

Abstract
An earlier linkage analysis conducted on a population derived from the Dahl salt-sensitive hypertensive (S) and the spontaneously hypertensive rat (SHR) identified 10 genomic regions linked to several renal and/or cardiovascular traits. In particular, loci on rat chromosomes (RNO) 8 and 13 were linked to proteinuria, albuminuria, and renal damage. At both loci, the S allele was associated with increased proteinuria and renal damage. The current study aimed to confirm the linkage analysis and to evaluate the effect of genetic background on the ability of each locus (either RNO8 or RNO13) to exert a phenotypic difference when placed on a genetic background either susceptible (S rat) or resistant (SHR) to the development of renal disease. Congenic strains developed to transfer genomic segments from either RNO8 or RNO13 from the SHR onto the S genetic background [S.SHR(8) or S.SHR(13)] demonstrated significantly reduced proteinuria and improved renal function. Both congenic strains demonstrated significantly reduced glomerular and tubular injury, with renal interstitial fibrosis as the predominant pathological difference compared with the S. In contrast, transfer of RNO8 or RNO13 genomic regions from the S onto the resistant SHR genetic background [SHR.S(8) or SHR.S(13)] yielded no significant difference in proteinuria or glomerular, tubular, or interstitial injury compared with SHR. These findings demonstrate that genetic context plays a significant and important role in the phenotypic expression of genes influencing proteinuria on RNO8 and RNO13.
Keywords: Dahl S, SHR, renal fibrosis, congenic strains
proteinuria is a significant predictor for progression to end-stage renal disease (ESRD) and also represents an important independent risk factor for the development of cardiovascular disease and overall mortality (36). It has been clearly established through familial aggregation studies, comparison of incidence between different racial and ethnic populations, and linkage analysis that ESRD has a substantial genetic component (4, 16). Additionally, age, gender, diet, and socioeconomic factors also play a role in the onset and progression of renal disease (1, 40, 60).
Hundreds of studies have utilized a candidate gene approach to identify genes involved in common forms of renal failure (4). However, there have been relatively few studies that have utilized the more systematic whole genome scan approach (5, 7, 10, 11, 15, 27, 28, 31, 61), and recently whole genome association studies (26). These genetic analyses have identified many genomic regions or single nucleotide polymorphisms (SNP) associated with renal disease and many interesting candidate genes. However, none have definitively established a causal role between the genetic variants and the disease. The difficulty in achieving this objective lies in the significant genetic heterogeneity within the patient population. Clinical symptoms and outcomes may be similar (e.g., ESRD), but the underlying genetic cause is different, limiting the power to identify disease-causing variants (35, 51). This is further complicated because some individuals may carry the same susceptibility allele(s) (at a particular locus), but one individual possesses a genetic background that compensates for the propensity to promote disease, while the other individual develops disease. Fortunately, the availability of several rat models of renal disease (6, 34, 39, 52–54, 57) offer hope not only in identifying genes involved in common forms of renal disease but also in understanding how genetic background and environmental factors determine occurrence and severity of renal damage.
The Dahl salt-sensitive rat (S) has been extensively used to study the genetics of hypertension (18, 20, 23, 29, 37). More recently, several studies have utilized this model to investigate the genetics of renal disease (17, 21, 43, 55). Previously, a genetic analysis using the S and the spontaneously hypertensive rat (SHR) identified quantitative trait loci (QTL) for proteinuria on rat chromosomes (RNO) 8 and 13 (17). For these loci, the SHR allele was associated with significantly less proteinuria and renal damage compared with the S allele. The advantage of this genetic cross was the ability to identify loci that influence proteinuria and renal injury on genetic backgrounds (from either S or SHR) permissive for hypertension.
The purpose of the current study was twofold: 1) confirm the linkage analysis and provide starting material for gene identification; and 2) evaluate the effect of genetic background on the ability of each locus (RNO8 or 13) to influence proteinuria by constructing reciprocal congenic strains. Specifically, the proteinuria-“resistant” SHR alleles were placed on the “susceptible” S background [S.SHR(8) or S.SHR(13)] or the proteinuria-susceptible S alleles were placed onto the resistant SHR background [SHR.S(8) or SHR.S(13)]. These strains were evaluated for proteinuria, various measures of renal function, as well as assessed for differences in kidney pathology.
MATERIALS AND METHODS
Animals
All experiments had approval of our Institutional Animal Care and Use Committee. The Dahl salt-sensitive (SS/Jr or S) and the spontaneously hypertensive rats (SHR/NHsd or SHR) were maintained in our animal facility at the University of Toledo, Health Science Campus (UTHSC, formerly the Medical University of Ohio). The S.SHR(8) or S.SHR(13) and SHR.S(8) or SHR.S(13) congenic strains were developed as previously described using the speed congenic approach (21). Briefly, female S rats were bred with male SHR to produce F1 rats. Female F1 rats were backcrossed to male S rats for S.SHR(8) or S.SHR(13), or male F1 rats were bred to female SHR rats for SHR.S(8) or SHR.S(13). This breeding scheme ensured that the mitochondrial genome for S.SHR congenics were from the S and the mitochondrial genome for SHR.S congenics were from the SHR. At the first backcross (BC1), animals were selected that retained the greatest amount of background genome while being heterozygous S/SHR on either RNO8 or RNO13. A total of 100–114 microsatellite markers were examined at each generation. The same process continued for BC2–BC5. At BC5, the background genome for each S.SHR strain was found to be homozygous for S alleles, while regions on RNO8 and RNO13 were heterozygous (S/SHR). Similarly, the background genome for each SHR.S strain was found to be homozygous for SHR alleles, while regions on RNO8 and RNO13 were heterozygous (S/SHR). The heterozygous animals for each region were intercrossed to fix the SHR alleles on the S background [S.SHR(8) or S.SHR(13)] or to fix the S alleles on the SHR background [SHR.S(8) and SHR.(13)]. The S.SHR(13) congenic was previously studied at an earlier time point and only for proteinuria (21).
At 4 wk of age, a group of age-matched male (n = 12/group) S, congenic, and SHR were weaned to a low-salt diet (0.3% NaCl; TD7034; Harlan Teklad, Madison, WI). The animals were studied for several renal and cardiovascular traits at 12 wk of age. Rats were killed by an overdose of pentobarbital sodium. Body (BW), heart (HW), and kidney weights (KW) were measured. Kidney samples were processed for histological examination, and serum samples were obtained from cardiac puncture to measure blood parameters.
Phenotyping
Urine and blood parameters.
To collect urine, animals were kept in metabolism cages (Lab Products, Seaford, DE) for 24 h with free access to water. Sodium azide was added as a preservative to the collection vials to a final concentration of ∼0.01%. Urinary protein excretion (UPE) and urine creatinine were determined as done previously (19, 21). No significant correlation was observed between UPE and BW. Thus it was not necessary to normalize UPE for differences in BW and it is reported as milligrams per 24 h. Blood parameters (Table 1) were determined by standard methods (e.g., creatinine was determined by the Jaffé method) using an Alfa Wassermann ACE automated chemistry analyzer (BioReliance, Rockville, MD).
Table 1.
Comparison of body weight, organ weight, and blood parameters for S, congenic strains, and SHR at week 12
| Strain | Weight |
Serum |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Body, g | Kidney, g | Heart, g | Total protein, g/dl | Albumin, g/dl | Uric acid, mg/dl | Cholesterol, mg/dl | Triglycerides, mg/dl | |||||||
| S | 375 (6.0) | 3.12 (0.049) | 1.22 (0.026) | 6.4 (0.11) | 2.9 (0.06) | 2.8 (0.11) | 119 (5.3) | 66 (4.1) | ||||||
| S.SHR (8) | 366 (4.3) | 2.70 (0.024)* | 1.19 (0.014) | 6.8 (0.08)* | 3.2 (0.04)* | 2.2 (0.08)* | 95 (2.8)* | 45 (1.4)* | ||||||
| S.SHR (13) | 397 (3.4)* | 2.79 (0.052)* | 1.22 (0.019) | 6.8 (0.08)* | 3.2 (0.04)* | 2.4 (0.12)* | 102 (3.3)* | 55 (3.4)* | ||||||
| SHR | 292 (3.6) | 2.13 (0.020) | 1.08 (0.011) | 7.4 (0.28) | 4.2 (0.17) | 3.6 (0.15) | 54 (2.4) | 51 (3.7) | ||||||
| SHR.S (8) | 306 (3.6)† | 2.07 (0.052) | 1.05 (0.019) | 7.0 (0.08) | 3.6 (0.03) | 3.4 (0.29) | 45 (1.1)† | 44 (2.4) | ||||||
| SHR.S (13) | 298 (3.6) | 2.14 (0.067) | 1.07 (0.017) | 6.9 (0.16) | 3.8 (0.03) | 2.8 (0.14) | 40 (1.8)† | 47 (5.3) | ||||||
Values are means ± SE with SE shown in parentheses; for weight, n = 12/group and for blood parameters, n = 8/group. SHR, spontaneously hypertensive rats. S, Dahl salt-sensitive rat. Heart and kidney weights were adjusted for differences in body weight when significantly different.
P < 0.01, S vs. S.SHR.
P < 0.01, SHR vs. SHR.S.
Histology.
Kidneys were fixed in 10% neutral buffered formalin and embedded in paraffin, cut into 3-μm sections, and stained with hematoxylin and eosin or Masson's trichrome. Two central longitudinal sections from each kidney (n = 12 for S and SHR, n = 8 for each congenic) were examined in a blinded fashion. Glomerular damage (glomerulosclerosis and mesangial expansion) was assessed as follows: grade 0, no changes; grade 1, lesions involving <25%; grade 2, lesions affecting 25- 50%; grade 3, lesions affecting 50–75%; and grade 4, lesions affecting >75% of glomeruli. Vascular, tubular, and degree of interstitial injury were evaluated separately on a semiquantitative scale from 0 (normal) to 4 (severe) (19, 21). Vascular compartments were assessed for vessel wall thickening, cell proliferation, and necrosis. Tubules were evaluated for the presence of necrosis, hydropic change, and/or tubular casts. Percent interstitial injury was determined by evaluation of slides stained with Masson's trichrome. Ten random regions of each slide were evaluated using MetaMorph image-analysis software (Downingtown, PA) to quantify percent fibrosis (blue staining) compared with background.
Genotyping
Genomic DNA was obtained by tail biopsy at each backcross generation during development of the congenic strains. The DNA was prepared using a Wizard SV 96 Genomic DNA kit (Promega, San Luis Obispo, CA). Genotyping was done as previously described using a fluorescent-based approach (19, 21).
Statistical Analysis
S and S.SHR or SHR and SHR.S strain data were evaluated by a one-way ANOVA followed by post hoc multiple comparisons using Dunnett's test. All data are presented as means ± SE.
RESULTS
To examine the effect of genetic background on QTL for proteinuria and renal function, congenic strains were developed by transferring a region of RNO8 or RNO13 from the SHR onto the susceptible S background [S.SHR(8) or S.SHR(13)] and S onto the resistant SHR background [SHR.S(8) or SHR.S(13)] (Fig. 1). In this case, the most relevant comparison to be made is between the S and S.SHR or between SHR and SHR.S. Figure 2 presents important renal parameters for each of these comparisons. The S.SHR(8) congenic demonstrated 75% lower UPE compared with the S parental (114 ± 9.3 vs. 32 ± 5.2 mg/24 h, P < 0.0001), whereas the S.SHR(13) demonstrated a twofold reduction (114 ± 9.3 vs. 59 ± 3.5 mg/24 h, P < 0.0001). Creatinine clearance (CrCl), normalized to kidney weight was higher in each S.SHR congenic compared with S, but this did not reach statistical significance when correcting for multiple comparisons. Blood urea nitrogen (BUN) and uric acid (UA) levels (Table 1) were also significantly lower compared with the S. However, the transfer of either the RNO8 or RNO13 region from the S onto the resistant SHR-background failed to significantly change any of the measured parameters. UPE for SHR.S(8) or SHR.S(13) was slightly elevated (10 ± 1.6 and 10 ± 1.4 mg/24 h, respectively) compared with the SHR parental (7.7 ± 1.0 mg/24 h), but this difference was not statistically significant. Additionally, no significant difference was observed for CrCl, BUN (Fig. 2), or UA levels (Table 1) among these strains.
Fig. 1.
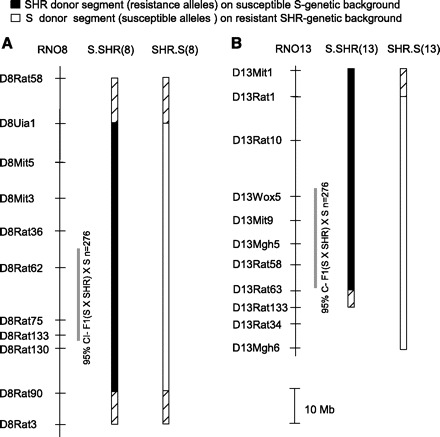
Physical map and schematic diagram of RNO8 and RNO13 congenic strains. RNO8 reciprocal congenic strains (A) and RNO13 reciprocal congenic strains (B) are shown. The grey bar designates ∼95% confidence interval for each quantitative trait loci (QTL) based on linkage analysis (17). The black bar designates the extent of introgressed spontaneously hypertensive rat (SHR alleles) on the S-susceptible background (i.e., S.SHR), and the open bar represents the extent of introgressed S alleles on the SHR-resistant background (i.e., SHR.S). The hatched region on each end of the congenic segment represents the recombination interval.
Fig. 2.
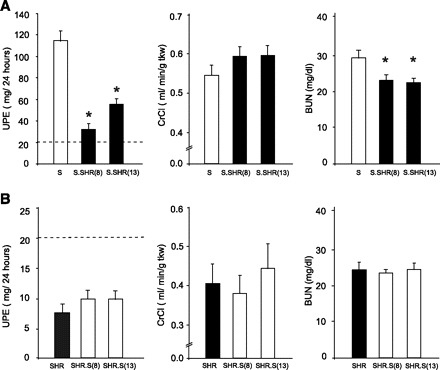
Comparison of key renal measures between strains. A: S vs. S.SHR(8) and S.SHR(13). B: SHR vs. SHR.S(8) and SHR.S(13). Urinary protein excretion (UPE), creatinine clearance (CrCl), and blood urea nitrogen (BUN) are shown. The dashed line under UPE represents the threshold for “proteinuria” (>20 mg/24 h). Rats were maintained on a low-salt diet (0.3% NaCl). Measurements were performed on n = 8–12 rats/group. Error bars are SE. *Significantly different from S at P < 0.05 by 1-way ANOVA followed by post hoc multiple comparisons using Dunnett's test.
Table 1 shows additional experimental parameters between the groups. For S and S.SHR(8), BW was similar, but S.SHR(13) was slightly heavier than S controls. No significant differences in HW among the groups were observed, but KW was significantly less in each S.SHR congenic strain (normalized for differences in BW). Both total cholesterol (TC) and triglyceride (TG) levels were also significantly decreased in the S.SHR compared with the S. There was a small, but significant difference in BW between SHR and SHR.S(8). However, once both HW and KW were corrected for differences in BW, no significant difference was observed among these strains.
Kidneys from each of the strains were examined and evaluated for glomerular, tubular, vascular, and interstitial damage (Fig. 3). At 12 wk of age, glomerular damage in S.SHR(8), but not the S.SHR(13) congenic strain was significantly lower than the S. Tubular changes were significantly attenuated in both S.SHR congenic strains compared with the S (Fig. 3A). No significant vascular differences were seen among the groups. The principal histological difference between the S and either S.SHR congenic strain was in the degree of interstitial injury (Fig. 3A). Figure 4 provides representative histological images for the degree of fibrosis observed among strains, as well as a quantitative assessment of interstitial fibrosis. The S rat kidney experienced significantly more interstitial fibrosis (13.1 ± 1.28%) compared with either the S.SHR(8) (4.6 ± 0.43%) or S.SHR(13) (5.9 ± 0.28%). For the SHR and SHR.S(8) or SHR.S(13) comparison, no significant difference in glomerular, tubular, or interstitial injury was observed. Interestingly, for both SHR.S congenic strains, the severity of vascular injury was significantly higher than the SHR (Fig. 3B). Figure 4B provides representative histological images of vascular damage observed in the SHR.S congenic strains compared with the SHR.
Fig. 3.
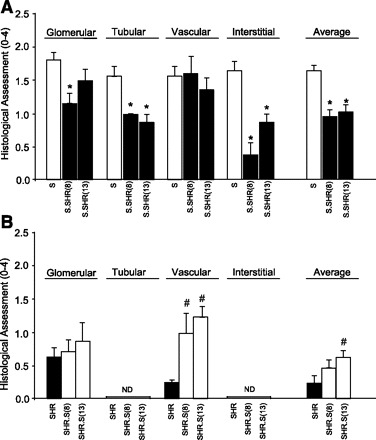
Histological assessment of glomerular, tubular, vascular, and interstitial injury between strains. A: S vs. S.SHR(8) and S.SHR(13). B: SHR vs. SHR.S(8) and SHR.S(13). Injury was assessed using a semiquantitative scale from 0 (normal) to 4 (severe). Kidneys from n = 8–12 animals were examined from each group. Error bars are SE. ND, not detectable. *Significantly different from S at P < 0.05. #Significantly different from SHR at P < 0.05 both by 1-way ANOVA followed by post hoc multiple comparisons using Dunnett's test.
Fig. 4.
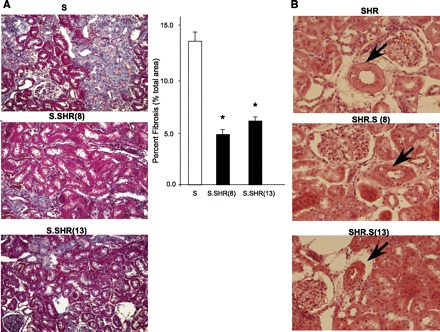
Representative images of key histological features. A: Masson's trichrome staining at ×20 for S vs. S.SHR(8) and S.SHR(13). B: hematoxylin and eosin staining at ×40 for SHR vs. SHR.S(8) and SHR.S(13). For quantitative assessment of interstitial fibrosis in S and S.SHR congenics, 10 random regions of each slide (×20) were evaluated using MetaMorph (Downingtown, PA). Arrows denote arterial vessels. The SHR.S congenics demonstrate vessel thickening and hypercellularity which in the not seen in the SHR parental. Error bars are SE. *Significantly different from S at P < 0.05 by 1-way ANOVA followed by post hoc multiple comparisons using Dunnett's test.
DISCUSSION
An important reason for developing a consomic or congenic strain is to provide a tool to investigate the effect of an individual locus separate from other loci that contribute to a particular quantitative trait (9, 46). The ultimate use of these strains (and subsequent congenic substrains) is usually to aid in identifying gene(s) that play a role in a particular disease process (9). More often than not, congenic strains have been developed to transfer a single locus from an animal exhibiting resistance onto the genetic background of the susceptible strain. The expectation is that the congenic strain will demonstrate improvement over the susceptible parental strain for any particular trait. Conversely, transfer of a region from the susceptible strain onto the genetic background of the resistant strain is expected to result in an animal worse off than the resistant parental. The main goal of this work, aside from confirming the proteinuria QTL on RNO8 and RNO13 identified in an earlier linkage analysis (17), was to investigate how genetic background plays a role in pathogenesis of proteinuria and renal injury between the S and SHR.
Our intention here was to test the ability of each locus to influence proteinuria and renal injury under conditions of moderate hypertension and not to severely increase BP by salt-loading (17, 19). Therefore, the strains were maintained on a low-salt diet to minimize the “salt-sensitive” component of blood pressure observed in the S rat. As expected, placement of the renal-protective SHR alleles on the susceptible S background [S.SHR(8) or S.SHR(13)] showed significant attenuation of proteinuria, serving to confirm the original linkage analysis (17). The current study did not observe a significant difference in CrCl between strains (at the age studied). However, improved renal function was demonstrated by significant changes in BUN and UA levels. The histological findings of reduced glomerular and tubular injury corroborated the reduction in proteinuria. In particular, renal interstitial fibrosis was the predominant histological improvement in the S.SHR compared with the S. This is important because renal fibrosis represents the pathological event most correlated with loss of renal function (32, 67). The fact that vascular injury was the same between strains (which can influence renal perfusion pressure and GFR) could partially explain why no difference in CrCl was observed.
TC and TG levels were also significantly elevated in proteinuric S rats compared with S.SHR congenic strains. which exhibited less renal injury. Several studies have demonstrated that dyslipidemia is a relatively common finding in patients with chronic kidney disease and/or ESRD (24, 33). Differences in TC and/or TG (between the strains) are likely due to enhanced production and accumulation of triglyceride-rich lipoproteins in the renal-impaired S rat. The observed differences in these measures are likely reflective of differences in renal injury (i.e., proteinuria, renal pathology, etc.) rather than the presence of QTL for TC and/or TG being on these chromosomes. However, the possibility that these loci are not also linked to TC and/or TG cannot be completely eliminated because several linkage analyses have identified QTL for these traits on RNO8 and 13 (25, 30, 42, 59).
The SHR.S(8) or SHR.S(13) congenic strains (i.e., susceptible locus on resistant genetic background) demonstrated no significant increase in proteinuria, decline in renal function, or increase in glomerular, tubular, or interstitial injury compared with the SHR. Significant differences in vascular injury were observed between the SHR and SHR.S strains. These vascular differences may be indicative of early renal injury that may eventually lead to proteinuria or impaired renal function (if studied in older animals). Additionally, the same locus (or set of loci) on each chromosome could be responsible for both renal vascular lesions and proteinuria (pleiotropy), such that the SHR genetic background allows the expression of the vascular component, and the S background allows expression of the proteinuria and tubular interstitial component.
In general, congenic strains developed on the SHR genetic background [SHR.S] demonstrated that proteinuria-resistant loci in the SHR (such as those on RNO1, 6, 9, 13, and 19) are able to resist pressure toward renal injury and proteinuria conferred by the susceptibility alleles donated by S on RNO8 or 13. This clearly suggests that multiple susceptibility genes are required for a pathological condition to manifest itself (at least in the SHR). For instance, in the earlier linkage analysis (17), several genetic interactions were observed among RNO2/8, RNO6/8, and RNO10/13. It is possible that the replacement of more than one resistance allele (e.g., RNO2/8) with prorenal injury loci from the S are required before significant renal injury and proteinuria would be exhibited on the SHR background. These interactions are possible to elucidate but would require detailed congenic analysis where single congenic strains (one locus substituted) are directly compared with double congenic strains (two loci substituted), along with appropriate parental controls (47).
There have been a number of studies that have utilized reciprocal congenic strains to investigate the genetic basis of various diseases, including hypertension (3, 13, 14, 58, 62), stroke (49), ethanol sensitivity (44), arthritis susceptibility (48), and proteinuria and glomerulosclerosis (63). In some instances, the reciprocal congenic strains demonstrated comparable effects on the phenotype, whereas in other studies the effect observed between the reciprocal strains was not comparable. For example, Frantz et al. (14) developed reciprocal congenic strains from the SHR (hypertensive) and Wistar-Kyoto (WKY; normotensive). The SHR.WHY-Sa demonstrated −11-mmHg blood pressure compared with SHR, and the WKY.SHR-Sa demonstrated +11 mmHg compared with WKY (14). In contrast, Yagil et al. (62) developed reciprocal congenic strains from the SBH (hypertensive) and SBN (normotensive). The SBH.SBN1 demonstrated a significant decrease in blood pressure compared with SBH, whereas the SBN-SBH1 showed no significant increase compared with SBN (62). A similar study found that SBN.SBH1 and SBH.SBN1 demonstrated a significant difference in proteinuria and glomerulosclerosis compared with control. However, there was about a twofold difference in the degree of the proteinuria effect between the SBH.SBN1 and SBN.SBH1 compared with their respective controls (63).
Studies involving hypertension appear to be variable in how genetic background can influence the degree of phenotypic effect between reciprocal congenic strains and their respective controls (e.g., susceptible on resistant or resistant on susceptible). For proteinuria, our data (and others) suggest that a genetic background resistant to renal injury (such as the SHR) is highly resilient to genetic pressure to promote injury. This may be partially explained from genetic crosses between high- and low-proteinuria strains, which consistently demonstrate that proteinuria is strongly recessive (i.e., F1 offspring exhibit proteinuria similar to the resistance strain) (6, 17, 34, 52, 64), whereas blood pressure appears to fit an additive model of inheritance (i.e., F1 offspring exhibit blood pressure midway between strains) (8, 41, 52, 66). There have been several studies using the mouse that have also demonstrated that genetic background can have a significant influence on disease occurrence and severity (56, 65). In particular, when cycloxygenase-2 deletion (cycloxygenase-2 deficient) was introduced into three genetic-backgrounds (129/Sv, C57/BL6, and BALB/c), there were remarkable strain-dependent differences in hypertension and proteinuria (BALB = no hypertension/proteinuria; C57 = no hypertension, but proteinuria; and 129 = severe hypertension/proteinuria) (65).
The present study did not address whether the proteinuria difference is independent from blood pressure changes. This was the case for the proteinuria locus on RNO2. No difference in blood pressure was observed, despite a large difference in proteinuria between strains (19). This was consistent with the linkage analysis because no association with blood pressure was found on RNO2. Similarly, no linkage to blood pressure was observed at either the RNO8 or RNO13 locus based on the linkage analysis (17). Regardless, even if significant differences in blood pressure were detected (along with proteinuria), the cause-effect relationship would be difficult to elucidate. The only way to dissect whether these traits are under independent control or linked would be through additional congenic analysis using progressively smaller congenic segments.
Genetic background plays an important role in the phenotypic expression of loci associated with proteinuria and renal injury. Congenic strains developed by placing the resistant SHR alleles onto the susceptible S-genetic background provided the most informative comparison for our work. However, the outcome of any congenic analysis is strongly dependent on the phenotype, genetic background, and/or the influence of each locus, and for these reasons constructing reciprocal congenic strains provides the best strategy to test the influence of any given QTL. In either case, the larger the phenotypic effect between the congenic and control animals, the greater the ability to track these differences when performing further congenic strain analysis (i.e., substrains). This is advantageous because the unfortunate reality is that QTL are likely to contain multiple loci that each contribute small-to-modest effects or interact with other loci, making the process of positional cloning difficult (22, 38, 50). Nevertheless, with persistence, the process from QTL to gene identification is valuable and achievable (2, 12, 45).
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant RO1-HL-066998 (M. R. Garrett).
Acknowledgments
The authors thank Dr. John Rapp for a critical reading of the manuscript.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Present address of Y. Saad: Univ. of Central Florida College of Medicine, Burnett School of Biomedical Sciences, Orlando, FL.
REFERENCES
- 1.Agodoa L, Norris K, Pugsley D. The disproportionate burden of kidney disease in those who can least afford it. Kidney Int Suppl: S1–S3, 2005 [DOI] [PubMed]
- 2.Aitman TJ, Dong R, Vyse TJ, Norsworthy PJ, Johnson MD, Smith J, Mangion J, Roberton-Lowe C, Marshall AJ, Petretto E, Hodges MD, Bhangal G, Patel SG, Sheehan-Rooney K, Duda M, Cook PR, Evans DJ, Domin J, Flint J, Boyle JJ, Pusey CD, Cook HT. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature 439: 851–855, 2006 [DOI] [PubMed] [Google Scholar]
- 3.Alemayehu A, Breen L, Krenova D, Printz MP. Reciprocal rat chromosome 2 congenic strains reveal contrasting blood pressure and heart rate QTL. Physiol Genomics 10: 199–210, 2002 [DOI] [PubMed] [Google Scholar]
- 4.Bowden DW. Genetics of kidney disease. Kidney Int Suppl: S8–S12, 2003 [DOI] [PubMed]
- 5.Bowden DW, Colicigno CJ, Langefeld CD, Sale MM, Williams A, Anderson PJ, Rich SS, Freedman BI. A genome scan for diabetic nephropathy in African Americans. Kidney Int 66: 1517–1526, 2004 [DOI] [PubMed] [Google Scholar]
- 6.Brown DM, Provoost AP, Daly MJ, Lander ES, Jacob HJ. Renal disease susceptibility and hypertension are under independent genetic control in the fawn-hooded rat. Nat Genet 12: 44–51, 1996 [DOI] [PubMed] [Google Scholar]
- 7.Chistiakov DA, Savost'anov KV, Shestakova MV, Chugunova LA, Samkhalova M, Dedov II, Nosikov VV. Confirmation of a susceptibility locus for diabetic nephropathy on chromosome 3q23-q24 by association study in Russian type 1 diabetic patients. Diabetes Res Clin Pract 66: 79–86, 2004 [DOI] [PubMed] [Google Scholar]
- 8.Clark JS, Jeffs B, Davidson AO, Lee WK, Anderson NH, Bihoreau M-T, Brosnan MJ, Devlin AM, Kelman AW, Lindpaintner K, Dominiczak AF. Quantitative trait loci in genetically hypertensive rats. Possible sex specificity. Hypertension 28: 898–906, 1996 [DOI] [PubMed] [Google Scholar]
- 9.Cowley AW Jr. The genetic dissection of essential hypertension. Nat Rev Genet 7: 829–840, 2006 [DOI] [PubMed] [Google Scholar]
- 10.DeWan AT, Arnett DK, Atwood LD, Province MA, Lewis CE, Hunt SC, Eckfeldt J. A genome scan for renal function among hypertensives: the HyperGEN study. Am J Hum Genet 68: 136–144, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeWan AT, Arnett DK, Miller MB, Peacock JM, Atwood LD, Province MA, Lewis CE, Hunt SC, Eckfeldt JH. Refined mapping of suggestive linkage to renal function in African Americans: the HyperGEN study. Am J Hum Genet 71: 204–205, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flint J, Valdar W, Shifman S, Mott R. Strategies for mapping and cloning quantitative trait genes in rodents. Nat Rev Genet 6: 271–286, 2005 [DOI] [PubMed] [Google Scholar]
- 13.Frantz S, Clemitson JR, Bihoreau MT, Gauguier D, Samani NJ. Genetic dissection of region around the Sa gene on rat chromosome 1: evidence for multiple loci affecting blood pressure. Hypertension 38: 216–221, 2001 [DOI] [PubMed] [Google Scholar]
- 14.Frantz SA, Kaiser M, Gardiner SM, Gauguier D, Vincent M, Thompson JR, Bennett T, Samani NJ. Successful isolation of a rat chromosome 1 blood pressure quantitative trait locus in reciprocal congenic strains. Hypertension 32: 639–646, 1998 [DOI] [PubMed] [Google Scholar]
- 15.Freedman BI, Beck SR, Rich SS, Heiss G, Lewis CE, Turner S, Province MA, Schwander KL, Arnett DK, Mellen BG. A genome-wide scan for urinary albumin excretion in hypertensive families. Hypertension 42: 291–296, 2003 [DOI] [PubMed] [Google Scholar]
- 16.Freedman BI, Bostrom M, Daeihagh P, Bowden DW. Genetic factors in diabetic nephropathy. Clin J Am Soc Nephrol 2: 1306–1316, 2007 [DOI] [PubMed] [Google Scholar]
- 17.Garrett MR, Dene H, Rapp JP. Time-course genetic analysis of albuminuria in Dahl salt-sensitive rats on low-salt diet. J Am Soc Nephrol 14: 1175–1187, 2003 [DOI] [PubMed] [Google Scholar]
- 18.Garrett MR, Dene H, Walder R, Zhang Q-Y, Cicila GT, Assadnia S, Deng AY, Rapp JP. Genome scan and congenic strains for blood pressure QTL using Dahl salt-sensitive rats. Genome Res 8: 711–723, 1998 [DOI] [PubMed] [Google Scholar]
- 19.Garrett MR, Gunning WT, Radecki T, Richard A. Dissection of a genetic locus influencing renal function in the rat and its concordance with kidney disease loci on human chromosome 1q21. Physiol Genomics 30: 322–334, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garrett MR, Joe B, Dene H, Rapp J.P. Identification of blood pressure QTL that differentiate two hypertensive strains. J Hypertens 20: 2399–2406, 2002 [DOI] [PubMed] [Google Scholar]
- 21.Garrett MR, Joe B, Yerga-Woolwine S. Genetic linkage of urinary albumin excretion in Dahl salt-sensitive rats: influence of dietary salt and confirmation using congenic strains. Physiol Genomics 25: 39–49, 2006 [DOI] [PubMed] [Google Scholar]
- 22.Garrett MR, Rapp JP. Multiple blood pressure QTL on rat chromosome 2 defined by congenic Dahl rats. Mammalian Genome 13: 41–44, 2002 [DOI] [PubMed] [Google Scholar]
- 23.Garrett MR, Saad Y, Dene H, Rapp JP. Blood pressure QTL that differentiate Dahl salt-sensitive and spontaneously hypertensive rats. Physiol Genomics 3: 33–38, 2000 [DOI] [PubMed] [Google Scholar]
- 24.Harper CR, Jacobson TA. Managing dyslipidemia in chronic kidney disease. J Am Coll Cardiol 51: 2375–2384, 2008 [DOI] [PubMed] [Google Scholar]
- 25.Herrera VL, Didishvili T, Lopez LV, Myers RH, Ruiz-Opazo N. Genome-wide scan identifies novel QTLs for cholesterol and LDL levels in F2[Dahl RxS]-intercross rats. Circ Res 94: 446–452, 2004 [DOI] [PubMed] [Google Scholar]
- 26.Hwang SJ, Yang Q, Meigs JB, Pearce EN, Fox CS. A genome-wide association for kidney function and endocrine-related traits in the NHLBI's Framingham Heart Study. BMC Med Genet 8, Suppl 1: S10, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Imperatore G, Hanson RL, Pettitt DJ, Kobes S, Bennett PH, Knowler WC. Sib-pair linkage analysis for susceptibility genes for microvascular complications among Pima Indians with type 2 diabetes. Pima Diabetes Genes Group. Diabetes 47: 821–830, 1998 [DOI] [PubMed] [Google Scholar]
- 28.Imperatore G, Knowler WC, Nelson RG, Hanson RL. Genetics of diabetic nephropathy in the Pima Indians. Curr Diab Rep 1: 275–281, 2001 [DOI] [PubMed] [Google Scholar]
- 29.Joe B, Garrett MR. Substitution mapping: ssing congenic strains to detect genes controlling blood pressure. In: Cardiovascular Genomics:Gene Mining for Pharmacogenomics and Gene Therapy, edited by Raizada MK PJ, Kasparov S, and Katovich MJ. Totowa, NJ: Humana, 2004, p. 39–56.
- 30.Klimes I, Weston K, Kovacs P, Gasperikova D, Jezova D, Kvetnansky R, Thompson JR, Sebokova E, Samani NJ. Mapping of genetic loci predisposing to hypertriglyceridaemia in the hereditary hypertriglyceridaemic rat: analysis of genetic association with related traits of the insulin resistance syndrome. Diabetologia 46: 352–358, 2003 [DOI] [PubMed] [Google Scholar]
- 31.Krolewski AS, Poznik GD, Placha G, Canani L, Dunn J, Walker W, Smiles A, Krolewski B, Fogarty DG, Moczulski D, Araki S, Makita Y, Ng DP, Rogus J, Duggirala R, Rich SS, Warram JH. A genome-wide linkage scan for genes controlling variation in urinary albumin excretion in type II diabetes. Kidney Int 69: 129–136, 2006 [DOI] [PubMed] [Google Scholar]
- 32.Liu Y. Renal fibrosis: new insights into the pathogenesis and therapeutics. Kidney Int 69: 213–217, 2006 [DOI] [PubMed] [Google Scholar]
- 33.Locatelli F, Pozzoni P, Tentori F, del Vecchio L. Epidemiology of cardiovascular risk in patients with chronic kidney disease. Nephrol Dial Transplant 18, Suppl 7: 2–9, 2003 [DOI] [PubMed] [Google Scholar]
- 34.Matsuyama M, Ogiu T, Kontani K, Yamada C, Kawai M, Hiai H, Nakamura T, Shimizu F, Toyokawa T, Kinoshita Y. Genetic regulation of the development of glomerular sclerotic lesions in the BUF/Mna rat. Nephron 54: 334–337, 1990 [DOI] [PubMed] [Google Scholar]
- 35.McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JP, Hirschhorn JN. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet 9: 356–369, 2008 [DOI] [PubMed] [Google Scholar]
- 36.McCullough PA, Jurkovitz CT, Pergola PE, McGill JB, Brown WW, Collins AJ, Chen SC, Li S, Singh A, Norris KC, Klag MJ, Bakris GL. Independent components of chronic kidney disease as a cardiovascular risk state: results from the Kidney Early Evaluation Program (KEEP). Arch Intern Med 167: 1122–1129, 2007 [DOI] [PubMed] [Google Scholar]
- 37.Moreno C, Dumas P, Kaldunski ML, Tonellato PJ, Greene AS, Roman RJ, Cheng Q, Wang Z, Jacob HJ, Cowley AW Jr. Genomic map of cardiovascular phenotypes of hypertension in female Dahl S rats. Physiol Genomics 15: 243–257, 2003 [DOI] [PubMed] [Google Scholar]
- 38.Moreno C, Kaldunski ML, Wang T, Roman RJ, Greene AS, Lazar J, Jacob HJ, Cowley AW Jr. Multiple blood pressure loci on rat chromosome 13 attenuate development of hypertension in the Dahl S hypertensive rat. Physiol Genomics 31: 228–235, 2007 [DOI] [PubMed] [Google Scholar]
- 39.Murayama S, Yagyu S, Higo K, Ye C, Mizuno T, Oyabu A, Ito M, Morita H, Maeda K, Serikawa T, Matsuyama M. A genetic locus susceptible to the overt proteinuria in BUF/Mna rat. Mamm Genome 9: 886–888, 1998 [DOI] [PubMed] [Google Scholar]
- 40.Norris KC, Agodoa LY. Unraveling the racial disparities associated with kidney disease. Kidney Int 68: 914–924, 2005 [DOI] [PubMed] [Google Scholar]
- 41.Ohno Y, Tanase H, Nabika T, Otsuka K, Sasaki T, Suzawa T, Morii T, Yamori Y, Saruta T. Selective genotyping with epistasis can be utilized for a major quantitative trait locus mapping in hypertension in rats. Genetics 155: 785–792, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okuno S, Watanabe TK, Ono T, Yamasaki Y, Goto Y, Miyao H, Asai T, Kanemoto N, Oga K, Mizoguchi-Miyakita A, Takagi T, Takahashi E, Nakamura Y, Tanigami A. Genetic determinants of plasma triglyceride levels in (OLETF x BN) x OLETF backcross rats. Genomics 62: 350–355, 1999 [DOI] [PubMed] [Google Scholar]
- 43.Poyan Mehr A, Siegel AK, Kossmehl P, Schulz A, Plehm R, de Bruijn JA, de Heer E, Kreutz R. Early onset albuminuria in Dahl rats is a polygenetic trait that is independent from salt loading. Physiol Genomics 14: 209–216, 2003 [DOI] [PubMed] [Google Scholar]
- 44.Radcliffe RA, Bludeau P, Asperi W, Fay T, Deng XS, Erwin VG, Deitrich RA. Confirmation of quantitative trait loci for ethanol sensitivity and neurotensin receptor density in crosses derived from the inbred high and low alcohol sensitive selectively bred rat lines. Psychopharmacology (Berl) 188: 343–354, 2006 [DOI] [PubMed] [Google Scholar]
- 45.Rangel-Filho A, Sharma M, Datta YH, Moreno C, Roman RJ, Iwamoto Y, Provoost AP, Lazar J, Jacob HJ. RF-2 gene modulates proteinuria and albuminuria independently of changes in glomerular permeability in the fawn-hooded hypertensive rat. J Am Soc Nephrol 16: 852–856, 2005 [DOI] [PubMed] [Google Scholar]
- 46.Rapp JP. Genetic analysis of inherited hypertension in the rat. Physiol Rev 80: 135–172, 2000 [DOI] [PubMed] [Google Scholar]
- 47.Rapp JP, Garrett MR, Deng AY. Construction of a double congenic strain to prove an epistatic interaction on blood pressure between rat chromosomes 2 and 10. J Clin Invest 101: 1591–1595, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ribbhammar U, Flornes L, Backdahl L, Luthman H, Fossum S, Lorentzen JC. High resolution mapping of an arthritis susceptibility locus on rat chromosome 4, and characterization of regulated phenotypes. Hum Mol Genet 12: 2087–2096, 2003 [DOI] [PubMed] [Google Scholar]
- 49.Rubattu S, Hubner N, Ganten U, Evangelista A, Stanzione R, Di Angelantonio E, Plehm R, Langanki R, Gianazza E, Sironi L, D'Amati G, Volpe M. Reciprocal congenic lines for a major stroke QTL on rat chromosome 1. Physiol Genomics 27: 108–113, 2006 [DOI] [PubMed] [Google Scholar]
- 50.Saad Y, Yerga-Woolwine S, Saikumar J, Farms P, Manickavasagam E, Toland EJ, Joe B. Congenic interval mapping of RNO10 reveals a complex cluster of closely-linked genetic determinants of blood pressure. Hypertension 50: 891–898, 2007 [DOI] [PubMed] [Google Scholar]
- 51.Schork NJ. Genetics of complex disease: approaches, problems, and solutions. Am J Respir Crit Care Med 156: S103–S109, 1997 [DOI] [PubMed] [Google Scholar]
- 52.Schulz A, Litfin A, Kossmehl P, Kreutz R. Genetic dissection of increased urinary albumin excretion in the Munich Wistar Fromter rat. J Am Soc Nephrol 13: 2706–2714, 2002 [DOI] [PubMed] [Google Scholar]
- 53.Schulz A, Standke D, Kovacevic L, Mostler M, Kossmehl P, Stoll M, Kreutz R. A major gene locus links early onset albuminuria with renal interstitial fibrosis in the MWF rat with polygenetic albuminuria. J Am Soc Nephrol 14: 3081–3089, 2003 [DOI] [PubMed] [Google Scholar]
- 54.Shiozawa M, Provoost AP, van Dokkum RP, Majewski RR, Jacob HJ. Evidence of gene-gene interactions in the genetic susceptibility to renal impairment after unilateral nephrectomy. J Am Soc Nephrol 11: 2068–2078, 2000 [DOI] [PubMed] [Google Scholar]
- 55.Siegel AK, Kossmehl P, Planert M, Schulz A, Wehland M, Stoll M, Bruijn JA, de Heer E, Kreutz R. Genetic linkage of albuminuria and renal injury in Dahl salt-sensitive rats on a high-salt diet: comparison with spontaneously hypertensive rats. Physiol Genomics 18: 218–225, 2004 [DOI] [PubMed] [Google Scholar]
- 56.Sommardahl C, Cottrell M, Wilkinson JE, Woychik RP, Johnson DK. Phenotypic variations of orpk mutation and chromosomal localization of modifiers influencing kidney phenotype. Physiol Genomics 7: 127–134, 2001 [DOI] [PubMed] [Google Scholar]
- 57.Stella P, Cusi D, Duzzi L, Bianchi G. Relations between hypertension and glomerulosclerosis in first-generation hybrid rats of the Milan strains. Child Nephrol Urol 11: 6–9, 1991 [PubMed] [Google Scholar]
- 58.Tripodi G, Florio M, Ferrandi M, Modica R, Zimdahl H, Hubner N, Ferrari P, Bianchi G. Effect of Add1 gene transfer on blood pressure in reciprocal congenic strains of Milan rats. Biochem Biophys Res Commun 324: 562–568, 2004 [DOI] [PubMed] [Google Scholar]
- 59.Ueno T, Tremblay J, Kunes J, Zicha J, Dobesova Z, Pausova Z, Deng AY, Sun YL, Jacob HJ, Hamet P. Rat model of familial combined hyperlipidemia as a result of comparative mapping. Physiol Genomics 17: 38–47, 2004 [DOI] [PubMed] [Google Scholar]
- 60.United States Renal Data System. 2003 Annual Data Report. In: Atlas of End-Stage Renal Disease in the United States. Bethesda, MD: National Institute of Diabetes and Digestive and Kidney Disease, 2005
- 61.Vardarli I, Baier LJ, Hanson RL, Akkoyun I, Fischer C, Rohmeiss P, Basci A, Bartram CR, Van Der Woude FJ, Janssen B. Gene for susceptibility to diabetic nephropathy in type 2 diabetes maps to 18q22.3-23. Kidney Int 62: 2176–2183, 2002 [DOI] [PubMed] [Google Scholar]
- 62.Yagil C, Hubner N, Kreutz R, Ganten D, Yagil Y. Congenic strains confirm the presence of salt-sensitivity QTLs on chromosome 1 in the Sabra rat model of hypertension. Physiol Genomics 12: 85–95, 2003 [DOI] [PubMed] [Google Scholar]
- 63.Yagil C, Sapojnikov M, Katni G, Ilan Z, Zangen SW, Rosenmann E, Yagil Y. Proteinuria and glomerulosclerosis in the Sabra genetic rat model of salt susceptibility. Physiol Genomics 9: 167–178, 2002 [DOI] [PubMed] [Google Scholar]
- 64.Yagil C, Sapojnikov M, Wechsler A, Korol A, Yagil Y. Genetic dissection of proteinuria in the Sabra rat. Physiol Genomics 25: 121–133, 2006 [DOI] [PubMed] [Google Scholar]
- 65.Yang T, Huang YG, Ye W, Hansen P, Schnermann JB, Briggs JP. Influence of genetic background and gender on hypertension and renal failure in COX-2-deficient mice. Am J Physiol Renal Physiol 288: F1125–F1132, 2005 [DOI] [PubMed] [Google Scholar]
- 66.Zagato L, Modica R, Florio M, Torielli L, Bihoreau MT, Bianchi G, Tripodi G. Genetic mapping of blood pressure quantitative trait loci in Milan hypertensive rats. Hypertension 36: 734–739, 2000 [DOI] [PubMed] [Google Scholar]
- 67.Zeisberg M, Strutz F, Muller GA. Renal fibrosis: an update. Curr Opin Nephrol Hypertens 10: 315–320, 2001 [DOI] [PubMed] [Google Scholar]