

Abstract
Poly(A) tail length is a readout of an mRNA’s translatability and stability, especially in developmental systems. PolyAdenylation Test (PAT) assays attempt to quickly measure the average poly(A) tail length of RNAs of experimental interest. Here we present sPAT, splint-mediated PAT, a procedure that uses a DNA splint to aid in the ligation of an RNA-tag to the poly(A) tail of an mRNA. In comparison to other PAT methodologies, including ePAT, sPAT is highly sensitive to low-abundance mRNAs, gives a more accurate profile of the poly(A) tail distribution, and requires little starting material. To demonstrate its strength, we calibrated sPAT on defined poly(A) tails of synthetic mRNAs, reassessed developmentally regulated poly(A) tail-length changes of known mRNAs from established model organisms, and extended it to the emerging evolutionary developmental nematode model Pristionchus pacificus. Lastly, we used sPAT to analyze the contribution of the two cytoplasmic poly(A) polymerases GLD-2 and GLD-4, and the deadenylase CCR-4, onto Caenorhabditis elegans gld-1 mRNA that encodes a translationally controlled tumor suppressor whose poly(A) tail length measurement proved elusive.
Keywords: CCR-4, GLD-2, GLD-4, deadenylases, ePAT, gld-1, poly(A) polymerases, poly(A) tail, sPAT
Introduction
The poly(A) tail is a defining feature of mRNAs and its metabolism is intimately linked to mRNA activities. Initially added in the nucleus as a uniform adenosine homopolymer of species-specific tail length, it is subject to further regulation in the cytoplasm, influencing mRNA stability and efficient translation (for recent reviews, see refs. 1 and 2). Deadenylases remove adenosine nucleotides, while poly(A) polymerases promote the addition of adenosines to free 3′ ends. RNA-modifying enzymes regulate specific mRNAs based on their ability to interact with sequence-specific mRNA-binding proteins that interpret the mRNA’s cis-regulatory sequence code (for recent reviews, see refs. 3–5). When transcriptional regulation is either reduced or turned off during development, or too slow and a faster pace of response is required for protein expression, post-transcriptional regulation becomes the dominant form of gene expression regulation and decides an mRNA’s fate.6 In these situations, mRNAs undergo translational inactivation and activation in a spatio-temporally coordinated manner. These waves of translational regulation are often registered in the length of the poly(A) tail of the message and a general trend has been observed: mRNAs with short poly(A) tails are translationally silent or poorly translated; mRNAs with long poly(A) tails are translationally active and efficiently translated (reviewed in refs. 7 and 8).
Accurately measuring and comparing the poly(A) tail lengths of developmental and physiologically relevant mRNAs is essential to our understanding of how biological processes are regulated. Several protocols exist for measuring the length of poly(A) tail length. All of these methods must balance a set of considerations that are often at odds with one another: First, how easy is the assay? Second, how sensitive is the assay? An early technique to measure poly(A) tail length involved isolating total RNA and subsequent northern blotting (e.g., refs. 9 and 10). To allow a better resolution of the poly(A) tail length, the 3′ end of the investigated mRNA is separated from its potentially large 5′ portion by a DNA oligo that hybridizes to the body of the mRNA and serves as a guide for RNase H-mediated cleavage.11 After blotting to a fixed matrix, the 3′ tail is visualized by hybridization with a radio-labeled probe specific to the mRNA tail downstream to the oligo used for RNase H digestion. Poly(A) tail length is assigned based upon size comparison with a radio-labeled DNA or RNA marker. An accurate assignment of poly(A) tail length relies upon knowledge of the site of poly(A) addition; to determine this, a second sample is run in an adjacent lane where an oligo-dT primer in addition to the gene-specific primer was added to the RNase H-treatment protocol. This results in the trimming of both the 5′ end of the message and the poly(A) tail. The strength of this approach is that the measurement of poly(A) tail length is taken directly off harvested RNA, ensuring accuracy of the tail sizes measured. Therefore, it gives a reliable and realistic view of poly(A) tail length distributions based upon the smear visible in the gel lane. However, this approach is labor intensive, technically challenging, and has many time-consuming steps. Importantly, it also requires large amounts of starting RNA, particularly for low-abundance messages that often require large samples of poly(A)-selected mRNA for visualization. Large RNA sample size can be particularly challenging in model organisms in which desirable mutant backgrounds limit source material due to developmental constrictions, such as sterility and lethality.
To combat the time restrictions, technical difficulties, and the requirement for large RNA samples with the RNase H/northern approach, subsequent techniques have been largely based upon polymerase chain reaction (PCR) amplification of cDNA sequences (reviewed in ref. 12). After gel separation, amplified tails are viewed in the gel itself. All of these poly(A) test assays (PATs) have the advantage of reduced experimental steps, rapid generation of results, and a reduction in starting RNA sample size. Here, a caveat is that poly(A) length measurement is not based on directly harvested RNA. Due to many PCR cycles, biases can result in sacrificing some accuracy and reliability in the reproduction of the poly(A) tail distribution for the tested mRNA. RACE-PAT uses a reverse transcription primer consisting of a 5′ PCR anchor sequence followed by an oligo-dT stretch at its 3′ end (hereafter, this type of DNA primer is referred to as an anchor primer) for annealing to the poly(A) tail.13 Ligation-mediated PAT (LM-PAT) includes an initial incubation step of the RNA sample with oligo-dT primers.14,15 The oligo-dT primers anneal to the poly(A) tail, in essence attempting to saturate potential internal binding sites for the anchor primer. After annealing, anchor primer is added to the mix and both the oligo-dT stretches and anchor primer are ligated together using DNA ligase before cDNA synthesis. Nevertheless, RACE-PAT and LM-PAT suffer from internal priming of the anchor primer during cDNA synthesis. When compounded by subsequent rounds of PCR-based amplification favoring the generation of smaller amplicons, this results in a non-representative population of amplicons biased toward small poly(A) tails. This effect is more pronounced for messages with long poly(A) tails and for low-abundance messages requiring a larger number of amplification rounds. The effect of skewed cDNA synthesis seems to be more substantial than those of the PCR amplification itself. Additionally, in LM-PAT, the ligation is not particularly efficient and reduces the size of the cDNA population, requiring larger RNA samples or more rounds of amplification.
Prior to cDNA synthesis, other techniques directly add an RNA extension 3′ to the poly(A) tail; i.e., circularization of mRNA,16,17 poly(G) tailing,18 and direct ligation of an RNA anchor oligonucleotide.19 As such, these techniques are not affected by internal priming of anchor primers to the poly(A) tail and yield a good representation of poly(A) tail distribution in cDNA synthesis. However, the catalytic reactions in these pre-PCR steps are often highly inefficient and involve expensive enzymes. Thus, these methods can require larger sample sizes, prior poly(A)-selection of mRNAs, increased PCR-amplification rounds, the use of radiolabeling, and increased technical complexity.
Recently, extension PAT (ePAT) has added an improved approach to address the trade-off between speed, cost, and accuracy.20 Like RACE-PAT, the RNA sample is mixed with an anchor primer. After annealing, Klenow polymerase is added to perform a fill-in reaction. Klenow will bind to the RNA–DNA duplex, and elongates the RNA with dNTPs, using the DNA anchor primer as a template. This generates RNAs with a 3′ DNA anchor sequence, providing a unique sequence tag at the 3′ end. Reverse transcription is then performed with the initially added anchor primer. To reduce internal priming by the anchor primer, the temperature is raised such that only the primers annealing to the extended 3′ end of the mRNA are likely to be retained. Thus, the initial cDNA population reflects a more realistic demographic than prior techniques. Nevertheless, ePAT still relies upon initial priming of an anchor primer containing dT stretches.
Here, we describe a newly developed poly(A) tail measurement protocol, sPAT. Our protocol incorporates a splint-mediated ligation technique,21 hence its abbreviation. In short, a single-stranded DNA splint serves as a bridge between the mRNA’s poly(A) tail and an RNA anchor tag. In our hands, sPAT is technically simple, requires little RNA sample with no prior poly(A) selection, requires minimal rounds of PCR amplification, and yields a more robust and accurate reflection of poly(A) tail populations with better detection of longer poly(A) tails than previous methods. We compare the sensitivity and accuracy of sPAT to other PAT methods using in vitro transcribed messages of known poly(A) tail length. In addition, we recapitulate and extend observations of messages that are subject to poly(A) metabolism in two well-known model organisms for the study of translational regulation in germ cells (Xenopus laevis and Caenorhabditis elegans). Moreover, we extend the use of sPAT to the evolutionary developmental nematode model Pristionchus pacificus as an indicator of likely translational regulation of the conserved germ cell tumor suppressor-encoding mRNA gld-1. Lastly, we applied sPAT to a developmentally regulated mRNA in C. elegans that has proven to be difficult to study in the past at the level of cytoplasmic poly(A) tail length metabolism.17 We show that the regulation of gld-1 mRNA by two non-canonical PAPs, GLD-2 and GLD-4, and the deadenylase CCR-4 is reflected in distinct changes of poly(A) tail length, which allows us to discriminate the specific functions of each enzyme to influence regulated cytoplasmic poly(A) tail metabolism.
Results
Experimental strategy for single-stranded DNA splint-mediated PAT assay
An overview of the splint-mediated ligation PolyAdenylation Test (sPAT) is depicted in Figure 1A. First, we describe the ligation of the RNA anchor tag sequence. There are two impediments to the ligation of the anchor oligonucleotide to the 3′ end of polyadenylated mRNA. First, the efficiency of the T4 RNA ligase to join two single-stranded RNA molecules is poor. Second, the majority (~99%) of total RNA isolated in a standard RNA extraction protocol is not polyadenylated. Thus, only a very small fraction of anchor oligonucleotide is ligated to polyadenylated mRNA, a much larger fraction is ligated to non-poly(A) tailed RNAs. We overcome these challenges by employing a splint-mediated ligation strategy.21 In brief, a donor RNA anchor with a 5′-monophosphate and an acceptor polyadenylated mRNA with a 3′ hydroxyl (OH) group are bridged via hybridization to a cDNA splint (Fig. 1A and B). To reduce non-specific inter- and intra-molecular ligation of the RNA anchor, a 3′ amino group (NH2) is added to the RNA anchor. Using a thermal cycler, the RNA anchor, DNA splint, and total RNA are incubated together at 70 °C followed by stepwise temperature reductions of 60 °C, 42 °C, 25 °C, and 15 °C. The RNA anchor and DNA splint anneal first in this process creating an RNA–DNA duplex with a nine-thymine nucleotide overhang. Next, as the temperature lowers further, the RNA–DNA duplex anchor anneals to the 3′ end of the poly(A) tail. This creates an exposed nick between the 3′ end of the poly(A) tail and the 5′ end of the RNA anchor (Fig. 1B). In comparison to the ligation of two single-stranded RNA molecules, the nick in the double-stranded complex is efficiently ligated using T4 RNA ligase 2 (Rnl2) overnight at 15 °C22 (Fig. 1B). The following day, DNase I-treatment digests the DNA splint and any residual cellular DNA. This prevents interference due to annealing of genomic DNA to the ligated mRNAs, unspecific amplification due to genomic DNA contamination, and prevents putatively interfering primer dimers caused by the DNA splint. Subsequently, standard phenol/chloroform extraction and ethanol washes remove any residual proteins, small molecule, and salt contaminants. Ultimately, this results in a purer population of mRNAs where a much higher fraction has been efficiently tagged than in more traditional approaches.

Figure 1. The splint-mediated poly(A) tail measurement (sPAT) assay. (A) A graphical representation of the sPAT assay. The starting point is total RNA. To establish a reference for mRNAs without a poly(A) tail, a fraction of the total RNA is hybridized to oligo-dT12-18 and RNase H treated. mRNAs are denoted with a filled circle at the mRNA’s 5′end, a filled box for the open reading frame, and a poly(A) tail of varying sizes in its 3′ end. Both RNA fractions are then separately hybridized to a DNA splint-mediated RNA anchor. The DNA splint positions the mRNA 3′ end and the 5′ phosphate of the RNA anchor for T4 RNA ligase-mediated ligation. The length of the DNA splint’s 5′ overhang increases the likelihood of annealing to mRNA and prevents non-favorable mRNA-to-RNA ligation artifacts. A DNase I treatment removes the splint and the RNA is cleaned up for reverse transcription, using a primer that preferentially hybridizes to the ligated RNA anchor. PCR amplifications are performed to detect the variation of the poly(A) tail on a specific mRNA, using a gene-specific forward primer and an RNA anchor reverse primer, which is identical to the RT primer. To enhance specificity and sensitivity, an additional PCR reaction can be performed using nested primer sets. (B) The RNA anchor and DNA splint combination is designed to support high annealing temperatures. The RNA anchor carries two synthesized modifications: a 5′ phosphorylation to aid ligation and a 3′ amino group to prevent further ligations by T4 RNA ligase 2 (Rnl2). The first 16 nts of the DNA splint are complementary to the last 16 nts of RNA anchor (sequence in capital case). Its remaining nucleotides are nine consecutive thymidines, which anneal to the end of the poly(A) tail forming a nicked double strand duplex of mRNA-3′OH: 5-P-Anchor RNA bridged by the DNA splint. Shorter overhangs as few as five nucleotides have been successfully used in testing analogous combinations.
For poly(A) tail amplification, anchor-tagged mRNAs are reverse transcribed using a unique primer complementary to the RNA anchor at a restrictive temperature of 50 °C to eschew non-specific priming. To ensure amplification of target mRNAs, we settled on an RNA anchor that is 32 nucleotides (nts) in length (Fig. 1B, data not shown), allowing two rounds of nested PCR using first an outer and then an internal anchor-specific primer (Fig. 1A and B), if necessary. Our nested RNA anchor-specific reverse primers are designed to be unique, universal primers that do not readily anneal to known nematode sequences deposited in the databases at NCBI (Fig. 1B). For the gene-specific forward primers, we design primers approximately 100–200 nts upstream of the cleavage and polyadenylation site. The sequential nested PCR steps are shown in Figure 1A (see also Table 1 for all sequences). PCR products are separated on a high-resolution agarose gel and visualized by ethidium bromide staining. The extent of polyadenylation is determined by comparison between RNase H/oligo-dT (RH/dT)-untreated samples to RH/dT-treated samples, where the poly(A) tails of the mRNAs have been trimmed.
Table 1. DNA and RNA oligonucleotides used in this study.
| Oligo ID | Oligonucleotide sequence (5′ → 3′) | Usage |
|---|---|---|
| RNA anchor | 5′-P-CAGCUGUAGC UAUGCGcacc gagucagauc ag-3′-NH2 | sPAT RNA anchor |
| DNA splint | CGCATAGCTA CAGCTGTTTT TTTTT | sPAT DNA splint |
| RT anchor primer | CTGATCTGAC TCGGTGCGCA | sPAT RT and PCR-reverse |
| anchor primer | TGCGCATAGC TACAGCTGTT TT | sPAT PCR-reverse, inner primer |
| ePAT anchor | GCGAGCTCCG CGGCCGCGTT TTTTTTTTTT | ePAT anchor |
| ePAT-i | GCGAGCTCCG CGGCCGCG | ePAT RT and PCR-reverse |
| egg-1-f | TCCCACTAAC AATCTCACCC C | PCR-forward |
| oma-2-f | CCCCATCTCT CGCAGGCTCT | PCR-forward |
| rps-6-f | CAGAAGCGCA ACCAGAAGAT C | PCR-forward |
| gld-1-o | TCTTCCCGGT TTGATGTAGT CATC | PCR-forward, outer primer |
| gld-1-i | AAACCCCTAC CCCTTCCCAA TC | PCR-forward, inner primer |
| gld-1-f | TGCAGTTCCT CTGCTCTCTC C | qPCR-forward |
| gld-1-r | CGTTAGATCC GAGAAGGTTG G | qPCR-reverse |
| Xl-actin type 5-o | CAAATGTTGC AGGTACACCT GT | PCR-forward, outer primer |
| Xl-actin type 5-i | GTATGTTGCC TTAATGTTCT CAC | PCR-forward, inner primer |
| Xl-Eg2-o | CCAGAGATTG GCCGGTGCAA T | PCR-forward, outer primer |
| Xl-Eg2-i | GCTGCTCGAA TACTGAGTGA AT | PCR-forward, inner primer |
| Ppa-gld-1-o | CCCCCATTTC TAGAATGTAC TAT | PCR-forward, outer primer |
| Ppa-gld-1-i | CAACCCGTTC CCCTCTTTCC | PCR-forward, inner primer |
sPAT captures correct poly(A) lengths and shows enhanced sensitivity using in vitro polyadenylated substrates
To demonstrate an enhanced detection sensitivity upon inclusion of splint DNA, we used in vitro transcribed artificial mRNAs with three defined poly(A) tail lengths of A15, A60, and A105 (Fig. 2A). Using these three substrates at total amounts of 100 pg, 10 pg, and 1 pg, we performed a direct ligation PAT assay without DNA splint (Fig. 2A, lanes 3, 6, and 9). A single band of the expected size was detected when 100 pg and 10 pg of substrates were used (lane 3, 6); however, no band was visible with 1 pg (lane 9). By contrast, the inclusion of a DNA splint in sPAT allows the detection of a single band at expected size down to 1 pg (lane 10). No band was generated when only DNA splint was incubated (lanes 2, 5, 8). By comparing the signal intensity of the amplified PCR products between direct ligation PAT and sPAT from the same amount of RNA substrates, we observed stronger intensities with sPAT in all the tests. Judging from the signal intensities, we estimate that the inclusion of DNA splint enhances detection levels by 10–20-fold.
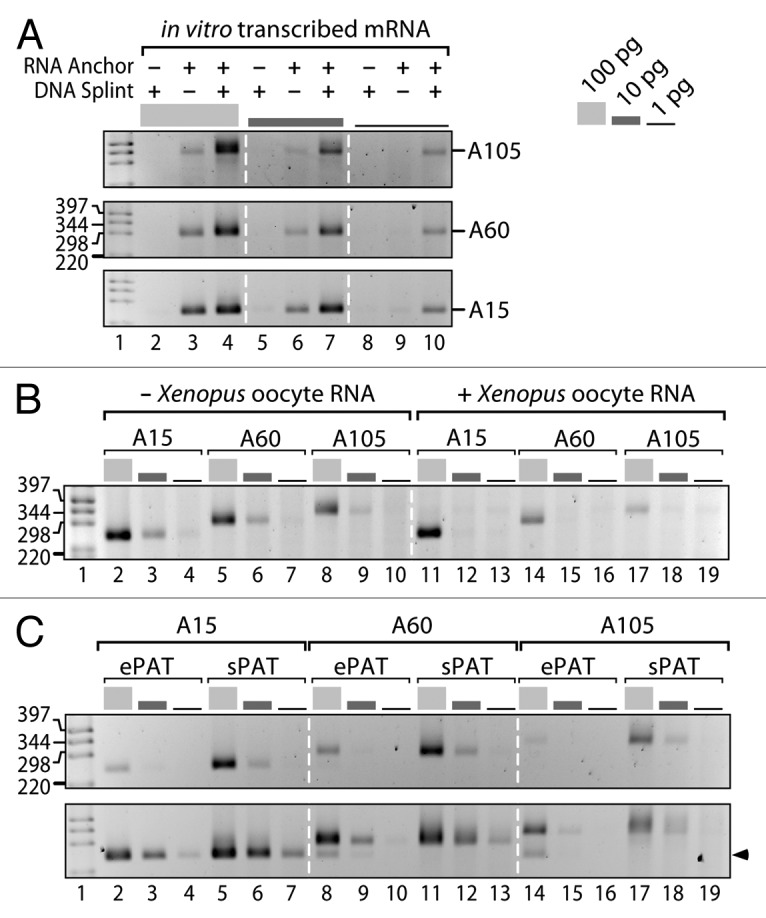
Figure 2. sPAT accurately measures poly(A) tail lengths of artificial mRNAs and requires a small RNA input. (A) The presence of a DNA splint enhances detection levels of artificial mRNAs with defined poly(A) lengths of 105 (top), 60 (middle), and 15 (bottom) adenosines. PAT assays were performed with the given mRNA amounts in the absence or presence of a DNA splint. A single primer pair targeting the 3′UTR and the RNA anchor was sufficient to measure accurately the length of the poly(A) tail; i.e., a second nested PCR was not required. (B) sPAT is accurate and sensitive. It detects accurate poly(A) tail lengths of in vitro transcribed artificial mRNAs in the absence (lanes 2–10) or presence (lanes 11–19) of 1 µg of Xenopus oocyte total RNA. Target mRNA amounts and PCR primer combination as in (A). (C) sPAT is more sensitive than ePAT and recapitulates poly(A) tail lengths without internal priming. PCR products from 22 (top) and 28 (bottom) PCR cycles are displayed. The arrowhead points at an truncated poly(A) tail product that occurred due to an internal priming event of the anchor primer, lanes 8 and 14. All products were verified by Sanger sequencing.
Ribosomal RNA (rRNA) constitutes a major fraction in standard total RNA preparation methods. The presence of overwhelming amounts of rRNA can result in anchor oligos ligating to rRNA as opposed to mRNA, thereby lowering detection levels. As our protocol is designed to preferentially select for ligations to poly(A) tails, we examined whether the presence of complex mixtures of RNA may interfere with the sPAT detection levels. Therefore, we performed sPAT using samples containing the artificial polyadenylated RNAs combined with 1 µg of total RNA from Xenopus laevis oocytes (Fig. 2B). This experimental set up mimics endogenous total RNA composition, as non-specific RNA was present at > 10 000-fold excess in comparison to the artificial target mRNA. Based on the signal intensity of the PCR product, the detection efficiency remains excellent even with the addition of Xenopus total RNA, strong bands comparable to those without the addition of total RNA are observed using 100 pg of target mRNA (Fig. 2B, compare lane 2 vs. 11, 5 vs. 14, 8 vs. 17). Nevertheless, transcript detection was reduced upon addition of Xenopus total RNA for the lower concentrations of 10 and 1 pg of target mRNA (Fig. 2B).
Recently, a novel protocol termed ePAT was described to have a better detection level in comparison with other PAT assays.20 The ePAT technique also tags the 3′ end of an mRNA, and is very similar to the TA-PAT technique.23 In brief, the technique anneals an anchor primer to the poly(A) tail and uses the Klenow DNA polymerase fragment to extend the mRNA’s 3′ end using the anchor primer as a template. We sought to compare the efficiency and accuracy between ePAT and sPAT using our synthetic polyadenylated mRNAs. With a low number of PCR cycles (Fig. 2C, top), ePAT-generated PCR products in the expected sizes from 100 pg of A15, A60, and A105 synthetic mRNAs (lane 2, 8, 14). However, smaller amounts of mRNA were insufficient to visualize amplicons (lane 3, 4, 9, 10, 15, 16). In parallel, sPAT generated both amplicons with stronger intensity from 100 pg samples compared with ePAT (lane 2 vs. 5, 8 vs. 11, and 14 vs. 17) and visible amplicons from less RNA. Thus, in our experiments, sPAT has a superior detection capacity to ePAT. Based on the PCR band intensity, we conclude that sPAT is approximately 10-times more sensitive in detecting our polyadenylated synthetic mRNAs. Using a higher number of amplification rounds, ePAT also produced detectable amplicons using less initial RNA (Fig. 2C, bottom).
ePAT may experience internal priming that we do not detect using sPAT. In our experiments using a higher number of amplification rounds, in addition to the expected PCR amplicon, ePAT produced a second smaller product from our A60 and A105 substrates (Fig. 2C, bottom, arrow). This lower band is comparable in size to the A15 amplicon. The reproducibility of the second band in intensity and size suggested to us a possible internal priming of DNA oligo anchor, which was confirmed by sequencing (data not shown). Internal priming of the anchor oligo can be an issue as higher PCR cycles are biased toward amplification of smaller PCR products. Importantly, we did not observe additional bands in sPAT (lane 11–13, 17–19), confirming that sPAT maintains high fidelity even with increased rounds of PCR amplification. Taken together, using in vitro transcribed artificial mRNAs with a defined poly(A) tail size, we found sPAT is superior to other standard PAT assays, sPAT maintains high specificity to target RNA in a complex mixture of RNA substrates, and sPAT showed higher detection levels compared with ePAT.
Using sPAT to capture in vivo poly(A) tail length in model organisms used for developmental and evolutionary studies
To test sPAT in a traditional in vivo setting, we first chose to look at oocyte maturation in Xenopus laevis. The two meiotic cell divisions in Xenopus are translationally regulated and well characterized. Control of the cell cycle must rely upon post-transcriptional regulation of maternally contributed RNAs in the maturing oocyte as transcription is silenced in the oocyte and early embryo.4,8 Eg2 encodes a kinase required for the phosphorylation of cell cycle regulators.24 Eg2 is present in the oocyte as a protected but silent mRNA with a short poly(A) tail. Upon progesterone treatment of oocytes to induce maturation, the Eg2 mRNA is activated by polyadenylation, i.e., the poly(A) tail is elongated.25 By contrast, the Actin Type 5 mRNA poly(A) tail shortens upon progesterone treatment.25 Hence, prior to RNA extraction, we subjected a fraction of isolated Xenopus oocytes to progesterone. Inclusion of RH/dT-treated RNA samples allow visualization of amplicons from poly(A)-trimmed mRNA (Fig. 3A lanes 3, 6), serving as a baseline by which poly(A) tail length can be measured. The PCR reverse primer has a stretch of four Ts at the 3′ end in order to increase specificity to ensure amplification of poly(A) ligated mRNAs (Table 1). Therefore, a minimum poly(A) tail size from RH/dT-treated samples was expected to have four adenosines. Consistent to the design, we observed four adenosine nucleotides in the sequence read (data not shown). In accordance with the previous studies, sPAT accurately recapitulated poly(A) tail metabolism during oocyte maturation. The poly(A) tail profile for Eg2 mRNA before progesterone treatment gave an upper poly(A) tail length of ~40 nts (Fig. 3A). Twelve hours after progesterone treatment, the poly(A) tail length had shifted upwards in the gel giving upper poly(A) tail lengths of ~80 nts (Fig. 3A). By contrast, the poly(A) tail of Actin Type 5 started as a large smear with upper lengths of ~100 nts (Fig. 3A). Twelve hours after progesterone treatment, the poly(A) tail had shifted downwards with upper poly(A) tail lengths of ~25 nts (Fig. 3A). In summary, sPAT accurately recapitulated the changes in poly(A) tail length due to temporal developmental regulation.

Figure 3. sPAT is versatile, sensitive, and less biased toward shorter poly(A) tails than ePAT. (A) sPAT recapitulates the known poly(A) changes in developmentally regulated mRNAs during Xenopus oocyte maturation. Upon progesterone-induced oocyte maturation, Eg2 mRNA is polyadenylated while Actin Type-5 mRNA is deadenylated. A nested PCR was performed to increase specificity. pA marks heterogeneic poly(A) tail lengths. A4 marks the remaining four nucleotides of adenosines after an RNase H (RH)/dT treatment. (B) Quantification of total RNA obtained from hand-selected C. elegans hermaphrodites. The amount of total RNA is plotted against 50, 100, and 200 animals. Approximately 1 μg of total RNA is extracted from 50 adult animals. (C) Comparison of ePAT and sPAT using three abundant mRNAs from C. elegans, egg-1, oma-2, and rps-6, The total RNA input values vary accordingly: 50 ng (black), 200 ng (dark gray), and 500 ng (light gray). A single PCR reaction of equal cycle number was performed. Labels as in (A). (D) Poly(A) tail length distributions of (C). Signal intensities were quantified by densitometry and displayed using the line-scan function of Fiji (ImageJ). The X axis is poly(A) tail length and the Y axis is the signal intensity shown in arbitrary units (a.u.). Additional labels as in (C).
Our own work focuses on translational regulation of developmentally regulated mRNAs in nematode models. In “worms” as in many systems, experiments are limited by the availability of tissue. This is particularly relevant as mutations in many of the regulators of poly(A) tail length in Caenorhabditis elegans germ cell development often result in sterility.26-29 Thus, collecting homozygous mutant animals en masse is often highly labor intensive. We therefore assessed the number of animals required to generate a total RNA sample sufficient for poly(A) tail-length analysis using sPAT. To this end, we prepared total RNA from 50, 100, and 200 handpicked, wild-type adult hermaphrodites and plotted the number of animals against the extracted total RNA (Fig. 3B). With our RNA extraction method, we obtained approximately 45 ng total RNA per adult hermaphrodite.
As sPAT demonstrated more robust detection of in vitro transcribed poly(A) tails in comparison to ePAT (Fig. 2C), we extended our finding to an in vivo model system using the nematode C. elegans. In accordance with the original ePAT literature,20 we selected two germ cell-enriched mRNAs, egg-1 (Fig. 3C lane 1–7) and oma-2 (lane 8–14). In addition, we chose rps-6 mRNA (lane 15–21) as a representative of a broadly expressed mRNA. Total RNA prepared from an age-matched population of adult C. elegans were subjected to ePAT (Fig. 3C lanes 1–3, 8–10, 15–17) and sPAT (Fig. 3C lanes 4–7, 11–14, 18–21). We tested three different amounts of total RNAs: 50 ng, 200 ng, and 500 ng. In addition, 500 ng of poly(A) tail-trimmed RNA was subjected to sPAT (lanes 7, 14, 21). While ePAT and sPAT produced similar poly(A) tail length profiles, differences in detection levels and the extent of polyadenylation exist. Especially the maximum length and distribution profile appeared biased toward shorter poly(A) tails in several ePAT measurements and depended on the investigated mRNAs and the amount of starting material. Consistently, egg-1, oma-2, and rps-6 mRNA poly(A) tail length was up to ~100, ~50, and ~60 nucleotide long from 200 ng of total RNA, respectively (Fig. 3C). To visualize the differences better, we also compared the two PAT assays by quantifying the intensity of the smears from analogous lanes based on the densitometer profile of electrophoresis gel images (Fig. 3D). In all three RNA amounts tested, sPAT produced stronger signals and detected longer poly(A) tail lengths for egg-1, oma-2, and rps-6 mRNA than ePAT (Fig. 3D). In sum, we found that sPAT performed superior in comparison to ePAT with respect to signal intensity and detection of longer poly(A) tails. Given that the sPAT assay allows us to measure poly(A) tail length from 50 ng of total RNA, investigators are able to concurrently measure poly(A) tail lengths of several mRNAs with repetitions from a very small amount of total RNA prepared from a small number or quantity of animals, cells, or tissues.
To demonstrate the use of sPAT to assess poly(A) tail length in a de novo system, we wanted to characterize a conserved mRNA of metazoans and selected a nematode model that is frequently used for comparative evolutionary developmental studies. We chose the RNA-binding protein-encoding gld-1 mRNA of Pristionchus pacificus (Ppa) (Fig. 4). In C. elegans (Cel), gld-1 mRNA is subject to developmental post-transcriptional regulation via cytoplasmic poly(A) tail metabolism (see below). Using Cel-gld-1, we identified a Ppa-gld-1 homolog based upon initial blast results from NCBI. The full-length cDNA was obtained through RT-PCR of total P. pacificus RNA (Fig. 4A). The protein sequence and domain structure was inferred from the cloned cDNA (Fig. 4B). GLD-1 is a member of the STAR protein family and nematodes contain multiple STAR proteins. Protein alignment and distance analysis placed our protein with other nematode GLD-1 proteins and not with the closest paralogous family of ASD-2 proteins (Fig. 4C). The Ppa-gld-1 cDNA is 2898 nucleotides long and predicted to encode a 553 amino acid long RNA-binding protein (Fig. 4B). The Ppa-gld-1 message is SL1 trans-spliced and as a mature message contains a 134 nucleotide 5′UTR, including the 22 nucleotide SL1 sequence (Fig. 4A). The 3′UTR is very large for a nematode gene, 1102 nt before poly(A) addition, and is even longer than that of Cel-gld-1 mRNA. Large 3′UTRs are often indicative of post-transcriptional control as they provide enough sequence space for cis-regulatory elements.30 In fact, when comparing the Ppa-gld-1 to Cel-gld-1 3′UTR, we notice a conserved stretch of sequence that resembles an FBF-binding element (FBE) that is known in Cel-gld-1 mRNA to confer post-transcriptional regulation (Fig. 4A).31 Unlike Cel-gld-1, Ppa-gld-1 contains two non-canonical polyadenylation sequences with two potential downstream sites for poly(A) addition (Fig. 4A). However, according to our sequencing results, only the most 3′ site is used. This is also in agreement with the more optimal sequence distance of 13 nucleotides between the 3′ most polyadenylation site to its cognate cleavage site (Fig. 4A).

Figure 4. De novo use of sPAT: Investigation of the Pristionchus pacificus gld-1 mRNA. (A) Ppa-1-gld-1 mRNA structure. A putative FBE element in a conserved sequence area to Cel-gld-1 is present in the 3′UTR. Seventeen consecutive cytosine nucleotides (C17) hampered efficient PCR amplification. Therefore, primers (arrow) downstream of C17 were used to detect Ppa-gld-1 poly(A) tail. Arrowheads indicate the position of two alternative polyadenylation sites (PAS), which are indicated in bold in the sequence below. The corresponding cleavage and PAS sites are indicated in bold and capital letters, respectively. (B) A comparison between C. elegans and P. pacificus GLD-1 protein sequences and its functional domains: Q1, Qua1; KH, K homology; gray box, Qua2 domain. (C) A phylogenetic analysis of GLD-1 and Quaking proteins across representative metazoan taxa. Distance based groupings. Ppa-GLD-1 clusters with the GLD-1 proteins from Caenorhabditis species. Dotted lines represent discontinuous branches, e.g., branches with either longer or shorter lengths than represented by the substitution scale shown at the bottom. (D) Ppa-gld-1 mRNA is polyadenylated (lane 5). Four different oligo-dT nucleotides (lane 1 to 4) are incubated to test efficiency of RNase H/dT cleavage. The dT6 shows an incomplete RNase H digestion, likely due to its inefficient annealing to long poly(A) tails. A nested round of PCR was performed.
Next, we used sPAT to measure the poly(A) tail distribution of Ppa-gld-1 mRNAs and observed a very broad smear with upper poly(A) tail lengths of ~150 nucleotides (Fig. 4D, compare lane 5 to lane 1). Lastly, in this experiment, we also tested the effectiveness of different lengths of oligo-dTs to trim the poly(A) tails and found that a size minimum of eight dTs is required for efficient poly(A) tail removal. However, only a stretch of 12 dTs prevented the artificial internal annealing to the five nucleotide-long adenosine stretch between the putative polyadenylation sites (Fig. 4A) and reduced the poly(A) tail to its minimum size of four nucleotides after the 3′ most polyadenylation site, which was confirmed by sequencing (data not shown). Taken together, we conclude that we have identified a P. pacificus homolog of gld-1 and that the molecular features of Ppa-gld-1 mRNA resembles that of Cel-gld-1 mRNA. sPAT efficiently demonstrates that the Ppa-gld-1 mRNA has a substantial poly(A) tail for nematodes, suggesting that Ppa-gld-1 mRNA may also be subject to developmental post-transcriptional regulation.
De novo use of sPAT assays to reveal dynamic changes in the poly(A) tail length as a consequence of cytoplasmic PAP and deadenylation activities
Genetic analysis suggests that poly(A) tail length of Cel-gld-1 mRNA is subject to post-transcriptional regulation during female germ cell development.17,28 However accurate readouts of such regulation through poly(A) tail length-modifying enzymes have been elusive. To investigate the roles of poly(A) addition via GLD-2 and GLD-4 cytoPAPs and removal via CCR-4 deadenylase, we extracted total mRNA from adult animals of wild-type and corresponding mutant backgrounds, and measured poly(A) tail-length distributions of Cel-gld-1 mRNA (Fig. 5).
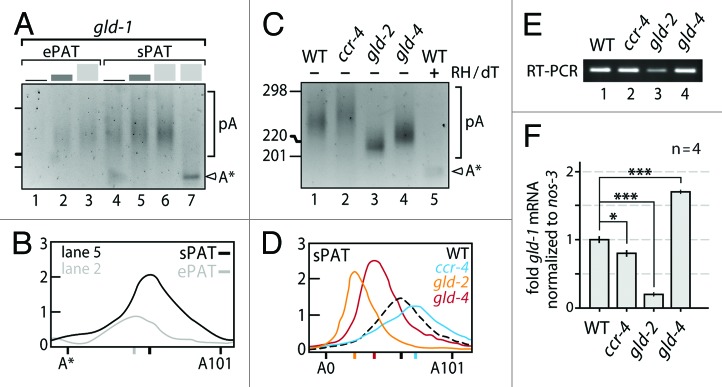
Figure 5. sPAT reveals functional insights into C. elegans gld-1 poly(A) tail metabolism. (A) Comparison of ePAT and sPAT using three total RNA input values: 50 ng (black), 200 ng (dark gray), and 500 ng (light gray). A single PCR reaction of equal cycle number (26x) was performed. Size markers as in (C). RNase H/dT12 cleavage (lane 7) generates a 3′ end truncated gld-1 mRNA (A*) that lacks an additional 14 nts from the original polyadenylation site (see text for details). (B) Example of a poly(A) tail length distributions from (A). Signal intensities of the 200ng RNA input lanes were quantified by densitometry and displayed using the line-scan function of Fiji (ImageJ). The X axis is poly(A) tail length and the Y axis is the signal intensity shown in arbitrary units. The position of each peak is indicated on the X axis. (C) sPAT visualizes poly(A) tail length changes in the absence of 3′ end RNA modifying enzymes. Twenty-four PCR cycles. WT, wild-type animals. Size marker given in nucleotides. Label as in (A). (D) Poly(A) tail length distributions of (C). Label as in (B). (E) Semi-quantitative RT-PCR as a reference of gld-1 mRNA levels among the different genetic backgrounds in B. (F) Normalized RT-qPCR values of gld-1 mRNA levels among the different genetic backgrounds in B. Note that the standard error of the mean is too small to visualize precisely. Students t test: ***, P > 0.001; **, P > 0.01; *, P > 0.05.
First, we compared poly(A) tail distributions of wild-type gld-1 mRNA in ePAT and sPAT assays, using different amounts of total RNAs: 50 ng, 200 ng, and 500 ng (Fig. 5A and B). Across all three concentrations, ePAT was a more sensitive and reflected a well quantifiable heterogeneous range of poly(A) tails. However, we also noticed that the size of the RH/dT-treated sample was somewhat lower than expected. Upon sequencing the ePAT product, we found that gld-1 mRNA possesses 10 nucleotides further upstream of the polyadenylation site an alternative 18 nt-long A-rich sequence stretch (78% adenosines) for a possible hybridization with the dT12 oligo. As a consequence, the mRNA is truncated by an additional 14 nucleotides further inward (Fig. 5, marked as A*), rather than leaving the usual four adenosines attached to the site of polyadenylation. This demonstrates once again that internal priming by oligo dT provides a potential caveat for accurate size measurements not only in amplification steps that use oligo dT as a primer, but also when trimming the poly(A) tail.
Next, we analyzed gld-1 poly(A) tail distributions in wild-type and single mutant animals of cytoplasmic PAPs and deadenylases by sPAT only (Fig. 5C and D). Compared with wild-type, removal of either GLD-2 or GLD-4 cytoPAP activity results in a poly(A) tail distribution skewed to shorter poly(A) tail lengths (Fig. 5C, compare lane 1 to lanes 3 and 4); albeit to different extents. Especially, the lack of GLD-2 has a more pronounced effect on the polyadenylation status of gld-1 mRNA than that of GLD-4 cytoPAP; the average poly(A) tail length is even shorter and its distribution appears more narrow (Fig. 5C and D). By contrast, animals lacking CCR-4 deadenylase activity reveal an extension in the poly(A) distribution of gld-1 mRNA (Fig. 5C, compare lanes 1 and 2). This shift toward slower-moving longer poly(A) tails is clearly eminent in the lane quantifications (Fig. 5D). We conclude that the deadenylase CCR-4 and the two cytoPAPs have opposing effects on gld-1 mRNA’s poly(A) tail length. Moreover, upon loss of either 3′ end-modifying enzyme, distinct poly(A) tail length changes were measured, suggesting that a tight balance between either activity must exist to set a defined poly(A) tail length among the gld-1 mRNA population.
Removal of the poly(A) tail is often the first step to exonucleolytic degradation of mRNAs.32 However, not all RNAs with shortened poly(A) tails are degraded, some are stored in an inactive, protected form for future activation via cytoplasmic polyadenylation.4,7 Two functions have been proposed for GLD-2-mediated poly(A) tail addition; enhancing germ cell mRNA translation,28,33 and the protection and storage of oogenic mRNAs.17,34 To assess these two effects upon the gld-1 mRNA, we measured gld-1 mRNA levels and compared them with the corresponding poly(A) tail profiles. Figure 5E shows the PCR products from semi-quantitative PCR of cDNA from animals with the genetic backgrounds given in Figure 5C. Figure 5F shows relative abundance of the gld-1 mRNA using quantitative PCR and the nos-3 mRNA as a normalization reference. nos-3 is a ubiquitous RNA not known to be translationally regulated in the germ line. Removal of GLD-2 function alone substantially reduces the level of the gld-1 mRNA consistent with the shifted and reduced poly(A) tail profile (Fig. 5F, compare column 1 and 3), which corroborates the previously proposed roles for GLD-2 in protection and storage.17,34 Interestingly, removal of GLD-4 alone gives an intriguing result; while the profile for the poly(A) tails shift to medium lengths, the amount of gld-1 mRNA increases (Fig. 5F, column 4), suggesting that GLD-4 may have a second, cytoPAP-independent role in gld-1 mRNA turnover (Fig. 5F, compare column 3 and 4). By contrast, removal of the CCR-4 activity has a minor, but statistically significant effect on gld-1 mRNA levels (Fig. 5D, column 2). Taken together, our results suggests that gld-1 mRNA turnover is not directly correlated with cytoplasmic poly(A) tail length regulation.
Discussion
sPAT, an improved PAT assay
In this study, we presented a novel protocol, sPAT, to measure poly(A) tail length. The inclusion of DNA splint oligonucleotides greatly enhanced the detection sensitivity and broadened PCR product size, reflecting a distribution of poly(A) tails for a given mRNA of interest to provide a better estimate of upper and median polyadenylation lengths. In our hands, other methods, such as direct ligation of an RNA anchor to RNA without a splint19 and ePAT,20 are more likely to underestimate the extent of an mRNA polyadenylation status. sPAT requires very little starting material to analyze even low abundance messages as demonstrated by mixing picogram amounts of artificial mRNAs with total RNA of X. laevis oocytes. Thus, sPAT provides a robust method to quantify the population of poly(A) tails of many different transcripts from a small amount of RNA sample in a single experiment. Furthermore, in every test we have performed using mRNAs from C. elegans, X. laevis, or with in vitro transcribed mRNAs, sPAT has accurately reproduced or outperformed the results obtained in previous studies.
The enhanced benefits of sPAT over other methods can substantially enhance post-transcriptional regulatory studies. This method allows, for example, full analysis of polysome-associated mRNAs (R. Minasaki and C.R. Eckmann, unpublished results). In addition, we have been able to study poly(A) tail changes of translational reporter mRNAs in transgenic animals; exploiting the nested PCR amplification setup, we were able to discriminate between endogenous mRNA and reporter mRNAs (R. Minasaki and C.R. Eckmann, unpublished results). Lastly, we also propose that variants of the sPAT assay may help in the discovery of different types of 3′ end modification. In recent years, many 3′ end modifications of RNA, such as mono/di/oligo-adenylation or uridylation, have been reported to correlate with RNA stability and functionality.35 For example, by changing the thymine nucleotides of the DNA splint to adenosines, the ligation of 3′ end-uridylated RNAs may be readily detected. Furthermore, it is also possible to vary the length of the single-stranded overhang that assists ligation. To date, we have successfully used also a five-thymidine nucleotide overhang (data not shown), making splint-mediated ligation a very versatile and adjustable, diagnostic tool for studying the extent of 3′ end modifications.
De novo use of sPAT to measure poly(A) tail lengths
We have used sPAT to demonstrate its use in a de novo system in which little is known about poly(A) tail length by looking at the P. pacificus gld-1 homolog. This model nematode is frequently used for comparative evolutionary developmental studies.36,37 It has a well-characterized genome and represents a significant amount of developmental and molecular genetic divergence while still allowing unambiguous comparison of homologous tissues and processes. We chose to characterize the poly(A) tail length of Ppa-gld-1 mRNA due to the broadly conserved nature of GLD-1 protein and the known post-transcriptional regulation of Cel-gld-1 mRNA. GLD-1 protein belongs to the evolutionary conserved STAR (for signal transduction and activation of RNA metabolism) protein family that contains a single maxi hnRNP K homology (KH) domain and two conserved flanking sequences (Qua1 and Qua2 domains) as its molecular hallmark.38 STAR proteins are sequence-specific mRNA-binding proteins implicated in diverse cellular processes, including gametogenesis.39 In C. elegans and in its closely related sister species C. briggsae, GLD-1 is essential to female germ cell development as a tumor suppressor.40 Based upon phylogenetic analysis, we identified a P. pacificus STAR protein that clearly groups with nematode GLD-1 proteins. We have used sequence analysis and sPAT as a mechanism to preliminarily predict this gene may be developmentally regulated at the post-transcriptional level. The Ppa-gld-1 mRNA has an exceptionally long 3′UTR for a worm gene of 1102 nucleotides and exceeds the length of C. elegans gld-1 by 285 nucleotides and contains a potential FBF-binding element; this suggests that translational regulation of the Ppa-gld-1 mRNA is likely and may therefore be conserved. This is an immensely intriguing result given its 300 million year divergence time from C. elegans and the large degrees of developmental genetic divergence observed in studies of the vulva and gonad development.36 Unfortunately, rapid means of efficient gene knockdown is not as straightforward in P. pacificus as C. elegans (i.e., RNAi), thus further testing of this hypothesis awaits meticulous mutagenesis or knockdown analysis beyond the scope of this work.
De novo use of sPAT to monitor poly(A) tail length dynamics
Cytoplasmic poly(A) tail-length metabolism is exemplified during germ cell development. In all sexually reproducing animals, germ cell differentiation starts post-mitotically, with entry into meiotic prophase I, and a concomitant differentiation according to the sexual fate of the germ cell.41 Due to the incompatibility of chromosome packaging rearrangements with effective transcription, many newly required proteins for gametogenesis and embryogenesis are synthesized from mRNAs that are produced during either germline stem cell proliferation or in the early post-mitotic stages of gamete production.42 To control efficient protein synthesis, these mRNAs are subject to translational repression and activation, which has been proposed to correlate with poly(A) tail length changes, mediated by deadenylases and PAPs.43
We used sPAT to gain functional insights into C. elegans gld-1 mRNA as a proxy for developmentally controlled cytoplasmic poly(A) metabolism and post-transcriptional mRNA regulation. Germ cell development in C. elegans depends heavily on translational control.43 In the adult hermaphrodite, female-fated germ cells are born in the proliferative zone and move away to start oogenesis.41 A key developmental regulator of commitment to meiosis is the germ cell-specific STAR protein GLD-1, which translationally represses proliferation and late differentiation genes. GLD-1 protein is poorly expressed in post-mitotic cells but abundantly expressed in cells that enter meiotic prophase.40 This sharp protein gradient is mediated by translational control of its ubiquitous mRNA and is the consequence of translational repressors and activators (Fig. 5B).17,28,31,44 Previous genetic work suggested that gld-1 mRNA stability and translation is negatively influenced by the deadenylase CCR-4.28 By contrast, GLD-1 protein accumulation is promoted by translational activation of gld-1 mRNA through the joint activities of the two cytoplasmic PAPs (cytoPAPs), GLD-2, and GLD-4.28 It has been proposed that this action involves changes in the poly(A) tail length of the gld-1 mRNA.17,28,44 However, convincing data for the individual roles of GLD-2, GLD-4, CCR-4 in gene-specific poly(A) metabolism remained a standing issue.
Our use of sPAT to analyze C. elegans gld-1 mRNA revealed distinct contributions of several 3′ end modifying enzymes, setting gld-1 mRNA’s poly(A) tail length, and govern its stability. While a removal of either cytoPAP caused a shortening of poly(A) tail length, a removal of the deadenylase CCR-4 extended the overall length of gld-1’s poly(A) tail. Thus, in agreement with our previous work that documented a translational enhancement of gld-1 mRNA via two redundantly acting cytoPAPs,28 GLD-2 and GLD-4 individually target the gld-1 message for polyadenylation. Moreover, we verified the predicted role of CCR-4 in gld-1 mRNA deadenylation, and find its moderate influence on gene-specific poly(A) tail reduction consistent with previous bulk poly(A) tail measurements.29 However, sPAT also revealed a yet unknown mechanistic difference between the two cytoPAPs. The contribution of GLD-2 cytoPAP activity on gld-1 poly(A) tail length is greater than that of GLD-4, and gld-1 mRNA stabilization depends on GLD-2 but not on GLD-4 presence. This discrepancy may reflect an additional role of GLD-2 in maintaining mRNAs that possess a shortened poly(A) tail and are kept translationally silent. For example, as an integral component of an mRNA-associated protein complex, GLD-2, may directly or indirectly protect developmentally regulated mRNAs from RNA turnover. Alternatively, GLD-4 may also contribute to enhanced gld-1 mRNA translation via an additional polyadenylation-independent mechanism. Surprisingly, CCR-4 removal resulted in a minor destabilization of gld-1 mRNA, despite a moderate extension of poly(A) tail length. This inverse correlation of poly(A) tail length extension and RNA abundance for the gld-1 mRNA reveals that poly(A) tail metabolism and RNA stability are not strictly connected and suggests that a complex interaction may exists among the enzymes of poly(A) tail metabolism and other RNA fate regulators. Our first foray into the characterization of the activities of cytoplasmic poly(A) polymerases and deadenylases on gld-1 demonstrates that sPAT offers an accurate, sensitive, and reproducible assay to study developmental mRNA regulation. Therefore, sPAT provides an enhanced tool in a laboratories’ repertoire of translational regulation assays and expands the experimental capabilities when the source material is limiting.
Materials and Methods
Animals and tissues used in the study
According to standard culture conditions,45 the following nematode strains were used: Caenorhabditis elegans wild-type N2 strain, and homozygote mutants of gld-2(q497), gld-4(ef15), ccr-4(tm1312); Pristionchus pacificus PS312. Adult C. elegans animals were staged by growing mid-L4 animals for 24 h at 20 °C. Xenopus laevis stage VI oocytes were treated with 10 μg/ml progesterone and harvested 12 h later.
Total RNA preparation and in vitro transcribed artificial mRNAs
All reagents were RNase-free or DEPC-treated. Nematode samples were hand-picked and typically frozen in 50 μl M9 solution. Total RNA was obtained through Trizol-mediated extraction as previously described.46 Artificial mRNAs of defined poly(A) tail length were transcribed for 3 h at 37 °C from a linearized plasmid template that encoded a firefly luciferase open reading frame, possessed the complete 3′UTR of C. elegans gld-1 mRNA, and ended in an adenosine homopolymer stretch of defined length (plasmid sequences are available upon request). The reaction volume of 100 μL contained: 1x transcription buffer (Epicenter), 10 mM DTT, 0.75 mM rATP, rCTP, rUTP, 0.15 mM rGTP, 0.5 mM Gppp-cap, 40 units (U) RNase inhibitor (Ribolock, Fermentas), 1 μg of DNA template, and 5 U T7 RNA polymerase (Epicenter). DNA templates were removed by addition of 20 U DNase I (Roche) for 30 min at 37 °C. The mRNAs were phenol/chloroform extracted, precipitated in a final concentration of 0.5 M NaOAc pH 5.2, 70% ethanol, 20 μg glycogen (Serva), washed with 70% ethanol, and dissolved in RNase-free water. mRNA quality and completeness of ~2200 nucleotides (nts) was assessed by denaturing agarose gel electrophoresis.
Trimming of poly(A) tail
To remove the poly(A) tail, 10 μg of total RNA was incubated with 20 mM TRIS-HCl pH7.8, 40 mM KCl, 8 mM MgCl2, 1 mM DTT, 4 μM oligo-dT12-18 in a reaction volume of 50 μl at 95 °C for 1 min, and annealed at 25 °C for 20 min. Next, 5 U RNase H (Fermentas) and 40 U RNase inhibitor (Ribolock, Fermentas) were added prior to incubating the mixture at 37 °C for 60 min. Poly(A)-trimmed RNA was phenol/chloroform treated, ethanol precipitated, and dissolved in 5 μl RNase-free water. We found that dT12-18 oligonucleotide mixtures efficiently truncate poly(A) tails. The use of dT-oligonucleotides shorter than eight nucleotides leave longer and quite heterogeneous poly(A) tails on mRNAs.
Ligation of RNA anchor oligonucleotide
To allow ligation to the mRNA, but block RNA anchor self-ligation, the anchor was synthesized with the typical 5′ phosphate group and an alternative 3′ amino group in comparison to a hydroxyl group (Metabion or biomers.net). In a 10 μl reaction volume, a mixture of total RNA, 2 μM DNA splint, and 3 μM RNA anchor were annealed in a thermo cycler (MJ Research) incubated at 70 °C for 5 min, 60 °C for 5 min, 42 °C for 5 min, 25 °C for 5 min, before storage at 15 °C. The ligation was performed in a reaction volume of 20 μl with 50 mM TRIS-HCl pH 7.5, 2 mM MgCl2, 1 mM DTT, 400 μM ATP, 10 U RNA ligase 2 (Rnl2, NEB), and 40 U RNase inhibitor (Ribolock, Fermentas) at 15 °C for 16 h. The DNA splint was removed by the addition of a 20 μl master mix that contains 80 mM TRIS-HCl pH7.9, 20 mM NaCl, 12 mM MgCl2, 2 mM CaCl2, 20 U DNase I (Roche), and 40 U Ribolock (Fermentas) at 37 °C for 3 h. The ligated RNA was phenol/chloroform extracted, ethanol precipitated, and dissolved in 20 μl water. The RNA anchor and DNA splint sequences are listed in Table 1.
Reverse transcription
Reverse transcription (RT) was performed in a 20 or 50 μl reaction volume that contains in 0.2 μM RT primer, 0.2 mM dNTPs, 50 mM TRIS-HCl pH 8.3, 50 mM KCl, 4 mM MgCl2, 10 mM DTT, 40 U Ribolock (Fermentas), 10 μl anchor-ligated RNA, and 200 U RevertAid Premium Reverse Transcriptase (Fermentas). The reaction was first incubated at 50 °C for 30 min, and then at 85 °C for 5 min. For an RT-minus control, the same reaction was performed omitting reverse transcriptase.
PCR, agarose gel electrophoresis, sequencing, and desitometric poly(A) tail measurements
The PCR reaction was typically performed in a 20 μl volume containing 50 μM dNTPs, 0.8 μM forward and reverse primers, 1x Phusion HF buffer (Finnzyme), 0.1 U Phusion DNA polymerase (Finnzyme), and 1 μl cDNA with following PCR conditions: 98 °C for 2 min, 18 cycles of 98 °C for 25 s, 60 °C for 25 s, 72 °C for 20 s, and a final extension of 72 °C for 1 min. As indicated, a second PCR reaction was performed identical to the first reaction conditions using 0.5–1 μl of the first PCR product as a DNA template. The entire PCR products were separated on a 3% high-resolution QA agarose gel (MP Biomedicals) in 1x TBE. The agarose gel was stained in 0.4 μg/L ethidium bromide for 15 min and destained with 1 mM MgSO4 for 30 min. Primers used for the PCR analysis are listed in Table 1. Expected PCR amplicons without poly(A) tail are as follows: egg-1 (143 nt for sPAT, 140 nt for ePAT), oma-2 (172 nt for sPAT, 169 nt for ePAT), rps-6 (229 nt for sPAT, 226 nt for ePAT), gld-1 (218 nt for sPAT, 215 nt for ePAT), Xl actin type 5 (143 nt), Xl Eg2 (163 nt), Ppa-gld-1 (199 nt). One μl of each second PCR product was diluted 1:5 and Sanger-sequenced with the forward primers of this PCR reaction. This confirmed the identity of every PCR product and allowed us to assess the extent/length of the poly(A) tail population with single nucleotide resolution. Line scans of the agarose-separated poly(A) tails were generated in ImageJ using the Fiji plug in “line width.” Graphs were processed in Illustrator CS5.1 (Adobe). It should be noted that endogenous poly(A) profiles are heterogeneous and appear as smears in the gel; though we attempt to be precise and equanimous in our interpretation of upper and lower poly(A) tail lengths, the values we state are approximations for comparison purposes. Also, our poly(A) tail length measurements in Figures 3D and 5B and D are inferred from the known sizes of the marker bands after line scanning.
Quantitative PCR (qPCR)
Total RNA was treated with DNase I (Roche) and reverse transcribed as described above. qPCR was performed using an Mx3000P qPCR machine (Stratagene) with ABsolute qPCR SYBR green mix (Thermo Scientific), and analyzed with MxPro qPCR software (Agilent Technologies) as instructed by the manufacturers. The conditions for the qPCR cycles were: initial activation at 95 °C for 15 min followed by 40 cycles of 95 °C for 30 s, 60 °C for 1 min, 72 °C for 30 s. Primers for the qPCR analysis are listed in Table 1.
Protein alignment and phylogenetic analysis
With the exceptions of P. pacificus GLD-1 and ASD-2, full-length proteins for C. elegans GLD-1 (NP_492143.1) and ASD-2 (NP_001021625.1), C. briggsae GLD-1 (XP_002638720.1) and ASD-2 (XP_002640228.1), C. remanei GLD-1 (XP_003100676.1) and ASD-2 (XP_003115053.1), Drosophila melanogaster (fruit fly) HOW-C (NP_001027203.1), Tribolium castaneum (flour beetle) HOW (NP_001164152.1), Danio rerio (zebrafish) Quaking A (NP_571299.1) and B (NP_957136.2), Xenopus laevis (frog) Quaking A (NP_001089857.1) and B (NP_001084987.1), Gallus gallus (chicken) Quaking (NP_989641.2), Mus musculus (mouse) Quaking (AAC99452.1), and human Quaking (AAF63414.1) were obtained from NCBI GenBank. A putative full-length Ppa-ASD-2 protein (PP:PP35693) was identified from WormBase (WS234). The complete Ppa-gld-1 sequence was obtained by RT-PCR of P. pacificus total RNA. Reciprocal blast searches using the Ppa-gld-1 sequence from the non-redundant data set at NCBI identify C. elegans gld-1 and asd-2 sequences as top hits. Protein sequence was inferred from the full-length cDNA. Complete protein sequences were used for alignments to take advantage of specific familial motifs and sequences that are not features common to all STAR proteins, the alignment is trimmed to remove terminal sequences with little homology. In particular, the estimated amino acid sequence for Ppa-GLD-1 is much longer than the other nematode STAR protein sequences. For the alignment, the following amino acid stretches were used for each protein: Ppa-GLD-1 (214–408), Cel-GLD-1 (144–336), Cbr-GLD-1 (144–339), Cre-GLD-1 (147–339), Ppa-ASD-2 (121–316), Cel-ASD-2 (25–219), Cbr-ASD-2 (25–219), Cre-ASD-2 (66–260), fruit fly HOW-C (72–267), tribolium HOW (10–205), zebrafish Quaking A (13–214) and B (12–213), frog Quaking A (12–213) and B (12–214), chicken Quaking (12–213), mouse Quaking (12–213), and human Quaking (34–235). Sequences were aligned using both the Clustal V and W methods in MegAlign version 8.1.4 (7) part of the Lasergene software (DNASTAR). The alignments were used to generate unrooted distance trees in MegAlign; both trees yielded the same topological relationships. The NCBI Genebank ID for Ppa-gld-1 mRNA sequence is KC491214.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Marco Nousch for the DNA templates of defined poly(A) tail lengths, Heino Andreas at the MPI-CBG for Xenopus laevis husbandry, and the MPI-CBG sequencing facility.
Funding
This work was supported by a start up grant from East Carolina University to Rudel D and by the Max Planck Society, and by a grant of the Deutsche Forschungsgemeinschaft DFG (grant numbers EC369-1/2 and EC369-2/1) to Eckmann CR.
Footnotes
Previously published online: www.landesbioscience.com/journals/rnabiology/article/27992
References
- 1.Barckmann B, Simonelig M. Control of maternal mRNA stability in germ cells and early embryos. Biochim Biophys Acta. 2013;1829:714–24. doi: 10.1016/j.bbagrm.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 2.Charlesworth A, Meijer HA, de Moor CH. Specificity factors in cytoplasmic polyadenylation. Wiley Interdiscip Rev RNA. 2013;4:437–61. doi: 10.1002/wrna.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldstrohm AC, Wickens M. Multifunctional deadenylase complexes diversify mRNA control. Nat Rev Mol Cell Biol. 2008;9:337–44. doi: 10.1038/nrm2370. [DOI] [PubMed] [Google Scholar]
- 4.Villalba A, Coll O, Gebauer F. Cytoplasmic polyadenylation and translational control. Curr Opin Genet Dev. 2011;21:452–7. doi: 10.1016/j.gde.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 5.Norbury CJ. Cytoplasmic RNA: a case of the tail wagging the dog. Nat Rev Mol Cell Biol. 2013;14:643–53. doi: 10.1038/nrm3645. [DOI] [PubMed] [Google Scholar]
- 6.Wickens M, Goodwin EB, Kimble J, Strickland S, Hentze MW. Translational Control of Developmental Decisions. In: Sonenberg N, Hershey JW, Mathews MB, eds. Translational Control of Gene Expression: New York, 2000:295-370. [Google Scholar]
- 7.Eckmann CR, Rammelt C, Wahle E. Control of poly(A) tail length. Wiley Interdiscip Rev RNA. 2011;2:348–61. doi: 10.1002/wrna.56. [DOI] [PubMed] [Google Scholar]
- 8.Mendez R, Richter JD. Translational control by CPEB: a means to the end. Nat Rev Mol Cell Biol. 2001;2:521–9. doi: 10.1038/35080081. [DOI] [PubMed] [Google Scholar]
- 9.Fox CA, Sheets MD, Wickens MP. Poly(A) addition during maturation of frog oocytes: distinct nuclear and cytoplasmic activities and regulation by the sequence UUUUUAU. Genes Dev. 1989;3(12B):2151–62. doi: 10.1101/gad.3.12b.2151. [DOI] [PubMed] [Google Scholar]
- 10.Ahringer J, Kimble J. Control of the sperm-oocyte switch in Caenorhabditis elegans hermaphrodites by the fem-3 3′ untranslated region. Nature. 1991;349:346–8. doi: 10.1038/349346a0. [DOI] [PubMed] [Google Scholar]
- 11.Zangar RC, Hernandez M, Kocarek TA, Novak RF. Determination of the poly(A) tail lengths of a single mRNA species in total hepatic RNA. Biotechniques. 1995;18:465–9. [PubMed] [Google Scholar]
- 12.Sallés FJ, Strickland S. Analysis of poly(A) tail lengths by PCR: the PAT assay. Methods Mol Biol. 1999;118:441–8. doi: 10.1385/1-59259-676-2:441. [DOI] [PubMed] [Google Scholar]
- 13.Sallés FJ, Richards WG, Strickland S. Assaying the polyadenylation state of mRNAs. Methods. 1999;17:38–45. doi: 10.1006/meth.1998.0705. [DOI] [PubMed] [Google Scholar]
- 14.Sallés FJ, Lieberfarb ME, Wreden C, Gergen JP, Strickland S. Coordinate initiation of Drosophila development by regulated polyadenylation of maternal messenger RNAs. Science. 1994;266:1996–9. doi: 10.1126/science.7801127. [DOI] [PubMed] [Google Scholar]
- 15.Sallés FJ, Strickland S. Rapid and sensitive analysis of mRNA polyadenylation states by PCR. PCR Methods Appl. 1995;4:317–21. doi: 10.1101/gr.4.6.317. [DOI] [PubMed] [Google Scholar]
- 16.Couttet P, Fromont-Racine M, Steel D, Pictet R, Grange T. Messenger RNA deadenylylation precedes decapping in mammalian cells. Proc Natl Acad Sci U S A. 1997;94:5628–33. doi: 10.1073/pnas.94.11.5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suh N, Jedamzik B, Eckmann CR, Wickens M, Kimble J. The GLD-2 poly(A) polymerase activates gld-1 mRNA in the Caenorhabditis elegans germ line. Proc Natl Acad Sci U S A. 2006;103:15108–12. doi: 10.1073/pnas.0607050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kusov YY, Shatirishvili G, Dzagurov G, Gauss-Müller V. A new G-tailing method for the determination of the poly(A) tail length applied to hepatitis A virus RNA. Nucleic Acids Res. 2001;29:E57–7. doi: 10.1093/nar/29.12.e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rassa JC, Wilson GM, Brewer GA, Parks GD. Spacing constraints on reinitiation of paramyxovirus transcription: the gene end U tract acts as a spacer to separate gene end from gene start sites. Virology. 2000;274:438–49. doi: 10.1006/viro.2000.0494. [DOI] [PubMed] [Google Scholar]
- 20.Jänicke A, Vancuylenberg J, Boag PR, Traven A, Beilharz TH. ePAT: a simple method to tag adenylated RNA to measure poly(A)-tail length and other 3′ RACE applications. RNA. 2012;18:1289–95. doi: 10.1261/rna.031898.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moore MJ. Joining RNA molecules with T4 DNA ligase. Methods Mol Biol. 1999;118:11–9. doi: 10.1385/1-59259-676-2:11. [DOI] [PubMed] [Google Scholar]
- 22.Bullard DR, Bowater RP. Direct comparison of nick-joining activity of the nucleic acid ligases from bacteriophage T4. Biochem J. 2006;398:135–44. doi: 10.1042/BJ20060313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.di Penta A, Mercaldo V, Florenzano F, Munck S, Ciotti MT, Zalfa F, Mercanti D, Molinari M, Bagni C, Achsel T. Dendritic LSm1/CBP80-mRNPs mark the early steps of transport commitment and translational control. J Cell Biol. 2009;184:423–35. doi: 10.1083/jcb.200807033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mendez R, Hake LE, Andresson T, Littlepage LE, Ruderman JV, Richter JD. Phosphorylation of CPE binding factor by Eg2 regulates translation of c-mos mRNA. Nature. 2000;404:302–7. doi: 10.1038/35005126. [DOI] [PubMed] [Google Scholar]
- 25.Charlesworth A, Cox LL, MacNicol AM. Cytoplasmic polyadenylation element (CPE)- and CPE-binding protein (CPEB)-independent mechanisms regulate early class maternal mRNA translational activation in Xenopus oocytes. J Biol Chem. 2004;279:17650–9. doi: 10.1074/jbc.M313837200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kadyk LC, Kimble J. Genetic regulation of entry into meiosis in Caenorhabditis elegans. Development. 1998;125:1803–13. doi: 10.1242/dev.125.10.1803. [DOI] [PubMed] [Google Scholar]
- 27.Eckmann CR, Crittenden SL, Suh N, Kimble J. GLD-3 and control of the mitosis/meiosis decision in the germline of Caenorhabditis elegans. Genetics. 2004;168:147–60. doi: 10.1534/genetics.104.029264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmid M, Küchler B, Eckmann CR. Two conserved regulatory cytoplasmic poly(A) polymerases, GLD-4 and GLD-2, regulate meiotic progression in C. elegans. Genes Dev. 2009;23:824–36. doi: 10.1101/gad.494009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nousch M, Techritz N, Hampel D, Millonigg S, Eckmann CR. The Ccr4-Not deadenylase complex constitutes the main poly(A) removal activity in C. elegans. J Cell Sci. 2013;126:4274–85. doi: 10.1242/jcs.132936. [DOI] [PubMed] [Google Scholar]
- 30.Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K, et al. modENCODE Consortium Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science. 2010;330:1775–87. doi: 10.1126/science.1196914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crittenden SL, Bernstein DS, Bachorik JL, Thompson BE, Gallegos M, Petcherski AG, Moulder G, Barstead R, Wickens M, Kimble J. A conserved RNA-binding protein controls germline stem cells in Caenorhabditis elegans. Nature. 2002;417:660–3. doi: 10.1038/nature754. [DOI] [PubMed] [Google Scholar]
- 32.Houseley J, Tollervey D. The many pathways of RNA degradation. Cell. 2009;136:763–76. doi: 10.1016/j.cell.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 33.Wang L, Eckmann CR, Kadyk LC, Wickens M, Kimble J. A regulatory cytoplasmic poly(A) polymerase in Caenorhabditis elegans. Nature. 2002;419:312–6. doi: 10.1038/nature01039. [DOI] [PubMed] [Google Scholar]
- 34.Kim KW, Wilson TL, Kimble J. GLD-2/RNP-8 cytoplasmic poly(A) polymerase is a broad-spectrum regulator of the oogenesis program Proceedings of the National Academy of Sciences of the United States of America 2010; ; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Minasaki R, Eckmann CR. Subcellular specialization of multifaceted 3’end modifying nucleotidyltransferases. Curr Opin Cell Biol. 2012;24:314–22. doi: 10.1016/j.ceb.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 36.Rudel D, Riebesell M, Sommer RJ. Gonadogenesis in Pristionchus pacificus and organ evolution: development, adult morphology and cell-cell interactions in the hermaphrodite gonad. Dev Biol. 2005;277:200–21. doi: 10.1016/j.ydbio.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 37.Sommer RJ, Bumbarger DJ. Nematode model systems in evolution and development. Wiley Interdiscip Rev Dev Biol. 2012;1:389–400. doi: 10.1002/wdev.33. [DOI] [PubMed] [Google Scholar]
- 38.Vernet C, Artzt K. STAR, a gene family involved in signal transduction and activation of RNA. Trends Genet. 1997;13:479–84. doi: 10.1016/S0168-9525(97)01269-9. [DOI] [PubMed] [Google Scholar]
- 39.Ryder SP, Massi F. Insights into the structural basis of RNA recognition by STAR domain proteins. Adv Exp Med Biol. 2010;693:37–53. doi: 10.1007/978-1-4419-7005-3_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee MH, Schedl T. C. elegans star proteins, GLD-1 and ASD-2, regulate specific RNA targets to control development. Adv Exp Med Biol. 2010;693:106–22. doi: 10.1007/978-1-4419-7005-3_8. [DOI] [PubMed] [Google Scholar]
- 41.Lehmann R. Germline stem cells: origin and destiny. Cell Stem Cell. 2012;10:729–39. doi: 10.1016/j.stem.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seydoux G, Braun RE. Pathway to totipotency: lessons from germ cells. Cell. 2006;127:891–904. doi: 10.1016/j.cell.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 43.Nousch M, Eckmann CR. Translational control in the Caenorhabditis elegans germ line. Adv Exp Med Biol. 2013;757:205–47. doi: 10.1007/978-1-4614-4015-4_8. [DOI] [PubMed] [Google Scholar]
- 44.Suh N, Crittenden SL, Goldstrohm A, Hook B, Thompson B, Wickens M, Kimble J. FBF and its dual control of gld-1 expression in the Caenorhabditis elegans germline. Genetics. 2009;181:1249–60. doi: 10.1534/genetics.108.099440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jedamzik B, Eckmann CR. Analysis of RNA-protein complexes by RNA coimmunoprecipitation and RT-PCR analysis from Caenorhabditis elegans. Cold Spring Harb Protoc. 2009;2009:t5300. doi: 10.1101/pdb.prot5300. [DOI] [PubMed] [Google Scholar]