

Abstract
Immune molecules, including complement proteins C1q and C3, have emerged as critical mediators of synaptic refinement and plasticity. Complement localizes to synapses and refines the developing visual system via C3-dependent microglial phagocytosis of synapses. Retinal ganglion cells (RGCs) express C1q, the initiating protein of the classical complement cascade, during retinogeniculate refinement; however, the signals controlling C1q expression and function remain elusive. Previous work implicated an astrocyte-derived factor in regulating neuronal C1q expression. Here we identify retinal TGF-β as a key regulator of neuronal C1q expression and synaptic pruning in the developing visual system. Mice lacking TGF-β receptor II (TGFβRII) in retinal neurons have reduced C1q expression in RGCs, reduced synaptic localization of complement, and phenocopy refinement defects observed in complement-deficient mice, including reduced eye specific segregation and microglial engulfment of RGC inputs. These data implicate TGF-β in regulating neuronal C1q expression to initiate complement- and microglia-mediated synaptic pruning.
Increasing evidence implicates immune molecules in synapse development and refinement. Several molecules best known for their functions in the immune system, including MHC class I1, neuronal pentraxins2, and complement3, mediate synaptic remodeling in the developing mouse brain, yet surprisingly little is known about the signals regulating the expression and function of these immune molecules at developing synapses.
Classical complement cascade proteins are components of the innate immune system that mediate developmental synaptic pruning, a process critical for the establishment of precise synaptic circuits. Complement proteins, C1q and C3, are expressed in the postnatal brain and localize to subsets of synapses during synaptic remodeling in the mouse retinogeniculate system3--a classic model for studying developmental synapse elimination. Early in postnatal development, retinal ganglion cells (RGCs) form transient functional synaptic connections with relay neurons in the dorsal lateral geniculate nucleus (dLGN). Prior to eye opening (~P14), many of these transient retinogeniculate synapses are eliminated while the remaining synaptic arbors are elaborated and strengthened4–6. C1q−/− and C3−/− mice exhibit sustained defects in synaptic refinement and elimination as shown by the failure to segregate into eye-specific territories and the retention of multi-innervated dLGN relay neurons3. However, the signals regulating complement-mediated pruning during development remain poorly understood.
As the initiator of the classical complement cascade, C1q is a critical point of regulation in this pathway. C1q, a large secreted protein composed of C1qA, C1qB, and C1qC peptide chains, is the recognition domain of the initiating protein, C1, in the classical complement cascade. In the immune system, binding of C1q to apoptotic cell membranes or pathogens triggers a proteolytic cascade of downstream complement proteins, resulting in C3 opsonization and phagocytosis by macrophages that express complement receptors. The function of complement proteins in the brain appears analogous to their immune system function: clearance of cellular material that has been “tagged” for elimination. Consistent with the well-ascribed role of complement proteins as opsonins or “eat me” signals, C1q and C3 localize to retinogeniculate synapses, and presynaptic terminals of retinal ganglion cells are similarly eliminated by phagocytic microglia expressing complement receptors. Genetic deletion of C1q, C3 or the microglia-specific complement receptor, CR3 (CD11b) results in sustained defects in eye-specific segregation7, suggesting that these molecules function in a common pathway to refine synaptic circuits3,7. Importantly, microglial engulfment of retinogeniculate inputs occurs during narrow window of postnatal development (P5–P8) coincident with retinal C1q expression3, suggesting that complement-dependent synaptic pruning may be initiated by C1q in the developing brain.
Remarkably little is known about the signals controlling C1q expression and function in the brain. In contrast to microglia, which express C1q continuously throughout development, C1q expression in RGCs is developmentally restricted to the early postnatal period when RGC axons undergo synaptic pruning3. Given the developmental expression of C1q in RGC neurons, we hypothesize RGC-derived C1q plays a key role in refinement of RGC synapses onto their target neurons in the dLGN. In the current study, we sought to identify the signals regulating C1q expression in RGCs and to determine whether retinal C1q is required to initiate downstream complement-dependent synaptic refinement and microglia-mediated synaptic pruning in the dLGN.
A screen investigating how astrocytes influence neuronal gene expression first identified the C1q genes among the few genes that were highly upregulated in developing RGCs in response to astrocytes3. The presence of immature astrocytes in the retina corresponds to C1q expression in RGCs in vivo, suggesting that the astrocyte-derived factor that upregulates C1q in RGC cultures may also regulate postnatal C1q expression in RGCs in vivo. Each of the three C1q genes (c1qa, c1qb, and c1qc) were found to be highly upregulated in purified RGCs upon exposure to a feeder layer of astrocytes3, implicating a secreted factor; however, the identity of the astrocyte-derived signal(s) that regulate C1q expression has remained unknown. In the immune system, expression of complement and other immune genes can be modulated by rapid cytokine signaling pathways that regulate the inflammatory response. In the developing brain, astrocytes are a major source of cytokines, several of which potently regulate synapse development and function8–11.
In the current study, we identified transforming growth factor beta (TGF-β) as the factor secreted by astrocytes necessary and sufficient for C1q expression in purified RGCs. TGF-β ligands are expressed in the retina during the refinement period and TGF-β receptors (TGFβRII) are developmentally expressed in the postnatal RGCs in vivo. Blocking TGF-β signaling in retinal neurons significantly reduced C1q expression in postnatal RGCs and decreased synaptic localization of complement in the dLGN in vivo. Moreover, specific disruption of TGFβRII in retinal neurons inhibited complement-and microglia-mediated synaptic pruning in the dLGN, suggesting that TGF-β-dependent regulation of neuronal C1q in the retina regulates downstream complement-dependent synapse elimination in the dLGN. Taken together, our data reveal a novel role for the TGF-β cytokine signaling pathway in regulating C1q expression in neurons and in initiating complement-dependent synaptic refinement in the developing CNS.
Results
C1q upregulation by a secreted factor is rapid and direct
Previous findings suggested that an astrocyte-derived factor triggers neuronal C1q expression3. Consistent with previous findings, we found that purified RGC neurons significantly upregulated the C1q genes (c1qa, c1qb, and c1qc) upon chronic exposure (6 days) to cortical astrocytes grown on tissue culture inserts suspended above cultured RGCs (Fig. 1a). To determine if C1q upregulation in RGCs was dependent on indirect, bidirectional signaling between astrocytes and neurons, we measured C1q mRNA levels in purified postnatal (P8) RGC cultures (DIV 4) treated with conditioned medium (ACM) collected from cortical astrocytes. Treating RGCs with ACM or astrocyte inserts resulted in a comparable 10–20 fold upregulation of C1q in RGCs (Fig. 1a), suggesting that an astrocyte-secreted factor directly upregulates neuronal C1q expression. Moreover, ACM collected from purified postnatal (P8) retinal astrocyte cultures12 stimulated a similar upregulation compared to purified or standard cortical astrocytes (Fig. 1a), suggesting that the astrocyte-derived factor that upregulates C1q in RGC cultures may also regulate C1q expression in RGCs in vivo.
Figure 1. C1q is rapidly upregulated in neurons in response to astrocyte-secreted factors.
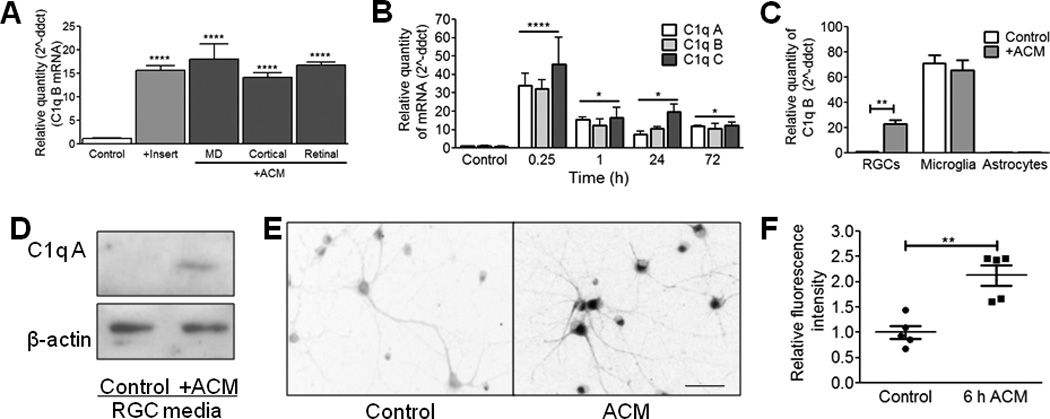
(A) QPCR results for c1qb showed increased c1qb expression relative to controls (RGCs treated with control media) after 6-day treatment with astrocyte insert or astrocyte conditioned medium (ACM). ACM prepared from purified cortical astrocytes (cortical), purified retinal astrocytes (retinal), or astrocytes prepared using the McCarthy and Devellis protocol (MD) all showed a similar upregulation (one-way ANOVA, n=3 experiments, p<0.0001, F(5,12)=75.41). (B) C1q upregulation timecourse for all three C1q genes (c1qa, c1qb, and c1qc) (two-way ANOVA, n=4 experiments,***p<0.001, *p<0.05, F(4,75)=34.37). (C) QPCR results for c1qb showed increased c1qb expression relative to control in RGC cultures but not in microglia or astrocyte cultures after treatment with ACM for 15 min. (two way ANOVA, n=3 experiments, p<0.005, F(2,18)=5.71). (D) Western blot showed an increase in C1q protein in RGC media after 6 days of treatment with ACM. Results shown are representative of 3 experiments. Full length blots are displayed in Supplemental Figure 6. (E) There is a corresponding increase in C1q protein within 6 hours of adding ACM to cultures detected by immunohistochemistry (rabbit anti-mouse C1qA14). Scale bar = 100um. (F) Quantification showed increased C1q levels in ACM treated cultures measured as a change in fluorescence intensity relative to control untreated cultures (t test, n=3 experiments, p<0.01, t(4)=37.48).
Our results support a direct astrocyte-to-neuron signaling pathway in C1q upregulation; however, chronic exposure to astrocytes promotes many developmental changes in neurons, including robust increases in synapse number and neuronal activity9,13. To determine the timecourse for C1q upregulation, we measured C1q mRNA levels by quantitative PCR (qPCR) following ACM treatment (15 min. – 3 days). Surprisingly, RGCs upregulated C1q after only 15 minutes of ACM treatment, further supporting that ACM directly triggers C1q upregulation (Fig. 1b). Moreover, RGCs rapidly upregulated C1r and C1s, the enzymes that associate with C1q to initiate the complement cascade (Supplemental Fig. 1a). This rapid upregulation occurred only in neurons and not in microglia or astrocytes treated with ACM for 15 minutes, suggesting differential regulatory mechanisms in neurons and glia (Fig. 1c). C1q upregulation was blocked by actinomycin, a transcriptional inhibitor, confirming that this effect was a result of transcription (Supplemental Fig. 1b). Furthermore, treating cells with boiled ACM prevented C1q upregulation, implicating a protein in ACM in neuronal C1q upregulation (Supplemental Fig. 1c).
In addition to the upregulation of C1q mRNA, we observed a corresponding increase in C1q protein levels as measured by immunocytochemistry for C1q (Rabbit anti-C1qA14) in purified RGC cultures treated with ACM (6 hours) (Fig. 1e,f) and by western blot analysis of media samples from ACM-treated RGC cultures (1 week) (Fig. 1d). Media samples of ACM alone or from RGCs treated with ACM acutely showed undetectable levels of C1q by western blot (data not shown), indicating that C1q produced by RGCs accumulates in the media over time in response to ACM. Thus, we used this purified culture system as a robust assay to screen for secreted signals that rapidly upregulate and sustain C1q expression in RGCs.
TGF-β is necessary and sufficient for C1q upregulation
To identify the astrocyte-derived factor responsible for C1q upregulation, we screened several candidate molecules for the ability to upregulate C1q in purified RGC neuronal cultures. Cytokines, potent modulators of immune and neural function, were the main class of molecule tested since astrocytes are a major source of cytokines in the brain and several cytokines elicit systemic increases in C1q in the bloodstream. We first measured the levels of several candidate cytokines in ACM by ELISA (MSD mouse 7-plex inflammatory cytokine assay and individual ELISAs for TGF-β1, 2, and 3) (Fig. 2a). Several cytokines were enriched in ACM including CXCL1, IL-12, and transforming growth factor beta (TGF-β) 1, 2, and 3. To determine which cytokines were required for astrocyte-dependent C1q upregulation in RGC neurons, we immunodepleted individual cytokines from ACM using specific antibodies. Two of the cytokines most enriched in ACM, CXCL1 and IL-12, showed no effect on C1q expression when added to cultures as recombinant protein (Supplemental Fig. 1d,e), thus immunodepletion of these cytokines was not performed. Although TNFα and IL-6 induced modest C1q upregulation when added directly to RGC cultures (Supplemental Fig. 1f), immunodepletion of these cytokines using neutralizing antibodies (R&D systems) did not affect ACM-induced C1q upregulation (Fig. 2b). In contrast, specific immunodepletion of TGF-β from ACM using an anti-pan TGF-β neutralizing antibody (1D11, R&D Systems) prevented ACM-induced C1q upregulation after 15 min. treatment as measured by qPCR (Fig. 2b). We verified TGF-β immunodepletion from ACM by ELISA (Fig. 2c). Together these data showed that TGF-β is necessary to upregulate neuronal C1q in purified RGC cultures.
Figure 2. TGF-β is necessary and sufficient for neuronal C1q upregulation in vitro.
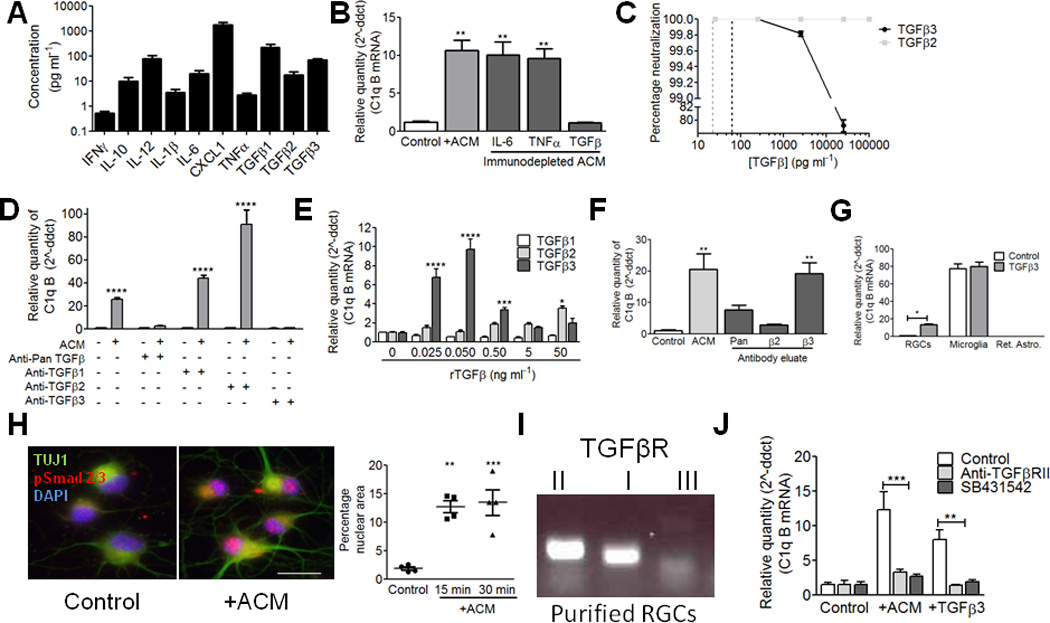
(A) ACM cytokine profiling by ELISA. N=3 independent ACM batches. (B) Immunodepletion of TGF-β, but not other cytokines, significantly reduced ACM-induced C1q upregulation (one-way ANOVA, n= 3 experiments, ***p<0.001, **p<0.01, F(4,12)=22.15). (C) Validation of immunodepletion. %Neutralization represents ([TGF-β]initial – [TGF-β]depletion)/(initial TGF-β concentration). N=3 experiments. (D) Immunodepletion of each TGF-β isoform showed that depletion of pan-TGF-β or TGF-β3 blocked C1q upregulation (two-way ANOVA, n=3 experiments, ***p<0.001, F(4,20)=79.52). (E) Concentration-response curves for TGF-β1, 2, and 3 (Two-way ANOVA, n= 3 experiments, ****p<0.0001, **p<0.01, F(10,36)=23.56). (F) Glycine elution showed that either anti-pan TGF-β or anti-TGF-β3 eluates upregulate C1q (one-way ANOVA, n= 3 experiments, ***p<0.001, F(4,10)=11.02). (G) QPCR results for c1qb in RGC cultures and in microglia or retinal astrocyte cultures after TGF-β3 treatment (50 pg/ml) for 15 min. (two way ANOVA, n=3 experiments, p<0.05, F(2,12)=415.96). (H) RGCs showed increased nuclear accumulation of pSmad2 (15–30 min. ACM treatment). Quantification showed a significant increase in pSmad (red) within the nuclear area (blue) (one-way ANOVA, n= 15 cells/condition, ***p<0.001, F(2,9)=19.83). Scale bar = 20um. (I) RT-PCR for tgfbr1, tgfbr2, and tgfbr3 in P5 retina. Data shown are representative of 4 samples tested. Full length gels are displayed in Supplemental Fig. 6. (J) Blocking TGFβRII signaling with neutralizing antibodies or with inhibitors of TGFβRI significantly reduced the effects of ACM or TGF-β3 (0.05 ng/ml) on C1q (two-way ANOVA, n= 3 experiments, **p<0.01, ***p<0.001, F(4,18)=35.06).
To determine which TGF-β isoform was upregulating C1q, we specifically depleted individual TGF-β isoforms from ACM using antibodies specific to TGF-β1, 2, or 3 (R&D Systems). Interestingly, only ACM depleted of TGF-β3 failed to upregulate C1q in RGCs, suggesting that this isoform may be key in regulating neuronal C1q (Fig. 2d). Furthermore, we generated concentration-response curves (50ng/ml-25pg/ml) in RGC cultures for each of the TGF-β isoforms and measured C1q expression by qPCR. TGF-β3 most effectively upregulated C1q in RGCs at concentrations similar to those measured in ACM, whereas TGF-β2 modestly upregulated C1q at high concentrations (Fig. 2e). In addition, glycine elution to release bound TGF-β from anti-pan or anti-TGF-β3 neutralizing antibodies produced an eluate which could upregulate C1q to the same extent as ACM when added to RGC cultures (Fig. 2f). We also found that retinal astrocytes show enrichment for TGF-β3 compared with RGCs, cortical astrocytes, and microglia (Supplemental Fig. 1g). However, treating retinal astrocyte or microglial cultures with recombinant TGF-β3 (50pg/ml, 15 min.) failed to induce C1q upregulation (Fig. 2g), consistent with our findings with ACM treatment (Fig. 1c). Taken together, these results demonstrate that, of the cytokines tested, TGF-β a major component in ACM responsible for C1q upregulation in RGCs, but not in microglia or astrocytes (Fig. 2g). Furthermore, our findings suggest that TGF-β3 may be the key isoform regulating retinal C1q expression (Fig. 2d,f).
We next addressed whether TGF-β receptor signaling in RGCs is required for C1q upregulation in RGCs. In the canonical TGF-β signaling pathway, TGF-β1, 2, or 3 binds to TGFβRII, which then phosphorylates TGFβRI. Once activated, TGFβRI phosphorylates a Smad transcription factor to alter gene expression. A third receptor, TGFβRIII, can also associate with TGFβRII and alters ligand affinity15. Purified RGCs express all of the components required for TGF-β signaling (Fig. 2h,i). Furthermore, treating RGCs with ACM stimulated rapid (15–30 min.) nuclear accumulation of phosphorylated Smads (pSmad 2/3), as measured by immunocytochemistry for phospho-Smad (Fig. 2h), consistent with the timing of C1q upregulation. In addition, transcripts for TGFβRII, TGFβRIII, and TGFβRI were present in purified RGCs (Fig. 2i). These data indicate that RGCs express functional TGF-β receptors that can be activated with a timecourse consistent with C1q upregulation.
To determine whether the TGF-β receptors, I and II, were responsible for this upregulation, we pre-incubated RGC cultures with specific TGFβRI and II inhibitors prior to acute treatment with either TGF-β3 (50 pg/ml) or ACM. Adding TGFβRII neutralizing antibodies (R&D Systems) or a TGFβRI-specific inhibitor16 (SB431542, Tocris, 30µM) blocked both TGF-β-induced and ACM-induced C1q upregulation (Fig. 2j). Thus, signaling through TGFβRI and II is required for ACM-induced C1q upregulation in purified RGCs.
TGF-β regulates developmental C1q expression in the retina
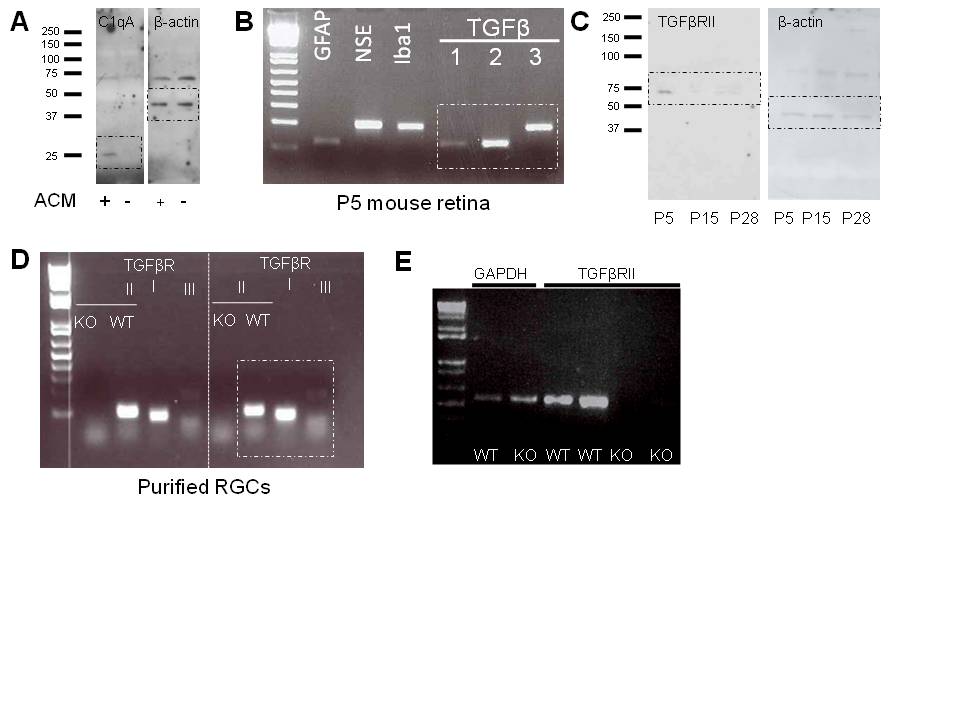
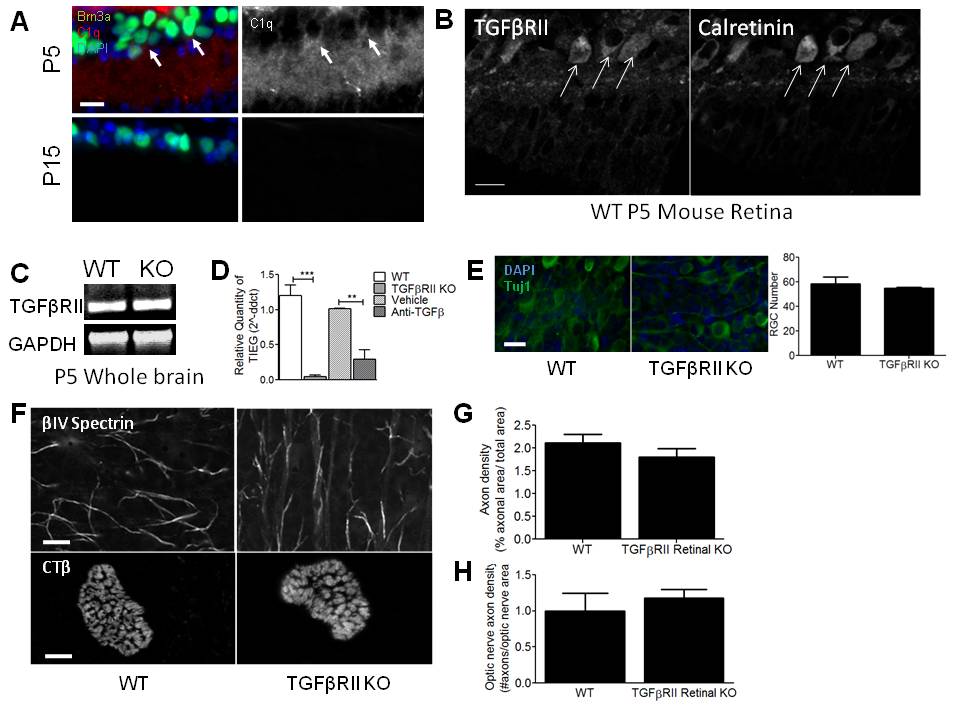
C1q is developmentally regulated in the postnatal retina, showing peak expression around P5 and sharply decreasing by P10–P153, corresponding to the synaptic refinement period in the retinogeniculate system and the presence of immature astrocytes throughout the developing CNS (Supplemental Fig. 2a). To determine if TGF-β signaling regulates developmental C1q expression in RGCs in vivo, we examined the expression of TGF-β signaling components during the refinement period. Expression of all three isoforms of TGF-β was detected by RT-PCR at P5 and in the mature retina (Fig. 3a). In addition, TGFβRII protein was detected in retinal lysates by western blot (Fig. 3b). Of the three ages examined, the level of TGFβRII protein was highest at P5 and sharply decreased by P15 and P28 (Fig. 3b,c), coinciding with C1q expression in the postnatal retina (Supplemental Fig. 2a). Consistent with western blot analysis, immunohistochemistry for TGFβRII showed TGFβRII localization to the RGC and inner plexiform layers (IPL) of the retina at P5 and TGFβRII levels decreased by P15 (Fig. 3d). Co-staining with anti-calretinin (Millipore), a marker of a subset of RGCs, confirmed TGFβRII localization to RGCs (Supplemental Fig. 2b). Taken together, these data show that TGF-β ligands and receptors are present at the right time in the retina to regulate C1q expression.
Figure 3. TGF-β expression corresponds to synaptic refinement period in the retinogeniculate system.
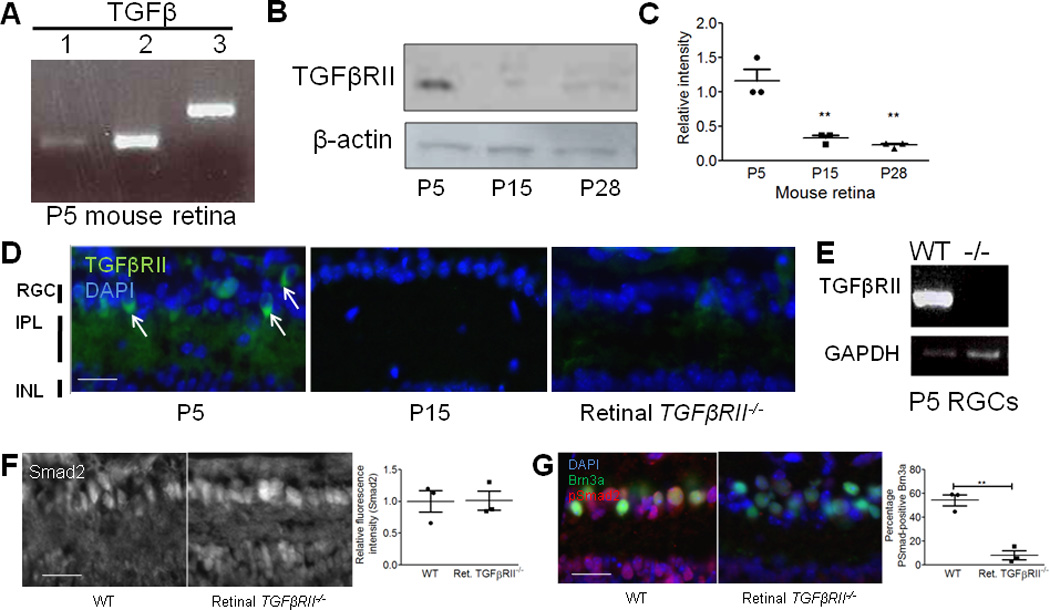
(A) RT-PCR showed expression of all three TGF-β isoforms in the P5 mouse retina. Data are representative of 4 mice. (B) Western blot for TGFβRII (goat anti-human TGFβRII, R&D systems) showed developmental expression of TGFβRII in the mouse retina. See Supplemental Fig. 6 for full length blot. (C) Relative intensity quantification normalized to beta-actin control for each age showed developmental TGFβRII expression in the postnatal mouse retina (one-way ANOVA, n=3 experiments, **p<0.01, F(2,6)=26.36). (D) Immunostaining with antibodies against TGFβRII (R&D systems, goat anti-TGFβRII) showed that the receptor localizes to the RGC layer and the IPL (arrows) and that staining intensity is dramatically reduced at P15 relative to P5. Antibody staining was confirmed for specificity by staining retinal TGFβRII−/− mice. All images were obtained with set exposure times. Scale bar = 50um. (E) RT-PCR confirmed the absence of tgfbr2 mRNA in RGCs acutely isolated from P5 mice using immunopanning. Data shown are representative of 4 animals tested. Full length gel in Supplemental Fig. 6. (F) Immunohistochemistry for total Smad2 showed no difference in relative fluorescence intensity (RGC layer) in WT littermates and retinal TGFβRII−/− mice (t test, n=3 mice/genotype, p=0.96 (ns), t(4)=0.053). Scale bar = 50µm. (G) Immunohistochemistry for phosphorylated Smad (pSmad). Co-staining for an RGC marker, Brn3a, and pSmad2/3 showed a significant reduction in pSmad levels quantified in RGCs specifically (t test, n= 3 mice/group, p<0.001, t(4)=13.18). Scale bar = 50µm.
To determine if TGF-β signaling regulates C1q expression in vivo during retinogeniculate refinement, we used genetic and pharmacological approaches to block TGF-β signaling and then measured C1q expression in vivo. Retina-specific TGFβRII−/− mice were generated by crossing floxed TGFβRII mice with CHX10-Cre mice that express Cre in all retinal neurons at E13.517,18. TGFβRII knockout was confirmed by immunostaining retinas (Fig. 3d) and performing RT-PCR using mRNA from WT and transgenic P5 whole brain (Supplemental Fig. 2c) and purified RGCs acutely isolated from P5 WT and transgenic mice (Fig. 3e). TIEG, a known TGF-β-dependent gene, also showed reduced expression in P5 acutely isolated RGCs (Supplemental Fig. 2d). Moreover, while total Smad2 levels were not significantly different between WT and retinal TGFβRII−/− mice (Fig. 3f), pSmad 2/3 levels, corresponding to active TGF-β signaling, were reduced in Brn3a-positive RGCs (Fig. 3g), further indicating that the TGFβRII retina-specific knockout was achieved. Since TGF-β signaling has many roles in development, we measured cell numbers, axon density, and axon caliber and found no significant difference from WT retinas (Supplemental Fig. 2e–h). Given that there were no gross abnormalities in retinal development, we used this mouse to analyze the role of TGF-β signaling in C1q regulation and synaptic refinement.
To determine if retinal TGF-β signaling is required for developmental C1q expression in RGCs, we assayed C1q expression levels in retinas and RGCs from retinal TGFβRII−/− mice relative to WT littermates, and from mice receiving intraocular injection of anti-TGF-β relative to vehicle treated littermates. In situ hybridization for c1qb revealed a decrease in signal for c1qb in the RGC layer in retinal TGFβRII−/− retinas (P5) compared to WT littermates (Fig. 4a; anti-TGF-β: Supplemental Fig. 3a). To verify reduction in C1q expression was in RGCs and not other cell types in the RGC layer, we acutely isolated RGCs and microglia from P5 retinas using established immunopanning techniques12,19. Acutely isolated RGCs from the retinal TGFβRII−/− showed a significant reduction in C1q expression by qPCR while acutely isolated retinal microglia showed no change in C1q levels (Fig. 4b), as expected given that microglia do not express the retinal neuron-specific gene, chx10. Similarly, RGCs acutely isolated from P5 mice receiving intraocular injection of anti-TGF-β (injected at P3) also showed significantly reduced C1q expression, while microglia again showed no change in C1q (Supplemental Fig. 3b). We verified cell purity after immunopanning using qPCR for the neuron-specific gene, nse, and the microglia-specific gene, iba1 (Supplemental Fig. 3c). Moreover, immunohistochemistry for C1q at P5 in retinal TGFβRII−/− (Fig. 4c,d) and anti-TGF-β injected mice (Supplemental Fig. 3d,e) also showed a 40–50% reduction in C1q immunostaining in the IPL and RGC layers of the retina, but showed no change in microglial staining. Taken together, these data show that TGF-β signaling is required for postnatal C1q expression in RGCs. These results also suggest that TGF-β regulation of C1q may be unique to neurons, as suggested by our earlier in vitro data (Fig. 1c).
Figure 4. TGF-β signaling is required for neuronal C1q expression in vivo.
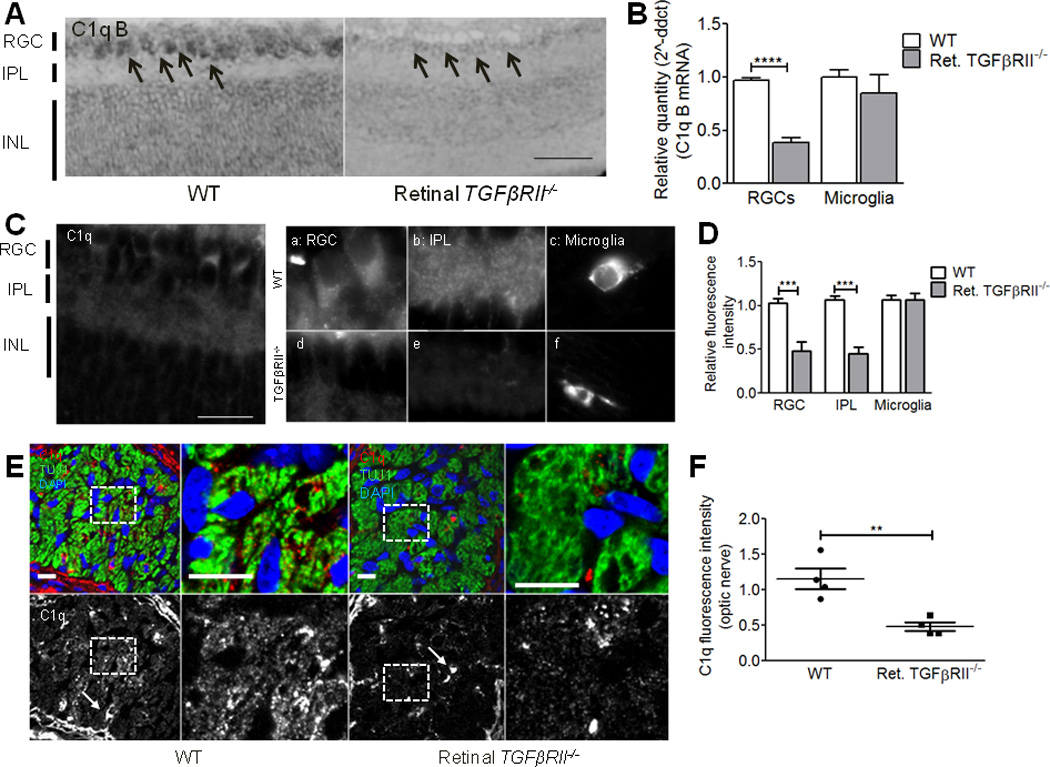
(A) In situ hybridization for c1qb showed significantly reduced C1q expression in the RGC layer (arrows) in retinal TGFβRII−/− mice. Scale bar = 100µm. (B) RGCs acutely isolated from P5 WT (white bar) and retinal TGFβRII−/− (grey bar) retinas using immunopanning showed significantly reduced C1q expression. Microglia acutely isolated using CD45 immunopanning showed no significant difference in C1q levels (two-way ANOVA, n= 5 mice/group, **p<0.01, F(1,16)=19.11). (C) Immunostaining for C1q in P5 retina. (Scale bar = 50µm). In retinal TGFβRII−/− mice, C1q localization to the RGC layer (inset a vs. d), the IPL (b vs. e) is reduced relative to WT animals. Retinal microglia showed no change in C1q levels (inset c and f). (D) Quantification of the relative fluorescence intensity in each retinal area in WT littermates and retinal TGFβRII−/− mice showed significantly reduced C1q localization to the RGC layer and IPL when TGF-β signaling was blocked (two-way ANOVA, n= 4 mice/group, *p<0.05, F(2,12)=11.69). (E) Immunohistochemistry for C1q in optic nerve cross sections showed C1q-immunopositive puncta co-localized with RGC axon fascicles labeled by TUJ1 (Green). C1q levels were significantly reduced in the retinal TGFβRII−/− mouse within the axon, as indicated by co-localization with TUJ1. C1q labeling of microglia remained unchanged (arrows). Scale bar = 10µm. (F) Quantification of the fluorescence intensity for C1q staining with axon bundles shows a significant decrease in C1q levels (t test, n=3 mice (two nerves per mouse), p=0.0055, t(6)=4.225).
Retinal TGF-β regulates complement levels in the thalamus
C1q and C3 localize to synapses in the dLGN during the period of synaptic refinement, but whether RGC neurons are a key source of C1q in the dLGN is not known. One possibility is that neuronal C1q is locally secreted from RGC axons into the dLGN to initiate complement-dependent synaptic pruning. Alternatively, microglia, although sparse in the dLGN at this time, may be a primary source of secreted C1q in the postnatal dLGN during refinement. To address this question, we examined C1q protein levels by immunohistochemistry in cross sections of postnatal (P5) optic nerve. We found that C1q protein could be detected in optic nerve fiber bundles, identified by TUJ1 staining. Moreover, C1q immunoreactivity in the optic nerve was significantly reduced in retinal TGFβRII−/− mice versus WT littermates (Fig. 4e,f), suggesting that TGF-β signaling in RGCs regulates the levels of C1q in RGC axons in the optic nerve.
If C1q in the dLGN is supplied by RGCs, we predicted that retinal TGFβRII−/− mice would have a reduction in C1q protein levels throughout the dLGN. Indeed, immunohistochemisy for C1q revealed a significant decrease in the intensity of C1q staining in the dLGN but not in primary visual cortex (V1) (Fig. 5a), which is not a direct target for RGCs. Quantification showed a significant reduction in fluorescence intensity in the retinal TGFβRII−/− as well as a significant decrease in the density of C1q puncta in the dLGN but not in V1 (Fig. 5b,c), indicating that RGC axons are a key source of C1q in the postnatal dLGN.
Figure 5. Retinal TGF-β signaling is required for complement localization in the dLGN.
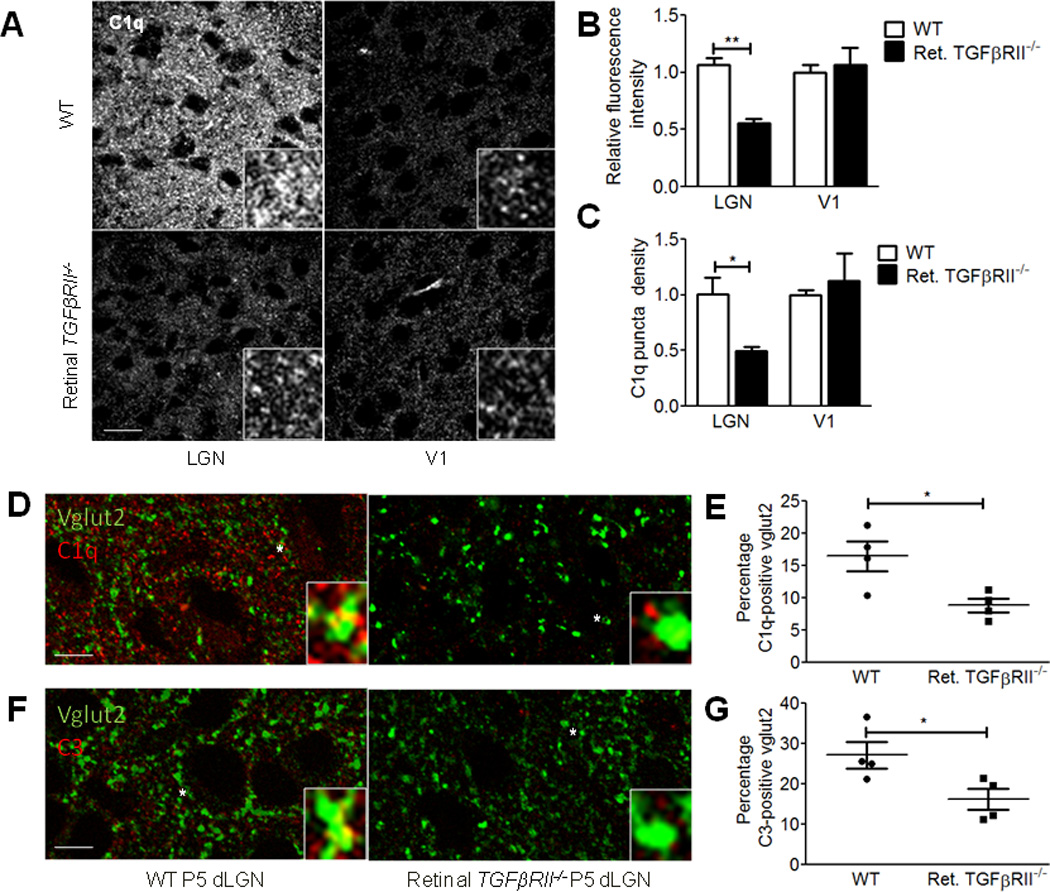
(A) C1q immunohistochemistry in the dLGN and primary visual cortex (V1) shows reduced C1q fluorescence intensity in the dLGN but not in V1. Scale bar = 20µm. Inset shows 3× magnification. (B) Quantifying relative fluorescence intensity showed significant reduced intensity in the dLGN of retinal TGFβRII−/− vs. WT mice (two-way ANOVA, n=4 mice/group, **p<0.01, F(1,12)=11.21). No differences were observed in V1. (C) C1q puncta density quantification showed significantly reduced C1q puncta density in the dLGN in retinal TGFβRII−/− mice vs. WT littermates (two-way ANOVA, n=4 d C1q expression. Microglia acutely isolated using CD45 immunopanning showed no significant difference in C1q levels (two-way ANOVA, n= 5 mice/group, *p<0.05, F(1,8)=7.48). No difference in C1q puncta density was observed in V1. (D) C1q localization to vglut2-positive RGC terminals is reduced in the retinal TGFβRII−/− mouse. Immunostaining for C1q and vglut2 in WT and retinal TGFβRII−/− P5 dLGNs showed a reduction in co-localization of C1q and vglut2. Scale bar = 20µm. Star indicates the enlarged synaptic puncta shown in the inset. (E) Quantification of C1q co-localization with vglut2 showed significantly reduced synaptic localization of C1q in retinal TGFβRII−/− mice. Co-localized puncta were identified and counted using ImageJ Puncta Analyzer (t test, n=4 mice, *p=0.0226, t(6)=3.045). (F) C3 localization to vglut2-positive RGC terminals is reduced in the retinal TGFβRII−/− mice, similar to what we observed for C1q. Scale bar = 20µm. (G) Co-localized C3 and vglut2 puncta were identified and counted using ImageJ Puncta Analyzer (t test, n=4 mice, *p<0.0395, t(6)=2.622).
Given previous findings that C1q and C3 localize to synapses in the dLGN, we examined synaptic localization of complement in the dLGN in retinal TGFβRII−/− mice and WT littermates to determine if synaptic localization of these proteins is dependent on retinal TGF-β signaling. Immunostaining in the dLGN using antibodies for C1q, C3, and vglut2 to label RGC terminals showed approximately 15% of vglut2-positive puncta co-labeled with C1q (Fig. 5d,e) and, ~25% of vglut2 puncta co-stained with the downstream complement protein, C3 (Fig. 5f,g). The increase in the number of RGC terminals labeled with C3 versus C1q is consistent with the placement of these molecules in the classical complement cascade, since multiple C3 molecules can be activated downstream of a single C1q molecule. Also, as expected given that C1q is upstream of C3 activation in the complement cascade, we found that C3 deposition in the dLGN was undetectable in the C1q−/− mouse (Supplemental Fig. 3f). In retinal TGFβRII−/− dLGNs, we also observed a significant reduction in synaptic localization of both C1q and C3 (Fig. 5d–g). Given that retinal TGFβRII−/− exhibit specific reduction in TGF-β signaling and C1q in the retina, a reduction in C1q and C3 synaptic localization in the dLGN suggests that a significant fraction of synaptically localized C1q is supplied by RGC terminals and that C1q and C3 localization to synapses is dependent on retinal TGF-β signaling. Thus, local microglia-derived C1q in the dLGN does not appear to localize to synapses to compensate for the loss of RGC-derived C1q.
TGF-β signaling is required for eye specific segregation
In C1q−/− mice, RGC axons do not segregate into eye specific territories normally as assessed by anterograde tracing techniques3. If C1q supplied by RGC axons is required for eye specific segregation and retinal TGF-β signaling regulates C1q expression, then retinal TGFβRII−/− mice should exhibit a phenotype similar to C1q−/− mice. To test this hypothesis, we used established anterograde tracing techniques to visualize the formation of eye specific territories in the dLGN and to assay eye specific segregation in retinal TGFβRII−/− mice and in mice injected with anti-TGF-β. Mice received intraocular injections of cholera toxin beta conjugated to Alexa 488 or 594 (CTB-488 or CTB-594) at P9 in the left and right eyes respectively and mice were sacrificed at P10. We assayed eye specific segregation using an unbiased assay in which the degree of segregation is represented by the variance of the distribution of the logarithm of the ratio of fluorescence intensity from each fluorescence channel (R value)20. Using this assay, high variance signifies a high degree of segregation in the dLGN, while low variance corresponds to increased overlap between contralateral and ipsilateral territories. Our results showed a significantly lower variance in C1q−/− mice compared to WT controls, as expected (Fig. 6a,b). Importantly, we observed a similar phenotype in retinal TGFβRII−/− (Fig. 6a,b) and mice injected with anti-TGF-β (Fig. 6a,c), suggesting a common pathway. Defects in eye-specific segregation were confirmed by quantifying the percent overlap using the original method used to identify the C1q−/− phenotype (Fig. 7a,b). No differences were observed in dLGN area (Fig. 7c) or in dLGN relay neuron number as determined by SMI32 staining (data not shown) in retinal TGFβRII−/− mice compared to WT.
Figure 6. TGF-β signaling and C1q are required for eye specific segregation and microglia-mediated pruning in the retinogeniculate system.
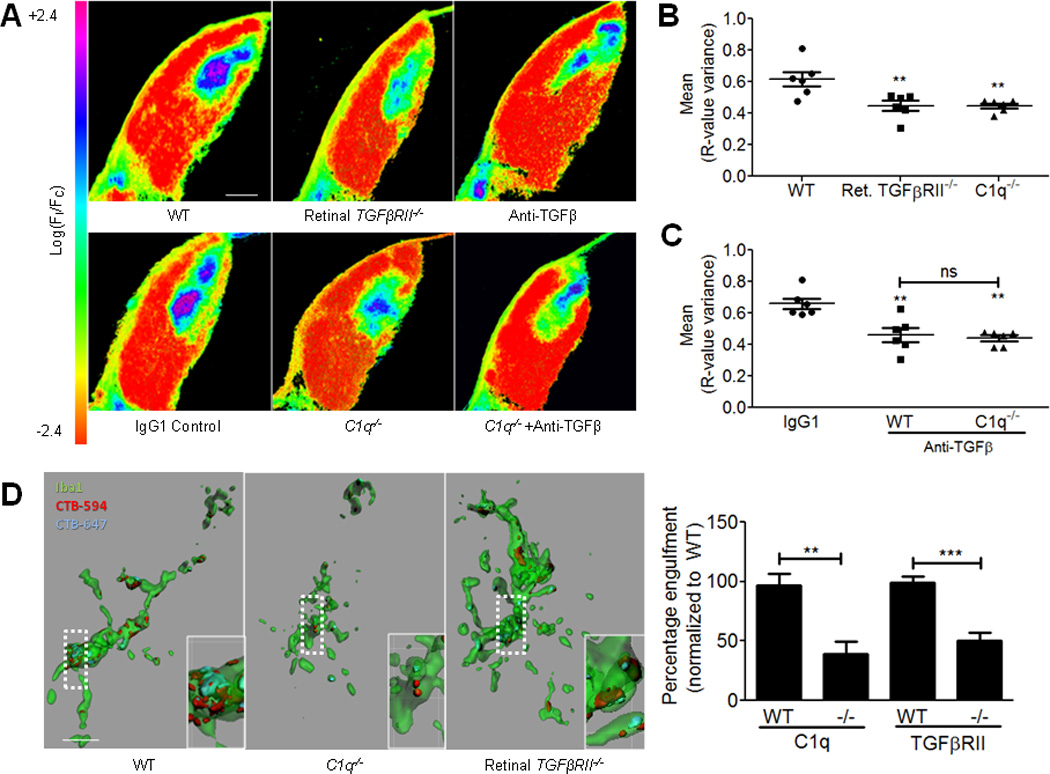
(A) Representative dLGN images for WT, retinal TGFβRII−/−, C1q−/−, IgG1 control, and anti-TGF-β injected WT and C1q−/− mice pseudocolored to show the R-value for each pixel. R=log10(FIpsi/FContra). Scale bar = 100µm. (B) Quantification of the mean variance of the R-value for each group. A significant reduction in the mean variance of the R value is seen in mice deficient in TGF-β signaling or C1q (one-way ANOVA, n= 6 animals/group, **p<0.01, F(2,15)=8.228). (C) There is no additional decrease in the R-value variance when TGF-β signaling is blocked in C1q−/− mice. Data shown as mean R-value variance +/− SEM (one-way ANOVA, n= 6 animals/group, *p<0.05, F(2,15)=8.567). (D) Microglia show reduced engulfment of RGC terminals in mice deficient in C1q or retinal TGF-β signaling. Volume of each microglia and the engulfed CTB was quantified in Imaris and the % engulfment defined as the volume of internalized CTB/volume of microglia. Results were normalized to WT engulfment levels and C1q−/− or retinal TGFβRII−/− mice both showed a significant reduction in % engulfment (one-way ANOVA, n= 6 mice/group, **p<0.01, F(3,20)=13.66). Scale bar = 10µm. Insets show enlargement of boxed area.
Figure 7. Mice deficient in C1q or retinal TGF-β signaling show increased overlap of contralateral and ipsilateral areas in the dLGN.
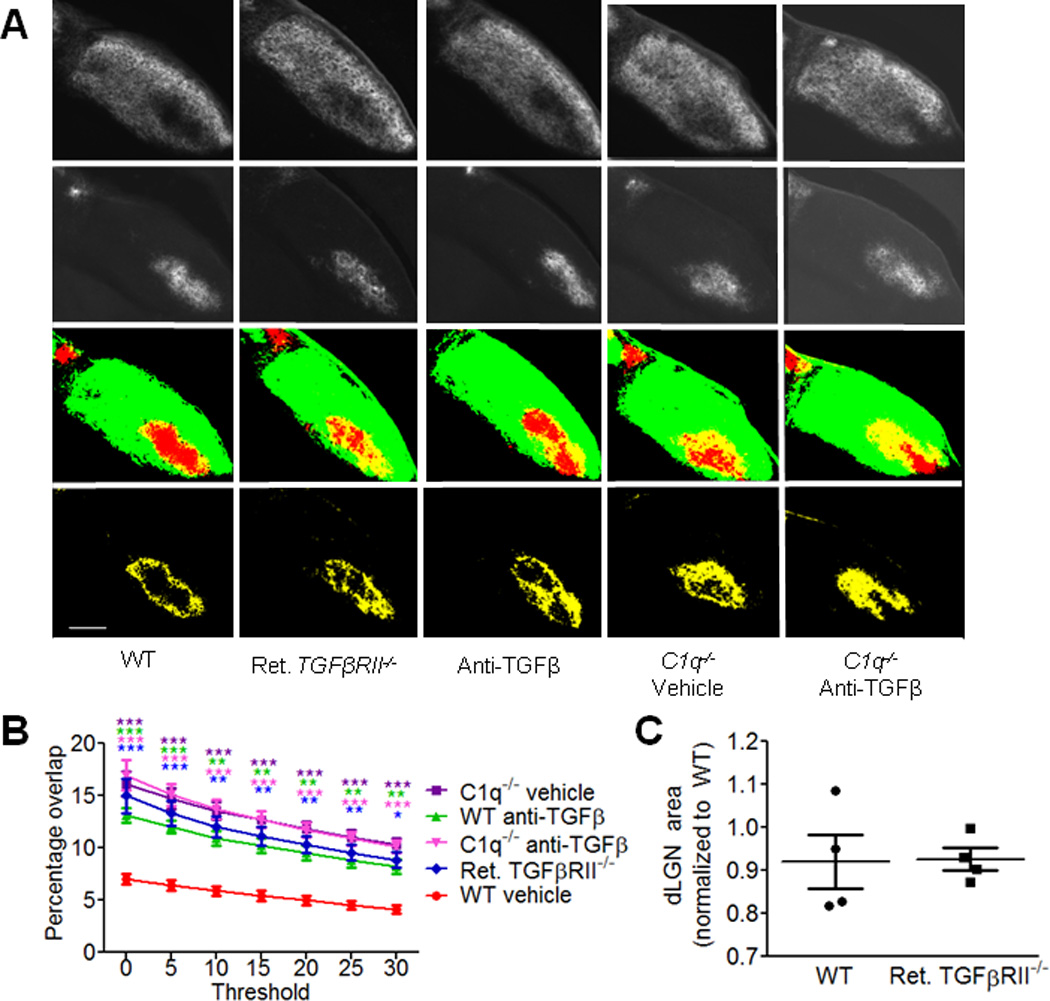
(A) Representative images of anterograde tracing (Alexa conjugated b-cholera toxin) of contralateral (top row) and ipsilateral (second row) retinogeniculate projections, merged channels (third row) and their overlap (yellow, bottom row) in the dLGN for WT, retinal TGFβRII−/−, C1q−/−, and anti-TGF-β injected WT and C1q−/− mice. Scale bar = 100µm. (B) Quantification of percentage of dLGN area receiving input from both contralateral and ipsilateral eyes (yellow area). Data shown as mean yellow area +/− SEM (two-way ANOVA, n= 6 animals/group, ***p<0.001, **p<0.01, *p<0.05, F(4,175)=101.00). (C) Measurements of dLGN area in P10 mice showed no significant difference between WT and retinal TGFβRII−/− mice. Results were normalized to WT dLGN area and 4 dLGNs were analyzed per mouse (t test, n=4 mice, p=0.9342(ns), t(6)=0.08611).
Our results showed that retinal TGF-β is required for eye specific segregation, but it was not clear whether this effect was through regulation of C1q. To determine if TGF-β could regulate synaptic refinement independently of C1q, we performed intraocular injection of anti-TGF-β neutralizing antibodies in WT and C1q−/− animals to determine if blocking TGF-β disrupted synaptic refinement in the absence of C1q. If TGF-β and C1q regulate pruning by different mechanisms, blocking TGF-β in C1q−/− mice should produce a more severe segregation defect, or increased overlap compared to vehicle-injected C1q−/− mice. If TGF-β and C1q are in the same pathway, blocking TGF-β signaling in the C1q−/− mouse should have no effect. Blocking TGF-β in the C1q−/− mouse did not result in a more severe phenotype, consistent with TGF-β exerting its effect on synaptic refinement via regulation of C1q (Fig. 6a–c; 7a,b). Taken together, these results show that retinal TGF-β signaling is required for eye specific segregation and that TGF-β and C1q likely work in the same pathway to regulate this process.
Retinal TGF-β and C1q regulate microglia-mediated pruning
Microglia engulfment of retinogeniculate synapses is thought to be the final step in complement-dependent synapse elimination. Recent work has demonstrated that microglia engulf RGC terminals in the dLGN during the pruning period in a complement (C3/CR3)-dependent manner7. Therefore, if RGC-derived C1q regulates this process, we predict retinal TGFβRII−/− mice to show a reduction in microglial engulfment of RGC terminals.
To test this hypothesis, we used an established microglia engulfment assay7 to quantify microglial phagocytosis of RGC inputs in C1q−/− mice and retinal TGFβRII−/− mice. We found a significant reduction in microglial engulfment of RGC inputs at P5 in C1q−/− and TGFβRII−/− mice as compared to littermate controls (Fig. 6d). We found no differences in the number, distribution, or activation state of microglia in the dLGN in WT vs. C1q−/− or TGFβRII−/− conditions (supplemental Fig. 4a–d). These results show that reduction of retinal TGF-β signaling, and consequently of C1q expression in RGCs, significantly reduced microglial phagocytosis of RGC terminals in the dLGN. Together these data support the hypothesis that RGC-derived C1q is required to initiate the process of complement-dependent synapse elimination by microglia.
Discussion
This study establishes that TGF-β signaling in RGC neurons plays a key role in refinement of RGC synapses on their target relay neurons in the dLGN by regulating expression of C1q, the initiating protein of the classical complement cascade. We demonstrated that TGF-β signaling is necessary and sufficient for transcriptional upregulation of C1q in purified RGCs (Fig. 2). Consistent with in vitro studies, conditional knockout of TGFβRII in retinal neurons reduced C1q expression in RGCs during the period of active refinement of retinogeniculate synapses in the thalamus (Fig. 4). Furthermore, our data show that C1q and C3 localization to synapses in the dLGN during retinogeniculate refinement is dependent on retinal TGF-β signaling (Fig. 5). Importantly, inhibition of TGF-β signaling in the postnatal retina resulted in significant defects in eye specific segregation in the dLGN, mimicking the phenotype observed in global C1q−/− mice (Fig. 6a–c). We also observed defects in microglia engulfment of RGC terminals (Fig. 6d), which phenocopied the C3−/− and CR3−/− mouse microglial engulfment defects7. Taken together, our findings support a model in which retinal TGF-β signaling controls C1q expression and local release in the dLGN to regulate microglia-mediated, complement-dependent synaptic refinement (Supplemental Fig. 5).
C1q regulates microglia-mediated pruning in the thalamus
Emerging evidence implicates microglia in developmental synaptic refinement7,21,22; however, the mechanisms controlling the timing and location of microglia-mediated synaptic refinement in the brain are poorly understood. Process-bearing, phagocytic microglia in the postnatal dLGN engulf RGC inputs during retinogeniculate refinement in a manner dependent on complement C3-CR3 signaling and neuronal activity7. Upon complement cascade activation, C3 is cleaved to form the potent opsonin, C3b, a ligand for the phagocytic complement receptor CR3 expressed in the brain by microglia exclusively. Thus, as the initiator of the classical complement cascade, C1q is a critical regulator of C3 activation and C3-dependent phagocytosis. Our results suggest that C3 localization to synapses (Fig. 5) and microglia-mediated pruning (Fig. 6) are dependent on retina-derived C1q. Specifically, we observed a significant decrease in C3 localization to synapses in C1q−/− and in the retinal TGFβRII−/− mice. We also found that microglial engulfment of RGC terminals is decreased in C1q−/− (Fig. 6d) and in the retinal TGFβRII−/− mice (Fig. 6d), placing retina-derived C1q and retinal TGF-β signaling upstream of this process.
Interestingly, recent work revealed that microglia-mediated pruning is regulated by neuronal activity7. During the retinogeniculate pruning period, microglia preferentially engulf less active RGC inputs in the dLGN7, suggesting that microglia can sense or read out local changes in synaptic activity. The engulfment defects observed in complement-deficient mice and the role for complement in the immune system suggest the intriguing possibility that complement proteins could be one cue guiding microglia to engulf less active synapses. Other immune molecules implicated in synaptic refinement, such as MHC class I23 and neural pentraxins24, show activity-dependent transcriptional regulation, but whether these molecules have any role in activity-dependent microglial engulfment is unknown. Interestingly, C1q expression corresponds to the onset of retinal wave activity, and the C1q−/− phenotype closely mimics the phenotype observed when spontaneous activity is disrupted in the retina. Whether and how neuronal activity regulates complement expression and function are open areas of investigation; however, based on our data, we envision a model in which C1q is secreted locally from RGC terminals in an activity-dependent manner to regulate C3-CR3 dependent microglial synaptic phagocytosis (Supplemental Fig. 5).
Retinal C1q is required for retinogeniculate refinement
In the postnatal retinogeniculate system, both RGCs and microglia express C1q. Although microglia express comparatively higher levels of C1q than neurons (Fig. 1c, 2g), several lines of evidence suggest that RGCs contribute significantly to C1q levels and complement-dependent synaptic refinement in the postnatal dLGN. In retinal TGFβRII−/− mice, which have reduced retinal C1q expression but normal microglial C1q expression, we observed defects in retinogeniculate refinement and microglial engulfment (Figs, 6 and 7), suggesting that microglial C1q expression cannot compensate functionally for the loss of RGC-derived C1q in complement-dependent synaptic refinement.
If RGC inputs are a critical source of C1q in the dLGN during synaptic refinement, how does C1q reach RGC terminals in the dLGN far from the cell bodies in the retina? C1q is a large glycoprotein packaged and secreted by macrophages via traditional secretory pathways in the golgi25. C1q then is secreted directly into the bloodstream by circulating macrophages. In RGCs, however, proteins that are produced and packaged in the soma in the retina have a long distance to travel to axon terminals in the dLGN. Given that in the retinal TGFβRII−/− mice we observed reduced C1q levels compared to WT in the retina (Fig. 4a–d), the optic nerve (Fig, 4e–f), and the dLGN (Fig. 5), we hypothesize that C1q is transported along RGC axons to the dLGN within secretory vesicles, based on its packaging and secretion in macrophages. Alternatively, local translation and synthesis of C1q within RGC axons in the optic nerve or the dLGN is possible, although there have been no previous reports of local translation of C1q. Given the importance of spontaneous retinal activity in driving synaptic refinement and that C1q regulation by TGF-β seems to be unique to neurons (Fig. 1c,4b), it is intriguing to speculate that C1q secretion from RGC inputs may be regulated by neuronal activity and act as a molecular cue linking information about the retina to the dLGN to drive proper circuit development.
TGF-β: regulating neuronal C1q and CNS development
In the immune system, TGF-β is characterized as an anti-inflammatory cytokine that dampens the immune response. In the mammalian nervous system, TGF-β signaling pathways regulate diverse developmental processes, including neuronal survival and programmed cell death, axon specification, and synaptogenesis. TGF-β signaling modulates embryonic and postnatal periods of programmed cell death in the mouse retina26 and also amplifies the efficacy of neurotrophic factors critical for cell survival, such as GDNF27. In the developing rodent neocortex, TGF-β signaling is necessary and sufficient for axon specification28. Recent work also has shown that TGF-β regulates synaptogenesis in cortical neurons in vitro29 and at the Drosophila and Xenopus NMJs30. At the Drosophila NMJ, glia produce the TGF-β ligand31 and they direct synaptogenesis via regulating a neuron-derived TGF-β family member and the downstream RacGEF Trio32. Also, in Drosophila, TGF-β directs large scale neuronal and axonal remodeling during metamorphosis33,34. TGF-β also affects synaptic function at excitatory and inhibitory synapses in the pre-Bötzinger complex in the mouse brainstem35 and modulates sensory neuron excitability and synaptic efficacy at sensorimotor synapses in the sea slug Aplysia36.
In the present study, we found that TGF-β signaling in postnatal RGCs is required for developmental synaptic refinement. Given the many important roles for TGF-β in development, we characterized other aspects of retinal development and found no significant deficits in the retinal TGFβRII−/− mouse. We found that disrupting TGFβRII in the retina did not dramatically affect the number of retinal neurons or retinal morphology. Moreover, despite the role for TGFβRII in axon specification in cortex, disruption of TGFβRII signaling in the retina does not alter axon specification in the retina or axon caliber in the optic nerve (Supplemental Fig. 2f–h), suggesting that other mechanisms underlie axon development and specification in RGCs. Although we have not quantified synapses in the dLGN of retinal TGFβRII−/− mice, it is unlikely that defective synaptogenesis could explain our phenotype, as we observed an increase in overlap between eye specific territories. Furthermore, previous studies have reported that TGF-β is not synaptogenic in RGC cultures10, further supporting that synaptogenesis is likely normal in these mice.
Together our data implicate TGFβ as a key regulator of synaptic refinement in the mammalian CNS. Interestingly, in Drosophila, a class of TGF-β family members together with an immunoglobulin superfamily member protein, Plum, has been implicated in the dramatic remodeling that occurs during metamorphosis in the CNS and at the NMJ33,34. This finding combined with our findings raises the question of whether TGF-β signaling represents an important signal for synaptic remodeling throughout the nervous system. Could TGF-β initiate a synaptic refinement gene program in RGCs or other neurons that undergo refinement? Interestingly, C1q and TGF-β expression are expressed in embryonic and postnatal spiral ganglion neurons37 around the time when spiral ganglion cell inputs onto outer hair cells are refined38. Little is known about the synaptic refinement mechanisms in the auditory system; however, this developmental expression of C1q and TGF-β suggests that the same mechanism for synapse elimination may drive refinement in both the auditory and visual systems. Importantly, in both of these systems, C1q expression in sensory neurons (RGCs and spiral ganglion neurons) is developmentally regulated and restricted to periods of developmental synaptic remodeling. These findings are consistent with our hypothesis that neuron-derived C1q plays a critical role in synaptic refinement, although it remains unclear if TGF-β regulates C1q or other refinement genes in other brain regions.
Implications of TGF-β regulation of C1q in disease
Our findings also have important implications for understanding mechanisms underlying synapse elimination in the diseased brain. Dysregulation of immune system components including complement proteins and cytokines has been demonstrated in many CNS disorders including epilepsy, schizophrenia, and neurodegenerative diseases such as glaucoma and Alzheimer’s disease. In particular, TGF-β localizes to beta-amyloid plaques and has been linked to the formation of these plaques in Alzheimer’s disease39 and blocking TGF-β and Smad2/3 signaling mitigates plaque formation in Alzheimer’s mouse models40. C1q associates with plaques in Alzheimer’s brains as well41, and in mouse models of Alzheimer’s, C1q-deficiency has been shown to be neuroprotective42. Synapse loss and/or dysfunction have emerged as early hallmarks of neurodegenerative diseases, suggesting that aberrant complement upregulation may reactivate the developmental synapse elimination pathway in disease to promote synapse loss. Our work demonstrating a new link between TGF-β signaling, complement, and synapse elimination opens up new avenues of investigation into the role of this regulatory mechanism for C1q in these disorders and in other regions of the healthy CNS during development.
ONLINE METHODS
Mice
Floxed TGFβRII mice (B6.129S6-TGF-βr2tm1Hlm) were obtained from the NCI Mouse Repository and crossed to the CHX10-Cre line, Tg(Chx10-EGFP/cre,-ALPP)2Clc/J (Jackson Lab), to generate retina-specific TGFβRII−/− mice. C1q−/− mice (C57BL6 background) were generously provided by M. Botto43. Experiments were approved by the institutional animal use and care committee in accordance with NIH guidelines for the humane treatment of animals.
Neuron and Astrocyte Cultures
Retinal ganglion cells were cultured from P8 Sprague-Dawley rats after serial immunopanning steps to yield >99.5% purity as described19. Cells were maintained in serum-free media as described44. Cortical astrocytes were prepared from P1–P2 rat cortices as previously described9. Retinal astrocytes were prepared from P8 Sprague Dawley rat or mouse retinas by adapting described methods for purification of cortical astrocytes12. After first immunopanning away microglia and RGCs, an anti-ITGB5-coated petri plate was used to isolate astrocytes from remaining cells in suspension. Purified cortical astrocytes were prepared as described12. Purified retinal and cortical astrocytes were maintained in a defined serum-free medium supplemented with hbEGF as described12. Astrocyte conditioned medium (ACM) was prepared as previously described9. Astrocytes were switched to minimal media (Neurobasal + glutamine, Pen/Strep, Sodium Pyruvate) once confluent and media was collected after 5 days and concentrated to 10× using Vivaspin columns (Sartorius).
qPCR
RGCs, microglia, and astrocytes were acutely isolated using immunopanning as described above from either mice or rats at indicated ages. For acute isolation experiments, RNA was collected directly from the immunopanning plate without culturing the cells. For cultured RGC experiments, total RNA was prepared, cDNA was synthesized, and qPCR performed using the Applied Biosystems Cells to Ct Power SYBR green kit as described by the manufacturer. Briefly, cell lysates were collected from 80,000 RGCs in provided lysis buffer and cDNA was synthesized directly from this lysate. QPCR reactions were assembled for the genes of interest (c1qa, c1qb, tieg, gapdh) using 4µL of cDNA per reaction and samples were run on the Rotogene qPCR machine (QIAGEN). Expression levels were compared using the ddCT method normalized to GAPDH.
Immunohistochemistry
Brains and eyes were harvested from mice after transcardial perfusion with 4% paraformaldehyde (PFA). Tissue was then immersed in 4% PFA for 2 hours following perfusion, cryoprotected in 30% sucrose, and embedded in a 2:1 mixture of OCT:20% sucrose PBS. Tissue was cryosectioned (12–14 microns), sections were dried, washed three times in PBS, and blocked with 2% BSA+ 0.2% Triton X in PBS for 1 hr. Primary antibodies were diluted in antibody buffer (+ 0.05% triton + 0.5% BSA) as follows: C1q (undiluted culture supernatant, Epitomics, validated in Stephan et al., 201314), C3 (Cappel,1:300), vglut2 (Millipore,1:2000), TGFβRII (R&D Systems goat antihuman, 1:200), pSmad (Millipore,1:200), Calretinin (Millipore,1:1000), Iba1 (Wako, 1:400), CD68 (Serotec,1:300), TUJ1 (Covance, 1:400), and incubated overnight at 4°C. Secondary Alexa-conjugated antibodies (Invitrogen) were added at 1:200 in antibody buffer for 2 hr at room temperature. Slides were mounted in Vectashield (+DAPI) and imaged using the Zeiss Axiocam, Zeiss LSM700, or Perkin Elmer Ultraview Vox Spinning Disk Confocal.
ELISA
The 7-plex Mouse Inflammatory Cytokine kit (MSD) was used to profile cytokines in ACM. Freshly prepared ACM was profiled according to manufacturer’s provided protocol. Standards were diluted in minimal media used to make ACM for greater accuracy. Plates were read and data were acquired and analyzed using the MSD Sector Imager 2400. ELISA kits were obtained for TGF-β1, 2, and 3 separately (R&D systems, MSD) and were performed according to manufacturer’s instructions.
Western Blot
RGC and whole retina lysates were collected and homogenized in RIPA buffer with complete protease inhibitors (Roche). Samples were boiled for 5 min in SDS sample buffer, resolved by SDS PAGE, transferred to PVDF membranes, and immunoblotted. Antibodies were diluted in 5% milk in PBS + 0.1% Tween. Antibodies: Rabbit anti-C1qA polyclonal (Epitomics, 1:5000, specificity validated in Stephan et al., 201314); Goat anti-TGFβRII (R&D Systems, 1:1000).
In situ hybridization
In situ hybridization for C1qb was performed on 12 um retinal sections as previously described3. Probes targeting the entire C1qb coding sequence (Open Biosystems clone: 5715633) were generated by digesting the plasmid with EcoRI, and performing in vitro transcription with T7 polymerase using the DIG RNA labeling kit (Roche Applied Science) as per the manufacturer’s instructions. 1.8kb probes were then cleaved to form 300 bp probes by alkaline hydrolysis before use.
LGN analysis
Mice received intraocular injection of cholera toxin-b subunit (CTB) and were sacrificed the following day. Tissue was processed and analyzed as previously described3,45. Mouse pups were anesthetized with inhalant isofluorane. Mice received intravitreal injections of cholera toxin-b subunit (CTB) conjugated to Alexa 488 (green label) in the left eye and CTB conjugated intraocular injection to Alexa 594 (red label) into the right eye as described2. Images were digitally acquired using the Zeiss Axiocam. All images were collected and quantified “blind,” and compared to age-matched littermate controls. Gains and exposures were established for each label. Raw images of the dLGN were imported to Photoshop (Adobe), and the degree of left and right eye axon overlap in dLGN was quantified using the multi-threshold protocol as previously described2 and using threshold independent R value analysis as described20. For threshold independent analysis, we performed background subtraction using a 200 pixel rolling ball radius filter and normalized the images. We then calculated the R value (log(FI/FC)) for each pixel and determined the variance of the R value distribution for each image (4 images/animal). Pseudocolored images representing the R value distribution were generated in ImageJ.
Retinal cell counts
Retinal flat mounts were prepared by dissecting out retinas whole from the eyecup and placing four relieving cuts along the major axis, radial to the optic nerve. Each retina was stained with DAPI (Vector Laboratories, Burlingame, CA) to reveal cell nuclei. Measurements of total cell density in the ganglion cell layer (which includes both ganglion cells and displaced amacrine cells) were carried out blind to genotype from matched locations in the central and peripheral retina for all four retinal quadrants of each retina. Quantification was limited to P30 retinas, which is an age subsequent to ganglion cell genesis and apoptosis in the mouse. For each retina (1 retina per animal; n=3 mice per treatment condition or genotype), 12 images of peripheral retina and 8 images of central retina were collected. For each field of view collected (20 per retina), Macbiophotonics ImageJ software (NIH) was used to quantify the total number of DAPI using the nuclei counter plugin and TUJ1-positive cells were counted using the cell counter plugin. All analyses were performed blind to genotype or drug treatment.
Z stack Image and Microglial Engulfment Analysis
In vivo microglia phagocytosis assay and analyses were performed as previously described in detail by Schafer et al., 2012. In brief, mice received intraocular injections of anterograde tracers at P4. All mice were sacrificed at P5 and brains were 4% PFA fixed overnight (4°C). Only those brains with sufficient dye fills were analyzed. For each animal, two sections of medial dLGN were chosen for imaging for reconstruction of RGC inputs and C1q staining as well as for microglia engulfment analysis. Images were acquired on a spinning disc confocal at 60× using 0.2 µm z-steps. For each dLGN, 4–8 fields were imaged in the ipsilateral territory and 4–8 fields were imaged in the contralateral territory (minimum of 8 fields per dLGN, 16 fields per animal). Subsequent images were processed and quantified using ImageJ (NIH) and Imaris software (Bitplane). For subsequent acquired z-stacks, ImageJ (NIH) was used to subtract background from all fluorescent channels (rolling ball radius=10) and a mean filter was used for the EGFP channel (stained for Iba1) of 1.5. Subsequently, Imaris software (Bitplane) was used to create 3D volume surface renderings of each z-stack. Surface rendered images were used to determine the volume of the microglia, all RGC inputs, and the volume of C1q staining. To visualize and measure the volume of engulfed inputs, any fluorescence that was not within the microglia volume was subtracted from the image using the mask function. The remaining engulfed/internal fluorescence was surface rendered using parameters previously determined for all RGC inputs/C1q and total volume of engulfed/internal inputs was calculated. To determine % engulfment (or %C1q-positive terminal), the following calculation was used: Volume of internalized RGC input (or volume of C1q) (µm3)/Volume microglial cell (or RGC inputs) (µm3). For all KO engulfment experiments, all analyses were performed blind.
Microglia Density Quantification
For quantification of cell density, 2 dLGN were imaged per animal (n=3 per treatment condition or genotype). To capture the entire dLGN, a 10× field was acquired. Microglia were subsequently counted from each 10× field. To calculate the density of microglia, the area of the dLGN was measured using ImageJ software (NIH). All analyses were performed blind to genotype or drug treatment.
Quantification of Microglial Activation State
Activation state was quantified based on established methods (Schafer et al, 2012). Immunohistochemistry was performed on 40um cryosections with antibodies against Iba1 and CD68. For each genotype, two 20× fields of view were analyzed on a spinning disk confocal using 2um z-steps. The activation state was then determined based on a maximum intensity projection. Iba1 staining was used to assess morphology based on the number of branches and the expression pattern of CD68 was analyzed and scored as 0 (no/scarce expression), 1 (punctate expression), or 2 (aggregated expression or punctate expression all over the cell). Scores from 0–3 for number of branches or 0–2 were assigned based on Iba1 and CD68 staining and these score were combined to give the most activated microglia an overall score of 5 and least activated scoring 0. For each genotype, 4 mice were analyzed and all analysis was performed blind.
Statistical Analysis
For all statistical analyses, data distribution was assumed to be normal but this was not formally tested. GraphPad Prism 5 software (La Jolla, CA) was used for all statistical tests. Analyses used include one-way ANOVA, two-way ANOVA, and Student’s t-test. For ANOVA analysis, Bonferonni post hoc tests were used. Igor was used to calculate the R value variance in Figure 6. No statistical methods were used to pre-determine sample sizes but our sample sizes are similar to those reported in previous publications3,7. Data collection and analysis were performed blind to the conditions of the experiments. Also, data for each experiment were collected and processed randomly and animals were assigned to various experimental groups randomly as well. All n and p values and statistical tests are indicated in figure legends. All error bars represent SEM and mean +/− SEM is plotted for all graphs except where noted.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank C. Chen, L. Benowitz, D.P. Schafer, and M. Buckwalter for helpful comments on the manuscript and critical discussion. In addition we thank Alexander Stephan, Ben Barres, and Andrea Tenner for assistance with anti-C1q antibody production and characterization. We also thank M. Rasband for the βIV spectrin antibody. Thank you to D.P. Schafer and E.K. Lehrman for technical expertise on the microglial engulfment assay and Imaris image analysis, T. Nelson for technical assistance, and the imaging core at Boston Children’s Hospital including T. Hill for technical support. This work was supported by grants from the Smith Family Foundation (B.S.), Dana Foundation (B.S.), the Ellison Foundation (B.S.), John Merck Scholars Program (B.S.), NINDS (RO1-NS-07100801; B.S.), NIDA (RO1-DA-15043; B.A.B.), NIH (P30-HD-18655; MRDDRC Imaging Core).
Footnotes
Author Contributions
A.R.B. conducted all experiments, performed data analysis, and wrote the manuscript.
B.S. advised on and supervised the project and co-wrote the manuscript.
References
- 1.Huh GS, et al. Functional requirement for class I MHC in CNS development and plasticity. Science. 2000;290:2155–2159. doi: 10.1126/science.290.5499.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bjartmar L, et al. Neuronal pentraxins mediate synaptic refinement in the developing visual system. J Neurosci. 2006;26:6269–6281. doi: 10.1523/JNEUROSCI.4212-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stevens B, et al. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 4.Campbell G, Shatz CJ. Synapses formed by identified retinogeniculate axons during the segregation of eye input. J Neurosci. 1992;12:1847–1858. doi: 10.1523/JNEUROSCI.12-05-01847.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sretavan D, Shatz CJ. Prenatal development of individual retinogeniculate axons during the period of segregation. Nature. 1984;308:845–848. doi: 10.1038/308845a0. [DOI] [PubMed] [Google Scholar]
- 6.Hooks BM, Chen C. Distinct roles for spontaneous and visual activity in remodeling of the retinogeniculate synapse. Neuron. 2006;52:281–291. doi: 10.1016/j.neuron.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 7.Schafer DP, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boulanger LM. Immune proteins in brain development and synaptic plasticity. Neuron. 2009;64:93–109. doi: 10.1016/j.neuron.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 9.Ullian EM, Sapperstein SK, Christopherson KS, Barres BA. 2001;Vol. 291:657–661. doi: 10.1126/science.291.5504.657. [DOI] [PubMed] [Google Scholar]
- 10.Christopherson KS, et al. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 11.Allen N, et al. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature. 2012;486:410–414. doi: 10.1038/nature11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foo LC, et al. Development of a Method for the Purification and Culture of Rodent Astrocytes. Neuron. 2011;71:799–811. doi: 10.1016/j.neuron.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ullian EM, Christopherson KS, Barres BA. Role for glia in synaptogenesis. Glia. 2004;47:209–216. doi: 10.1002/glia.20082. [DOI] [PubMed] [Google Scholar]
- 14.Stephan AH, et al. A Dramatic Increase of C1q Protein in the CNS during Normal Aging. The Journal of Neuroscience. 2013;33:13460–13474. doi: 10.1523/JNEUROSCI.1333-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Massague J. How cells read TGF-beta signals. Nat Rev Mol Cell Biol. 2000;1:169–178. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 16.Inman GJ, et al. SB-431542 Is a Potent and Specific Inhibitor of Transforming Growth Factor-β Superfamily Type I Activin Receptor-Like Kinase (ALK) Receptors ALK4, ALK5, and ALK7. Molecular Pharmacology. 2002;62:65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- 17.de Melo J, Qiu X, Du G, Cristante L, Eisenstat DD. Dlx1, Dlx2, Pax6, Brn3b, and Chx10 homeobox gene expression defines the retinal ganglion and inner nuclear layers of the developing and adult mouse retina. The Journal of Comparative Neurology. 2003;461:187–204. doi: 10.1002/cne.10674. [DOI] [PubMed] [Google Scholar]
- 18.Rowan S, Cepko CL. Genetic analysis of the homeodomain transcription factor Chx10 in the retina using a novel multifunctional BAC transgenic mouse reporter. Developmental biology. 2004;271:388–402. doi: 10.1016/j.ydbio.2004.03.039. [DOI] [PubMed] [Google Scholar]
- 19.Barres BA, Silverstein BE, Corey DR, Chun LLY. Immunological, morphological, and electrophysiological variation among retinal ganglion cells purified by panning. Neuron. 1988;1:791–803. doi: 10.1016/0896-6273(88)90127-4. [DOI] [PubMed] [Google Scholar]
- 20.Torborg CL, Feller MB. Unbiased analysis of bulk axonal segregation patterns. Journal of neuroscience methods. 2004;135:17–26. doi: 10.1016/j.jneumeth.2003.11.019. [DOI] [PubMed] [Google Scholar]
- 21.Schafer DP, Lehrman EK, Stevens B. The “quad-partite” synapse: Microglia-synapse interactions in the developing and mature CNS. Glia. 2012 doi: 10.1002/glia.22389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tremblay ME, et al. The role of microglia in the healthy brain. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:16064–16069. doi: 10.1523/JNEUROSCI.4158-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corriveau RA, Huh GS, Shatz CJ. Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron. 1998;21:505–520. doi: 10.1016/s0896-6273(00)80562-0. [DOI] [PubMed] [Google Scholar]
- 24.Tsui CC, et al. Narp, a novel member of the pentraxin family, promotes neurite outgrowth and is dynamically regulated by neuronal activity. Journal of Neuroscience. 1996;16:2463–2478. doi: 10.1523/JNEUROSCI.16-08-02463.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuzaki M. Synapse formation and maintenance by C1q family proteins: a new class of secreted synapse organizers. The European journal of neuroscience. 2010;32:191–197. doi: 10.1111/j.1460-9568.2010.07346.x. [DOI] [PubMed] [Google Scholar]
- 26.Beier M, Franke A, Paunel-Görgülü AN, Scheerer N, Dünker N. Transforming growth factor beta mediates apoptosis in the ganglion cell layer during all programmed cell death periods of the developing murine retina. Neuroscience research. 2006;56:193–203. doi: 10.1016/j.neures.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 27.Peterziel H, Unsicker K, Krieglstein K. TGFβ induces GDNF responsiveness in neurons by recruitment of GFRα1 to the plasma membrane. The Journal of cell biology. 2002;159:157–167. doi: 10.1083/jcb.200203115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yi JJ, Barnes AP, Hand R, Polleux F, Ehlers MD. TGF-[beta] Signaling Specifies Axons during Brain Development. Cell. 2010;142:144–157. doi: 10.1016/j.cell.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diniz LP, et al. Astrocyte-induced Synaptogenesis Is Mediated by Transforming Growth Factor β Signaling through Modulation of d-Serine Levels in Cerebral Cortex Neurons. Journal of Biological Chemistry. 2012;287:41432–41445. doi: 10.1074/jbc.M112.380824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feng Z, Ko C-P. Schwann Cells Promote Synaptogenesis at the Neuromuscular Junction via Transforming Growth Factor-beta1. J. Neurosci. 2008;28:9599–9609. doi: 10.1523/JNEUROSCI.2589-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fuentes-Medel Y, et al. Integration of a Retrograde Signal during Synapse Formation by Glia-Secreted TGF-β Ligand. Current Biology. 2012 doi: 10.1016/j.cub.2012.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ball R, et al. Retrograde BMP signaling controls synaptic growth at the NMJ by regulating trio expression in motor neurons. Neuron. 2010;66:536–585. doi: 10.1016/j.neuron.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 33.Awasaki T, Huang Y, O'Connor MB, Lee T. Glia instruct developmental neuronal remodeling through TGF-[beta] signaling. Nature neuroscience. 2011;14:821–823. doi: 10.1038/nn.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu XM, et al. Plum, an Immunoglobulin Superfamily Protein, Regulates Axon Pruning by Facilitating TGF-β Signaling. Neuron. 2013;78:456–468. doi: 10.1016/j.neuron.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heupel K, et al. Loss of transforming growth factor-beta 2 leads to impairment of central synapse function. Neural Dev. 2008;3:25. doi: 10.1186/1749-8104-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chin J, Angers A, Cleary LJ, Eskin A, Byrne JH. Transforming growth factor beta 1 alters synapsin distribution and modulates synaptic depression in Aplysia. J. Neurosci. 2002:20026363. doi: 10.1523/JNEUROSCI.22-09-j0004.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu CC, Appler JM, Houseman EA, Goodrich LV. Developmental profiling of spiral ganglion neurons reveals insights into auditory circuit assembly. The Journal of Neuroscience. 2011;31:10903–10918. doi: 10.1523/JNEUROSCI.2358-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang L-C, Thorne PR, Housley GD, Montgomery JM. Spatiotemporal definition of neurite outgrowth, refinement and retraction in the developing mouse cochlea. Development. 2007;134:2925–2933. doi: 10.1242/dev.001925. [DOI] [PubMed] [Google Scholar]
- 39.Wyss-Coray T, et al. Amyloidogenic role of cytokine TGF-β1 in transgenic mice and in Alzheimer's disease. Nature. 1997;389:603–606. doi: 10.1038/39321. [DOI] [PubMed] [Google Scholar]
- 40.Town T, et al. Blocking TGF-β–Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nature medicine. 2008;14:681–687. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Afagh A, Cummings BJ, Cribbs DH, Cotman CW, Tenner AJ. Localization and cell association of C1q in Alzheimer's disease brain. Experimental neurology. 1996;138:22–32. doi: 10.1006/exnr.1996.0043. [DOI] [PubMed] [Google Scholar]
- 42.Fonseca MI, Zhou J, Botto M, Tenner AJ. Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer's disease. The Journal of Neuroscience. 2004;24:6457–6465. doi: 10.1523/JNEUROSCI.0901-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Botto M, et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nature genetics. 1998;19:56–59. doi: 10.1038/ng0598-56. [DOI] [PubMed] [Google Scholar]
- 44.Meyer-Franke A, Kaplan MR, Pfrieger FW, Barres BA. Characterization of the signaling interactions that promote the survival and growth of developing retinal ganglion cells in culture. Neuron. 1995;15:805–820. doi: 10.1016/0896-6273(95)90172-8. [DOI] [PubMed] [Google Scholar]
- 45.Jaubert-Miazza L, et al. Structural and functional composition of the developing retinogeniculate pathway in the mouse. Vis Neurosci. 2005;22:661–676. doi: 10.1017/S0952523805225154. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.