

Abstract
Context:
Adrenocortical carcinoma (ACC) is a rare malignancy with a poor prognosis in need of more effective treatment options. Published evidence indicates many ACCs express the vascular endothelial growth factor receptor (VEGFR), suggesting inhibiting vascular endothelial growth factor signaling could potentially impact tumor growth.
Objective:
The objective of the study was to determine the antitumor efficacy of axitinib (AG-013736), a potent, selective inhibitor of VEGFR1, -2, and -3.
Design:
This was a phase II, open-label trial using a two-stage design.
Patients:
Thirteen patients with metastatic ACC previously treated with at least one chemotherapy regimen with or without mitotane participated in the study.
Intervention:
Starting axitinib dose was 5 mg orally twice daily. Dose escalations were permitted if the administered dose was tolerable.
Results:
Thirteen patients were enrolled. Dose escalation was possible in seven patients, but the majority could not tolerate a dose higher than the starting 5 mg, twice-daily dose for prolonged periods of time. All patients experienced known grade 1/2 toxicities, and 10 of 13 patients had at least one grade 3/4 adverse event. No patient tumor could be scored as a Response Evaluation Criteria in Solid Tumors response, although the growth rate on therapy compared with that prior to starting axitinib was reduced in 4 of the 13 patients. The median progression-free survival was 5.48 months, and the median overall survival was longer than 13.7 months.
Conclusion:
Axitinib has limited effectiveness in ACC. Together with 48 patients previously reported who received either sorafenib or sunitinib, a total of 61 ACC patients have now been treated with a VEGFR tyrosine kinase inhibitor without an objective Response Evaluation Criteria in Solid Tumors response. Future trials in ACC should look to other targets for possible active agents.
Adrenocortical carcinoma (ACC) is a rare malignancy with a poor prognosis (1–6). Standard treatment options include surgery, radiotherapy, and chemotherapy including mitotane (7–10). More effective treatment approaches are urgently needed. As with many other human tumors, expression of vascular endothelial growth factor (VEGF) receptor (VEGFR) and evidence of angiogenesis has been found in many ACCs, raising the possibility that inhibiting VEGF signaling in patients with ACC may have antitumor activity (11–13). Axitinib (AG-013736) is an oral, potent, and selective inhibitor of VEGFR-1, -2, and -3 that was approved by the US Food and Drug Administration in January 2012 “for the treatment of advanced renal cell carcinoma after failure of one prior systemic therapy” (14). We conducted a clinical trial to determine the utility, if any, of axitinib in ACC.
Materials and Methods
Clinical trial design and analysis
Eligibility criteria included a pathologically confirmed diagnosis of ACC by the Laboratory of Pathology, National Cancer Institute. Patients could have a diagnosis of recurrent, metastatic, or primary unresectable ACC and needed to have measurable disease at diagnosis. Patients who had received prior therapy with a VEGFR tyrosine kinase inhibitor (TKI) were excluded. The Institutional Review Board of the National Cancer Institute approved the study. All patients signed institutional review board-approved informed consent forms prior to receiving treatment and participating in the trial. The primary objective of the trial was to evaluate the response rate to axitinib (AG-013736) in patients with recurrent, metastatic, or primary unresectable ACC. The secondary objectives were to determine progression-free survival (PFS) and to explore the relationship of potential biomarkers of axitinib activity with clinical outcomes, provided measurable activity was recorded.
This was a phase II, open-label, nonrandomized trial, whereby patients took axitinib twice daily by mouth in 4-week cycles. Patients were evaluated for response every 8 weeks using Response Evaluation Criteria in Solid Tumors (RECIST) criteria (15). The statistical design of the trial allowed for the enrollment of an initial 12 patients, with anticipated expansion to 40 patients if one patient experienced either a complete response or a partial response. The objective of the trial was to determine whether treatment with axitinib could be associated with responses (partial response + complete response) that could rule out an estimated response rate of 5% or less (P = .05) in favor of a more desirable 20% or greater response rate.
The regression-growth equation
We modeled data sets of tumor quantity using a set of equations that describe tumor regression after classical exponential decay kinetics and the concomitant exponential growth of tumor that is (relatively) resistant to the therapy (16–19). This equation was developed on the premise that a change in the size of a tumor during therapy is due to two separate processes: an exponential (first order kinetics) regression and an exponential regrowth of tumor. The equation is:
where exp is the base of the natural logarithm, e = 2.7182 …, and f(t) is the tumor measurement at time t in days, normalized to (divided by) the tumor measurement at day 0, the point at which treatment is commenced. Rate constant, d (decay; in days−1) represents the exponential decrease/regression of tumor during therapy. Rate constant, g (growth; also in days−1) represents the exponential growth/regrowth of the tumor during treatment. We have noted a strong correlation between the rate of growth (log g) and overall survival in previous analyses.
Immunohistochemistry
Immunohistochemistry was performed on formalin-fixed, paraffin-embedded specimens using an avidin-biotin-peroxidase complex immunoperoxidase procedure as previously described (20, 21). A rabbit monoclonal antibody specific for VEGFR2 (clone 55B11; Cell Signaling Technologies) and a rabbit polyclonal antibody that recognizes phospho-VEGFR2-Tyr951 (Santa Cruz Biotechnology) were used. Primary antibodies were applied to tissue sections at 1:600 (VEGFR2) and 1:50 (phospho-VEGFR2-Tyr951) dilutions. A breast cancer and a colon cancer specimen known to stain positive were used as VEGFR2 and phospho-VEGFR2-Tyr951-positive controls, respectively. Breast cancer and colon cancer specimens without expression of the antigens of interest, stained with rabbit isotype immunoglobulins, were used as negative controls. For quantitative analysis, slides were scanned by an automated Cellular Imaging System (ACIS III; Dako Automation). Digital images were reviewed by an investigator (S.X.Y.) using a ×40 magnification and a free-scoring tool to score six areas of tumor. VEGFR2 and phospho-VEGFR2-Tyr951 expression are reported as a staining index calculated by multiplying the percentage of tumor cells by the intensity of staining (after subtracting the tissue readouts of the corresponding negative control) divided by 100 (22). Staining for vessels was performed using monoclonal CD31 (clone JC70A; Dako). Microvessel density (MVD) was determined using computer software that enumerated the number of CD31-positive vessels in each of six tumor areas using a ×20 magnification. The mean number of vessels was recorded for each sample (20).
Results
Thirteen patients were enrolled because the twelfth and thirteenth patients were screened at the same time and both were eligible for the study. Supplemental Table 1 and Supplemental Table 2, published on The Endocrine Society's Journals Online web site at http://jcem.endojournals.org, summarize the clinical characteristics of the patients and their prior therapies. All 13 presented with evidence of metastases, with a median of three different sites involved. With a median of 2.59 years since their original diagnosis, most had received multiple lines of chemotherapy, three had previously enrolled in the First International Randomized Trial in Locally Advanced and Metastatic Adrenocortical Carcinoma Treatment (FIRM-ACT) trial, a trial that enrolled and randomized 304 patients with advanced adrenocortical carcinoma to receive mitotane plus either a combination of etoposide, doxorubicin, and cisplatin (EDP) or streptozocin (23). Ten of the 13 patients in this trial had undergone an original resection and at least one metastasectomy, with seven having undergone more than three metastasectomies, reflecting our aggressive surgical approach to managing metastatic disease.
The starting dose of axitinib was 5 mg administered orally twice a day, and the protocol guidelines allowed the dose to be increased to 7 mg twice daily if the starting dose was tolerated for at least 8 weeks and then to 10 mg twice a day if the 7 mg dose was tolerated for at least 8 additional weeks. However, only seven patients were able to advance to a dose of 7 mg twice a day, and only five patients were able to advance to the dose of 10 mg twice a day; but no patient was able to sustain a dose of 10 mg for more than 16 weeks. With a cycle defined as 4 weeks, a total of 67 cycles were administered. Of these 67 cycles, 38 cycles were administered at the 5 mg, twice-a-day dose, whereas only 17 and 12 cycles were administered at 7 mg and 10 mg twice a day, respectively. A wide range of toxicities tabulated in Supplemental Table 3 prevented further dose escalation. All patients had at least one grade 1/2 toxicity, whereas 10 patients had at least one grade 3/4 adverse event. Because this was a phase 2 study using the recommended doses of axitinib, serum levels of axitinib were not obtained. However, the high incidence of known toxicities, which in all patients precluded advancing the dose or sustaining administration of a higher dose, was interpreted as indirect pharmacodynamic evidence of adequate blood levels.
Although the occurrence of toxicity was common, the activity that was observed was very limited and not felt to be clinically meaningful to the patients. No patient achieved a partial or a complete response, and a designation of stable disease for greater than 3 months could be assigned to only eight patients. For 11 of the 13 patients, we had adequate preenrollment tumor measurements (dating back a minimum of 6 months before enrollment) and could evaluate the effect of axitinib on the regression and growth rates of their tumor using a novel assessment approach we have developed (16–19). This approach uses the same tumor measurements that are obtained as part of the RECIST assessments and plots these over time with the starting quantity set to 1. The curve drawn through the tumor measurements can be described by the equation, f(t) = exp (−d · t) + exp (g · t) − 1, where d is the rate of regression or decay and g is rate of tumor growth. The analysis recognizes that in patients receiving therapy, tumor regression and growth can occur concurrently because the fraction of tumor that is sensitive to a therapy regresses or disappears and that which is (relatively) resistant continues to grow. And it describes this simultaneous regression and growth in terms of rates of regression and growth. Effective therapies can both impact the rates of regression and growth, accelerating the regression rate and reducing the growth rate. We have shown that effective therapies can affect the rate of growth of tumor that, although not sufficiently sensitive to be killed by the therapy, can have its rate of growth slowed.
Figures 1 and 2 demonstrate the effect axitinib had on the regression and growth rates. Figure 1 shows graphs for four patients that depict the quantity of tumor measured radiographically with the curve fits guided by the equation above. The two upper panels are examples of two patients in whom the rates of tumor growth (g) while on axitinib decreased compared with the rates before receiving axitinib, whereas the two lower panels are examples of two patients in whom the rates of tumor growth (g) increased. Some of the patients who enrolled on the study had been receiving another therapy prior to their enrollment and began the axitinib study because their tumor was growing. In some, the tumors grew faster while on axitinib than on their prior therapies, explaining the observed increases in the rates of growth. Figure 2 depicts prestudy and on-study (post) growth rates (g) in 11 of the 13 patients to illustrate the effect of axitinib on the rate of tumor growth. Modest decreases in the rates of growth can be seen in four patients, whereas in the other seven patients, a reduction could not be demonstrated. Note that Figure 2 does not depict tumor quantity but rather the rate of growth of tumor. So that despite the reduction of the tumor growth rates seen in some patients, their tumor quantity did not decrease, but rather increased, albeit at a rate that was slower than that before the administration of axitinib. With regard to survival, for the 13 patients enrolled on the axitinib trial, the median PFS was 5.48 months (1.8–10.92 months), the same as the time to treatment failure. The median overall survival (OS) was longer than 13.7 months.
Figure 1.
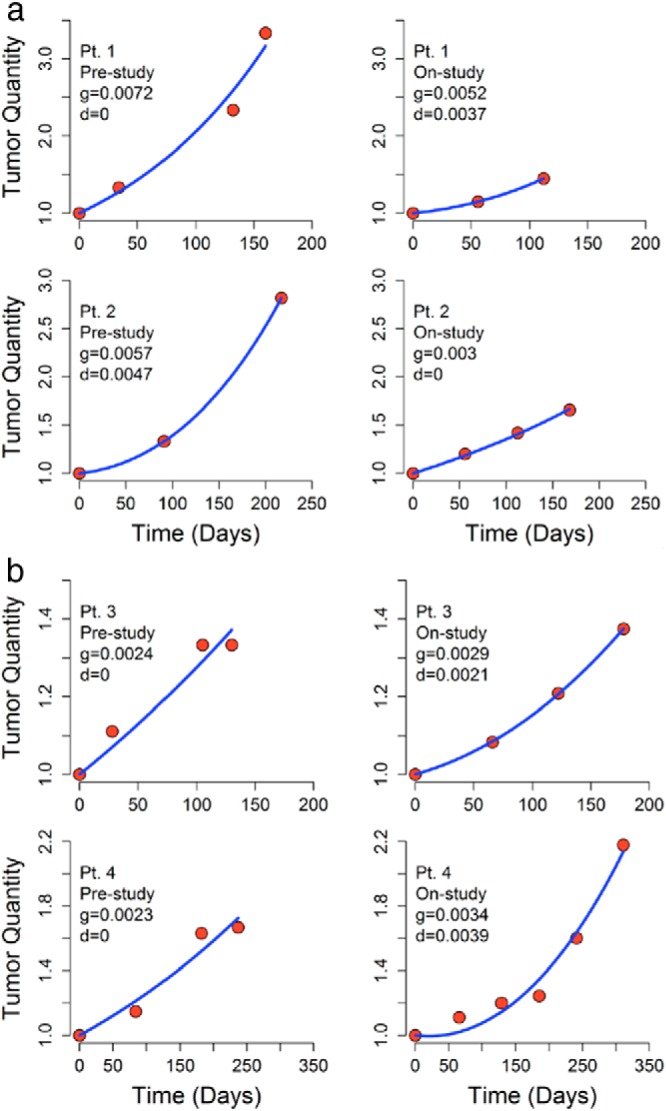
Graphical presentation of tumor quantities over time in four patients enrolled in the axitinib trial. The data represent the sum of the longest diameters of the same tumor masses, both before the study and on-study. The curve fits allow one to calculate the growth (g) and regression (d) rate constants that describe the simultaneous occurrence of growth of resistant tumor cells and regression of sensitive cells. In the two upper panels, one can see the results in two patients (patient 7 above and patient 9 below) in whom the rate of tumor growth decreased while on axitinib. The two in the lower panels (patient 4 above and patient 11 below) did not experience a decrease in the rate of their tumor growth. Note that in all cases the quantity of tumor has continued to increase. There is only a difference in the rate of growth, not in the amount of tumor. Note also the x-axes are different.
Figure 2.
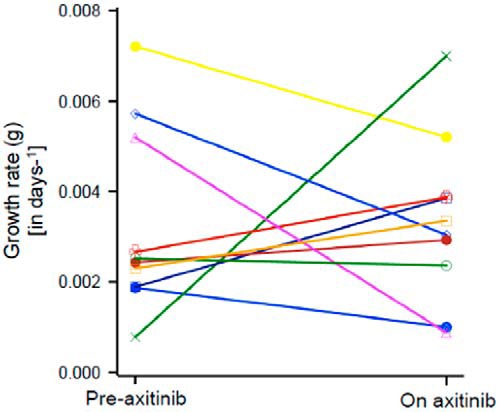
Summary of data such as that depicted in Figure 1 for 10 of the 13 patients enrolled on the axitinib study. The growth rate (in days−1) is shown. The rate fell in four patients but in the other six it remained unchanged or even increased. Because not all patients were being followed up without treatment, an increase can occur if the prior therapy was able to slow tumor growth better than axitinib. Again, note this is the rate of growth, not the absolute tumor quantity.
Finally, to confirm that in the tumors of the patients enrolled on study evidence of VEGF-mediated angiogenesis could be demonstrated, 11 of the 13 samples were stained with antibodies against CD31, a marker of new blood vessels as well as antibodies recognizing VEGFR2 and phospho-VEGFR2-Tyr951. These results together with additional clinical information regarding mitotane and axitinib are summarized in Supplemental Table 4. Examples of the staining results are also depicted in Figure 3. In this small sample, there was no correlation between the staining intensity for any of the proteins examined and the effect of axitinib on the rate of tumor growth. Excluding one outlier, there was a correlation between phosphorylated VEGFR2 and MVD.
Figure 3.
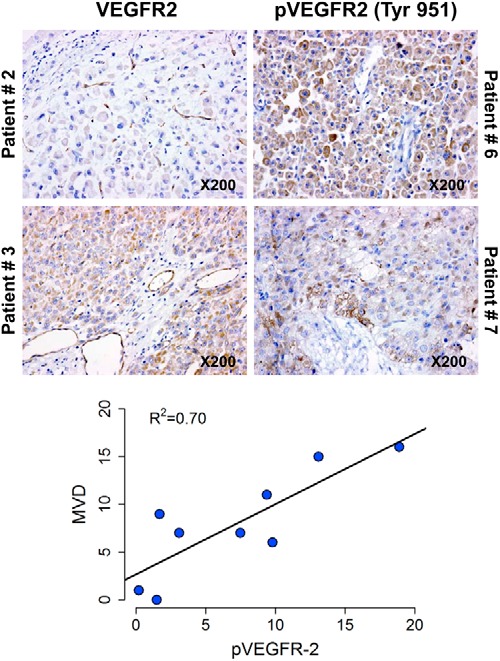
Examples of immunohistochemistry results. Excluding one outlier a correlation between phosphorylated VEGFR2 and MVD was observed.
Discussion
ACC is a rare malignancy, with an estimated incidence of 1 per million to 2 per million people per year (1–4). Typically, women are more commonly affected than men, at a ratio of 1.5:1 (6, 24, 25). Clinical presentations are variable; however, approximately two thirds of patients present with signs and symptoms of adrenal steroid excess, such as Cushing's syndrome with or without virilization (5, 8, 10). Mitotane continues to be used in the treatment of ACC both in the adjuvant and metastatic settings. In patients with advanced disease who are not surgical candidates, EDP plus mitotane, or streptozocin plus mitotane are commonly used chemotherapy regimens (7). The FIRM-ACT trial showed that response rates and PFS were better with EDP and mitotane than with streptozocin plus mitotane as first-line therapy; however, there was no difference in OS or toxicities (23). And despite data showing clear efficacy of the EDP regimen, the 5-year survival for ACC ranges from 16% to 44% (5, 6, 9, 24, 26–30). Therefore, new treatment approaches are urgently needed.
Folkman (31), in considering the need for tumors to have blood vessels to survive, proposed that targeting angiogenesis would be an effective therapeutic strategy. Signaling through the VEGFR is reported to stimulate angiogenesis. Of the three VEGFRs, VEGFR1 (fms-like tyrosine kinase-1), VEGFR2 (fetal liver kinase-1/kinase insert domain receptor), and VEGFR3 (fms-like tyrosine kinase-4), VEGFR2 has the most significant role in mediating the growth of blood vessels. Sunitinib and sorafenib are multikinase inhibitors that target VEGFRs as well as other kinases. Axitinib is an oral, potent selective inhibitor of VEGFR1, VEGFR2, and VEGFR3. In ACC, as in nearly all other tumors, expression of VEGFR has been reported, providing support for studies targeting these receptors (11, 12, 32).
Unfortunately, we must conclude that axitinib has limited activity in ACC despite evidence demonstrating a slowing of growth in some patients. And although investigators evaluating sunitinib in ACC could not convincingly rule out serum drug levels may have been reduced by mitotane-induced cytochrome P450–3A4 activity (33), raising the possibility of a significant drug interaction, we concluded it was unlikely this had occurred in this study for several reasons. First, the clinical toxicities observed while patients were treated with axitinib were frequent and precluded sustained escalation of doses above the starting 5 mg administered orally twice a day. We were persuaded this indicated adequate exposure to axitinib. Second, as summarized in Supplemental Table 4, in most patients mitotane had never been given or had been discontinued more than 2 months before receiving axitinib and hence 4–6 months before an escalation of axitinib to 7 and 10 mg, respectively, was attempted. Three patients had never been treated with mitotane, whereas 10 discontinued mitotane including five who stopped more than 2 months before receiving axitinib. Although three of the four patients who were able to attempt a dose of 10 mg axitinib twice daily had discontinued mitotane less than 1 month before starting axitinib, they had been off mitotane 4–5 months when the dose was increased to 10 mg, and this included two of the four whose rate of tumor growth decreased. Finally, 6 of 13 patients (42%) attempted an escalation of the axitinib dose. This percentage is similar to that in the original phase 3 study comparing axitinib and sorafenib in patients with advanced renal cell carcinoma, in which the dose was increased above 5 mg twice daily in 132 patients receiving axitinib (37%) (23).
This study is the third study in ACC using a small-molecule TKI targeting the VEGFRs that has failed to demonstrate benefit (33, 34). The totality of the data suggests that targeting the VEGFRs in ACC is largely an ineffective treatment strategy. The first reported study administered a combination of iv paclitaxel at a dose of 60 mg/m2 every week and 400 mg of sorafenib orally twice a day. No patient had benefit (34). Tumor assessment at the first evaluation, 8 weeks into therapy, demonstrated progression in the first 10 patients enrolled, appropriately leading to premature termination of the trial. Importantly, progression was marked, with all patients exceeding the 120% RECIST cutoff. The quantity of the tumor as per RECIST was 136%–252% that of the entry amount of 100%, with a median of 161% and a mean of 171%, indicating progression sufficient to qualify as progressive disease by RECIST had occurred rapidly in most patients, well before the 8-week assessment period. The second study, evaluating oral sunitinib administered at a standard dose of 50 mg for 4 weeks followed by a 2-week rest period was equally ineffective (33). Although, as noted above, some concerns were raised as to whether mitotane administration might have accelerated sunitinib metabolism, this was unlikely to have impacted outcomes substantially. The observation that among the five patients with stable disease, only one was receiving mitotane likely reflects their indolent disease course rather than any efficacy. And as with sorafenib, no responses were seen among 38 patients enrolled on the study with a diagnosis of ACC. The median PFS of 83 days, exactly the time of the first evaluation, reflects the lack of activity. Indeed, 23 patients had experienced disease progression and six had died by the time of the first evaluation at 12 weeks, and as with sorafenib, the data show that with a median tumor size of 132%, progression had in fact occurred rapidly, earlier than 12 weeks.
The investigators conducting the FIRM-ACT study concluded that “the poor overall survival rates in our study confirm … the need for improved treatment options” (23). The published data together with the 13 patients reported here bring to 61 (10 sorafenib + 38 sunitinib + 13 axitinib) the total number of patients with ACC treated with a TKI targeting the VEGFRs, and in no patient has an objective RECIST response to therapy been recorded. The median PFS of 8 and 12 weeks with sorafenib and sunitinib, respectively, with values likely shorter as discussed above, are inferior to those of the EDP regimen and comparable with or inferior to single-agent streptozocin. Thus, the use of sorafenib and sunitinib for ACC cannot be seen as an improved treatment option. The median PFS with axitinib of 5.48 months (confidence interval 3.67 months, not estimable; Figure 4) most likely reflects the fact that these were patients with more indolent disease as reflected by a median survival prior to enrollment of 2.59 years and an overall survival of 26.92 months (confidence interval 9.05 months, not estimable; Figure 5); we do not interpret this as indicative of a better outcome than achieved with streptozocin. The existing data suggest instead the following: 1) to date, VEGFR has not been shown to be a valuable target in ACC; 2) targeting VEGFR is a strategy with limited effectiveness, and further evaluation of TKIs targeting VEGFR in ACC should be considered with caution; and 3) consideration should be given to directing future efforts elsewhere if we are to find improved treatment options that are effective and so desperately needed in the therapy of ACC.
Figure 4.
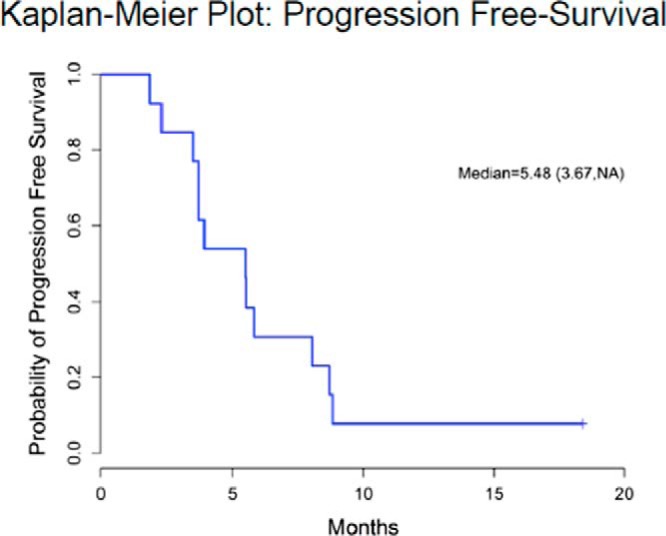
Kaplan-Meier curve showing PFS outcomes in patients.
Figure 5.
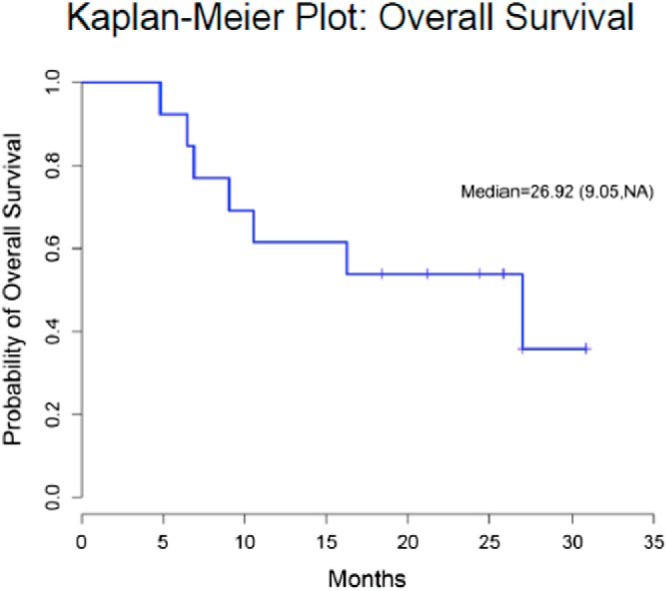
Kaplan-Meier curve showing OS outcomes in patients.
Acknowledgments
Footnotes
- ACC
- adrenocortical carcinoma
- EDP
- etoposide, doxorubicin, and cisplatin
- FIRM-ACT
- First International Randomized Trial in Locally Advanced and Metastatic Adrenocortical Carcinoma Treatment
- MVD
- microvessel density
- OS
- overall survival
- PFS
- progression-free survival
- RECIST
- Response Evaluation Criteria in Solid Tumors
- TKI
- tyrosine kinase inhibitor
- VEGF
- vascular endothelial growth factor
- VEGFR
- VEGF receptor.
References
- 1. Wajchenberg BL, Albergaria Pereira MA, Medonca BB, et al. Adrenocortical carcinoma: clinical and laboratory observations. Cancer. 2000;88(4):711–736 [PubMed] [Google Scholar]
- 2. Dackiw AP, Lee JE, Gagel RF, Evans DB. Adrenal cortical carcinoma. World J Surg. 2001;25(7):914–926 [DOI] [PubMed] [Google Scholar]
- 3. Allolio B, Fassnacht M. Clinical review: adrenocortical carcinoma: clinical update. J Clin Endocrinol Metab. 2006;91(6):2027–2037 [DOI] [PubMed] [Google Scholar]
- 4. Third National Cancer Survey: incidence data. Natl Cancer Inst Monogr. 1975:i-x, 1–454 [PubMed] [Google Scholar]
- 5. Abiven G, Coste J, Groussin L, et al. Clinical and biological features in the prognosis of adrenocortical cancer: poor outcome of cortisol-secreting tumors in a series of 202 consecutive patients. J Clin Endocrinol Metab. 2006;91(7):2650–2655 [DOI] [PubMed] [Google Scholar]
- 6. Luton JP, Cerdas S, Billaud L, et al. Clinical features of adrenocortical carcinoma, prognostic factors, and the effect of mitotane therapy. N Engl J Med. 1990;322(17):1195–1201 [DOI] [PubMed] [Google Scholar]
- 7. Schteingart DE, Doherty GM, Gauger PG, et al. Management of patients with adrenal cancer: recommendations of an international consensus conference. Endocr Relat Cancer. 2005;12(3):667–680 [DOI] [PubMed] [Google Scholar]
- 8. Koschker AC, Fassnacht M, Hahner S, Weismann D, Allolio B. Adrenocortical carcinoma—improving patient care by establishing new structures. Exp Clin Endocrinol Diabetes. 2006;114(2):45–51 [DOI] [PubMed] [Google Scholar]
- 9. Pommier RF, Brennan MF. An eleven-year experience with adrenocortical carcinoma. Surgery. 1992;112(6):963–970; discussion 970–971 [PubMed] [Google Scholar]
- 10. Libe R, Fratticci A, Bertherat J. Adrenocortical cancer: pathophysiology and clinical management. Endocr Relat Cancer. 2007;14(1):13–28 [DOI] [PubMed] [Google Scholar]
- 11. Xu YZ, Zhu Y, Shen ZJ, et al. Significance of heparanase-1 and vascular endothelial growth factor in adrenocortical carcinoma angiogenesis: potential for therapy. Endocrine. 2011;40(3):445–451 [DOI] [PubMed] [Google Scholar]
- 12. Feige JJ. Angiogenesis in adrenocortical physiology and tumor development. Ann Endocrinol (Paris). 2009;70(3):153–155 [DOI] [PubMed] [Google Scholar]
- 13. Lee JO, Lee KW, Kim CJ, et al. Metastatic adrenocortical carcinoma treated with sunitinib: a case report. Jpn J Clin Oncol. 2009;39(3):183–185 [DOI] [PubMed] [Google Scholar]
- 14. [Accessed January 5, 2014]. http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/202324lbl.pdf.
- 15. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–247 [DOI] [PubMed] [Google Scholar]
- 16. Stein WD, Gulley JL, Schlom J, et al. Tumor regression and growth rates determined in five intramural NCI prostate cancer trials: the growth rate constant as an indicator of therapeutic efficacy. Clin Cancer Res. 2011;17(4):907–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stein WD, Figg WD, Dahut W, et al. Tumor growth rates derived from data for patients in a clinical trial correlate strongly with patient survival: a novel strategy for evaluation of clinical trial data. Oncologist. 2008;13(10):1046–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stein WD, Yang J, Bates SE, Fojo T. Bevacizumab reduces the growth rate constants of renal carcinomas: a novel algorithm suggests early discontinuation of bevacizumab resulted in a lack of survival advantage. Oncologist. 2008;13(10):1055–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stein WD, Wilkerson J, Kim ST, et al. Analyzing the pivotal trial that compared sunitinib and IFN-α in renal cell carcinoma, using a method that assesses tumor regression and growth. Clin Cancer Res. 2012;18(8):2374–2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wedam SB, Low JA, Yang SX, et al. Antiangiogenic and antitumor effects of bevacizumab in patients with inflammatory and locally advanced breast cancer. J Clin Oncol. 2006;24(5):769–777 [DOI] [PubMed] [Google Scholar]
- 21. Yang SX, Kummar S, Steinberg SM, et al. National Cancer Institute Phase 0 Working Group. Immunohistochemical detection of poly(ADP-ribose) polymerase inhibition by ABT-888 in patients with refractory solid tumors and lymphomas. Cancer Biol Ther. 2009;8(21):2004–2009 [DOI] [PubMed] [Google Scholar]
- 22. Yang SX, Costantino JP, Kim C, et al. Akt phosphorylation at Ser473 predicts benefit of paclitaxel chemotherapy in node-positive breast cancer. J Clin Oncol. 2010;28(18):2974–2981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fassnacht M, Terzolo M, Allolio B, et al. Combination chemotherapy in advanced adrenocortical carcinoma. N Engl J Med. 2012;366(23):2189–2197 [DOI] [PubMed] [Google Scholar]
- 24. Icard P, Goudet P, Charpenay C, et al. Adrenocortical carcinomas: surgical trends and results of a 253-patient series from the French Association of Endocrine Surgeons study group. World J Surg. 2001;25(7):891–897 [DOI] [PubMed] [Google Scholar]
- 25. Wooten MD, King DK. Adrenal cortical carcinoma. Epidemiology and treatment with mitotane and a review of the literature. Cancer. 1993;72(11):3145–3155 [DOI] [PubMed] [Google Scholar]
- 26. Haak HR, Hermans J, van de Velde CJ, et al. Optimal treatment of adrenocortical carcinoma with mitotane: results in a consecutive series of 96 patients. Br J Cancer. 1994;69(5):947–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Venkatesh S, Hickey RC, Sellin RV, Fernandez JF, Samaan NA. Adrenal cortical carcinoma. Cancer. 1989;64(3):765–791 [DOI] [PubMed] [Google Scholar]
- 28. Soreide JA, Brabrand K, Thoresen SO. Adrenal cortical carcinoma in Norway, 1970–1984. World J Surg. 1992;16(4):663–667; discussion 668 [DOI] [PubMed] [Google Scholar]
- 29. Schulick RD, Brennan MF. Adrenocortical carcinoma. World J Urol. 1999;17(1):26–34 [DOI] [PubMed] [Google Scholar]
- 30. Bellantone R, Ferrante A, Boscherini M, et al. Role of reoperation in recurrence of adrenal cortical carcinoma: results from 188 cases collected in the Italian National Registry for Adrenal Cortical Carcinoma. Surgery. 1997;122(6):1212–1218 [DOI] [PubMed] [Google Scholar]
- 31. Folkman J. Tumor angiogenesis: a possible control point in tumor growth. Ann Intern Med. 1975;82(1):96–100 [DOI] [PubMed] [Google Scholar]
- 32. Sasano H, Ohashi Y, Suzuki T, Nagura H. Vascularity in human adrenal cortex. Mod Pathol. 1998;11(4):329–333 [PubMed] [Google Scholar]
- 33. Kroiss M, Quinkler M, Johanssen S, et al. Sunitinib in refractory adrenocortical carcinoma: a phase II, single-arm, open-label trial. J Clin Endocrinol Metab. 2012;97(10):3495–3503 [DOI] [PubMed] [Google Scholar]
- 34. Berruti A, Sperone P, Ferrero A, et al. Phase II study of weekly paclitaxel and sorafenib as second/third-line therapy in patients with adrenocortical carcinoma. Eur J Endocrinol. 2012;166(3):451–458 [DOI] [PubMed] [Google Scholar]