

Abstract
Merkel Cell Carcinoma (MCC) is a rare and highly aggressive neuroendocrine skin cancer for which no effective treatment is available. MCC represents a human cancer with the best experimental evidence for a causal role of a polyoma virus. Large T antigens (LTA) encoded by polyoma viruses are oncoproteins, which are thought to require support of cellular heat shock protein 70 (HSP70) to exert their transforming activity. Here we evaluated the capability of MAL3-101, a synthetic HSP70 inhibitor, to limit proliferation and survival of various MCC cell lines. Remarkably, MAL3-101 treatment resulted in considerable apoptosis in 5 out of 7 MCC cell lines. While this effect was not associated with the viral status of the MCC cells, quantitative mRNA expression analysis of the known HSP70 isoforms revealed a significant correlation between MAL3-101 sensitivity and HSC70 expression, the most prominent isoform in all cell lines. Moreover, MAL3-101 also exhibited in vivo antitumor activity in an MCC xenograft model suggesting that this substance or related compounds are potential therapeutics for the treatment of MCC in the future.
Introduction
Merkel Cell Carcinoma (MCC) is a highly aggressive neuroendocrine skin cancer, which primarily affects elderly or immunocompromised individuals [1], [2]. MCC is a rare disease, but its incidence is rapidly increasing [3], [4]. Treatment of primary tumors includes surgical resection and adjuvant radiotherapy [5], [6]. Therapeutic options for advanced disease are of limited efficacy with no proven benefit on overall survival [7]–[11]. MCC is associated in the majority of cases with Merkel cell polyoma virus (MCPyV). Indeed, MCC represents the human cancer with the best experimental evidence for a causal role of a polyoma virus, and expression of the T antigens by MCPyV is required for growth of MCC cells in cell culture and in xenografts [12], [13]. In particular, MCC cells depend on large T antigen (LTA) and its ability to interact with Retinoblastoma protein (Rb) [12]. It is believed that this interaction requires the activity of a cellular heat shock protein 70, or HSP70 [14].
Members of the HSP70 superfamily are highly expressed in many cancers [15], [16]. Notably, high HSP70 expression is associated with poor prognosis and resistance to chemotherapy while low HSP70 levels correlate with reduced tumorigenicity [15], [17]. The HSP70 superfamily is evolutionary highly conserved and consists of 17 isoforms [18]. Besides stress-inducible variants the family also includes the constitutively expressed HSC70 (HSPA8) [19]. HSP70 proteins are ATP-dependent molecular chaperones that regulate diverse cellular functions, including folding and assembly of newly synthesized as well as refolding of misfolded proteins, transport of proteins across intracellular membranes and maintenance of protein homeostasis within the cell [20]. Moreover, HSPs can interfere with cell death at different stages by blocking apoptosis in a caspase-dependent or independent manner [15], [21]. While the precise mechanisms by which HSP70 exerts its anti-apoptotic function are not yet fully understood, inactivation of HSP70 may hold great therapeutic value as HSP70-inactivating antisense oligonucleotides efficiently triggered cell death and cell cycle arrest in cancer cells [19], [22], [23].
MAL3-101 is a small molecule HSP70 inhibitor and exerts anti-proliferative and pro-apoptotic effects on cell lines derived from various cancers, including small cell lung carcinoma [24], [25]. MAL3-101 is a membrane permeable dihydropyrimidine analog that modulates the ATPase activity of HSP70 proteins and, in particular, inhibits the ATPase activity induced by simian virus 40 LTA, which interacts with HSP70 proteins via its J-domain [26]. LTAs promote G1/S cell cycle progression by inactivating proteins from the Rb family [27]–[29]. Notably, both the J-domain of LTA and HSC70-dependent ATP hydrolysis is required for Rb inactivation [30]–[32].
Although it has not yet been established whether binding of HSC70 by MCPyV LTA is required to support proliferation of MCC cells, it has been demonstrated that MCPyV LTA binds HSC70 via the J-domain, and that this interaction facilitates MCPyV replication [14]. As HSP70 proteins generally support growth and survival of tumor cells and may be particularly critical for MCPyV-transformed MCC cells, we evaluated the impact of MAL3-101 on MCC cell lines. These experiments revealed apoptosis induction in vitro as well as significant MCC tumor inhibition in vivo in a xenograft murine model. Strikingly, the efficiency of MAL3-101 correlated with HSC70 expression, but did not require the presence of MCPyV LTA in the analyzed cells.
Material and Methods
Ethics statement
The presented work was conducted according to the principles expressed in the Declaration of Helsinki. The generation and characterization of MCC cell lines was approved by the Institutional Review Board of University Hospital Würzburg (sequential study number 124/05). All the animal experiments were approved by the local authorities (government of Unterfranken; animal experiment request AZ: 55.2-2531.01-59/06) according to the legal requirements.
Cell lines, cultures and reagents
The human MCPyV+ cell lines WaGa, BroLi, MKL-1 and MKL-2 as well as the MCPyV- MCC cell lines UISO, MCC13 and MCC26 have been described previously [13], [33]–[35]. In addition, this cell panel was complemented by the melanoma cell line FM88 [36], the T-cell line Jurkat [37] and HaCat keratinocytes [38], primary human keratinocytes and primary human fibroblasts. Primary human keratinocytes were obtained from foreskin and cultured as described [39]. Primary human fibroblasts were isolated from skin biopsies as described by Green et. al [40]. Recently, it was suggested that UISO might be not representative of Merkel cell carcinoma [41].
All MCC cell lines, as well as the FM88 and Jurkat line were grown in RPMI1640 (PAN Biotech, Aidenbach, Germany) supplemented with 10% (v/v) FCS (Biochrom AG, Berlin, Germany), 100 U/ml penicillin, and 100 μg/ml streptomycin (Sigma Aldrich, Munich, Germany), and maintained in a humidified atmosphere with 5% (v/v) CO2.
Trypan-blue exclusion assay
Cell proliferation and viability under MAL3-101 treatment was measured using the trypan blue exclusion assay. After treatment, the cells from each group were stained with 0.4% trypan blue (Sigma Aldrich GmbH) and the number of dye-excluding (living) cells and positively blue stained (dead) cells was counted using a haemocytometer.
Annexin V assay and cell cycle analysis
Apoptosis analysis was performed using the PE Annexin-V Apoptosis Detection Kit (BD Biosciences, Heidelberg, Germany) according to the manufacturer's instructions. Early apoptotic cells are identified as Annexin-V-positive and 7-AAD-negative, while late apoptotic cells are positive for both.
For cell cycle analysis, DNA staining was performed following overnight fixation with ethanol (90%). Cells were then pelleted and resuspended in PBS supplemented with 1% FCS, 0.05 mg/ml propidium iodide (Sigma Aldrich GmbH), and 0.1 mg/ml RNase A (Fermentas GmbH, St. Leon-Rot, Germany). After a one hour incubation at 37°C, cells were measured on a FACSCanto flow cytometer (BD Biosciences). Data were evaluated using FlowJo analysis software (Tree Star, Inc., Ashland, OR, USA).
Real time polymerase chain reaction (PCR) for HSP70 isoforms
Relative expression levels of the HSP70 isoforms were determined by SybrGreen real time PCR applying the comparative ΔΔ−CT method. Total cellular RNA was isolated using the RNAeasy kit (Qiagen, Hilden, Germany) with a subsequent DNaseI digestion step according to the manufacturer's instructions followed by cDNA synthesis with the Superscript II RT First Strand Kit (Invitrogen GmbH, Karlsruhe).
Real time PCR was conducted in the ABI 7500 Fast Real-Time PCR cycler (Applied Biosystems Inc., Foster City, CA, USA). The standard PCR reactions (20 μl) contained 1 μl cDNA and 10 μl 2× SybrGreen I Low Rox Mastermix (Eurogentec GmbH, Cologne, Germany) for detection. The thermal cycling conditions comprised an initial denaturation step at 95 °C for 10 min, followed by 40 cycles of three-step PCR including 15 sec at 95 °C, 60 sec at 60 °C and 30 sec at 95 °C. The CT levels of the investigated HSP70 isoforms were normalized to GAPDH (Δ-CT level). Δ-CT was calculated as CT HSP isoform, sample - CT GAPDH, sample.
Primer pairs for the different HSP70 isoforms were selected from NCI qPrimerDepot (Table 1) (http://primerdepot.nci.nih.gov/).
Table 1. Primer pairs for HSP70 isoform quantification by real-time PCR.
| primer sequence | ||
| HSP70 isoform | Forward | Reverse |
| HSPA1B | 5‘-TGAAGCAGCAAAGAGCTGAA-3‘ | 5‘-GTGGATTAGGGGCCTTTGTT-3‘ |
| HSPA2 | 5‘-AAAGTTTGCTGATGATGGGG-3‘ | 5‘-TCGACAAGTGTCAGGAGGTG-3‘ |
| HSPA4 | 5‘-TACCTGGCTTTTAGCTGCTG-3‘ | 5‘-CGCTAATGAGTATAGCGACCG-3‘ |
| HSPA5 | 5‘-TGATTGTCTTTTGTCAGGGGT-3‘ | 5‘-CACAGTGGTGCCTACCAAGA-3‘ |
| HSPA6 | 5‘-GATAAGTCAGCTGTGACTGTCAGG-3‘ | 5‘-TTATTTGAAGCAGAAGAGGATGAA-3‘ |
| HSPA7 | 5‘-TTCCATGAAGTGGTTCACGA-3‘ | 5‘-TTGACGCTGGTGTCTTTGAG-3‘ |
| HSPA8 | 5‘-TGGAAAACACCCACACAAGA-3‘ | 5‘-TCCTTCGTTATTGGAGCCAG-3‘ |
| HSPA9B | 5‘-ATTGAGCACGGGTCAACTTC-3‘ | 5‘-ATGGCACTTCAGAGGGTACG-3‘ |
| HSPA12A | 5‘-CAGGAATAACGCCTCTGTCC-3‘ | 5‘-CACCACGAGAAATGACTGCT-3‘ |
| HSPA14 | 5‘-ATTCGAGAAGACCCTCCACA-3‘ | 5‘-GCAATGTGTCCAGAGCAAGA-3‘ |
| APG-1 | 5‘-CATTTCCAATGGCTCGAGTT-3‘ | 5‘-CATTGCTGTCGCGAGAAGT-3‘ |
| STCH | 5‘-GAGCCAACATATTCTGGGGA-3‘ | 5‘-GGCTGAAATTGGCAGATACC-3‘ |
| HSPH1 | 5‘-CGGCCATGAAATCTTTTGAA-3‘ | 5‘-AGACCATCGCCAATGAGTTC-3‘ |
| HYOU-1 | 5‘-CTGCTCATTGAAAAGCCCA-3‘ | 5‘-CTGCAGATCCGGGGAGTAG-3‘ |
Xenograft model, tumor induction and treatment protocols
Female NOD.CB17/JHliHsd-Prkdc scid mice were purchased from Harlan Laboratories GmbH (Eystrup, Germany) at the age of 4-6 weeks. All mice were housed under specific pathogen-free conditions according to the animal care guidelines in the Department of Dermatology of the University Hospital Würzburg (Germany).
Tumors were established by injection of 5×106 WaGa cells (mixed 1∶2 with MatriGel, BD Biosciences) in a final volume of 100 μl subcutaneously into the lateral flank. The volume of the tumors was determined by measuring the diameter in two dimensions with a slide gauche and applying the following formula: V = π/6 × a2 × b (a: length; b: width) twice weekly. When the tumors reached a volume of approximately 100 mm3, the mice were randomly divided into two groups (n = 6): i.e., the vehicle control group (mice injected intraperitoneally with 200 μl of PBS/20%DMSO every second day) and the MAL3-101 group (mice injected intraperitoneally with 40 mg/kg MAL3-101 every other day). The mice were closely monitored. On day 38, the experiment was terminated due to tumor size in the control group.
Immunohistochemistry (IHC)
To measure the levels of HSP70 expression in vitro and in vivo, either cultured cells embedded in a bovine plasma/thrombin (5∶1) clot prior to formalin fixation, archived formalin-fixed and paraffin-embedded (FFPE) tumor samples from MCC patients, or FFPE samples from xenografted tumors were used. Antigen retrieval was achieved by incubation of the de-paraffinized sections with Dako Target Retrieval Solution (Dako, Hamburg, Germany), pH 6.0. For inactivation of endogenous peroxidases, slides were incubated with peroxidase blocking solution (Dako, Hamburg, Germany). Subsequently, sections were incubated overnight with the primary antibody (HSPA4/8, 1∶100, mouse monoclonal antibody, clon N27F3-4, Abnova GmbH, Heidelberg, Germany; Cleaved Caspase-3 (Asp175), 1∶1000, rabbit monoclonal antibody, Cell Signaling Technology, Inc., Danvers, MA, USA) in a humidified chamber at 4°C. For detection, the EnVision system-polymer HRP-labelled anti-rabbit or anti-mouse secondary antibodies (Dako) were applied followed by incubation with the Nova Red substrate kit (Linearis, Wertheim, Germany) according to the manufacturer's protocol. Finally, slides were counterstained with Mayer's hematoxylin (Dako).
Statistical analysis
All quantitative assays were performed in triplicate. To evaluate the correlation between HSC70 expression levels and MAL3-101 sensitivity, relative gene expression levels were compared with the percentage of living cells after MAL3-101 treatment. Gaussian distribution of the data was confirmed by using the Shapiro-Wilk normality test. The correlation was determined using Spearman's correlation coefficient. Tumor volumes are expressed as means +/− SD. The related 2-way ANOVA was performed to determine significance of differences in tumor volume between vehicle and MAL3-101 treated animals. The statistical analysis was performed with GraphPad Prism 5.03 software (GraphPad Software, Inc. La Jolla, CA, USA). A value of p < 0.05 was considered significant.
Results
HSC70 is the most prominent HSP70 isoform in MCC cell lines
Since HSP70s are frequently overexpressed in cancer cells, we determined mRNA expression levels of all known human HSP70 genes (excluding pseudo genes) in seven MCC cell lines using SybrGreen based quantitative PCR assays (Fig. 1). To this end, the mean expression of the isoforms HSPA1B, HSPA4, HSPA14, APG1, STCH and HYOU-1 was low (high Δ−CT), although expression varied widely among the different cell lines, in particular for HSPA2. For all other HSP70 isoforms, mRNA expression levels were high, i.e. equal to or above the mRNA expression of the housekeeping gene GAPDH. Notably, HSC70 was the most abundantly expressed mRNA in all MCC cell lines (low Δ−CT) and relative HSC70 transcript levels in the MCC cells were higher than those in all non-transformed control cells (HaCat, primary keratinocytes, primary fibroblasts). To confirm that HSC70 mRNA levels correlated with protein expression, we performed immunohistochemistry with cells of the respective MCC cell lines embedded in a bovine plasma/thrombin clot prior to formalin fixation. This analysis revealed a strong staining for HSC70 in the cell lines WaGa and MCC13 (relative mRNA expression compared to primary fibroblasts, the cell line with the lowest expression: 22.6 and 64.0, respectively, data not shown); however, under the same staining conditions, HSC70 was hardly detectable in the BroLi and MKL-1 cell lines (relative mRNA expression: 4.0 and 3.5, respectively, data not shown) (Fig. 2A). Moreover, we stained patient derived tumor tissues for HSC70 expression, demonstrating its presence in MCC cells in situ as well (Fig. 2B).
Figure 1. HSC70 (HSPA8) is the most prominent HSP70 isoform in MCC cell lines.

Δ−CT levels of the known HSP70 isoforms were determined applying SybrGreen real time PCR (red: MCPyV-positive; blue: MCPyV-negative; black: controls).
Figure 2. HSC70 protein expression in vitro and in vivo.

HSC70 expression was assayed by immunohistochemistry applied to MCC cell lines embedded in a bovine plasma/thrombin clot prior to formalin fixation (A) and FFPE MCC metastasis of a patient (B). The depicted scale bar measures 20 μm.
The HSP70 inhibitor MAL3-101 induces apoptosis in MCC cell lines irrespective of their viral status
HSC70 is required for SV40-mediated cellular transformation through its interaction with the LTA [42]. Therefore HSC70 or other HSP70 isoforms may – in addition to other tumor-promoting abilities – be of particular relevance for MCPyV associated MCC by functioning with the MCPyV LTA. Consequently, we studied the impact of the HSP70 inhibitor MAL3-101 on MCPyV-positive (Fig. 3A) and MCPyV-negative tumor cell lines (Fig. 3B). The MCPyV-positive cohort included four MCC cell lines that all strictly require MCPyV LTA expression for maintenance. The MCPyV-negative cell lines investigated included three putative MCC cell lines, one melanoma cell line (FM88), and one T cell leukemia cell line (Jurkat). In either group, some cell lines responded very sensitively to MAL3-101 and demonstrated loss of viability, whereas others were hardly affected by the inhibitor (Fig. 3). These observations indicate that the presence of the polyoma virus is not critical for sensitivity towards MAL3-101, a notion that is in agreement with the effect of MAL3-101 in small cell lung cancer and myeloma [24], [25]. Nevertheless, five out of seven (72%) MCC cell lines responded to MAL3-101, suggesting that MCC can be targeted by HSC70 inhibition. The Annexin-V assay revealed a phosphatidylserine translocation in a large proportion of sensitive cells after treatment, and DNA degradation was evident (Fig. 4A and B). These data suggest that cell death induced by MAL3-101 is via an apoptotic pathway.
Figure 3. Heterogeneous MAL3-101 sensitivity of MCPyV-positive and MCPyV-negative tumor cell lines.

Cell lines were seeded with 10000 cells in 96-well plates and incubated for 24 h before they were treated for 72 h with the indicated concentrations of MAL3-101. Cell viability was determined by the trypan blue exclusion assay (A, B). Given are mean values (+/- SD) of three independent experiments. FM88 is a melanoma cell line, Jurkat cells are derived from a T-cell leukemia, while all other cell lines have been established from primary or metastatic MCCs. The MCPyV status was determined by real time PCR and by immunohistochemistry for the MCPyV Large T antigen. MCPyV-positive cell lines are grouped in A and C, and MCPyV-negative in B and D. The dashed line indicates an arbitrary threshold (80% viability) allowing the discrimination between MAL3-101 sensitive and resistant cell lines.
Figure 4. MAL3-101 induces apoptosis in cell lines with high HSC70 expression.

(A) DNA staining of MAL3-101 treated WaGa cells (relative HSC70 mRNA level compared to primary fibroblasts: 22.6) reveals a strong increase in the sub-G1 population upon 20 h MAL3-101 treatment. In contrast, treatment of MKL-1 cells (relative HSC70 mRNA level compared to primary fibroblasts: 3.5) did not result in induction of a sub-G1 population. (B) Annexin-V/7AAD staining demonstrates increased early apoptosis (quadrant 3) as well as increased cell death (quadrant 4) in WaGa cells caused by MAL3-101 treatment (17 μM).
Significant correlation of MAL3-101 sensitivity with HSC70 expression
Since the apoptotic response of tumor cells to MAL3-101 was not dependent on the presence of MCPyV, we tested whether MAL3-101 sensitivity would correlate with the level of HSP70 expression. Indeed, the viability of the treated MCC cell lines displayed a significant correlation with HSC70 mRNA expression (p = 0.0433, Spearman R = −0.7029) (Figure 5).
Figure 5. MAL3-101 sensitivity of MCC cell lines correlates with HSC70 expression levels.
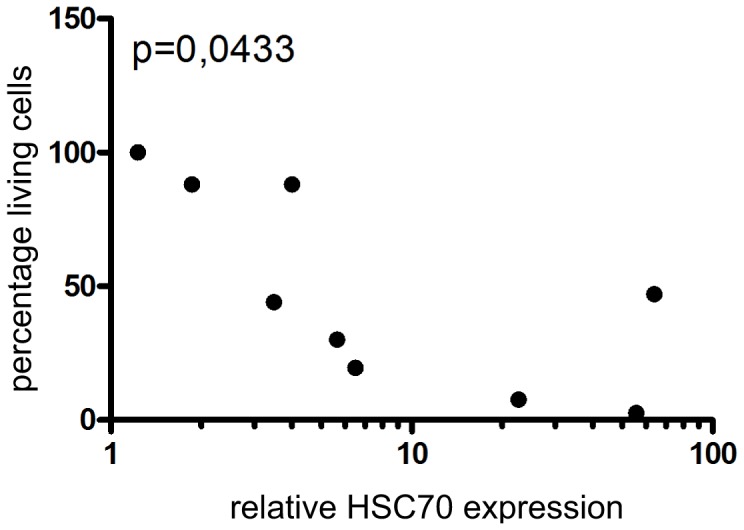
The relative HSC70 mRNA expression levels (with the lowest value arbitrarily set to 1) of the investigated cell lines were blotted against viability following 72 h of treatment with 17 μM MAL3-101. Gaussian distribution of the data was confirmed by using the Shapiro-Wilk normality test. Spearman's correlation coefficient as statistical test was applied.
MAL3-101 induces apoptosis in vivo and inhibits tumor growth
To test whether MAL3-101 can affect MCC in vivo, we used a recently established xenotransplantation model [12]. Following subcutaneous injection of WaGa cells in NOD/Scid mice, animals were treated with the inhibitor once palpable tumors reached a size of approximately 100 mm3. Animals receiving 10 doses of 40 mg/kg MAL3-101 systemically over 21 days showed significantly reduced tumor growth compared to the vehicle control group (Fig. 6A and B). Tumor tissue was stained for cleaved caspase III, revealing that the MAL3-101 treated tumors showed a significantly higher presence of the cleaved form of caspase III compared to the control group. Thus, MAL3-101 also induces apoptosis in sensitive MCC cells in vivo (Figure 6C). Moreover, we could not detect any abnormalities or limitations in the animal behaviour or toxic side effects by MAL3-101 treatment suggesting that systemic delivery of MAL3-101 was well tolerated.
Figure 6. MAL3-101 treatment in an MCC xenotransplantation model demonstrated induction of apoptosis and reduced tumor growth.

WaGa cells embedded in MatriGel were injected s.c. in NOD.CB17-Prkdcscid/NCrHsd mice. Intraperitoneal injection of MAL3-101 (40 mg/kg) was started (arrow) on day 17 when the tumor volume had reached approximately 100 mm3 and was repeated every second day. (A) Mean values (± SEM, N = 6) of the tumor volume are depicted in the graph. The p-value was calculated using the 2-way ANOVA statistical test. (B) Representative macroscopic photographs depict the respective tumors at day 38. (C) IHC for cleaved caspase III in FFPE tumors excised on day 38 indicating caspase III dependent apoptosis induction in the MAL3-101 treated group compared to control group. The depicted scale bar measures 20 μm.
Discussion
The five-year overall survival rate for patients with MCC is 40% and the relative survival rate (compared to an age- and sex-matched population) is 54% [39]. High mortality is largely due to the high propensity even of small primary MCCs to metastasize, and the resulting metastatic disease is treated by various combinations of chemotherapeutics [43]. However, although good initial response rates are achievable, the duration of these responses is generally short and it has not been conclusively demonstrated that any measure improves overall survival; thus, the median survival is less than a year [6], [11]. Therefore, there is a great medical need for alternative therapeutic approaches to treat MCC.
The discovery of a virus as a likely molecular cause for most MCCs will facilitate the rational design of targeted therapeutics. MCPyV was first described in 2008, followed by identification of the viral oncogenes, i.e. the early genes or T antigens, and the demonstration of an oncogene addiction of most MCPyV-positive MCC cell lines to these early genes [13]. Based on this notion and on the assumption that in analogy to the SV40 encoded LTA, MCPyV LTA may also critically depend on the HSP70 activity for interference of Rb-E2F interactions, we wanted to test if HSP70 inhibition would be detrimental for MCPyV-positive MCC cell lines.
HSP70 family members have been suggested as possible targets for therapy of different cancer entities apart from MCC [44]–[46] based on the following observations: HSP70 proteins (i) are overexpressed in cancer cells and/or are induced upon chemotherapy, (ii) possess several cytoprotective and anti-apoptotic functions [47], and (iii) have been demonstrated to be essential for cancer cell survival [19]. In the case of MCpyV-positive MCC, the essential role of HSP70 in the inactivation of the tumor suppressor protein Rb by polyoma LTA [48]–[50] suggests that HSP70 may be particularly crucial for this cancer type.
Here, we demonstrate that treatment with MAL3-101, a specific inhibitor of HSP70 proteins, induces apoptosis in sensitive MCC cells. Indeed, five out of seven MCC cell lines responded to MAL3-101 at low micromolar concentration. Although we cannot unequivocally conclude that survival of the MCC cells is affected by the compounds ability to inhibit HSP70, these results suggest that HSP70 might be a suitable target for therapy of some MCCs and in particular that MAL3-101 is a candidate for treatment of MCC. This notion is further substantiated by the effects of MAL3-101 in a preclinical mouse model for MCC. Specifically, we discovered that MAL3-101 is well tolerated by the mice and leads to significantly reduced growth of the xeno-transplanted MCC tumors. However, not all MCC cell lines were sensitive to the HSP70 inhibitor; conspicuously, the variable response of MCC cell lines to MAL3-101 did not correlate with the virus status of these cells; i.e. both MCPyV-positive and -negative MCC cell lines may be sensitive or resistant to MAL3-101. Instead, the sensitivity of the MCC cell lines towards HSP70 inhibitor appears to correlate with the expression level of HSC70 suggesting that a high expression of this protein results in increased sensitivity to HSP70 inhibition.
The observation that one of the MCPyV-positive MCC cell lines (BroLi) is almost completely insensitive to MAL3-101 may be due to MCPyV LT - at least in this cell line - lacking HSP70 dependency as we have previously demonstrated that this cell line is sensitive to MCPyV T antigen knock down [13]. Until recently, it has only been shown that MCPyV LT binds HSC70 and that an intact HSC70 binding site – which is present in MCPyV sT and LT – is required for T antigen driven viral replication [14]. Whether HSC70 activity is necessary for the ability of MCPyV to inactivate Rb and to support growth of MCPyV-positive MCC cells has not yet been demonstrated. It is worth noting that select functional differences between the MCPyV T antigens and their SV40 homologs have been noted [51]–[53].
It was recently suggested that all MCCs are MCPyV associated [54], putting into question whether the MCPyV-negative cell lines that were all established before the identification of MCPyV are really MCC cells. Indeed, these cells lack expression of many of the MCC markers, in particular of cytokeratin-20 [13]. However, the strongest response towards MAL3-101 among the MCC derived cell lines is observed in two of the virus-positive cell lines. Therefore, irrespective of the question of the identity of the MCPyV-negative MCC derived cell lines, our data suggest that inhibition of HSP70 by small molecule inhibitors such as MAL3-101 may represent a novel therapeutic strategy for the treatment of MCC either alone or in combination with other emerging therapeutic options [11], [55], [56].
Acknowledgments
We thank Claudia Siedel, Susanne Schüler and Lena Völkert for excellent technical assistance and Marc Schmidt for proofreading of the manuscript.
Funding Statement
This study was supported by the Wilhelm-Sander-Stiftung (grant number: 2007.057.3) (http://www.wilhelm-sander-stiftung.de). This publication was funded by the German Research Foundation (DFG) and the University of Würzburg in the funding programme Open Access Publishing. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Howard RA, Dores GM, Curtis RE, Anderson WF, Travis LB (2006) Merkel cell carcinoma and multiple primary cancers. Cancer Epidemiol Biomarkers Prev 15: 1545–1549. [DOI] [PubMed] [Google Scholar]
- 2. Wieland U, Kreuter A (2011) Merkel cell polyomavirus infection and Merkel cell carcinoma in HIV-positive individuals. Curr Opin Oncol 23: 488–493. [DOI] [PubMed] [Google Scholar]
- 3. Agelli M, Clegg LX, Becker JC, Rollison DE (2010) The etiology and epidemiology of merkel cell carcinoma. Curr Probl Cancer 34: 14–37. [DOI] [PubMed] [Google Scholar]
- 4. Albores-Saavedra J, Batich K, Chable-Montero F, Sagy N, Schwartz AM, et al. (2010) Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol 37: 20–27. [DOI] [PubMed] [Google Scholar]
- 5. Duprat JP, Landman G, Salvajoli JV, Brechtbuhl ER (2011) A review of the epidemiology and treatment of Merkel cell carcinoma. Clinics (Sao Paulo) 66: 1817–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schrama D, Ugurel S, Becker JC (2012) Merkel cell carcinoma: recent insights and new treatment options. Curr Opin Oncol 24: 141–149. [DOI] [PubMed] [Google Scholar]
- 7. Davids MS, Charlton A, Ng SS, Chong ML, Laubscher K, et al. (2009) Response to a novel multitargeted tyrosine kinase inhibitor pazopanib in metastatic Merkel cell carcinoma. J Clin Oncol 27: e97–100. [DOI] [PubMed] [Google Scholar]
- 8. Fakiha M, Letertre P, Vuillez JP, Lebeau J (2010) Remission of Merkel cell tumor after somatostatin analog treatment. J Cancer Res Ther 6: 382–384. [DOI] [PubMed] [Google Scholar]
- 9. Meier G, Waldherr C, Herrmann R, Maecke H, Mueller-Brand J, et al. (2004) Successful targeted radiotherapy with 90Y-DOTATOC in a patient with Merkel cell carcinoma. A Case Report. Oncology 66: 160–163. [DOI] [PubMed] [Google Scholar]
- 10. Samlowski WE, Moon J, Tuthill RJ, Heinrich MC, Balzer-Haas NS, et al. (2010) A phase II trial of imatinib mesylate in merkel cell carcinoma (neuroendocrine carcinoma of the skin): A Southwest Oncology Group study (S0331). American journal of clinical oncology 33: 495–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miller NJ, Bhatia S, Parvathaneni U, Iyer JG, Nghiem P (2013) Emerging and mechanism-based therapies for recurrent or metastatic merkel cell carcinoma. Current treatment options in oncology 14: 249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Houben R, Adam C, Baeurle A, Hesbacher S, Grimm J, et al. (2012) An intact retinoblastoma protein-binding site in Merkel cell polyomavirus large T antigen is required for promoting growth of Merkel cell carcinoma cells. International journal of cancer Journal international du cancer 130: 847–856. [DOI] [PubMed] [Google Scholar]
- 13. Houben R, Shuda M, Weinkam R, Schrama D, Feng H, et al. (2010) Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. Journal of virology 84: 7064–7072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kwun HJ, Guastafierro A, Shuda M, Meinke G, Bohm A, et al. (2009) The minimum replication origin of merkel cell polyomavirus has a unique large T-antigen loading architecture and requires small T-antigen expression for optimal replication. Journal of virology 83: 12118–12128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Garrido C, Brunet M, Didelot C, Zermati Y, Schmitt E, et al. (2006) Heat shock proteins 27 and 70: anti-apoptotic proteins with tumorigenic properties. Cell cycle 5: 2592–2601. [DOI] [PubMed] [Google Scholar]
- 16. Gurbuxani S, Schmitt E, Cande C, Parcellier A, Hammann A, et al. (2003) Heat shock protein 70 binding inhibits the nuclear import of apoptosis-inducing factor. Oncogene 22: 6669–6678. [DOI] [PubMed] [Google Scholar]
- 17. Myung JK, Afjehi-Sadat L, Felizardo-Cabatic M, Slavc I, Lubec G (2004) Expressional patterns of chaperones in ten human tumor cell lines. Proteome science 2: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brocchieri L, Conway de Macario E, Macario AJ (2008) hsp70 genes in the human genome: Conservation and differentiation patterns predict a wide array of overlapping and specialized functions. BMC evolutionary biology 8: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nylandsted J, Brand K, Jaattela M (2000) Heat shock protein 70 is required for the survival of cancer cells. Annals of the New York Academy of Sciences 926: 122–125. [DOI] [PubMed] [Google Scholar]
- 20. Young JC, Agashe VR, Siegers K, Hartl FU (2004) Pathways of chaperone-mediated protein folding in the cytosol. Nature reviews Molecular cell biology 5: 781–791. [DOI] [PubMed] [Google Scholar]
- 21. Ravagnan L, Gurbuxani S, Susin SA, Maisse C, Daugas E, et al. (2001) Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nature cell biology 3: 839–843. [DOI] [PubMed] [Google Scholar]
- 22. Rohde M, Daugaard M, Jensen MH, Helin K, Nylandsted J, et al. (2005) Members of the heat-shock protein 70 family promote cancer cell growth by distinct mechanisms. Genes Dev 19: 570–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schmitt E, Maingret L, Puig PE, Rerole AL, Ghiringhelli F, et al. (2006) Heat shock protein 70 neutralization exerts potent antitumor effects in animal models of colon cancer and melanoma. Cancer research 66: 4191–4197. [DOI] [PubMed] [Google Scholar]
- 24. Braunstein MJ, Scott SS, Scott CM, Behrman S, Walter P, et al. (2011) Antimyeloma Effects of the Heat Shock Protein 70 Molecular Chaperone Inhibitor MAL3-101. Journal of oncology 2011: 232037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rodina A, Vilenchik M, Moulick K, Aguirre J, Kim J, et al. (2007) Selective compounds define Hsp90 as a major inhibitor of apoptosis in small-cell lung cancer. Nature chemical biology 3: 498–507. [DOI] [PubMed] [Google Scholar]
- 26. Fewell SW, Smith CM, Lyon MA, Dumitrescu TP, Wipf P, et al. (2004) Small molecule modulators of endogenous and co-chaperone-stimulated Hsp70 ATPase activity. J Biol Chem 279: 51131–51140. [DOI] [PubMed] [Google Scholar]
- 27. Campbell KS, Mullane KP, Aksoy IA, Stubdal H, Zalvide J, et al. (1997) DnaJ/hsp40 chaperone domain of SV40 large T antigen promotes efficient viral DNA replication. Genes Dev 11: 1098–1110. [DOI] [PubMed] [Google Scholar]
- 28. Sheng Q, Denis D, Ratnofsky M, Roberts TM, DeCaprio JA, et al. (1997) The DnaJ domain of polyomavirus large T antigen is required to regulate Rb family tumor suppressor function. Journal of virology 71: 9410–9416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gewirtz DA, Di YM, Randolph JK, Jain PT, Valerie K, et al. (2001) Rb dephosphorylation and suppression of E2F activity in human breast tumor cells exposed to a pharmacological concentration of estradiol. Archives of biochemistry and biophysics 388: 243–252. [DOI] [PubMed] [Google Scholar]
- 30. Kim HY, Ahn BY, Cho Y (2001) Structural basis for the inactivation of retinoblastoma tumor suppressor by SV40 large T antigen. The EMBO journal 20: 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Moens U, Van Ghelue M, Johannessen M (2007) Oncogenic potentials of the human polyomavirus regulatory proteins. Cell Mol Life Sci 64: 1656–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sullivan CS, Cantalupo P, Pipas JM (2000) The molecular chaperone activity of simian virus 40 large T antigen is required to disrupt Rb-E2F family complexes by an ATP-dependent mechanism. Molecular and cellular biology 20: 6233–6243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ronan SG, Green AD, Shilkaitis A, Huang TS, Das Gupta TK (1993) Merkel cell carcinoma: in vitro and in vivo characteristics of a new cell line. Journal of the American Academy of Dermatology 29: 715–722. [DOI] [PubMed] [Google Scholar]
- 34. Shuda M, Feng H, Kwun HJ, Rosen ST, Gjoerup O, et al. (2008) T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proceedings of the National Academy of Sciences of the United States of America 105: 16272–16277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Van Gele M, Leonard JH, Van Roy N, Van Limbergen H, Van Belle S, et al. (2002) Combined karyotyping, CGH and M-FISH analysis allows detailed characterization of unidentified chromosomal rearrangements in Merkel cell carcinoma. International journal of cancer Journal international du cancer 101: 137–145. [DOI] [PubMed] [Google Scholar]
- 36. Bakker AB, Phillips JH, Figdor CG, Lanier LL (1998) Killer cell inhibitory receptors for MHC class I molecules regulate lysis of melanoma cells mediated by NK cells, gamma delta T cells, and antigen-specific CTL. Journal of immunology 160: 5239–5245. [PubMed] [Google Scholar]
- 37. Gillis S, Watson J (1980) Biochemical and biological characterization of lymphocyte regulatory molecules. V. Identification of an interleukin 2-producing human leukemia T cell line. The Journal of experimental medicine 152: 1709–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, et al. (1988) Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol 106: 761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pincelli C, Haake AR, Benassi L, Grassilli E, Magnoni C, et al. (1997) Autocrine nerve growth factor protects human keratinocytes from apoptosis through its high affinity receptor (TRK): a role for BCL-2. J Invest Dermatol 109: 757–764. [DOI] [PubMed] [Google Scholar]
- 40. Green D, Ryan C, Malandruccolo N, Nadler HL (1971) Characterization of the coagulant activity of cultured human fibroblasts. Blood 37: 47–51. [PubMed] [Google Scholar]
- 41. Daily k, Coxon A, Williams JS, Lee C, Coit DG, et al. (2013) UISO cell line is not representative of Merkel cell carcinoma tumors. Journal of Investigative Dermatology 133: S71. [Google Scholar]
- 42. Sawai ET, Butel JS (1989) Association of a cellular heat shock protein with simian virus 40 large T antigen in transformed cells. J Virol 63: 3961–3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tai PT, Yu E, Winquist E, Hammond A, Stitt L, et al. (2000) Chemotherapy in neuroendocrine/Merkel cell carcinoma of the skin: case series and review of 204 cases. J Clin Oncol 18: 2493–2499. [DOI] [PubMed] [Google Scholar]
- 44. Liu T, Daniels CK, Cao S (2012) Comprehensive review on the HSC70 functions, interactions with related molecules and involvement in clinical diseases and therapeutic potential. Pharmacol Ther 136: 354–374. [DOI] [PubMed] [Google Scholar]
- 45. Tutar Y (2011) Hsp70 in oncology. Recent Pat DNA Gene Seq 5: 214–218. [DOI] [PubMed] [Google Scholar]
- 46. Wang RE (2011) Targeting heat shock proteins 70/90 and proteasome for cancer therapy. Curr Med Chem 18: 4250–4264. [DOI] [PubMed] [Google Scholar]
- 47. Schmitt E, Gehrmann M, Brunet M, Multhoff G, Garrido C (2007) Intracellular and extracellular functions of heat shock proteins: repercussions in cancer therapy. Journal of leukocyte biology 81: 15–27. [DOI] [PubMed] [Google Scholar]
- 48. DeCaprio JA (1999) The role of the J domain of SV40 large T in cellular transformation. Biologicals 27: 23–28. [DOI] [PubMed] [Google Scholar]
- 49. Sullivan CS, Tremblay JD, Fewell SW, Lewis JA, Brodsky JL, et al. (2000) Species-specific elements in the large T-antigen J domain are required for cellular transformation and DNA replication by simian virus 40. Mol Cell Biol 20: 5749–5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zalvide J, Stubdal H, DeCaprio JA (1998) The J domain of simian virus 40 large T antigen is required to functionally inactivate RB family proteins. Mol Cell Biol 18: 1408–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cheng J, Rozenblatt-Rosen O, Paulson KG, Nghiem P, Decaprio JA (2013) Merkel cell polyomavirus large T antigen has growth-promoting and inhibitory activities. J Virol 87: 6118–6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Houben R, Dreher C, Angermeyer S, Borst A, Utikal J, et al.. (2013) Mechanisms of p53 Restriction in Merkel Cell Carcinoma Cells Are Independent of the Merkel Cell Polyoma Virus T Antigens. J Invest Dermatol. [DOI] [PubMed]
- 53. Shuda M, Kwun HJ, Feng H, Chang Y, Moore PS (2011) Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J Clin Invest 121: 3623–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rodig SJ, Cheng J, Wardzala J, Dorosario A, Scanlon JJ, et al.. (2012) Improved detection suggests all Merkel cell carcinomas harbor Merkel polyomavirus. J Clin Invest. [DOI] [PMC free article] [PubMed]
- 55. Arora R, Shuda M, Guastafierro A, Feng H, Toptan T, et al. (2012) Survivin is a therapeutic target in Merkel cell carcinoma. Sci Transl Med 4: 133ra156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Willmes C, Adam C, Alb M, Volkert L, Houben R, et al. (2012) Type I and II IFNs inhibit Merkel cell carcinoma via modulation of the Merkel cell polyomavirus T antigens. Cancer research 72: 2120–2128. [DOI] [PubMed] [Google Scholar]