

Abstract
We have examined the use of our bis(sulfonamide) diol ligand (1) in the asymmetric addition of phenyl groups to cyclic α,β-unsaturated ketones. Good to excellent enantioselectivities have been obtained with cyclic enones bearing alkyl substituents in the 2 position (71–97% enantiomeric excess). Furthermore, excellent enantioselectivities have been observed in the asymmetric phenylation of cyclic enones with 2-iodo and 2-bromo substituents. The results of this study broaden the scope of the asymmetric additions to ketones promoted by the titanium catalyst derived from ligand 1.
Keywords: alkylation, tertiary alcohols, zinc, titanium, asymmetric catalysis
We recently developed a highly enantioselective titanium catalyst based on the bis(sulfonamide) diol ligand 1 (Fig. 1) for the asymmetric addition of alkylzinc reagents to ketones (Eq. 1; ref. 1). When cyclic α,β-unsaturated ketones were used as substrates, we found that the asymmetric addition reaction could be coupled with a diastereoselective epoxidation by using molecular oxygen as the oxidant to provide a one-pot synthesis of epoxy alcohols with high enantio- and diastereoselectivity (Eq. 2; ref. 2). These studies provide a solution to a long-standing problem in synthetic organic chemistry: the catalytic synthesis of tertiary alcohols from ketones with high enantioselectivity. Unlike related additions of dialkylzinc reagents to aldehyde substrates, which are promoted by hundreds of catalysts (3, 4), only a few systems will catalyze alkyl additions to ketones (1, 2, 5–8), and most of them use high catalyst loadings and long reaction times (6–8).
Fig. 1.

Structure of ligand 1.
 |
 |
The successful use of ligand 1 in the alkyl additions to ketones led our group (9) and Yus and coworkers (10) to examine the enantioselective phenylation of acyclic ketones with ZnPh2. The only previous results in the catalytic asymmetric phenylation of ketones were reported by Dosa and Fu in 1998 (11). In this pioneering work, these authors used Noyori's amino alcohol ligand, (+)-3-exo-(dimethylamino)isoborneol [(+)-DAIB] (12, 13), and diphenylzinc (3.5 eq). Initially, the reaction gave low yields and moderate enantioselectivities. It was found, however, that addition of 1.5 eq of methanol to the diphenylzinc resulted in formation of a mixed alkoxy phenyl zinc reagent that led to increased yields and enantioselectivities (Eq. 3) (11). Under these conditions, excellent enantioselectivities were obtained with substituted propiophenones [86–91% enantiomeric excess (ee)], and acetophenone derivatives afforded enantioselectivities as high as 80%. In the case of cyclohexyl methyl ketone and 3-methyl-2-butanone, the reaction proceeded with 75% and 60% enantioselectivities, respectively. Yields in the phenylation reaction with DAIB ranged from 53% to 91%. In contrast to the phenylation of ketones, several highly enantioselective catalysts have been developed for the asymmetric addition of phenyl groups to aldehydes (14–20).
 |
In our initial study on the addition of diphenylzinc to acyclic ketones with ligand 1, we found that enantioselectivities peaked when 0.6 eq of titanium tetraisopropoxide was used (9). Using 1.6 eq of diphenylzinc, 0.6 eq of titanium tetraisopropoxide, 10 mol % ligand 1, and a toluene/hexane solvent system, reactions reached completion at room temperature (RT) after 6–24 h (Eq. 4). Propiophenone and acetophenone derivatives were excellent substrates for our catalyst, and products were generated with 88–92% and 95–96% enantioselectivities, respectively. Cyclohexyl methyl ketone and 3-methyl-2-butanone also were good substrates, providing the tertiary alcohols in 87% and 75% enantioselectivity, respectively.
 |
In the present work, we examined the reactions of a variety of cyclic α,β-unsaturated ketones. Such substrates with substitution α to the carbonyl group resulted in good to excellent enantioselectivities. We also explored cyclic enones bearing a halogen at the 2 position, imagining that the resulting vinyl halide could be elaborated further through cross-coupling reactions. These substrates also gave excellent enantioselectivities and good yields.
Experimental Procedures
General Method for the Asymmetric Phenylation of Cyclic Enones. The bis(sulfonamide) ligand 1 (5.4 mg, 10 mol %) was weighed into the well dried Schlenk flask and put under a nitrogen atmosphere. Diphenylzinc (43.9 mg, 0.2 mmol) was added, as a solution in 1 ml of toluene, followed by distilled titanium tetraisopropoxide (50 μl, 1.2 M hexanes solution, 0.06 mmol). The homogeneous reaction mixture was stirred at RT for 15 min and 2,4,4-trimethyl-2-cyclohexenone (15 μl, 0.1 mmol) was added. The reaction mixture was stirred at RT until TLC showed complete consumption of the ketone. The reaction mixture was quenched with a few drops of water, dried by using anhydrous MgSO4, and filtered, and the solvent was removed under vacuum. The residue was purified by flash silica gel column chromatography (95:5 hexanes/EtOAc) to give the resulting tertiary alcohol (20 mg, 92% yield, 97% ee) as a white solid: m.p. 37–39°C; 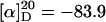 (c 0.88, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 1.10 (s, 3H), 1.11 (s, 3H), 1.40–1.44 (m, 1H), 1.52–1.57 (m, 4H), 2.00–2.04 (m, 2H), 5.51 (s, 1H), 7.26–7.29 (m, 1H), 7.36–7.39 (m, 2H), 7.46–7.48 (m, 2H) ppm; 13C NMR (CDCl3, 125 MHz) δ 14.5, 29.5, 30.0, 32.0, 33.9, 38.7, 76.1, 126.3, 127.1, 128.4, 134.3, 138.1, 146.7 ppm; IR (film) 3442, 2950, 1446, 1359 cm–1. High-resolution MS calcd for C15H20O (M)+: 216.1514. Found: 216.1508.
(c 0.88, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 1.10 (s, 3H), 1.11 (s, 3H), 1.40–1.44 (m, 1H), 1.52–1.57 (m, 4H), 2.00–2.04 (m, 2H), 5.51 (s, 1H), 7.26–7.29 (m, 1H), 7.36–7.39 (m, 2H), 7.46–7.48 (m, 2H) ppm; 13C NMR (CDCl3, 125 MHz) δ 14.5, 29.5, 30.0, 32.0, 33.9, 38.7, 76.1, 126.3, 127.1, 128.4, 134.3, 138.1, 146.7 ppm; IR (film) 3442, 2950, 1446, 1359 cm–1. High-resolution MS calcd for C15H20O (M)+: 216.1514. Found: 216.1508.
Other Procedures. Experimental procedures for the synthesis of ketones and full characterization of all compounds are provided in Appendix 1, which is published as supporting information on the PNAS web site.
Results and Discussion
The products of the asymmetric phenylation of cyclic enones are cyclic allylic alcohols that can be functionalized easily at the double bond in a diastereoselective step by taking advantage of the directing ability of the hydroxyl group (21). We are unaware of previous reports of catalytic asymmetric phenylation of this class of substrates.
In our initial reactions involving the phenylation of α,β-unsaturated ketones, we used conditions used in the phenylation of simple ketones (Eq. 4). These reactions were conducted by combining a toluene solution of diphenylzinc with 10 mol % of ligand 1 under a nitrogen atmosphere followed by addition of titanium tetraisopropoxide. After stirring for 15 min, the substrate was added, and the reaction was stirred at RT. Reaction progress was monitored by TLC. After completion of the reaction, work-up was performed with 15% aqueous tartaric acid or saturated NH4Cl followed by flash chromatography on silica.
Under these conditions, however, yields and enantioselectivities were surprisingly low. Nonpolar products derived from elimination were isolated from the reaction mixtures. We suspected that the tertiary alcohols derived from the cyclic enone substrates would exhibit a greater propensity to undergo elimination and carbocation formation and that these processes were likely responsible for the reduced enantioselectivities and yields. The work-up procedure, therefore, was modified to ensure that the product was maintained under basic conditions. After the asymmetric addition, the reactions were quenched only with water. Gratifyingly, these new work-up conditions allowed isolation of the tertiary alcohols with higher yields and enantioselectivities. The results of our additions to cyclic enones are listed in Table 1.
Table 1. Asymmetric phenylation of cyclic enones.
| Entry | Substrate | Product | Time, h | Yield, % | ee,* % |
|---|---|---|---|---|---|
| 1 |

|
14 | 81 | 1 | |
| 2 | 19 | 92 | 97 | ||
| 3 | 31 | 64 | 80 | ||
| 4 | 25 | 60 | 84 | ||
| 5 | 19 | 74 | 97 | ||
| 6 | 23 | 94 | 84 | ||
| 7 | 22 | 81 | 71 | ||
See Appendix 1 for ee determination protocol.
 |
Use of 2-cyclohexenone as a substrate in the asymmetric phenylation reaction provided the product in high yield. Unfortunately, the resulting tertiary alcohol was nearly racemic (Table 1, entry 1). In the asymmetric alkylation of 2-cyclohexenone with the same catalyst (2), the enantioselectivity was significantly higher, albeit only 52%. The lower enantioselectivities for this challenging substrate are attributed to the similarity of the lone-pair environments of the carbonyl oxygen. As shown in Scheme 1, binding of cyclohexenone (R = H) to the titanium center is likely to give two isomeric adducts. Presumably, these isomers both undergo the asymmetric addition reaction to form enantiomeric products, resulting in reduced enantioselectivities. When R is a larger substituent, the formation of the anti-adduct will be favored for steric reasons, and higher enantioselectivity is expected. It is likely that the enantioselectivity with 2-cyclohexenone is much lower in the diphenylzinc addition than in the diethylzinc addition because of the greater reactivity and reduced selectivity of diphenylzinc.
Scheme 1.

Substrates bearing a substituent at the 2 position did give significantly improved results in the phenyl addition chemistry. Use of 2,4,4-trimethyl-2-cyclohexenone in Eq. 5 afforded the allylic alcohol in excellent yield and enantioselectivity (Table 1, entry 2). The more functionalized cyclohexenone derivative in Table 1, entry 3, gave 64% yield of allylic alcohol with 80% ee. It is not clear why the product of this reaction was not obtained with higher ee, because the bulky tert-butyldimethylsilyl group will disfavor coordination of the pendent oxygen to the titanium catalyst. Cyclopentenone derivatives also proved to be good substrates for the asymmetric phenylation reaction. Use of 2-methyl-2-cyclopentenone afforded the product in 60% yield and 84% ee, whereas 2-pentyl-2-cyclopentenone gave 74% yield and 97% ee (Table 1, entries 4 and 5). The results in Table 1, entries 2–5 suggest that the catalyst enantioselectivity is sensitive to the size of the substituent at the 2 position. The benzocycloheptenone derivative (Table 1, entry 6) provided the product in 94% yield and 84% enantioselectivity. Finally, the fused furan derivative underwent the addition reaction in 81% yield and 71% ee (Table 1, entry 7). In this case, the enantioselectivity of the product is lower than anticipated, because it begins to racemize under the purification procedure.
We next wanted to examine the reactivity of 2-halo substituted enones in Eq. 5. These substrates are among the most attractive, because the resulting vinyl halide products will be useful cross-coupling partners for subsequent carbon–carbon bond-forming reactions. The results of this survey are located in Table 2. The yields of the addition products are variable, ranging from 46% to 77%. Importantly, however, the enantioselectivities are uniformly very high (>93%).
Table 2. Asymmetric phenyl addition to 2-halo-2-enones.
| Entry | Substrate | Product | Time, h | Yield, % | ee,* % |
|---|---|---|---|---|---|
| 1 |  |
60 | 46 | 93 | |
| 2 | 28 | 66 | 94 | ||
| 3 | 60 | 77 | 93 | ||
See Appendix 1 for ee determination protocol.
Two additional substrates were examined that did not participate in the asymmetric addition chemistry. Both 2-tributylstannyl- and 2-trimethylsilyl-2-cyclohexenone exhibited no reaction at RT and resulted in formation of decomposition products when heated to 45°C.
In summary, an effective protocol for the enantioselective addition of phenyl groups to cyclic conjugated enones is reported. Good to excellent enantioselectivities have been obtained with cyclic enones bearing substituents in the 2 position. Furthermore, excellent enantioselectivities have been found in the asymmetric phenylation of cyclic α,β-unsaturated ketones with 2-iodo and 2-bromo substituents. The results of this study broaden the scope of the asymmetric additions to ketones promoted by our titanium catalyst derived from ligand 1. Given that 1 can be synthesized in two steps from commercially available materials and can be used in a variety of operationally simple asymmetric additions to ketones, we believe that it has significant potential in catalytic asymmetric carbon–carbon bond-forming reactions.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (National Institute of General Medical Sciences Grant GM58101).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ee, enantiomeric excess; RT, room temperature.
References
- 1.García, C., LaRochelle, L. K. & Walsh, P. J. (2002) J. Am. Chem. Soc. 124, 10970–10971. [DOI] [PubMed] [Google Scholar]
- 2.Jeon, S.-J. & Walsh, P. J. (2003) J. Am. Chem. Soc. 125, 9544–9545. [DOI] [PubMed] [Google Scholar]
- 3.Soai, K. & Niwa, S. (1992) Chem. Rev. (Washington, D.C.) 92, 833–856. [Google Scholar]
- 4.Pu, L. & Yu, H.-B. (2001) Chem. Rev. (Washington, D.C.) 101, 757–824. [DOI] [PubMed] [Google Scholar]
- 5.Yus, M., Ramón, D. J. & Prieto, O. (2002) Tetrahedron Asymmetry 13, 2291–2293. [Google Scholar]
- 6.Ramón, D. J. & Yus, M. (1998) Tetrahedron Lett. 39, 1239–1242. [Google Scholar]
- 7.Ramón, D. J. & Yus, M. (1998) Tetrahedron 54, 5651–5666. [Google Scholar]
- 8.Yus, M., Ramón, D. J. & Prieto, O. (2003) Tetrahedron Asymmetry 14, 1103–1114. [Google Scholar]
- 9.García, C. & Walsh, P. J. (2003) Org. Lett. 5, 3641–3644. [DOI] [PubMed] [Google Scholar]
- 10.Prieto, O., Ramón, D. J. & Yus, M. (2003) Tetrahedron Asymmetry 14, 1955–1957. [Google Scholar]
- 11.Dosa, P. I. & Fu, G. C. (1998) J. Am. Chem. Soc. 120, 445–446. [Google Scholar]
- 12.Kitamura, M., Suga, S., Kawai, K. & Noyori, R. (1986) J. Am. Chem. Soc. 108, 6071–6072. [DOI] [PubMed] [Google Scholar]
- 13.Noyori, R. (1994) Asymmetric Catalysis in Organic Synthesis (Wiley, New York).
- 14.Dosa, P. I., Ruble, J. C. & Fu, G. C. (1997) J. Org. Chem. 62, 444–445. [DOI] [PubMed] [Google Scholar]
- 15.Bolm, C., Hermanns, N., Hildebrand, J. P. & Muñiz, K. (2000) Angew. Chem. Int. Ed. Engl. 39, 3465–3467. [DOI] [PubMed] [Google Scholar]
- 16.Rudolph, J., Rasmussen, T., Bolm, C. & Norrby, P.-O. (2003) Angew. Chem. Int. Ed. Engl. 42, 3002–3005. [DOI] [PubMed] [Google Scholar]
- 17.Huang, W.-S. & Pu, L. (2000) Tetrahedron Lett. 41, 145–148. [Google Scholar]
- 18.Zhao, G., Lib, X.-Z. & Wang, X.-R. (2001) Tetrahedron Asymmetry 12, 399–403. [Google Scholar]
- 19.Ko, D.-H., Kim, K. H. & Ha, D.-C. (2002) Org. Lett. 4, 3759–3762. [DOI] [PubMed] [Google Scholar]
- 20.Bolm, C., Hildebrand, J. P., Muñiz, K. & Hermanns, N. (2001) Angew. Chem. Int. Ed. Engl. 40, 3284–3308. [DOI] [PubMed] [Google Scholar]
- 21.Hoveyda, A. H., Evans, D. A. & Fu, G. C. (1993) Chem. Rev. (Washington, D.C.) 93, 1307–1370. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.