of Medication Treatment for Depression, 1987 to 2001. Prim Care Companion J Clin Psychiatry 3:232–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stancer HC, Reed KL. (1986) Desipramine and 2-hydroxydesipramine in human breast milk and the nursing infant’s serum. Am J Psychiatry 143:1597–1600 [DOI] [PubMed] [Google Scholar]
- Steer RA, Scholl TO, Hediger ML, Fischer RL. (1992) Self-reported depression and negative pregnancy outcomes. J Clin Epidemiol 45:1093–1099 [DOI] [PubMed] [Google Scholar]
- Stephansson O, Kieler H, Haglund B, Artama M, Engeland A, Furu K, Gissler M, Nørgaard M, Nielsen RB, Zoega H, et al. (2013) Selective serotonin reuptake inhibitors during pregnancy and risk of stillbirth and infant mortality. JAMA 309:48–54 [DOI] [PubMed] [Google Scholar]
- Stewart CW, Scalzo FM, Valentine J, Holson RR, Ali SF, Slikker W., Jr (1998) Gestational exposure to cocaine or pharmacologically related compounds: effects on behavior and striatal dopamine receptors. Life Sci 63:2015–2022 [DOI] [PubMed] [Google Scholar]
- Stowe ZN, Cohen LS, Hostetter A, Ritchie JC, Owens MJ, Nemeroff CB. (2000) Paroxetine in human breast milk and nursing infants. Am J Psychiatry 157:185–189 [DOI] [PubMed] [Google Scholar]
- Stowe ZN, Hostetter AL, Owens MJ, Ritchie JC, Sternberg K, Cohen LS, Nemeroff CB. (2003) The pharmacokinetics of sertraline excretion into human breast milk: determinants of infant serum concentrations. J Clin Psychiatry 64:73–80 [DOI] [PubMed] [Google Scholar]
- Stowe ZN, Owens MJ, Landry JC, Kilts CD, Ely T, Llewellyn A, Nemeroff CB. (1997) Sertraline and desmethylsertraline in human breast milk and nursing infants. Am J Psychiatry 154:1255–1260 [DOI] [PubMed] [Google Scholar]
- Su S-W, Cherng C-FG, Lin YC, Yu L. (2007) Prenatal exposure of bupropion may enhance agitation, anxiety responses, and sensitivity to cocaine effects in adult mice. Chin J Physiol 50:1–8 [PubMed] [Google Scholar]
- Suri R, Altshuler L, Hendrick V, Rasgon N, Lee E, Mintz J. (2004) The impact of depression and fluoxetine treatment on obstetrical outcome. Arch Women Ment Health 7:193–200 [DOI] [PubMed] [Google Scholar]
- Suri R, Hellemann G, Stowe ZN, Cohen LS, Aquino A, Altshuler LL. (2011) A prospective, naturalistic, blinded study of early neurobehavioral outcomes for infants following prenatal antidepressant exposure. J Clin Psychiatry 72:1002–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerts CAS, Costa AMDD, Esteves A, Borato CES, Swerts MSO. (2010) Effects of fluoxetine and imipramine in rat fetuses treated during a critical gestational period: a macro and microscopic study. Rev Bras Psiquiatr 32:152–158 [DOI] [PubMed] [Google Scholar]
- Talvenheimo J, Nelson PJ, Rudnick G. (1979) Mechanism of imipramine inhibition of platelet 5-hydroxytryptamine transport. J Biol Chem 254:4631–4635 [PubMed] [Google Scholar]
- Thase ME. (2008) Do antidepressants really work? A clinicians’ guide to evaluating the evidence. Curr Psychiatry Rep 10:487–494 [DOI] [PubMed] [Google Scholar]
- Toh S, Mitchell AA, Louik C, Werler MM, Chambers CD, Hernández-Díaz S. (2009) Antidepressant use during pregnancy and the risk of preterm delivery and fetal growth restriction. J Clin Psychopharmacol 29:555–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonge SR. (1972) Permanent changes in brain monoamine metabolism induced by the administration of psychotropic drugs during the pre- and neonatal periods in rats. J Pharm Pharmacol 24 (Suppl):149P–150P [PubMed] [Google Scholar]
- Tonge SR. (1973) Catecholamine concentrations in discrete areas of the rat brain after the pre- and neonatal administration of phencyclidine and imipramine. J Pharm Pharmacol 25:164–166 [DOI] [PubMed] [Google Scholar]
- Trivedi MH, Rush AJ, Armitage R, Gullion CM, Grannemann BD, Orsulak PJ, Roffwarg HP. (1999) Effects of fluoxetine on the polysomnogram in outpatients with major depression. Neuropsychopharmacology 20:447–459 [DOI] [PubMed] [Google Scholar]
- Tronick E, Reck C. (2009) Infants of depressed mothers. Harv Rev Psychiatry 17:147–156 [DOI] [PubMed] [Google Scholar]
- Tuccori M, Montagnani S, Testi A, Ruggiero E, Mantarro S, Scollo C, Pergola A, Fornai M, Antonioli L, Colucci R, et al. (2010) Use of selective serotonin reuptake inhibitors during pregnancy and risk of major and cardiovascular malformations: an update. Postgrad Med 122:49–65 [DOI] [PubMed] [Google Scholar]
- Tuccori M, Testi A, Antonioli L, Fornai M, Montagnani S, Ghisu N, Colucci R, Corona T, Blandizzi C, Del Tacca M. (2009) Safety concerns associated with the use of serotonin reuptake inhibitors and other serotonergic/noradrenergic antidepressants during pregnancy: a review. Clin Ther 31:1426–1453 [DOI] [PubMed] [Google Scholar]
- Van den Hove DLA, Blanco CE, Scheepens A, Desbonnet L, Myint A-M, Leonard BE, Prickaerts J, Steinbusch HWM. (2008) Prenatal maternal paroxetine treatment and neonatal mortality in the rat: a preliminary study. Neonatology 93:52–55 [DOI] [PubMed] [Google Scholar]
- Vartazarmian R, Malik S, Baker GB, Boksa P. (2005) Long-term effects of fluoxetine or vehicle administration during pregnancy on behavioral outcomes in guinea pig offspring. Psychopharmacology (Berl) 178:328–338 [DOI] [PubMed] [Google Scholar]
- Vedernikov Y, Bolanos S, Bytautiene E, Fulep E, Saade GR, Garfield RE. (2000) Effect of fluoxetine on contractile activity of pregnant rat uterine rings. Am J Obstet Gynecol 182:296–299 [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. (2001) The sequence of the human genome. Science 291:1304–1351 [DOI] [PubMed] [Google Scholar]
- Verbeeck RK, Ross SG, McKenna EA. (1986) Excretion of trazodone in breast milk. Br J Clin Pharmacol 22:367–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ververs T, Kaasenbrood H, Visser G, Schobben F, de Jong-van den Berg L, Egberts T. (2006) Prevalence and patterns of antidepressant drug use during pregnancy. Eur J Clin Pharmacol 62:863–870 [DOI] [PubMed] [Google Scholar]
- Vohr BR, Wright LL, Dusick AM, Mele L, Verter J, Steichen JJ, Simon NP, Wilson DC, Broyles S, Bauer CR, et al. (2000) Neurodevelopmental and functional outcomes of extremely low birth weight infants in the National Institute of Child Health and Human Development Neonatal Research Network, 1993-1994. Pediatrics 105:1216–1226 [DOI] [PubMed] [Google Scholar]
- Wang JS, Newport DJ, Stowe ZN, Donovan JL, Pennell PB, DeVane CL. (2007) The emerging importance of transporter proteins in the psychopharmacological treatment of the pregnant patient. Drug Metab Rev 39:723–746 [DOI] [PubMed] [Google Scholar]
- Watson RE, Desesso JM, Hurtt ME, Cappon GD. (2006) Postnatal growth and morphological development of the brain: a species comparison. Birth Defects Res B Dev Reprod Toxicol 77:471–484 [DOI] [PubMed] [Google Scholar]
- Webster PA. (1973) Withdrawal symptoms in neonates associated with maternal antidepressant therapy. Lancet 2:318–319 [DOI] [PubMed] [Google Scholar]
- Weissman AM, Levy BT, Hartz AJ, Bentler S, Donohue M, Ellingrod VL, Wisner KL. (2004) Pooled analysis of antidepressant levels in lactating mothers, breast milk, and nursing infants. Am J Psychiatry 161:1066–1078 [DOI] [PubMed] [Google Scholar]
- Wessler I, Herschel S, Bittinger F, Kirkpatrick CJ. (2007) Release of non-neuronal acetylcholine from the isolated human placenta is affected by antidepressants. Life Sci 80:2210–2213 [DOI] [PubMed] [Google Scholar]
- Whitaker-Azmitia PM. (2001) Serotonin and brain development: role in human developmental diseases. Brain Res Bull 56:479–485 [DOI] [PubMed] [Google Scholar]
- Wilson JG. (1973) Environment and Birth Defects, Academic Press, Inc, New York [Google Scholar]
- Wisner KL, Perel JM. (1991) Serum nortriptyline levels in nursing mothers and their infants. Am J Psychiatry 148:1234–1236 [DOI] [PubMed] [Google Scholar]
- Wisner KL, Perel JM, Blumer J. (1998) Serum sertraline and N-desmethylsertraline levels in breast-feeding mother-infant pairs. Am J Psychiatry 155:690–692 [DOI] [PubMed] [Google Scholar]
- Wisner KL, Perel JM, Findling RL, Hinnes RL. (1997) Nortriptyline and its hydroxymetabolites in breastfeeding mothers and newborns. Psychopharmacol Bull 33:249–251 [PubMed] [Google Scholar]
- Wogelius P, Nørgaard M, Gislum M, Pedersen L, Munk E, Mortensen PB, Lipworth L, Sørensen HT. (2006) Maternal use of selective serotonin reuptake inhibitors and risk of congenital malformations. Epidemiology 17:701–704 [DOI] [PubMed] [Google Scholar]
- Yaris F, Kadioglu M, Kesim M, Ulku C, Yaris E, Kalyoncu NI, Unsal M. (2004) Newer antidepressants in pregnancy: prospective outcome of a case series. Reprod Toxicol 19:235–238 [DOI] [PubMed] [Google Scholar]
- Yonkers KA, Gotman N, Smith MV, Forray A, Belanger K, Brunetto WL, Lin H, Burkman RT, Zelop CM, Lockwood CJ. (2011) Does antidepressant use attenuate the risk of a major depressive episode in pregnancy? Epidemiology 22:848–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Smith B, Craggs M, Kumar RC. (1997) Investigation of pharmacokinetics and of possible adverse effects in infants exposed to tricyclic antidepressants in breast-milk. J Affect Disord 43:225–237 [DOI] [PubMed] [Google Scholar]
- Zajicek E. (1981) Psychiatric problems during pregnancy, in Pregnancy: A Psychological and Social Study (Wolkind S, Zajicek E, eds) pp 57–73, Academic, London [Google Scholar]
- Zeskind PS, Stephens LE. (2004) Maternal selective serotonin reuptake inhibitor use during pregnancy and newborn neurobehavior. Pediatrics 113:368–375 [DOI] [PubMed] [Google Scholar]
- Zuckerman B, Amaro H, Bauchner H, Cabral H. (1989) Depressive symptoms during pregnancy: relationship to poor health behaviors. Am J Obstet Gynecol 160:1107–1111 [DOI] [PubMed] [Google Scholar]
- Zuckerman B, Bauchner H, Parker S, Cabral H. (1990) Maternal depressive symptoms during pregnancy, and newborn irritability. J Dev Behav Pediatr 11:190–194 [PubMed] [Google Scholar]