

Abstract
Galactosidases are widespread enzymes that are used for manifold applications, including production of prebiotics, biosynthesis of different transgalactosylated products, improving lactose tolerance and in various analytical approaches. The nature of these applications often require galactosidases to be present in a purified form with clearly defined properties, including precisely determined substrate specificities, low sensitivity to inhibitors, and high efficiency and stability under distinct conditions. In this study, we present the recombinant expression and purification of two previously uncharacterized β-galactosidases from Aspergillus nidulans as well as one β-galactosidase from Aspergillus niger. All enzymes were active toward p-nitrophenyl-β-d-galactopyranoside as substrate and displayed similar temperature and pH optima. The purified recombinant galactosidases digested various complex substrates containing terminal galactose β-1,4 linked to either N-acetylglucosamine or fucose, such as N-glycans derived from bovine fibrin and Caenorhabditis elegans. In our comparative study of the recombinant galactosidases with the commercially available galactosidase from Aspergillus oryzae, all enzymes also displayed various degrees of activity toward complex oligosaccharides containing β-1,3-linked terminal galactose residues. All recombinant enzymes were found to be robust in the presence of various organic solvents, temperature variations, and freeze/thaw cycles and were also tested for their ability to synthesize galactooligosaccharides. Furthermore, the use of fermentors considerably increased the yield of recombinant galactosidases. Taken together, we demonstrate that purified recombinant galactosidases from A. niger and from A. nidulans are suitable for various glycobiological and biotechnological applications.
Electronic supplementary material
The online version of this article (doi:10.1007/s00253-013-5192-3) contains supplementary material, which is available to authorized users.
Keywords: Aspergillus, Galactosidase, Recombinant protein, N-glycans, Biotechnology, Pichia pastoris
Introduction
β-Galactosidases (β-d-galactohydrolases, EC 3.2.1.23) are ubiquitous enzymes capable of hydrolyzing terminal β-d-galactosyl moieties from substrates such as disaccharides, diverse glycoconjugates, and polysaccharides. They belong to the glycoside hydrolase clan A (GH-A), which encompasses, among others, β-galactosidase families 35 and 42. Under distinct reaction conditions, β-galactosidases can also catalyze transglycosylation reactions using various acceptor molecules, a capability used for the synthesis of, e.g., galactooligosaccharides (GOS) (Park and Oh 2010). β-Galactosidases have been widely used to hydrolyze lactose and render dairy products consumable for lactose-intolerant individuals. Further applications of these enzymes range from analytical studies (Titz et al. 2009) to glycan remodeling (Iskratsch et al. 2009) and include various processes of biotechnological and medical importance (Husain 2010).
The β-galactosidases in general use are mainly derived from microbial sources, which include both prokaryotic and eukaryotic organisms. They can be purified at high yields directly from fungal organisms (Tanaka et al. 1975; Husain 2010; Sen et al. 2012) or produced in a recombinant form at high levels in different expression systems (Wang et al. 2009; Oliveira et al. 2011). Other commonly used sources of β-galactosidases include bacteria (e.g., bifidobacteria (Hung et al. 2001; Hsu et al. 2005) and Streptococcus pneumonia (Hughes and Jeanloz 1964)), yeasts (e.g., Kluyveromyces lactis (Dickson and Markin 1978)), plants (e.g., from jack bean (Arakawa et al. 1974; Li et al. 1975)), and animals (e.g., bovine testes β-galactosidase (Distler and Jourdian 1973)).
One of the most widely used fungal β-galactosidase originates from Aspergillus oryzae. As such, detailed data on its characteristics and many applications have been reported (Scheckermann et al. 1997; Zeleny et al. 1997; Torres et al. 2003; Vera et al. 2011). The β-galactosidase from Aspergillus niger was previously used to overcome the Lac− phenotype of Saccharomyces cerevisiae (Kumar et al. 1992) and its heterologous expression in various yeast hosts as well as its use in lactose hydrolysis and transgalactosylation reactions was reviewed recently (Oliveira et al. 2011). A closely related recombinant β-galactosidase from A. niger van Tiegh has been recently described as a highly acid-stable enzyme with prospective use in the treatment of lactose intolerance (Hu et al. 2010) and a purified native β-galactosidase from this strain possesses a rather low pH optimum (O’Connell and Walsh 2010). In 2008, two β-galactosidases secreted by Aspergillus carbonarius ATCC6276 were purified to homogeneity and were shown to be resilient to simulated gastric conditions (O’Connell and Walsh 2008). Most recently, a β-galactosidase was purified from Aspergillus alliaceus and partially characterized; this enzyme, unlike various other fungal galactosidases, has a pH optimum in the neutral range, an enzymatic property suitable for the preparation of low-lactose milk (Sen et al. 2012).
Since the Aspergillus spp. are obviously an established source of robust but different β-galactosidases, we sought to thoroughly characterize pure forms of recombinant β-galactosidases using various substrates and conditions. In addition, as the A. oryzae β-galactosidase only digests terminal β-1,3-linked galactose residues at a very slow rate (Zeleny et al. 1997), we examined whether any of the galactosidases from other Aspergillus spp. display higher activity toward these residues, a feature interesting for glycobiological analyses. In this study, we report on the expression and characterization of two β-galactosidases from Aspergillus nidulans and one β-galactosidase from A. niger. We have expressed C-terminally His-tagged versions of these previously putative β-galactosidases in Pichia pastoris as an expression host and purified the recombinant enzymes to apparent homogeneity. In-depth characterization of the recombinant proteins and comparison with the commercially available, native galactosidase from A. oryzae were performed. Additionally, we emphasized the evaluation of the recombinant enzymes as an analytical tool, such as for glycobiological analyses, which is an important application of β-galactosidases rarely evaluated for novel enzymes. The obtained data show that these recombinant β-galactosidases are promising as reagents for a variety of glycobiological and biotechnological applications.
Materials and methods
Cloning of sequences encoding Aspergillus β-galactosidases
Open reading frames (ORF) from A. niger (lacA, gene ID 4977988) and A. nidulans (lacA, gene ID 2876531; lacB, gene ID 2876757) were polymerase chain reaction (PCR) amplified (Expand Polymerase) from cDNA templates (A. niger ATCC1015 and A. nidulans A713 strain (Pisanelli et al. 2010)) using the following primers: KpnI_Aniger_lacA_fw (AGGTACCGCGTCCATTAAGCATCGAATCAAT), NotI_Aniger _lacA_rv (AGCGGCCGCGTATGCACCCTTCCGCTTCTTGTA), EcoRI_Anidulans_lacA_fw (TGAATTCGTTGCTCTTACCCACAAGCTCAAT), NotI_Anidulans_lacA_rv (TGCGGCCGCATAGACACCCTTCCTAGACTGATAACG), EcoRI_Anidulans_lacB_fw (AGAATTCCAGAATAGTTCCCAATCCGAATG), and NotI_Anidulans_lacB_rv (AGCGGCCGCTAGTTGAAGACATTCGAATATGTAGACG). RNA samples, extracted with TRIzol reagent (Invitrogen) according to the manufacturer’s protocol from standard A. niger and A. nidulans cultures, were kindly provided by Matthias Steiger and Clemens Peterbauer (both from the University of Natural Resources and Life Sciences, Vienna, Austria). cDNA was produced from 500 ng total RNA using the SuperScript III (Invitrogen) reverse transcriptase according to the manufacturer’s guidelines. The PCR products, which lacked the region encoding the original signal peptide and native stop codon, were digested with the respective enzymes (New England Biolabs), ligated (NEB T4 Ligase) in frame with the digested pGAPZαA vector (Invitrogen), and used to transform E. coli Top10F′. Positive clones were selected on LBZeo (25 μg mL−1) plates and the insert sequence was verified by Sanger sequencing.
Expression in P. pastoris
pGAPZα vectors carrying the ORF of the galactosidases were linearized with DraI and transformed into P. pastoris X-33 for secreted expression mediated by the S. cerevisiae α-factor leader sequence. Positive clones were selected on YPDZeo (2 % (w/v) peptone, 1 % (w/v) yeast extract, 2 % (w/v) agar, 2 % (w/v) glucose, 100 μg mL−1 zeocin, pH 7.4) plates and used for an initial screening in YPDZeo (100 μg mL−1) medium. From 8 to 12 clones, the one with the highest activity in the culture supernatant was chosen for cultivation in 200 mL YPD medium (2 % (w/v) peptone, 1 % (w/v) yeast extract, 2 % (w/v) glucose, pH 7.4) at 25 °C on an orbital shaker at 180 rpm for a total of 48 h. Cultures were supplemented with an additional 1 % glucose after 24 h.
For large-scale expression of A. niger and A. nidulans rlacA in fermentors, the cultivation conditions and growth medium were essentially as described previously (Gasser et al. 2010). In short, batch cultivations in 1.5 L minimal mineral medium with glycerol as carbon source were performed in Minifors (Infors HT) bioreactor units. After a batch phase of approximately 25 h (25 g L−1 yeast dry mass), a constant glucose feed at a rate of 0.269 g min−1 was initiated and performed for a total process time of 100 h (100 g L−1 final yeast dry mass). Temperature was set to 25 °C, diluted oxygen concentration was kept at 20 % via stirrer speed, and pH was kept at pH 5.0 by the addition of ammonia throughout the cultivations. Samples for biomass determination and activity assays were taken in regular intervals. For biomass determination, 5 to 10 mL of culture samples were collected by centrifugation, washed twice with RO-H2O, and dried to constant weight at 80 °C.
Protein purification
Galactosidase-expressing P. pastoris clones were cultivated in 10 mL YPGZeo (2 % (w/v) peptone, 1 % (w/v) yeast extract, 1 % (w/v) glycerol, 100 μg mL−1 zeocin, pH 7.4) at 28 °C, 200 rpm on an orbital shaker overnight. From these cultures, main cultures in 200 mL YPD medium were inoculated at an OD600 = 0.05. Cultures were grown at 25 °C, 200 rpm for a total of 48 h, including feeding with additional 1 % glucose after 24 h. Cells were harvested by centrifugation at 4 °C and culture supernatants were collected for protein purification. All subsequent steps were performed at 4 °C. Culture supernatants were concentrated using an Amicon ultrafiltration unit (Millipore) with a 30 kDa NMWL filter (Millipore), followed by buffer exchange to Ni-NTA column loading buffer (20 mM Tris, 20 mM NaCl, 5 mM Imidazole, pH 8.5). Protein-containing solutions were mixed with pre-equilibrated 1 mL of Ni-NTA agarose slurry (Qiagen) and incubated on a rotator for 1 h. Then, the mixture was transferred to a plastic column and, after three washing steps with loading buffer containing 20 mM imidazole (two column volumes per step), recombinant proteins were eluted with 500 mM imidazole (one column volume per elution fraction). Purity of the recombinant enzymes was evaluated by Tris–glycine sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE; resolving gel: T12.5/C1, stacking gel: T5.7/C2.2). Pure fractions were pooled and imidazole was removed by buffer exchange to the storage buffer (20 mM Tris–Cl, 25 mM NaCl, pH 7.5) using Vivaspin columns (30 kDa NMWL; Millipore). All three recombinant proteins were stored at 4 °C and, as such, remained active with only minor loss of activity (<15 %) for at least 18 months.
Determination of protein concentration
Protein concentration of purified products was measured after the removal of imidazole using a bicinchoninic acid protein quantification kit (Sigma) with bovine serum albumin (BSA) as standard protein.
Product verification by peptide mass fingerprinting
Tryptic digests were essentially performed as described previously (Paschinger et al. 2012a). Briefly, protein bands were cut from Coomassie Brilliant Blue G-250-stained SDS-polyacrylamide gels and, after several washing steps, “in-gel” tryptic digestion was performed at 37 °C overnight. Peptides were released from the gel, dried in a SpeedVac, and resuspended in 10 μL 50 % acetonitrile/0.1 % trifluoroacetic acid prior to analysis in positive ion mode on an Autoflex Speed matrix-assisted laser desorption/ionization–time of flight mass spectrometer (MALDI-TOF/TOF MS; Bruker Daltonics) using 1 % α-cyano-4-hydroxycinnamic as matrix. The obtained list of different m/z ratios was used to verify protein identity using MS-Fit (ProteinProspector, UCSF; http://prospector.ucsf.edu/prospector/mshome.htm).
Removal of N-glycans using PNGase F
Approximately 2 μg of purified recombinant protein was supplemented with 1 % SDS and incubated at 95 °C for 5 min. After cooling on ice, 0.5 μL PNGase F (glycosidase F, NEB) or water was added as well as 1× G7 buffer (50 mM sodium phosphate, pH 7.5) and 1 % NP-40 (Nonidet P-40, octylphenoxypolyethoxyethanol) in a total volume of 20 μL. Reactions were incubated at 37 °C for 48 h before Tris–glycine SDS-PAGE (resolving gel: T12.5/C1, stacking gel: T5.7/C2.2) and Coomassie Brilliant Blue G-250 staining were performed.
Enzyme activity assays using p-nitrophenyl-monosaccharides as substrate
Standard enzymatic activity assays were performed in triplicate with either P. pastoris culture supernatants or purified products using p-nitrophenyl-β-d-galactopyranoside (PNPG; Sigma) as substrate. Standard assays were performed at a substrate concentration of 5 mM in McIlvaine buffer (citrate–phosphate buffer (McIlvaine 1921)), pH 4.5, at 37 °C in a reaction volume of 40 μL for 2 h. All purified enzymes were also tested for additional activities using a range of p-nitrophenyl-sugars as substrates (Table 2). After the addition of 200 μL of 0.4 M glycine/NaOH pH 10.4, A 405 nm was determined on a Tecan Infinite M200 microtiter plate reader (Tecan, Austria). Enzymes were appropriately diluted in order to measure reactions within the linear range of the assay. Enzyme units are given in micromoles of product (nitrophenol) per minute and specific activity as units per milligram of purified protein.
Table 2.
Activity of recombinant galactosidase preparations toward substrates not having a terminal β-galactose residue
| Percent activity | A. niger rlacA | A. nidulans rlacA | A. nidulans rlacB |
|---|---|---|---|
| β-Galactose | 100 | 100 | 100 |
| α-Galactose | <0.01 | <0.1 | <0.5 |
| α-Glucose | <0.1 | 0 | <2.0 |
| β-Glucose | <0.1 | <0.1 | <2.0 |
| α-Xylose | <0.05 | 0 | <2.0 |
| β-Xylose | <0.1 | <0.05 | <2.0 |
| α-GlcNAc | 0 | <0.05 | <0.15 |
| β-GlcNAc | 0 | <0.05 | <0.1 |
| α-GalNAc | 0 | <0.01 | 0 |
| β-GalNAc | ND | 0 | <1.0 |
| α-Mannose | 0 | 0 | <0.5 |
Activities were determined with different p-nitrophenyl substrates in standard reaction conditions for 4 h. Values are given in percent, with PNPG substrate defined as 100 % activity
ND not determined, GlcNAc N-acetylglucosamine, GalNAc N-acetylgalactosamine
Determination of the pH optimum, temperature optimum, temperature stability, and metal ion susceptibility
Tests were performed as described above at 37 °C with 5 mM PNPG as substrate. McIlvaine buffers with a pH range from 3 to 8 were used. Tests for the determination of temperature optimum were performed with 5 mM PNPG at different temperatures in a Gradient PCR Thermocycler (Bio-Rad) using the McIvaine buffer at the pH determined to be optimal for the specific galactosidase. For the determination of temperature stability, aliquots of 10 μL of the respective enzyme, diluted in storage buffer (20 mM Tris–Cl, 25 mM NaCl, pH 7.5), were incubated at 4, 30, 45, 55, 65, and 75 °C for 2 h. Remaining activity was determined using standard PNPG assays at 37 °C. For the reactions supplemented with BSA, the final concentration of BSA was 500 μg mL−1. The effect of divalent metal ions was tested using the standard PNPG assay mixtures supplemented with either 1 or 10 mM of the respective metal chloride salts at 37 °C.
Impact of organic solvents on activity and freeze/thaw experiments
Diluted enzyme was used in the setup with 5 mM PNPG as described previously, with the exception that McIlvaine buffer was partially replaced by acetonitrile, acetone, isopropanol (IPA), ethanol (EtOH), methanol (MeOH), or RO-H2O to yield the final concentrations of 0, 10, 25, 50, and 75 % (v/v) of the added solvents. The pH of the reaction mixture was adjusted to the optimal pH of the tested enzyme. For freeze/thaw experiments, aliquots of galactosidases with a concentration of 100 μg mL−1 (±1 mg mL−1 BSA) were stored in storage buffer (20 mM Tris–Cl, 25 mM NaCl, pH 7.5) at −20 °C. After each freeze/thaw cycle, activity was measured using the standard PNPG assay as described previously.
Determination of Km, specific activity, kcat, and Ki
Standard assays were performed at 37 °C in McIlvaine buffers of the optimal pH for the respective enzyme. Substrate concentrations in the range of 0.1 to 10 mM PNPG were used. Michaelis constant (K m), specific activity, and k cat were calculated using the nonlinear curve fitting function available in the OriginPro 8 software package. For K i determination of galactose, PNGP assays were supplemented with galactose (1–1,000 mM) and IC50 was calculated based on exponential curve fitting using OriginPro. K i was calculated based on the Cheng–Prusoff equation: K i = IC50/(1 + [S]/K m) with IC50 being the half maximal inhibitory concentration of galactose, [S] the PNPG concentration (5 mM), and K m the Michaelis–Menten constant. For Dixon plots (Dixon 1953), standard assays were performed at 37 °C with PNPG concentrations of 5, 2.5, and 0.5 mM and supplemented with galactose in the range of 0 to 50 mM.
Determination of α-mannosidase side activity
2-Aminopyridine (PA)-labeled Man9 (Takara) was used. Ten picomoles (1 μL) of substrate was mixed with 1 μL MES pH 4.5 or 6.9 and 0.25 μL 0.2 M CaCl2 and combined with water and 0.5 μg purified enzyme (1.5 μg in case of A. oryzae galactosidase) to a total reaction volume of 5 μL. Reactions were incubated for 16 h prior to analysis by reverse-phase high-performance liquid chromatography (HPLC) using a Shimadzu HPLC system equipped with a fluorescence detector (either RF 10AXL or RF 20AX) and a Hypersil ODS Column (5 μm, 4 × 250 mm) equilibrated with 0.1 M ammonium acetate, pH 4.0 (solvent A). Runs were performed at a flow rate of 1.5 mL min−1 with a 1 % increase of solvent B (30 % MeOH) per minute for a total of 25 min.
Enzyme activity assays using dabsylated-GalGal glycopeptide as substrate
Dabsylated-GalGal [Gal-β-1,4-GlcNAc-β-1,2-Man-α-1,6(Gal-β-1,4-GlcNAc-β-1,2-Man-α-1,3)Man-β-1,4-GlcNAc-β-1,4-GlcNAc-β-1-Asn] glycopeptide (0.25 nmol) derived from bovine fibrin and possessing the common peptide sequence GENR (Gly-Glu-Asn-Arg) (Altmann et al. 1995) was used in a total reaction volume of 10 μL with galactosidases in McIlvaine buffer pH 4.5. Purified enzyme (0.2 μg) was added in 24-h intervals. Assays were performed at 37 °C (or at 50 °C for A. nidulans rlacB) for a total of 120 h (or 240 h for A. nidulans rlacB). Removal of galactose moieties was monitored in regular intervals on an Autoflex Speed MALDI-TOF/TOF MS in positive ion mode using 1 % α-cyano-4-hydroxycinnamic as matrix.
Enzyme activity assays using PA-labeled N-glycan structure carrying the Galβ1,4Fuc epitope as substrate
PA-labeled N-glycan structure carrying the Galβ1,4Fuc epitope derived from Caenorhabditis elegans (Yan et al. 2012) was used as substrate. Reactions were performed in McIlvaine buffer (pH 4 to 5, i.e., at the optimal pH for each galactosidase tested) at 37 °C for 24 h with 0.5 μg purified enzyme per assay and analyzed using an Autoflex Speed MALDI-TOF/TOF MS in positive ion mode using 0.3 % 6-aza-2-thiothymine (Sigma) as matrix.
Determination of β-1,3 and β-1,4 specificity
Fifty micrograms of lacto-N-tetraose [Gal-β-1,3-GlcNAc-β-1,3-Gal-β-1,4-Glc] and lacto-N-neotetraose [Gal-β-1,4-GlcNAc-β-1,3-Gal-β-1,4-Glc] (Dextra Laboratories) were labeled with PA as described previously (Paschinger et al. 2012b). PA-labeled substrates (final concentration, 40 μM) were used for enzymatic assays in McIlvaine buffer pH 4.5, 37 °C with 0.5 μg purified enzyme in a total reaction volume of 10 μL for a total of 48 h. Samples were taken at 2, 24, and 48 h and analyzed by using an Autoflex Speed MALDI-TOF/TOF MS in positive ion mode using 0.3 % 6-aza-2-thiothymine (Sigma) as matrix.
Lactose substrate assays and galactooligosaccharide formation
Lactose was used as substrate at a concentration of 20 % (w/v) in McIlvaine buffer optimal for the respective galactosidase activity (pH 4 to 5). Four micrograms of purified galactosidase enzyme was added and reactions were performed in a total volume of 50 μL for 24 h. Samples were taken at 4 and 24 h for analysis by HPLC (Shimadzu) and an RI detector (Knaur) using a Rezex RHM column (Phenomenex) with 4 mM H2SO4 as solvent at a flow rate of 1 mL min−1 at 60 °C. Monosaccharides and disaccharides used as HPLC standards were purchased from Sigma. GOS were obtained from Agrana Stärke GmbH, Austria.
A. oryzae β-galactosidase was purchased from Sigma and purified by cation-exchange chromatography (Zeleny et al. 1997). The DNA sequences of the recombinant galactosidases, as they were obtained in this study, will be available under the GenBank accession numbers KC175324 (A. niger lacA), KC145279 (A. nidulans lacA), and KC145280 (A. nidulans lacB).
Results
Expression and purification of recombinant galactosidases
For the A. niger lacA and the A. nidulans lacA genes, the obtained sequences1 matched the sequences deposited in the NCBI database (GenBank gene IDs 4977988 and 2876531). Sequence analysis of the PCR products amplified from an A. nidulans cDNA corresponding to the ORF of the A. nidulans lacB gene identified in this study revealed small differences between the amplified transcript1 and the predicted ORF deposited in the NCBI database (GenBank gene ID 2876757). The observed differences are most probably due to improper in silico prediction of introns and intron splicing sites as the relevant database entries currently contain only predicted open reading frames based on the genome of A. nidulans. All three enzymes contain conserved sequence motifs that are typical for glycoside hydrolase family 35, a member of the glycoside hydrolase clan A (GH-A) (Henrissat and Bairoch 1996; Cantarel et al. 2009). The protein sequence alignment of the three enzymes (Online Resource 1, Supplemental Fig. S1), as well as a phylogenetic comparison with other galactosidases (Fig. 1a), showed that the sequence similarity between the translated A. niger lacA and A. nidulans lacA ORF is higher, to each other as compared with the A. nidulans lacB gene product. After cloning of the three ORF and initial expression screening, proteins of approximately 130 kDa in size (denoted as A. niger and A. nidulans rlacA and rlacB hereafter) were secreted into the supernatant of P. pastoris cultures and could be detected by Western blotting (via C-terminal Myc/His-tag; data not shown) and by Coomassie Brilliant Blue staining. Furthermore, β-galactosidase activity was confirmed in P. pastoris supernatants using PNPG as substrate, whereas β-galactosidase activity was absent in the supernatants of control “empty plasmid” clones. After selection of the best expressing clone for each recombinant galactosidase, the P. pastoris supernatants were subject to a single-step His-tag affinity batch purification using Ni-NTA agarose. As judged by SDS-PAGE analysis, pure proteins were obtained (Fig. 1b). Peptide mass fingerprinting (tryptic digests followed by MALDI-TOF/TOF MS analysis) was performed in order to verify the identity of the recombinant proteins. Sixty-seven to eighty percent of the tryptic peptides covering 40–51 % protein sequence could be identified (Online Resource 1, Supplemental Fig. S2). None of the three recombinant proteins contained tryptic peptides that would match the secretion signal peptide. This indicates that signal peptide cleavage by the Kex2 protease occurred correctly. For A. niger rlacA, we observed peptides that contain the double glutamine–alanine repeat sequence that follows the Kex2 site. Although not analyzed further, this may indicate that the cleavage at the glutamine–alanine repeats by the Ste13 protease in P. pastoris is not efficient in the case of A. niger rlacA, a phenomenon that was reported to occur in a protein-dependent manner in P. pastoris (Ghosalkar et al. 2008; Invitrogen 2010). Additionally, each galactosidase contains several potential N-glycosylation sites (A. niger rlacA has 13, A. nidulans rlacA has 7, and A. nidulans rlacB has 10 putative N-glycosylation sites; Online Resource 1, Supplemental Fig. S2) and SDS-PAGE analysis shows a smear suggestive of heterogeneous N-glycosylation of the recombinant proteins (Fig. 1b). Thus, we performed a PNGase F digest with all three enzymes in order to remove potential high mannose N-glycan structures typical for P. pastoris (Hamilton et al. 2006). As expected for glycosylated proteins, digestion with PNGase F led, due to removal of the N-glycans, to an increased mobility and a considerable reduction in heterogeneity of the protein bands as judged by SDS-PAGE (Fig. 1b).
Fig. 1.

Galactosidase phenogram and SDS-PAGE analysis of recombinant enzymes. a Protein alignment based on the ClustalW method was performed in MegAlign 7.2.1 (DNAstar Lasergene software package) using full-length protein sequences of galactosidases as obtained from the GenBank database. The phenogram was prepared using the MegAlign’s built-in phylogenetic tree function. The enzymes described in this study are indicated by a gray background (including the A. oryzae enzyme used for comparative purposes). The amino acid sequences with the following IDs were used: A. niger lacA ID XP_001389622, A. niger ID A2QL84, A. nidulans lacA ID XP_658360, A. nidulans lacB ID XP_658584, A. clavatus ID XP_001268843, A. fumigatus ID XP_752787, A. fumigatus ID XP_748360, A. oryzae ID XP_001727461, Penicilium sp. ID Q700S9, Trichoderma reesei CAD70669, Homo sapiens ID NP_000395, K. lactis XP_452194, K. lactis 3OB8_A, and Escherichia coli ID NP_414878. b SDS-PAGE (Tris–glycine; resolving gel: T12.5/C1, stacking gel: T5.7/C2.2) analysis of purified recombinant enzymes. Each galactosidase was obtained free from visible contaminations as judged by Coomassie Brilliant Blue staining. Purified recombinant proteins were incubated with (+) or without (−) PNGase F to verify whether they carry N-glycans. MW molecular weight in kilodaltons
Enzyme kinetics and characteristics of recombinant galactosidases
We determined the basic enzyme kinetic parameters of the purified recombinant enzymes and compared them with the native A. oryzae galactosidase (Zeleny et al. 1997), which is widely used in glycobiotechnological research (Scheckermann et al. 1997; Torres et al. 2003; Vera et al. 2011). The K m value of the A. niger rlacA and A. nidulans rlacA galactosidases toward PNPG was very similar and just below 2 mM, whereas the other two enzymes (A. nidulans rlacB and A. oryzae galactosidase) appear to have slightly lower affinity toward this substrate (Table 1). We also observed up to 30-fold difference in specific activity among the studied enzymes, with A. niger rlacA displaying the highest specific activity followed by A. nidulans rlacA. The low activity of A. nidulans rlacB was already observed in the initial screening of multiple P. pastoris clones that expressed the enzyme, as well as in multiple batches of purified enzyme. Similarly, different batches of the other two recombinant galactosidases showed consistent K m values and specific activities (data not shown). Generally, the kinetic values obtained in this study, and shown in Table 1, are comparable to those previously reported for other β-galactosidases (Park and Oh 2010).
Table 1.
Properties of Aspergillus β-galactosidases
| K m [mM] | specific activity [μmol min−1 mg−1] | k cat [s−1] | k cat/K m [mM−1 s−1] | |
|---|---|---|---|---|
| A. niger rlacA | 1.84 ± 0.33 | 89.3 ± 5.6 | 195.9 ± 3.6 | 105.3 ± 15.2 |
| A. nidulans rlacA | 1.85 ± 0.21 | 29.2 ± 1.3 | 63.4 ± 0.9 | 34.1 ± 1.5 |
| A. nidulans rlacB | 3.33 ± 0.29 | 2.9 ± 0.1 | 6.6 ± 0.9 | 1.9 ± 0.3 |
| A. oryzae β-galactosidasea | 3.02 ± 0.61 | 19.3 ± 1.4 | 46.4 ± 1.2 | 14.6 ± 0.6 |
K m, specific activity, k cat, and k cat/K m (as determined with standard reaction conditions at 37 °C) of purified enzymes using PNPG as substrate are shown. Values represent averages of three measurements ± standard error
aNative A. orzyae galactosidase was obtained commercially and is presented for comparative purposes
Since various galactosidases were shown to be sensitive to the presence of divalent metal cations (Tanaka et al. 1975; Sen et al. 2012), we evaluated the influence of selected metal ions on the activity of recombinant Aspergillus galactosidases. Using PNPG assays supplemented with different metal ions, we found no clear impact of these ions on the activity of the studied galactosidases (Online Resource 1, Supplemental Fig. S3).
For enzyme activity tests, diluted enzymes were used in order to ensure measurements in the linear range. We observed that the supplementation of the diluted enzymes with BSA (for all galactosidases) leads to enhanced activity ranging from two times for A. nidulans rlacB to ten times for A. niger rlacA (data not shown). The most probable explanation for this observation are molecular crowding effects of proteins and solvents, which can have a stabilizing effect on diluted enzymes in solution (Homouz et al. 2008; Shen et al. 2009). The following tests were performed without and with BSA supplementation in order to account for this observation.
In terms of optimal temperature, we observed unexpected differences among recombinant galactosidases (Fig. 2). Without BSA supplementation, A. niger rlacA showed a temperature optimum of about 35 °C with a very broad temperature range of activity, showing 60 % activity at temperatures as high as 70 °C (Fig. 2a). However, the addition of BSA as a stabilizing agent resulted in a shift of the temperature optimum to 65 °C; this stabilizing effect was also evident when testing the temperature stability of different batches of A. niger rlacA. Similar to A. niger rlacA, recombinant lacA from A. nidulans also showed a rather low temperature optimum of about 30 °C (Fig. 2b), a result comparable to the previous study on a purified β-galactosidase from A. nidulans (Díaz et al. 1996); the addition of BSA also had a stabilizing effect on enzyme activity. A. nidulans rlacB and A. oryzae galactosidase showed a rather sharp temperature optimum between 45 and 55 °C irrespective of the presence of BSA (Fig. 2c, d). We also tested the thermal stability of the galactosidases and observed that BSA supplementation also had a stabilizing effect on the storage of enzymes at low temperatures, whereas at higher temperatures, BSA does not enhance the stability of the enzymes (Online Resource 1, Supplemental Fig. S4).
Fig. 2.

Temperature and pH optima of Aspergillus galactosidases. The activity of the Aspergillus galactosidases was measured at various temperatures: a A. niger rlacA, b A. nidulans rlacA, c A. nidulans rlacB, and d A. oryzae galactosidase. Black squares represent data obtain without BSA supplementation and open circles represent data obtained with BSA supplementation (500 μg mL−1 BSA). The activity of the galactosidases was also measured at various pH values (e): black squares A. niger rlacA, black circles A. nidulans rlacA, open circles A. nidulans rlacB, open squares A. oryzae galactosidase. Reactions were performed at 37 °C (unless otherwise indicated) with 5 mM PNPG as substrate and 0.02 U μL−1 of galactosidase per reaction. All values represent averages ± standard error
For all recombinant enzymes, the pH optimum was between pH 4 and 5.5 with both A. nidulans galactosidases retaining 80 to 90 % of their maximal activity at pH 6 (Fig. 2e). These findings are in contrast with recently reported lower pH optimum for the homologous protein derived from A. niger van Tiegh (O’Connell and Walsh 2010) and with data acquired on a purified A. nidulans β-galactosidase, which was shown to have a pH optimum of 7.5 (Díaz et al. 1996). The addition of BSA had no significant effect on the pH optima of the analyzed galactosidases.
For industrial applications in which large amounts of products accumulate, the tolerance of recombinant enzymes toward high product concentrations is a critical factor (Vera et al. 2011). We used standard PNPG assays, supplemented with different amounts of either glucose or galactose (as products from, e.g., lactose digestion), in order to investigate product inhibition. We found that all enzymes, including A. oryzae galactosidase, were strongly inhibited by galactose but not by glucose (Online Resource 1, Supplemental Table S1). A. nidulans rlacA proved to be the least inhibited by galactose, as it is also indicated by the calculated K i values for galactose: K i = 3.0, 10.2, 1.4, and 0.6 mM for A. niger rlacA, A. nidulans rlacA, A. nidulans rlacB, and A. oryzae galactosidase, respectively. In order to further analyze the mode of enzyme inhibition by galactose, additional enzymatic assays with different substrate (PNPG) and galactose concentrations were performed. As expected, Dixon plots indicated that galactose acted as competitive inhibitor on all enzymes in this study (data not shown).
Substrate specificity of recombinant galactosidase preparations
p-Nitrophenyl derivatives also serve as good substrates in quickly screening enzyme activity. Thus, we used ten different p-nitrophenyl substrates in order to test the recombinant enzyme preparations for potential activities toward other terminal monosaccharide residues, which, for example, could derive from contaminations with P. pastoris proteins. We show that the enzyme preparations exhibited extremely weak or negligible activity toward other substrates than PNPG and thus possess very high purity (Table 2).
In order to appraise the analytical value of these enzymes, we also tested several potential “natural” substrates: a glycopeptide containing a biantennary N-glycan with two terminal galactose residues, two tetrasaccharides having either β-1,3-linked or β-1,4-linked terminal galactose residues (lacto-N-tetraose and lacto-N-neotetraose) and an N-glycan structure with β-1,4-linked galactose attached to a core α1,6-linked fucose (the N-glycan containing the “Galβ1,4Fuc” epitope). In addition, we tested whether the preparations of recombinant galactosidase act on an oligomannosidic structure with nine mannose residues.
N-glycans as substrates for Aspergillus galactosidases
The dabsylated-GalGal glycopeptide derived from bovine fibrin carries two terminal galactose residues. The assays with the dabsylated-GalGal glycopeptide show that A. niger rlacA can be used to trim the galactose moieties from the substrate. Approximately 5 % of the galactose residues, as estimated from the MALDI-TOF/TOF MS data, were not cleaved, although assays were incubated for up to 96 h with additional enzyme being added every 24 h (Fig. 3a). Since it has been estimated that roughly 95 % of the galactose on the glycopeptide are β-1,4-linked, whereas the residual 5 % are β-1,3-linked (Pabst et al. 2007), the assays indicate a strict β-1,4 specificity of A. niger rlacA under these conditions. The residual 5 % of the galactose residues could be removed by addition of bovine testes β-galactosidase known to trim β-1,3-linked galactose residues (Distler and Jourdian 1973). Similar to A. niger rlacA, A. nidulans rlacA and rlacB could also be used for the removal of galactose from the fibrin-derived dabsylated-GalGal glycopeptide. However, in contrast to A. niger rlacA, A. nidulans rlacA removed almost all galactose residues (0.4 % remaining) within 96 h, indicating a β-1,3 hydrolyzing activity of this galactosidase (Fig. 3b). Due to the low efficiency of A. nidulans rlacB, higher temperatures and longer incubation times were necessary to achieve the same degree of substrate conversion and, similar to A. niger rlacA, no cleavage of β-1,3-galactose residues from the fibrin-derived dabsylated-GalGal glycopeptide occurred (data not shown).
Fig. 3.

Complex N-glycans as substrates for recombinant galactosidases. Dabsylated-GalGal glycopeptide derived from bovine fibrin was used as substrate for a A. niger rlacA and b A. nidulans rlacA. Additional enzyme (0.2 μg) was added every 24 h. An approximate amount of 5 % non-cleavable galactose, which corresponds to β-1,3-linked galactose remained in the A. niger rlacA reactions (a) and could be successfully removed upon the addition of bovine testes galactosidase at 96 h. The glycans are depicted following the glycan nomenclature of the Consortium for Functional Glycomics (http://www.functionalglycomics.org)
β-1,3 hydrolyzing activity of recombinant and native galactosidases
To further address the β-1,4 and β-1,3 specificity of the recombinant enzymes, pyridylaminated forms of lacto-N-tetraose [Gal-β-1,3-GlcNAc-β-1,3-Gal-β-1,4-Glc] and lacto-N-neotetraose [Gal-β-1,4-GlcNAc-β-1,3-Gal-β-1,4-Glc] were tested as substrates. As judged by MALDI-TOF/TOF MS analyses, all three recombinant enzymes, as well as the native galactosidase from A. oryzae, accept both linear substrates, although with different efficiencies (Fig. 4 and Supplemental Table S2). Although in most studies this aspect of family 35 β-galactosidases is not addressed, these findings are in line with the data on purified β-galactosidase from A. niger, which was shown to act, among others, on Gal-β-1,3-Gal-β-PNP, Gal-β-1,3-Gal-α-PNP, and to lesser extent, Gal-β-1,3-GalNAc/GlcNAc substrates (Sykes et al. 1983).
Fig. 4.

β-1,3 activity of recombinant galactosidases. PA-labeled lacto-N-neotetraose (contains terminal β-1,4-Gal) (a) and lacto-N-tetraose (contains terminal β-1,3-Gal) (b) were used as substrates for recombinant and native galactosidases (0.5 μg of each enzyme was used). After 24 h incubation, reactions were analyzed by MALDI-TOF/TOF MS. Hydrogen (m/z 786.3 and 624.2) as well as sodium adducts (m/z 808.3 and 646.2) were detected. The removal of galactose resulted in ∆m/z 162 Da. The glycans are depicted following the glycan nomenclature of the Consortium for Functional Glycomics (http://www.functionalglycomics.org)
Other tests on pyridylaminated N-glycans
In the experiments described previously, we could show that the recombinant galactosidases remove terminal galactose residues linked to N-acetylglucosamine (either from a branched biantennary N-glycan or a linear tetrasaccharide). Studies on some invertebrate N-glycans clearly demonstrate the existence of structures carrying galactose residues also directly β-1,4-linked to fucose (Galβ1,4Fuc epitope (Zhang et al. 1997b; Yan et al. 2012)). Using a PA-labeled substrate and MALDI-TOF/TOF MS analysis of the assays, we show that all three recombinant galactosidases were able to catalyze the removal of the galactose residue from the Galβ1,4Fuc epitope (Fig. 5 and Online Resource 1, Supplemental Fig. S5). In agreement with all previous tests, despite equal amounts of protein used, A. nidulans rlacB showed incomplete conversion due to lower efficiency of this enzyme.
Fig. 5.

Unusual galactose-carrying N-glycan structures as substrates. A PA-labeled N-glycan structure carrying Galβ1,4Fuc epitope was tested as a substrate for recombinant galactosidases (a no enzyme, b A. niger rlacA, c A. nidulans rlacA, and d A. nidulans rlacB; 0.5 μg of each enzyme was used). The substrate has an m/z of 1,500.5 (hydrogen adduct) and 1,522.5 (sodium adduct), whereas the product has an expected m/z of 1,338.5 (hydrogen adduct) and 1,360.5 (sodium adduct). Additional MS/MS spectra show that the 162-Da loss attributes to the loss of the galactose moiety and not a mannose moiety (Online Resource 1, Supplemental Fig. S5). The glycans are depicted following the glycan nomenclature of the Consortium for Functional Glycomics (http://www.functionalglycomics.org)
As a final test of purity, we also tested preparations of both native and recombinant enzymes for potential mannosidase activity using PA-labeled oligomannosidic Man9, since p-nitrophenyl-mannoside is often poorly accepted by α1,2-mannosidases as a substrate (Forsee et al. 1989). Any mannosidase activity present in galactosidase preparations would be detrimental for the potential use in mass spectrometric N-glycan analyses, in which the analyzed glycans contain both mannose and galactose as terminal residues, since these monosaccharides have the same mass (162.14 Da when bound to a glycan). Indeed, class I mannosidase activity was observed when using commercially available, native A. oryzae galactosidase (Online Resource 1, Supplemental Fig. S6). In contrast, α1,2-mannosidase activity was not observed in the preparations of recombinantly produced and purified Aspergillus enzymes (Online Resource 1, Supplemental Fig. S6), indicating the successful removal of endogenous P. pastoris mannosidases during protein purification.
Lactose as substrate and galactooligosaccharide formation by galactosidases
We tested whether lactose is accepted as substrate and whether using lactose as a substrate could lead to GOS formation. At a lactose concentration of 20 %, all three recombinant galactosidases could be successfully used to hydrolyze lactose. After 4 h of incubation, additional peaks were detected, which co-eluted with trigalactooligosaccharides and tetragalactooligosaccharides in a commercial GOS preparation. Prolonged incubation for 24 h leads to degradation of GOS and lactose to glucose and galactose. The conversion efficiency to monosaccharides of the three recombinant enzymes was in agreement with the specific activities that were also observed for PNPG as substrate; A. niger rlacA showed the highest efficiency, followed by A. nidulans rlacA and rlacB (Online Resource 1, Supplemental Fig. S7).
Stability of recombinant galactosidases
For certain industrial applications such as transgalactosylation other than GOS formation, substantial amounts of organic solvents might be present in a reaction mixture in order to dissolve the substrate (Scheckermann et al. 1997). We examined the influence of organic solvents including acetonitrile, acetone, IPA, EtOH, and MeOH on the enzymatic activity using PNPG assays. The data obtained show that all three recombinant galactosidases retained high activity with considerably high amounts of these solvents; A. niger rlacA displayed the highest tolerance (Online Resource 1, Supplemental Fig. S8). Interestingly, in some cases, the highest enzymatic activity was observed at intermediate organic solvent concentrations. This effect, similar to the addition of BSA to the diluted enzyme, can be explained by refolding of partly misfolded protein and its subsequent stabilization induced either by molecular crowding effects or simply by altered properties of the solution (Homouz et al. 2008; Shen et al. 2009). In contrast, the addition of pure water to dilute the buffer, instead of organic solvent, had a minor effect on enzyme activity (Online Resource 1, Supplemental Fig. S8).
As described in the “Materials and methods” section, all three preparations of recombinant, purified galactosidases could be stored as concentrated enzyme solutions at 4 °C for 18 months without major loss of activity (<15 %), indicating high stability. In order to further investigate the stability of the enzymes, we performed freeze/thaw experiments and show that all Aspergillus galactosidases, including the nonrecombinant A. oryzae galactosidase, could withstand at least ten freeze/thaw cycles without major loss of enzymatic activity (Online Resource 1, Supplemental Fig. S9). The enzymes were stable at a concentration of 100 μg mL−1 without the addition of glycerol as a cryoprotective agent. Furthermore, no significant positive effect of BSA supplementation to the storage solution for protein stability was observed.
Scale-up of A. niger rlacA and A. nidulans rlacB expression
The use of P. pastoris as an expression host followed by one-step His-tag purification was found to be an excellent choice for recombinant expression and subsequent purification of fungal β-galactosidases. Efficient low-cost production of recombinant proteins with high yields is of great importance, especially for enzymes of potential industrial use. As estimated by the specific activity of the purified enzymes, yeast culture supernatants contained recombinant protein in a concentration of more than 100 μg mL−1 after 48 h of growth in shaker flasks without optimization of the culture conditions at low cell densities in complex medium. For P. pastoris as expression host, high cell densities can be achieved in fermentors in defined minimal medium; however, cultivation in minimal medium can also come at the cost of lower yields of recombinant protein as compared with complex medium (Macauley-Patrick et al. 2005). To examine this, we performed pilot expressions with A. niger rlacA and A. nidulans rlacA expressing P. pastoris clones in fermentors. We did not perform expression of A. nidulans rlacB in fermentors, since the characterization of this enzyme indicated limited value for glycobiotechnological and biotechnological applications. A simple linear glucose feed protocol aiming at a final yeast dry mass of 100 g L−1 and a total process time of 100 h resulted in very high recombinant protein titers with up to approximately 3.5 and 0.2 g L−1 of recombinant protein for A. niger rlacA and A. nidulans rlacA, respectively (Online Resource 1, Supplemental Fig. S10).
Discussion
In this study, we cloned, expressed, purified, and analyzed properties of two previously uncharacterized β-galactosidases from A. nidulans and of the homologous enzyme from A. niger previously expressed in various systems optimized for high-yield protein production (Oliveira et al. 2011) and used to study Lac− phenotype of S. cerevisiae (Kumar et al. 1992). The previously uncharacterized aspects of these three enzymes are compared to those of the commercially available, native enzyme from A. oryzae. We demonstrate that one of the recombinant enzymes, A. nidulans rlacA, has a number of exceptional properties, including reduced inhibition by galactose (K i = 10.2 mM) and an increased activity toward β-1,3-linked terminal galactose (Fig. 3 and Online Resource 1, Supplemental Table S2). The enzymes were essentially free from other glycosidase activities (including α1,2-mannosidase activity) and performed up to ten times better when incubated in presence of BSA. As such, these enzymes are suitable for a wide range of applications. On the other hand, the A. nidulans rlacB underperformed in most aspects tested in this study.
To date, none of the β-galactosidases were shown to have absolute specificity toward either terminal β-1,3-linked, β-1,4-linked, or β1,6-linked galactose. Most β-galactosidases (the ones from E. coli, A. oryzae, Canavalia ensiformis (jack beans), and S. pneumoniae) strongly prefer terminal β-1,4-linked galactose; these enzymes were also shown to process terminal β-1,3-linked galactose, albeit at a very low rate (Zeleny et al. 1997). On the other hand, bovine testes β-galactosidase prefers terminal β-1,3-linked galactose, but can also process β-1,4-linked galactose efficiently (Distler and Jourdian 1973). Accordingly and similar to A. niger rlacA described in this study (Fig. 3), A. oryzae β-galactosidase activity toward β-1,3-galactose residues has proven insufficient for complete degalactosylation of a bovine fibrin-derived dabsylated-GalGal glycopeptide (Mucha et al. 2004). However, A. nidulans rlacA was shown here to have increased preference toward β-1,3-linked galactose and to possess the ability to remove β-1,3-linked galactose residues from the dabsylated-GalGal glycopeptide (Fig. 3 and Online Resource 1, Supplemental Table S2). In addition, this enzyme has already been used for the removal of terminal β-1,3-linked galactose from oyster (hybrid) N-glycans in our laboratory (K. Paschinger, personal communication).
The β-galactosidases also differ in their ability to process terminally galactosylated structures according to an N-glycan backbone. Recently, it was shown that C. ensiformis galactosidase is not able to remove galactose β-1,4-linked to a core fucose residue, a structure found in many species belonging to Protostomia, whereas S. pneumoniae galactosidase could digest that epitope (Takeuchi et al. 2011). All recombinant enzymes tested in the current study were shown to be able to remove galactose β-1,4-linked to a core fucose residue. The activities displayed by recombinant and native Aspergillus β-galactosidases in comparison to other commonly used β-galactosidases in glycan analyses are summarized in Table 3.
Table 3.
Substrate preference of commonly used β-galactosidases in glycomic studies
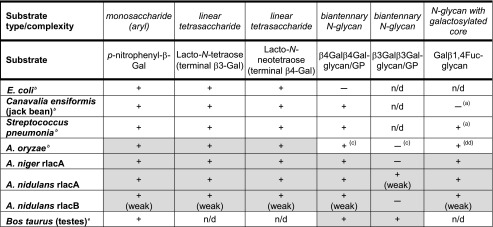
Using high lactose concentrations for our assays to test the ability for lactose hydrolysis, we could show that all three recombinant enzymes were able to accept lactose as a substrate, although with large differences in terms of their efficiency. The commercial A. oryzae β-galactosidase has already been used in multiple studies on lactose hydrolysis and GOS production (Iwasaki et al. 1996; Park and Oh 2010; Guerrero et al. 2013). The kinetics of the GOS production process demand that the amount of enzyme, the lactose concentration, and the reaction conditions, such as temperature, pH, and reaction time, are tightly controlled in order to allow for efficient synthesis (Vera et al. 2011; Palai et al. 2012). In line with the dynamics of GOS production, we observed the appearance of GOS after 4 h of incubation with an initial lactose concentration of 20 % and their subsequent degradation after prolonged incubation for 24 h (Online Resource 1, Supplemental Fig. S7) It is beyond the scope of the current study to optimize the GOS production conditions; however, these initial results highlight that A. niger rlacA and A. nidulans rlacA are suitable for such a process in principle and may also serve for other transglycosylation reactions that are mediated by β-galactosidases such as, for example, antibiotics and PEG transglycosylation (Scheckermann et al. 1997; Fang et al. 2012) as these enzymes tolerate fairly high amounts of organic solvents.
In order to improve the cost-effectiveness of large-scale industrial applications for lactose hydrolysis and GOS production, various immobilization strategies, such as the immobilization on amino-epoxy beads, magnetic beads, and cotton cloth, were already applied for β-galactosidases (Albayrak and Yang 2002; Torres et al. 2003; Zhang et al. 2010); altogether, these immobilization strategies have been successful as they increase the operational, thermal, and pH robustness of the enzymes (Klein et al. 2013) and are, as such, beneficial for continuous bioprocesses (Liu et al. 2012). Considering the generally high thermal stability of A. niger and A. nidulans rlacA, it will also be of great interest to explore different immobilization strategies for these novel enzymes in future projects.
Moreover, β-galactosidases have also been successfully engineered in order to achieve decreased product inhibition by galactose (Hu et al. 2010; Kim et al. 2011). In this regard, A. nidulans rlacA may be, due to its generally high K i for galactose, a very interesting and suitable candidate for future protein engineering studies. The availability of crystal structures of closely related enzymes, including the recently reported structure of A oryzae β-galactosidase (Maksimainen et al. 2013) may further facilitate not only the directed engineering of fungal galactosidases toward decreased product inhibition but also the engineering toward other relevant improved activities such as glucuronidase (Xiong et al. 2007), fucosidase (Zhang et al. 1997a), or glycosynthase activity (Shim et al. 2012).
Large-scale processes demand large quantities of enzyme, and the use of immobilized enzymes proved successful in order to improve the economics of such processes. In the current study, high yields of these prospective enzymes could be achieved by using the P. pastoris GAP promoter system and fermentor cultivation (Online Resource 1, Supplemental Fig. S10). Whereas we only used minimal medium, yields may be further increased by growth medium optimization and the addition of proper feed supplements (Pal et al. 2006) or altered process conditions and advanced feed strategies that already proved successful for GAP promoter-based protein production processes in P. pastoris (Maurer et al. 2006; Baumann et al. 2008). Nevertheless, according to our fermentor experiments, cost-effective large-scale production of these novel recombinant galactosidases is certainly feasible.
All three enzymes in the current study share predicted β-galactosidase protein domains, and we could verify this hydrolytic activity. A. niger and A. nidulans rlacA could be successfully applied in order to process lactose and N-glycan substrates. Nevertheless, A. nidulans rlacB showed the weakest performance on all substrates that were tested. As the ORF used for expression did not completely match the predicted one, we cannot exclude the possibility that this discrepancy influences the catalytic activity of this enzyme. In their natural habitat, fungi subsist to some extent on the degradation of plant material and are a valuable source for polysaccharide-degrading enzymes (van den Brink and de Vries 2011). Furthermore, filamentous fungi also produce β-galactosidases when grown on substrates such as polygalacturonic acid (McKay 1991). In this context, it is possible that neither of the tested compounds represents a natural substrate for the A. nidulans rlacB enzyme.
In conclusion, with the potential of low-cost and efficient large-scale production and ease of purification, recombinant galactosidases from A. niger and A. nidulans (rlacA) enrich the series of valuable β-galactosidase enzymes available for various biotechnological and analytical procedures and may also serve as potential targets for glycoenzyme engineering studies.
Electronic supplementary material
Below is the link to the electronic supplementary material.
The electronic supplementary file contains supplemental tables and figures. (PDF 3080 kb)
Acknowledgments
We thank the members of the research group for their help and useful comments on the manuscript. Special thanks to Shi Yan for supplying the N-glycan with Galβ1,4Fuc epitope, to Rhiannon Stanton for some HPLC analysis, and to Katharina Paschinger for the help with the MALDI-TOF/TOF MS and for critically reading the manuscript. We would also like to thank Hans Marx for his assistance during the GOS HPLC assays, Michael Maurer for his assistance with fermentations, and Barbara Amon from the Agrana Stärke AG for providing the GOS samples. This work was funded by the Austrian Science Fund (FWF) [TRP127 to D.R.].
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
References
- Albayrak N, Yang ST. Production of galacto-oligosaccharides from lactose by Aspergillus oryzae β-galactosidase immobilized on cotton cloth. Biotechnol Bioeng. 2002;77:8–19. doi: 10.1002/bit.1195. [DOI] [PubMed] [Google Scholar]
- Altmann F, Schweiszer S, Weber C. Kinetic comparison of peptide: N-glycosidases F and A reveals several differences in substrate specificity. Glycoconj J. 1995;12:84–93. doi: 10.1007/BF00731873. [DOI] [PubMed] [Google Scholar]
- Arakawa M, Ogata S, Muramatsu T, Kobata A. β-Galactosidases from jack bean meal and almond emulsin. Application for the enzymatic distinction of Galβ1→4GlcNAc and Galβ1→3GlcNAc linkages. J Biochem. 1974;75:707–714. doi: 10.1093/oxfordjournals.jbchem.a130443. [DOI] [PubMed] [Google Scholar]
- Baumann K, Maurer M, Dragosits M, Cos O, Ferrer P, Mattanovich D. Hypoxic fed-batch cultivation of Pichia pastoris increases specific and volumetric productivity of recombinant proteins. Biotechnol Bioeng. 2008;100:177–183. doi: 10.1002/bit.21763. [DOI] [PubMed] [Google Scholar]
- Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 2009;37:D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz M, Pedregosa AM, de Lucas JR, Torralba S, Monistrol IF, Laborda F. Purification and properties of β-galactosidase from Aspergillus nidulans. Microbiologia. 1996;12:585–592. [PubMed] [Google Scholar]
- Dickson RC, Markin JS. Molecular cloning and expression in E. coli of a yeast gene coding for β-galactosidase. Cell. 1978;15:123–130. doi: 10.1016/0092-8674(78)90088-0. [DOI] [PubMed] [Google Scholar]
- Distler JJ, Jourdian GW. The purification and properties of β-galactosidase from bovine testes. J Biol Chem. 1973;248:6772–6780. [PubMed] [Google Scholar]
- Dixon M. The determination of enzyme inhibitor constants. Biochem J. 1953;55:170–171. doi: 10.1042/bj0550170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Xu W, Wu J, Xu ZK. Enzymatic transglycosylation of PEG brushes by β-galactosidase. Chem Commun (Camb) 2012;48:11208–11210. doi: 10.1039/c2cc35369e. [DOI] [PubMed] [Google Scholar]
- Forsee WT, Palmer CF, Schutzbach JS. Purification and characterization of an α-1,2-mannosidase involved in processing asparagine-linked oligosaccharides. J Biol Chem. 1989;264:3869–3876. [PubMed] [Google Scholar]
- Gasser B, Dragosits M, Mattanovich D. Engineering of biotin-prototrophy in Pichia pastoris for robust production processes. Metab Eng. 2010;12:573–580. doi: 10.1016/j.ymben.2010.07.002. [DOI] [PubMed] [Google Scholar]
- Ghosalkar A, Sahai V, Srivastava A. Secretory expression of interferon-α 2b in recombinant Pichia pastoris using three different secretion signals. Protein Expr Purif. 2008;60:103–109. doi: 10.1016/j.pep.2008.02.006. [DOI] [PubMed] [Google Scholar]
- Guerrero C, Vera C, Illanes A. Optimisation of synthesis of oligosaccharides derived from lactulose (fructosyl-galacto-oligosaccharides) with β-galactosidases of different origin. Food Chem. 2013;138:2225–2232. doi: 10.1016/j.foodchem.2012.10.128. [DOI] [PubMed] [Google Scholar]
- Hamilton SR, Davidson RC, Sethuraman N, Nett JH, Jiang Y, Rios S, Bobrowicz P, Stadheim TA, Li H, Choi BK, Hopkins D, Wischnewski H, Roser J, Mitchell T, Strawbridge RR, Hoopes J, Wildt S, Gerngross TU. Humanization of yeast to produce complex terminally sialylated glycoproteins. Science. 2006;313:1441–1443. doi: 10.1126/science.1130256. [DOI] [PubMed] [Google Scholar]
- Henrissat B, Bairoch A. Updating the sequence-based classification of glycosyl hydrolases. Biochem J. 1996;316:695–696. doi: 10.1042/bj3160695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homouz D, Perham M, Samiotakis A, Cheung MS, Wittung-Stafshede P. Crowded, cell-like environment induces shape changes in aspherical protein. Proc Natl Acad Sci U S A. 2008;105:11754–11759. doi: 10.1073/pnas.0803672105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu CA, Yu RC, Chou CC. Production of β-galactosidase by Bifidobacteria as influenced by various culture conditions. Int J Food Microbiol. 2005;104:197–206. doi: 10.1016/j.ijfoodmicro.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Hu X, Robin S, O’Connell S, Walsh G, Wall JG. Engineering of a fungal β-galactosidase to remove product inhibition by galactose. Appl Microbiol Biotechnol. 2010;87:1773–1782. doi: 10.1007/s00253-010-2662-8. [DOI] [PubMed] [Google Scholar]
- Hughes RC, Jeanloz RW. The extracellular glycosidases of Diplococcus pneumoniae. I. Purification and properties of a neuraminidase and a β-galactosidase. Action on the α-1-acid glycoprotein of human plasma. Biochemistry. 1964;3:1535–1543. doi: 10.1021/bi00898a025. [DOI] [PubMed] [Google Scholar]
- Hung MN, Xia Z, Hu NT, Lee BH. Molecular and biochemical analysis of two β-galactosidases from Bifidobacterium infantis HL96. Appl Environ Microbiol. 2001;67:4256–4263. doi: 10.1128/AEM.67.9.4256-4263.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husain Q. β Galactosidases and their potential applications: a review. Crit Rev Biotechnol. 2010;30:41–62. doi: 10.3109/07388550903330497. [DOI] [PubMed] [Google Scholar]
- Invitrogen (2010) Pichia expression kit. For expression of recombinant proteins in Pichia pastoris. User manual. Available at http://tools.invitrogen.com/content/sfs/manuals/pich_man.pdf
- Iskratsch T, Braun A, Paschinger K, Wilson IBH. Specificity analysis of lectins and antibodies using remodeled glycoproteins. Anal Biochem. 2009;386:133–146. doi: 10.1016/j.ab.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Iwasaki K, Nakajima M, Nakao S. Galacto-oligosaccharide production from lactose by an enzymic batch reaction using β-galactosidase. Process Biochem. 1996;31:69–76. doi: 10.1016/0032-9592(94)00067-0. [DOI] [Google Scholar]
- Kim YS, Yeom SJ, Oh DK. Reduction of galactose inhibition via the mutation of β-galactosidase from Caldicellulosiruptor saccharolyticus for lactose hydrolysis. Biotechnol Lett. 2011;33:353–358. doi: 10.1007/s10529-010-0445-z. [DOI] [PubMed] [Google Scholar]
- Klein MP, Fallavena LP, Schöffer J, Ayub MA, Rodrigues RC, Ninow JL, Hertz PF. High stability of immobilized β-d-galactosidase for lactose hydrolysis and galactooligosaccharides synthesis. Carbohydr Polym. 2013;95:465–470. doi: 10.1016/j.carbpol.2013.02.044. [DOI] [PubMed] [Google Scholar]
- Kumar V, Ramakrishnan S, Teeri TT, Knowles JK, Hartley BS. Saccharomyces cerevisiae cells secreting an Aspergillus niger β-galactosidase grow on whey permeate. Biotechnology (N Y) 1992;10:82–85. doi: 10.1038/nbt0192-82. [DOI] [PubMed] [Google Scholar]
- Li SC, Mazzotta MY, Chien SF, Li YT. Isolation and characterization of jack bean β-galactosidase. J Biol Chem. 1975;250:6786–6791. [PubMed] [Google Scholar]
- Liu H, Liu J, Tan B, Zhou F, Qin Y, Yang R. Covalent immobilization of Kluyveromyces fragilis β-galactosidase on magnetic nanosized epoxy support for synthesis of galacto-oligosaccharide. Bioprocess Biosyst Eng. 2012;35:1287–1295. doi: 10.1007/s00449-012-0716-2. [DOI] [PubMed] [Google Scholar]
- Macauley-Patrick S, Fazenda M, McNeil B, Harvey L. Heterologous protein production using the Pichia pastoris expression system. Yeast. 2005;22:249–270. doi: 10.1002/yea.1208. [DOI] [PubMed] [Google Scholar]
- Maksimainen MM, Lampio A, Mertanen M, Turunen O, Rouvinen J. The crystal structure of acidic β-galactosidase from Aspergillus oryzae. Int J Biol Macromol. 2013;60:109–115. doi: 10.1016/j.ijbiomac.2013.05.003. [DOI] [PubMed] [Google Scholar]
- Maurer M, Kühleitner M, Gasser B, Mattanovich D. Versatile modeling and optimization of fed batch processes for the production of secreted heterologous proteins with Pichia pastoris. Microb Cell Fact. 2006;5:37. doi: 10.1186/1475-2859-5-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlvaine T. A buffer solution for colorimetric comparison. J Biol Chem. 1921;49:183–186. [Google Scholar]
- McKay M. Extracellular β-galactosidase production during growth of filamentous fungi on polygalacturonic acid. Lett Appl Microbiol. 1991;12:75–77. doi: 10.1111/j.1472-765X.1991.tb00508.x. [DOI] [Google Scholar]
- Mucha J, Domlatil J, Lochnit G, Rendić D, Paschinger K, Hinterkörner G, Hofinger A, Kosma P, Wilson IBH. The Drosophila melanogaster homologue of the human histo-blood group Pk gene encodes a glycolipid-modifying α1,4-N-acetylgalactosaminyltransferase. Biochem J. 2004;382:67–74. doi: 10.1042/BJ20040535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell S, Walsh G. Application relevant studies of fungal β-galactosidases with potential application in the alleviation of lactose intolerance. Appl Biochem Biotechnol. 2008;149:129–138. doi: 10.1007/s12010-007-8098-7. [DOI] [PubMed] [Google Scholar]
- O’Connell S, Walsh G. A novel acid-stable, acid-active β-galactosidase potentially suited to the alleviation of lactose intolerance. Appl Microbiol Biotechnol. 2010;86:517–524. doi: 10.1007/s00253-009-2270-7. [DOI] [PubMed] [Google Scholar]
- Oliveira C, Guimarães PM, Domingues L. Recombinant microbial systems for improved β-galactosidase production and biotechnological applications. Biotechnol Adv. 2011;29:600–609. doi: 10.1016/j.biotechadv.2011.03.008. [DOI] [PubMed] [Google Scholar]
- Pabst M, Bondili JS, Stadlmann J, Mach L, Altmann F. Mass + retention time = structure: a strategy for the analysis of N-glycans by carbon LC-ESI-MS and its application to fibrin N-glycans. Anal Chem. 2007;79:5051–5057. doi: 10.1021/ac070363i. [DOI] [PubMed] [Google Scholar]
- Pal Y, Khushoo A, Mukherjee KJ. Process optimization of constitutive human granulocyte–macrophage colony-stimulating factor (hGM-CSF) expression in Pichia pastoris fed-batch culture. Appl Microbiol Biotechnol. 2006;69:650–657. doi: 10.1007/s00253-005-0018-6. [DOI] [PubMed] [Google Scholar]
- Palai T, Mitra S, Bhattacharya PK. Kinetics and design relation for enzymatic conversion of lactose into galacto-oligosaccharides using commercial grade β-galactosidase. J Biosci Bioeng. 2012;114:418–423. doi: 10.1016/j.jbiosc.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Park AR, Oh DK. Galacto-oligosaccharide production using microbial β-galactosidase: current state and perspectives. Appl Microbiol Biotechnol. 2010;85:1279–1286. doi: 10.1007/s00253-009-2356-2. [DOI] [PubMed] [Google Scholar]
- Paschinger K, Gonzalez-Sapienza GG, Wilson IBH. Mass spectrometric analysis of the immunodominant glycan epitope of Echinococcus granulosus antigen Ag5. Int J Parasitol. 2012;42:279–285. doi: 10.1016/j.ijpara.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschinger K, Hykollari A, Razzazi-Fazeli E, Greenwell P, Leitsch D, Walochnik J, Wilson IBH. The N-glycans of Trichomonas vaginalis contain variable core and antennal modifications. Glycobiology. 2012;22:300–313. doi: 10.1093/glycob/cwr149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisanelli I, Kujawa M, Gschnitzer D, Spadiut O, Seiboth B, Peterbauer C. Heterologous expression of an Agaricus meleagris pyranose dehydrogenase-encoding gene in Aspergillus spp. and characterization of the recombinant enzyme. Appl Microbiol Biotechnol. 2010;86:599–606. doi: 10.1007/s00253-009-2308-x. [DOI] [PubMed] [Google Scholar]
- Scheckermann C, Wagner F, Fischer L. Galactosylation of antibiotics using the β-galactosidase from Aspergillus oryzae. Enzyme Microb Technol. 1997;20:629–634. doi: 10.1016/S0141-0229(96)00211-6. [DOI] [Google Scholar]
- Sen S, Ray L, Chattopadhyay P. Production, purification, immobilization, and characterization of a thermostable β-galactosidase from Aspergillus alliaceus. Appl Biochem Biotechnol. 2012;167:1938–1953. doi: 10.1007/s12010-012-9732-6. [DOI] [PubMed] [Google Scholar]
- Shen VK, Cheung JK, Errington JR, Truskett TM. Insights into crowding effects on protein stability from a coarse-grained model. J Biomech Eng. 2009;131:071002. doi: 10.1115/1.3127259. [DOI] [PubMed] [Google Scholar]
- Shim JH, Chen HM, Rich JR, Goddard-Borger ED, Withers SG. Directed evolution of a β-glycosidase from Agrobacterium sp. to enhance its glycosynthase activity toward C3-modified donor sugars. Protein Eng Des Sel. 2012;25:465–472. doi: 10.1093/protein/gzs045. [DOI] [PubMed] [Google Scholar]
- Sykes DE, Abbas SA, Barlow JJ, Matta KL. Substrate specificity and other properties of the β-d-galactosidase from Aspergillus niger. Carbohydr Res. 1983;116:127–138. doi: 10.1016/S0008-6215(00)90960-1. [DOI] [PubMed] [Google Scholar]
- Takeuchi T, Sugiura K, Nishiyama K, Takahashi H, Natsugari H, Arata Y, Natsuka S, Kasai K. β-Galactosidases from jack bean and Streptococcus have different cleaving abilities towards fucose-containing sugars. Biol Pharm Bull. 2011;34:567–569. doi: 10.1248/bpb.34.567. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Kagamiishi A, Kiuchi A, Horiuchi T. Purification and properties of β-galactosidase from Aspergillus oryzae. J Biochem. 1975;77:241–247. [PubMed] [Google Scholar]
- Titz A, Butschi A, Henrissat B, Fan YY, Hennet T, Razzazi-Fazeli E, Hengartner MO, Wilson IBH, Künzler M, Aebi M. Molecular basis for galactosylation of core fucose residues in invertebrates: identification of Caenorhabditis elegans N-glycan core α1,6-fucoside β1,4-galactosyltransferase GALT-1 as a member of a novel glycosyltransferase family. J Biol Chem. 2009;284:36223–36233. doi: 10.1074/jbc.M109.058354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres R, Mateo C, Fernández-Lorente G, Ortiz C, Fuentes M, Palomo JM, Guisan JM, Fernández-Lafuente R. A novel heterofunctional epoxy-amino sepabeads for a new enzyme immobilization protocol: immobilization-stabilization of β-galactosidase from Aspergillus oryzae. Biotechnol Prog. 2003;19:1056–1060. doi: 10.1021/bp025771g. [DOI] [PubMed] [Google Scholar]
- van den Brink J, de Vries RP. Fungal enzyme sets for plant polysaccharide degradation. Appl Microbiol Biotechnol. 2011;91:1477–1492. doi: 10.1007/s00253-011-3473-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera C, Guerrero C, Illanes A. Determination of the transgalactosylation activity of Aspergillus oryzae β-galactosidase: effect of pH, temperature, and galactose and glucose concentrations. Carbohydr Res. 2011;346:745–752. doi: 10.1016/j.carres.2011.01.030. [DOI] [PubMed] [Google Scholar]
- Wang H, Luo H, Bai Y, Wang Y, Yang P, Shi P, Zhang W, Fan Y, Yao B. An acidophilic β-galactosidase from Bispora sp. MEY-1 with high lactose hydrolytic activity under simulated gastric conditions. J Agric Food Chem. 2009;57:5535–5541. doi: 10.1021/jf900369e. [DOI] [PubMed] [Google Scholar]
- Xiong AS, Peng RH, Zhuang J, Li X, Xue Y, Liu JG, Gao F, Cai B, Chen JM, Yao QH. Directed evolution of a β-galactosidase from Pyrococcus woesei resulting in increased thermostable beta-glucuronidase activity. Appl Microbiol Biotechnol. 2007;77:569–578. doi: 10.1007/s00253-007-1182-7. [DOI] [PubMed] [Google Scholar]
- Yan S, Bleuler-Martinez S, Plaza DF, Künzler M, Aebi M, Joachim A, Razzazi-Fazeli E, Jantsch V, Geyer R, Wilson IBH, Paschinger K. Galactosylated fucose epitopes in nematodes: increased expression in a Caenorhabditis mutant associated with altered lectin sensitivity and occurrence in parasitic species. J Biol Chem. 2012;287:28276–28290. doi: 10.1074/jbc.M112.353128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeleny R, Altmann F, Praznik W. A capillary electrophoretic study on the specificity of β-galactosidases from Aspergillus oryzae, Escherichia coli, Streptococcus pneumoniae, and Canavalia ensiformis (jack bean) Anal Biochem. 1997;246:96–101. doi: 10.1006/abio.1996.9973. [DOI] [PubMed] [Google Scholar]
- Zhang JH, Dawes G, Stemmer WP. Directed evolution of a fucosidase from a galactosidase by DNA shuffling and screening. Proc Natl Acad Sci U S A. 1997;94:4504–4509. doi: 10.1073/pnas.94.9.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Iwasa T, Tsuda M, Kobata A, Takasaki S. A novel monoantennary complex-type sugar chain found in octopus rhodopsin: occurrence of the Gal β1→4Fuc group linked to the proximal N-acetylglucosamine residue of the trimannosyl core. Glycobiology. 1997;7:1153–1158. doi: 10.1093/glycob/7.8.1153. [DOI] [PubMed] [Google Scholar]
- Zhang S, Gao S, Gao G. Immobilization of β-galactosidase onto magnetic beads. Appl Biochem Biotechnol. 2010;160:1386–1393. doi: 10.1007/s12010-009-8600-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The electronic supplementary file contains supplemental tables and figures. (PDF 3080 kb)