

Abstract
Helically chiral polymers from achiral monomers containing N and P atoms have been shown to be ligands for transition metals such as Pd and Rh. The Rh complex of the phosphane-containing polyisocyanate p(18-co-17) was an active albeit hardly enantioselective catalyst in the asymmetric hydrogenation of the dehydro amino acid N-acetamidocinnamic acid (15% enantiomeric excess). The most active catalyst obtained until now was the Pd-complexed polymethacrylate Pd-p12, which catalyzes the allylic substitution reaction of 1,3-diphenylprop-2-enyl acetate with dimethyl malonate even at –20°C in quantitative yield, although again the enantioselectivity was unsatisfactory. The most successful application of a helically chiral polymer in asymmetric catalysis with respect to both reactivity and enantioselectivity is the polymethacrylate p(5-co-8). Its palladium complex catalyzes the above-mentioned reaction at 0°C with quantitative yield and 60% enantiomeric excess.
Keywords: polymethacrylate, polyisocyanate, helicity, polymeric ligand, chiral catalyst
Soluble chiral polymers may be promising ligands in asymmetric transition metal catalysis for a number of reasons. The reisolation of the mostly expensive chiral catalyst by precipitation or ultrafiltration should be easy, while all of the analytical and kinetic advantages of a reaction in homogeneous phase should be maintained. Moreover, there might be a number of beneficial effects related to the macromolecular state, for example cooperativity or chiral amplification as observed in polyisocyanates (1). These advantages may allow for the synthesis of novel catalysts with properties not achievable with micromolecular systems.
The most obvious way to prepare macromolecular metal catalysts is to attach well known, catalytically active chiral components to soluble polymers. To get equal asymmetric inductions from each active site, the microenvironments of the complexed metal atoms must be uniform. This uniformity can be achieved either by attaching only one metal-binding site per polymer chain or by complexing several transition metals by a stereoregular polymer. The former strategy was realized rather successfully in the asymmetric dihydroxylation reaction with MeO-polyethylene glycol-bound chiral ligands described by Bolm and Gerlach (2) and Han and Janda (3). However, the major disadvantage of this approach is the very low density of reactive centers per unit mass. A successful example for the second strategy is the application of polybinaphthol complexes as catalysts as exemplified by Pu (4). Problems associated with the latter ligands include the necessity to prepare the enantiomerically pure monomers and the question of potential counterproductive interactions of the different sources of chirality (axial chirality of the monomers and helical chirality of the polymer). Indeed, we think that for the phosphane-modified helical chiral dodecapeptides developed by Gilbertson and Wang (5) the major reason for their failure to achieve good enantioselectivities in asymmetric hydrogenation reactions is such a counterproductive interaction. Guided by that analysis, we developed the idea of synthesizing donor atom-containing stereoregular polymers devoid of any elements of chirality except their helically chiral superstructure.
Several types of synthetic polymers that are optically active only or at least mainly because of their helical secondary structure are known. Depending on the barrier for helix–helix inversion in solution, these polymers can be divided into two classes. The first class comprises sterically congested polymethacrylates (6, 7) or polyquinoxalines (8, 9) with their helical conformations in solution being stabilized by repulsion of sterically demanding lateral groups. Polymers of that type with single-handed helical structures can be obtained by helix sense-selective polymerization with chiral initiators (6, 7). A different type of helically chiral polymers features dynamic helical structures, for example, polyisocyanates (PICs) (10) or polyacetylenes (11). Each polymer chain may contain (P)- and (M)-helical segments, which are separated by helix-reversal points (1, 12). The most striking feature of these polymers is their nonlinear behavior toward chiral stimulation. For example, PICs with single-handed helical sense can be obtained by copolymerization of achiral isocyanates (soldiers) with small amounts of chiral monomers (sergeants) (13, 14). As a consequence, the centrochiral elements are diluted along the polymer chain and the optical activity is mainly a result of the helical secondary structure.
In a recent article (15) we described the synthesis and characterization (NMR, CD) of monodentate palladium complexes Pd-p1 with (+)-, (–)-, and rac-poly(3PyDBSMA) (Scheme 1) as helically chiral ligands.
Scheme 1.

Complexation of poly(3PyDBSMA) (p1) with allyl palladium chloride (15). r.t., room temperature.
These complexes are active catalysts for the allylic substitution reaction (16–19) with 1,3-diphenylprop-2-enyl acetate (DPPA; 2) as substrate and dimethyl malonate (DMM; 3) as nucleophile (Scheme 2). The observed enantioselectivities were a direct consequence of the helical chiral nature of the polymeric ligand. Although the enantiomeric excesses (ee) were only moderate (33%), this was the “proof of principle” for the validity of our concept.
Scheme 2.

Allylic substitution of 1,3-diphenylprop-2-enyl acetate (DPPA; 2) catalyzed by palladium complexes of p1, yielding 1,3-diphenylprop-2-enyl dimethyl malonate (DPPMM; 4) (15).
In this article we report on our studies to improve these polymethacrylate-based ligands with respect to both catalytic activity and enantioselectivity. Further, efforts to synthesize helically chiral metal-containing PICs as well as some preliminary results concerning their catalytic properties are described.
Materials and Methods
Materials and General Procedures. 1,3-Diphenylprop-2-enyl acetate (DPPA; 2) (20), phenyl[bis(2-pyridyl)]methyl methacrylate (PB2PyMA; 5) (21), poly(PB2PyMA) (p5) (21), N,N′-diphenylethylenediamine (DPEDA; 6) (22), (+)-(S)-1-(2-pyrrolidinomethyl)pyrrolidine [(+)-PMP; 7] (23, 24), poly(PB2PyMA-co-TrMA) [p(5-co-8)] (25), and 2,6-dimethylheptyl isocyanate (DMHIC; 17) (26) were prepared according to published procedures (see also supporting information on the PNAS web site). All other reagents and solvents were purchased from commercial sources and used as received unless otherwise noted.
Diethyl ether (Et2O), tetrahydrofuran (THF), toluene, and benzene were distilled from sodium/benzophenone, and dichloromethane (CH2Cl2) was distilled from calcium hydride (all under argon) just before use. If necessary, they were degassed either by pump-thaw and freeze or by bubbling argon through the solvent for half an hour. Argon (99.998%) was passed over a Cu catalyst to remove oxygen and then through 4-Å molecular sieve, blue gel, concentrated sulfuric acid, phosphorus pentoxide on silica (SicaPent, Fluka), and finally over potassium hydroxide to remove water and acidic contamination. Preparations of air-sensitive materials were carried out by using either standard Schlenk and vacuum line techniques or a glove box.
Spectroscopic Techniques. NMR spectra were recorded on a Bruker DRX 500 or an ARX 300. Gel-permeation chromatography was carried out at 30°C, with JASCO HPLC pumps, two columns with exclusion molecular weights of 70,000 and 4,000,000; flow 0.6 ml/min [pressure: 10 bar (1 MPa)], JASCO UV-detector and JASCO OR-detector OR-990. Specific optical rotations were determined on a Perkin–Elmer Polarimeter 241 with Haake D8 thermostat in 1-dm cuvettes.
Complexation of PB2PyMA (5) with Allyl Palladium Chloride. A solution of 69.4 mg (210 μmol, 1.05 eq) of PB2PyMA (5) and 36.6 mg (100 μmol, 0.50 eq) of [Pd(η3-C3H5)Cl]2 in 2.5 ml of dry and degassed CH2Cl2 was stirred for 1 hr at room temperature under argon. Then, 51.8 mg (205 μmol, 1.025 eq) of AgPF6 was added to the yellow mixture. After stirring for 1 hr in the dark, the gray precipitate was separated by filtration through Celite and the solvent was removed under reduced pressure. Slow diffusion of Et2O into a cold CH2Cl2 solution of the yellow residue produced colorless needles suitable for x-ray structural analysis. NMR data of Pd-5 are published as supporting information on the PNAS web site.
Allylic Substitution Reaction (Typical Procedure, 25 mol % Palladium, 0°C). In a Schlenk flask under argon, a solution of 8.0 mg (21.8 μmol, 0.125 eq) of [Pd(η3-C3H5)Cl]2 in 1 ml of dry and degassed CH2Cl2 was added dropwise into a 0°C solution of 34.6 mg (52.3 μmol, 0.3 eq, according to repeating units) of poly(PB2PyMA-co-TrMA) [p(5-co-8); TrMA = triphenylmethyl methacrylate] in 2 ml of CH2Cl2. After stirring for 1 hr at 0°C, 44.2 mg (175 μmol, 1.0 eq) of DPPA (2), 69.4 mg (525 μmol, 3.0 eq) of dimethyl malonate (DMM; 3), 106.8 mg (525 μmol, 3.0 eq) of N,O-bis(trimethylsilyl)acetamide (BSA), and ≈5 mg of KOAc were added in this order. The reaction mixture was stirred at 0°C until complete conversion (checked by TLC analysis). The reaction mixture was filtered through silica, the solvent was removed under reduced pressure, and the residue was purified by flash-chromatography on silica. Optical purity of the resulting malonate 4 (57 mg, 99%) was determined to be 60% ee by 1H-NMR spectroscopic analysis in the presence of 20 mol % of the chiral shift reagent Eu(hfc)3 [hfc = 3-(heptafluoropropylhydroxymethylene)-d-camphorate].
NMR data of 4 are published as supporting information on the PNAS web site.
Poly(D4[DPB]MA) (p12). The synthesis of diphenyl-4-[(diphenylphosphanylborane)phenyl]methyl methacrylate (D4[DPB]MA; 12) from 1,4-dibromobenzene (9; see Scheme 4) is described in detail in the supporting information. The polymerization was carried out analogously to the preparation of poly(PB2PyMA) (p5; see ref. 21 and supporting information) by adding the chiral base mixture of 142.0 mg (0.67 mmol, 1.0 eq) of DPEDA (6), 372.0 mg (0.67 mmol, 1.0 eq) of nBuLi, and 123.8 mg (131 μl, 0.80 mmol, 1.2 eq) of (+)-PMP (7) to a solution of 4.600 g (8.74 mmol, 13.0 eq) of D4[DPB]MA (12) in 180 ml of absolutely dry toluene. The reaction mixture was stirred for 72 hr at –78°C and then for another 10 hr at +10°C. The polymerization was quenched by adding 0.3 ml of methanol, and the polymer was precipitated in 1 liter of ice-cold methanol, separated by centrifugation, and dried under reduced pressure. The obtained polymer/oligomer mixture was dissolved in toluene and reprecipitated in 400 ml of benzene/hexane (1:1, vol/vol). Subsequent drying of the residue in vacuum yielded 1.395 g (29%) of p12 as a white solid. A 60-mg portion of this polymer was converted into the corresponding poly(methyl methacrylate) (PMMA) by acidic hydrolysis (in refluxing methanol containing hydrochloric acid) and subsequent methylation of the resulting poly(acrylic acid) with diazomethane. The obtained PMMA was used to analyze the tacticity (by 1H-NMR spectroscopy) as well as the degree of polymerization (DP) and molecular weight distribution (by gel-permeation chromatography). 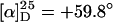 (c = 1in 9:1 CHCl3/CF3CH2OH). Tacticity: >99% meso meso (mm). Gel-permeation chromatography (THF, PMMA standards): DP = 19, weight-average molecular weight/number-average molecular weight (Mw/Mn) = 1.10. 1H-NMR (500 MHz, CD2Cl2, 300 K, tetramethylsilane reference): δ = 0.706–1.766, 1.882–2.748 (br m, CH, CH2, CH3, BH3), 5.7–7.8 (m, aromatic-H24) ppm. 31P-NMR (202 MHz, CD2Cl2, 300 K): δ = 18.52 (P–BH3), [24.93 (m, P—O)] ppm.
(c = 1in 9:1 CHCl3/CF3CH2OH). Tacticity: >99% meso meso (mm). Gel-permeation chromatography (THF, PMMA standards): DP = 19, weight-average molecular weight/number-average molecular weight (Mw/Mn) = 1.10. 1H-NMR (500 MHz, CD2Cl2, 300 K, tetramethylsilane reference): δ = 0.706–1.766, 1.882–2.748 (br m, CH, CH2, CH3, BH3), 5.7–7.8 (m, aromatic-H24) ppm. 31P-NMR (202 MHz, CD2Cl2, 300 K): δ = 18.52 (P–BH3), [24.93 (m, P—O)] ppm.
Scheme 4.

Synthesis, helix sense selective polymerization, and subsequent complexation of methacrylate 12 with palladium (p12: n = 19, Mw/Mn = 1.10, >99% isotactic, 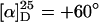 ) (see supporting information for details).
) (see supporting information for details).
Complexation of Poly(D4[DPB]MA) (p12) with Allyl Palladium Chloride. Under argon, 20.0 mg (0.04 mmol, 1 eq, according to the monomer units) of p12 and 12.8 mg (0.08 mmol, 3 eq) of diazabicyclo[2.2.2]octane (DABCO) were dissolved in 5 ml of dry and degassed toluene. After cooling to –20°C, a solution of 6.9 mg (0.02 mmol, 0.5 eq) of [Pd(η3-C3H5)Cl]2 in 2 ml of dry and degassed THF was added dropwise. The reaction mixture was allowed to warm to room temperature during 1 hr [(Pd-p12)a] or overnight [(Pd-p12)b]. Then the solvent was removed under reduced pressure and the yellow residue was suspended in 1 ml of dry CD2Cl2 (degassed) under treatment with ultrasound for 5 min. The insoluble fraction (DABCO-borane) was separated by filtration under an argon atmosphere and the filtrate was studied by 1H- and 31P-NMR spectroscopy. Alternatively, CH2Cl2 was used instead of CD2Cl2. In this case, the properties of (Pd-p12)a/b as a potential catalyst for the allylic substitution reaction of 2 (see above) were examined.
Spectroscopic data of the η3-complex (Pd-p12)a. 1H-NMR (500 MHz, CD2Cl2, 300 K, tetramethylsilane): δ = 0.65–1.65, 2.126 (br m, CH, CH2, CH3, BH3), 3.223 (br s, 2 × 1-Ht), 4.508 (br s, 2 × 1-Hc), 5.394 (m, 2-H), 6.36–7.78 (m, aromatic-H24) ppm. 31P-NMR (202 MHz, CD2Cl2, 300 K): δ = 20.28 (br s, P-Pd) ppm.
Spectroscopic data of the η1-complex (Pd-p12)b. 1H-NMR (500 MHz, CD2Cl2, 300 K, tetramethylsilane): δ = 0.65–1.68, 2.088 (br m, CH, CH2, CH3, BH3), 4.238 (d, 3-H2), 5.636 (d, 1-Hc), 5.698 (d, 1-Ht), 5.881 (ddt, 2-H), 6.35–7.77 (m, aromatic-H24) ppm. J2,3 = 7.5 Hz, 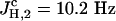 ,
, 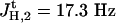 . 31P-NMR (200 MHz, CD2Cl2, 300 K): δ = 25.06 (br s, P-Pd) ppm.
. 31P-NMR (200 MHz, CD2Cl2, 300 K): δ = 25.06 (br s, P-Pd) ppm.
Poly(3DPBPIC-co-DMHIC) [p(16-co-17)] (Typical Procedure). 3-(Diphenylphosphanylborane)propylisocyanate (3DPBPIC; 16) was synthesized from amine 13 (see Scheme 5; see also supporting information). For the copolymerization with (R)-DMHIC (17) (26), 0.300 g (1.060 mmol, 0.98 eq) of 3DPBPIC (16) was placed in a long-necked flask, equipped with a rubber septum and a gas inlet, and freeze-dried from benzene. DMHIC (17) (0.190 g, 1.122 mmol, 1.04 eq) and 13 ml of THF were added by means of syringe and the solution was cooled to –95°C. Then, 1.5 ml (0.22 mmol, 0.2 eq) of a NaBPh4 solution (50 mg/ml in THF) was added and after 5 min 0.53 ml (0.022 mmol, 0.02 eq) of the initiator solution (0.041 M NaCN in dimethylformamide). After stirring for 16.5 hr at –95°C, the polymerization was terminated by adding 2.2 ml (0.22 mmol, 10 eq) of anhydrous HCl (0.1 M in methanol), and the mixture was poured into 60 ml of methanol. After separation of the precipitate by centrifugation, the residue was dissolved in 15 ml of CH2Cl2 and reprecipitated in 60 ml of methanol. Subsequent freeze-drying of the residue from benzene yielded 0.275 g (56%) of p(16-co-17) as a white solid. For analysis of the molar ratios of the incorporated monomers as well as the distribution of the comonomers along the polymer chain, a portion of the polymer was transformed into the corresponding cyclic trimers by base-induced degradation with sodium methanolate (see supporting information). 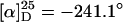 (c = 0.5 in CH2Cl2). [3DPBPIC]:[DMHIC] calculated: 49:51, found: 76:24. Gel-permeation chromatography (THF, polystyrene standards): (x + y) = 90, Mw/Mn = 2.34. 1H-NMR (300 MHz, CDCl3, 300 K): δ = 0.2–4.3 (br m), 6.91–8.00 (br m) ppm. 13C-NMR (75.475 MHz, CDCl3, 300 K): δ = 16.1, 17.47, 17.64, 22.68, 24.62, 27.92, 34.43, 34.59, 39.18, 48.50, 127.9–132.7, 155.6 ppm.
(c = 0.5 in CH2Cl2). [3DPBPIC]:[DMHIC] calculated: 49:51, found: 76:24. Gel-permeation chromatography (THF, polystyrene standards): (x + y) = 90, Mw/Mn = 2.34. 1H-NMR (300 MHz, CDCl3, 300 K): δ = 0.2–4.3 (br m), 6.91–8.00 (br m) ppm. 13C-NMR (75.475 MHz, CDCl3, 300 K): δ = 16.1, 17.47, 17.64, 22.68, 24.62, 27.92, 34.43, 34.59, 39.18, 48.50, 127.9–132.7, 155.6 ppm.
Scheme 5.

Synthesis of 3DPBPIC (16; see supporting information for details). Boc, t-butoxycarbonyl.
Deprotection of p(16-co-17) to Poly(3DPPIC-co-DMHIC) [p(18-co-17)]. First, 0.072 g (0.182 mmol, 1.0 eq, according to phosphanylborane) of p(16-co-17) and 0.113 g (1.007 mmol, 5.5 eq) of DABCO were suspended in 7.5 ml of toluene/THF (4:1) in an atmosphere of argon. Then, 0.18 ml of a 0.1 M solution of dry HCl in methanol was added. After 69 hr, the polymer was precipitated in 32 ml of degassed methanol, isolated by centrifugation, and dried under reduced pressure to yield 0.064 g (92%) of p(18-co-17) as a white solid. 1H-NMR (500 MHz, CDCl3, 300 K): δ = 0.3–2.5 (br m, ≈15.5 H), 2.8–4.2 (br m, ≈2.9 H), 7.00–7.51 (br m, ≈9.0 H, phosphane Ph-H), 7.52–7.78 (br, ≈1.0 H, phosphane-borane Ph-H). 31P-NMR (202 MHz, CDCl3, 300 K): δ = –16.2 (br, 78%, phosphane), 16.7 (br, 22%, phosphane-borane) ppm.
Asymmetric Hydrogenation. In a Schlenk flask under argon, 15.9 mg (46.99 μmol, 3 eq according to phosphorus) of p(18-co-17) was dissolved in 4 ml of dry benzene, and a solution of 7.4 mg (15.80 μmol, 1 eq) of [Rh(COD)2]OTf in 1 ml of CH2Cl2 was added. After precipitation of the polymeric complex, the solvent was removed by lyophilization and replaced by 11 ml of CH2Cl2/methanol (9:1) to yield a yellow solution. Upon addition of 0.327 g (1.593 mmol, 101 eq) of N-acetamidocinnamic acid (19), the atmosphere was changed to hydrogen. After complete conversion (checked by 1H-NMR) the solvent was removed under reduced pressure and the residue was suspended in 30 ml of a 0.5 M NaOH solution. After filtration, the solution was acidified with 2 M HCl to pH 2 and extracted with ethyl acetate (3 times with 15 ml). The collected organic layers were dried over MgSO4 and the solvent was removed under reduced pressure, yielding 20 as a white solid. Spectroscopic data were in accordance with those reported in ref. 27. Optical purity was determined as 14.5% ee by treatment of a portion of the acid in THF with diazomethane and 1H-NMR spectroscopic analysis of the resulting methyl ester in the presence of 15 mol % of the chiral shift reagent Pr(hfc)3 [hfc = 3-(heptafluoropropylhydroxymethylene)-d-camphorate).
Results and Discussion
Polymethacrylates. In the course of the catalyses with poly(3PyDBSMA) (p1) we observed the precipitation of palladium black, indicating insufficient complexation of the intermediate Pd(0) species by the monodentate ligand. Following published procedures (21), we therefore synthesized PB2PyMA (5), which offers two pyridine side groups. To check the ability of this ligand to form bidentate complexes with palladium, we prepared the monomeric palladium complex by using [Pd(η3-C3H5)Cl]2 as the palladium source. After changing the counterion to 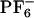 , we were able to obtain single crystals suitable for an x-ray structural analysis (Fig. 1). Although the resolution is not too good (R factor = 13.3%), the structure reveals successful bidentate complexation.
, we were able to obtain single crystals suitable for an x-ray structural analysis (Fig. 1). Although the resolution is not too good (R factor = 13.3%), the structure reveals successful bidentate complexation.
Fig. 1.

Formula (Left) and x-ray structure (Right) of the PB2PyMA complex with palladium (Pd-5) (counterion 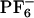 not shown).
not shown).
After this encouraging result we synthesized the known poly(PB2PyMA) (p5) (21) by helix sense-selective anionic polymerization with a mixture of DPEDA monolithium amide (6-Li; DPEDA-Li) and (+)-7 [(+)-PMP] as an initiator (Scheme 3). In accordance with ref. 21, the macromolecule was not perfectly isotactic and the optical activity of p5 (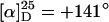 ) was considerably lower than that of p1 (ref. 15:
) was considerably lower than that of p1 (ref. 15: 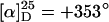 ), indicating that the helix is not single-handed. Moreover, the specific optical rotation in solution decreased very fast, showing that the helix is not conformationally stable (see supporting information). Nevertheless, we prepared a catalyst from p5 and allyl palladium chloride and found it to be much more active in the model substitution reaction than the corresponding complex with p1 as a ligand (Scheme 2, Table 1). This time the reaction was quantitative even at room temperature and with only 5 mol % palladium (Table 1, no. 3). Not unexpectedly, considering the low optical activity of p5, the enantioselectivities were rather low (5–6% ee).
), indicating that the helix is not single-handed. Moreover, the specific optical rotation in solution decreased very fast, showing that the helix is not conformationally stable (see supporting information). Nevertheless, we prepared a catalyst from p5 and allyl palladium chloride and found it to be much more active in the model substitution reaction than the corresponding complex with p1 as a ligand (Scheme 2, Table 1). This time the reaction was quantitative even at room temperature and with only 5 mol % palladium (Table 1, no. 3). Not unexpectedly, considering the low optical activity of p5, the enantioselectivities were rather low (5–6% ee).
Scheme 3.

Helix sense selective anionic polymerization to poly(PB2PyMA) (p5) (30% yield, n = x + y = 40, Mw/Mn = 1.42, 73% isotactic, 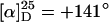 ) and poly(TrMA-co-PB2PyMA) p(5-co-8) (77% yield, (x + y) = 65, x/y = 1:1, Mw/Mn = 1.26, 99% isotactic,
) and poly(TrMA-co-PB2PyMA) p(5-co-8) (77% yield, (x + y) = 65, x/y = 1:1, Mw/Mn = 1.26, 99% isotactic, 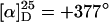 ) (see ref. 21; see also supporting information for details).
) (see ref. 21; see also supporting information for details).
Table 1. Allylic alkylation of 1,3-diphenylprop-2-enyl acetate (2) with dimethyl malonate and p5 or p(5-co-8) as ligand.
| No. | Ligand | Pd,* mol% | Temp., °C | Time, hr | Yield,† % | ee,‡ % |
|---|---|---|---|---|---|---|
| 1 | (+)-p5 | 25 | 40 | 1 | 91§ | 6.4 |
| 2 | (+)-p5 | 10 | 25 | 3 | 85§ | 4.7 |
| 3 | (+)-p5 | 5 | 25 | 6 | 87§ | 6.4 |
| 4 | (+)-p5 | 2 | 25 | 150 | 20 | 5.8 |
| 5 | (+)-p(5-co-8) | 10 | 25 | 4.5 | 83§ | 38 |
| 6 | (+)-p(5-co-8) | 25 | 25 | 1 | 84§ | 44 |
| 7 | (+)-p(5-co-8) | 25 | 0 | 16 | 99 | 60 |
| 8 | (+)-p(5-co-8) | 25 | -10 | 64 | 72 | 55 |
Reaction was in CH2Cl2,[Pd]/[repeating units] = 1:1.2.
Based on substrate 2.
Yields of isolated product.
Determined by 1H-NMR in the presence of 20 mol % of the chiral shift reagent [Eu(hfc)3], hfc = 3-(heptafluoropropylhydroxymethylene)-d-camphorate. All products were (R)-configured (determined by comparison with ref. 28).
Quantitative conversion according to TLC.
To maintain the possibility of a bidentate complexation we then thought about a stabilization of the helical structure of poly(PB2PyMA). Following a publication of Ren et al. (25), we prepared the statistically 1:1-copolymer p(5-co-8) (Scheme 3). A nearly perfectly isotactic backbone and a high optical activity (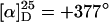 ) account for a single-handed helical configuration. The value of specific optical rotation decreased only very slowly with time, proving an almost stable conformation in solution (see supporting information). Obviously, this copolymer combines the advantages of both monomers, i.e., chance of bidentate complexation and a stable helical conformation. The reactivity of the catalyst with p(5-co-8) as ligand was indeed similar to that derived from the homopolymer (Table 1, compare nos. 5 and 2). In accordance with our expectation the complexed copolymer gave a far better enantioselectivity. With 25 mol % palladium at 0°C the substitution product was isolated with 60% ee (no. 7).
) account for a single-handed helical configuration. The value of specific optical rotation decreased only very slowly with time, proving an almost stable conformation in solution (see supporting information). Obviously, this copolymer combines the advantages of both monomers, i.e., chance of bidentate complexation and a stable helical conformation. The reactivity of the catalyst with p(5-co-8) as ligand was indeed similar to that derived from the homopolymer (Table 1, compare nos. 5 and 2). In accordance with our expectation the complexed copolymer gave a far better enantioselectivity. With 25 mol % palladium at 0°C the substitution product was isolated with 60% ee (no. 7).
At this point we should note that the investment of chiral nonracemic material [(+)-PMP; (+)-7] is extremely low. In the ideal case, a polymerization reaction in which 1 mol of chiral initiator is used produces n mol of homochiral coordination sites in the resulting polymer. The whole process can thus be interpreted as an autoinductive multiplication of chiral units. Furthermore, different helical ligands exhibiting different chiral environments can be synthesized by using the same cheap auxiliary.
Nevertheless, even with polymeric PB2PyMA (5) as palladium ligand, precipitation of palladium black was not completely suppressed at the end of the substitution reaction. To coordinate palladium even more strongly to the ligand, we changed the donor atom from nitrogen to phosphorus. Beside the potential catalysis-related advantages, the beneficial NMR-spectroscopic properties of 31P should facilitate the characterization of the polymeric transition metal complexes. After several unsuccessful attempts to synthesize phosphinine-modified methacrylates, we succeeded in preparing the methacrylate 12 from 1,4-dibromobenzene (9) in three steps (66% overall yield, Scheme 4).
Methacrylate 12 was polymerized with the chiral base mixture 6-Li/(+)-7, yielding p12 in 30% yield and with a narrow molecular weight distribution (Mw/Mn = 1.10). Unfortunately, the optical rotation (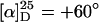 ) was rather low compared with that of p1 (ref. 15:
) was rather low compared with that of p1 (ref. 15: 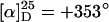 ), indicating that the secondary structure was helically chiral, but probably not single handed. Palladium complexes of p12 were obtained by in situ deboranation with DABCO and complexation with allyl palladium chloride (Scheme 4). The 31P-NMR spectra of Pd-p12 show that, depending on the reaction time, two different phosphorus species emerge (Fig. 2). The primarily formed species was identified as the η3 complex (Pd-p12)a. This was derived from a comparison of the 1H-NMR spectra of [Pd(η3-C3H5)Cl]2 with the spectrum of (Pd-p12)a (Fig. 2 D and E). Assignment of the signals was confirmed by a (1H,1H)-COSY spectrum (not shown). In case of a longer reaction time, we suspect the formation of the corresponding η1 complex (Pd-p12)b, as can be deduced from the 1H-NMR spectrum (Fig. 2F). Especially 1-Hc and 1-Ht are shifted downfield significantly, indicating their olefinic character. We think that this unusual η1 complex is a result of the high steric congestion exerted by the polymeric ligand.
), indicating that the secondary structure was helically chiral, but probably not single handed. Palladium complexes of p12 were obtained by in situ deboranation with DABCO and complexation with allyl palladium chloride (Scheme 4). The 31P-NMR spectra of Pd-p12 show that, depending on the reaction time, two different phosphorus species emerge (Fig. 2). The primarily formed species was identified as the η3 complex (Pd-p12)a. This was derived from a comparison of the 1H-NMR spectra of [Pd(η3-C3H5)Cl]2 with the spectrum of (Pd-p12)a (Fig. 2 D and E). Assignment of the signals was confirmed by a (1H,1H)-COSY spectrum (not shown). In case of a longer reaction time, we suspect the formation of the corresponding η1 complex (Pd-p12)b, as can be deduced from the 1H-NMR spectrum (Fig. 2F). Especially 1-Hc and 1-Ht are shifted downfield significantly, indicating their olefinic character. We think that this unusual η1 complex is a result of the high steric congestion exerted by the polymeric ligand.
Fig. 2.

(A–C) 31P-NMR spectra (CD2Cl2, 202 MHz, 300 K) of p12 (A), (Pd-p12)a, 1 hr after deprotection and complexation (B, compare Scheme 4), and (Pd-p12)b, 12 hr after deprotection and complexation (C, compare Scheme 4). (D–F) 1H-NMR spectra (CD2Cl2, 500 MHz, 300 K) of [Pd(η3-C3H5)Cl]2 (D), (Pd-p12)a (E), and (Pd-p12)b (F).
To study the catalytic properties of (Pd-p12)a and (Pd-p12)b we again chose the asymmetric allylic substitution reaction (Table 2).
Table 2. Allylic alkylation of 1,3-diphenylprop-2-enyl acetate (2) with dimethyl malonate and (Pd-p12)a or (Pd-p12)b as ligand.
| No. | Catalyst | Pd,* mol % | Temp., °C | Time, hr | Yield,† % | ee,‡ % |
|---|---|---|---|---|---|---|
| 1 | (Pd-p12)a | 25 | 25 | 0.02 | 95§ | 1.8 |
| 2 | (Pd-p12)a | 25 | 0 | 2 | 99 | 4.5 |
| 3 | (Pd-p12)a | 25 | -20 | 12 | 99 | 2.6 |
| 4 | (Pd-p12)a | 25 | -40 | 72 | 0¶ | — |
| 5 | (Pd-p12)b | 25 | 25 | 0.02 | 82§ | 3.0 |
Reaction was in CH2Cl2, 30 mol % repeating units (based on substrate 2).
Based on substrate 2.
Yields of isolated product.
Determined by 1H-NMR in the presence of 20 mol % of the chiral shift eagent [Eu(hfc)3], hfc = 3-(heptafluoropropylhydroxymethylene)-d-camphorate. All products were (R)-configured (determined by comparison with ref. 28).
Quantitative conversion according to TLC.
Insolubility of KOAc in CH2Cl2 at -40°C prevented reaction.
We were very pleased to see that the palladium complexes (Pd-p12)a as well as (Pd-p12)b were substantially more active catalysts than the corresponding pyridine derivatives p1 and even p(5-co-8) (compare Scheme 2, Table 1). Now the conversion with 25 mol % catalyst was quantitative within 12 hr even at –20°C (Table 2, no. 3), but in accordance with the low optical activities of these ligands the enantioselectivities were rather low (2–5% ee).
With these highly reactive catalysts at hand we might be able to achieve higher levels of enantioselectivity by changing both the polymerization conditions and the monomeric units. Furthermore, it may be possible to apply these polymethacrylate-based ligands in other asymmetric catalyses, such as hydrogenation, Diels–Alder, and cyclopropanation reactions.
PICs. Besides our studies related to polymethacrylates we examined the suitability of helically chiral PICs as ligands in asymmetric transition metal catalysis. As already mentioned, the helical backbones of these macromolecules are dynamic, and the energy barriers for helix–helix inversions are low. To induce and to maintain a preferred helical sense a certain amount of a chiral, nonracemic comonomer is needed. Therefore, a common method to get single-handed helical PICs is to prepare statistically copolymers of achiral monomers (soldiers) and chiral nonracemic monomers (sergeants) (1, 13). These polymers are characterized by a large number of achiral units taking part in the overall helical sense dictated by a small number of chiral units, entailing the fascinating possibility of generating helically chiral polymeric ligands by using only small amounts of chiral units, which even must not necessarily be enantiomerically pure (1, 29). Thus, if the donor atom-containing units are achiral and in large excess, asymmetric induction should mainly be caused by the stereoregular secondary structure induced by a minority of chiral units. Contrasting the behavior of polymethacrylates, which racemize irreversibly, the chiral and structure-determining perturbation is always present in sergeants and soldiers PICs. This circumstance makes them configurationally stable even in solution. Another striking feature of this type of polymer is that several PICs are known that switch their helical sense under UV-irradiation or under the influence of circularly polarized light (12, 30). By means of such effects it could be possible to develop chiral catalysts with completely new properties such as the ability to invert the asymmetric induction by external stimuli.
Based on our encouraging results with the borane-phosphane-modified polymethacrylates such as p12, we studied the synthesis and catalytic activities of PICs with phosphorus donor atoms. Therefore, we prepared 3DPBPIC (16) from amine 13 in 44% overall yield (Scheme 5).
The anionic polymerization of 16 with different amounts of the known (R)-DMHIC (17) (26) as a sergeant was carried out with NaCN as an initiator and NaBPh4 to prevent “back-biting” (31) (Scheme 6, Table 3).
Scheme 6.

Copolymerization of 16 and 17 followed by deprotection with DABCO. DMF, dimethylformamide.
Table 3. Copolymers of alkylisocyanates 16 and 17.
| No. | Batch [16]/[17]* | Polymer† [16]/[17] | Time, hr | Yield,‡ % | DP§ | Mw/Mn§ |
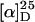 ¶ ¶
|
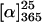 ¶ ¶
|
|---|---|---|---|---|---|---|---|---|
| 1 | 50:50 | 76:24 | 17 | 56 | 90 | 2.34 | -241 | -906 |
| 2 | 30:70 | 71:29 | 20 | 23 | 51 | 1.73 | -242 | -906 |
| 3 | 25:75 | 60:40 | 20 | 30 | 61 | 1.78 | -280 | -1048 |
All polymerizations were carried out with 3-5 mmol of monomers (total amounts) in THF at -95°C. The monomer:initiator ratio was 100:1. Polymerizations were quenched by adding 10 eq of HCl in MeOH.
Initial ratio (mol %) of monomers.
Ratio (mol %) of monomer units incorporated in the polymer. Determined by complete degradation of the polymer with sodium methanolate and 1H-NMR spectroscopic analysis of the resulting cyclic trimers.
Fraction insoluble in MeOH.
DP, degree of polymerization. Determined by gel-permeation chromatography (polystyrene standard).
c = 0.5, CH2Cl2.
The copolymers p(16-co-17) were isolated in moderate to good yields (23–56%) and with acceptable molecular weight distributions (Mw/Mn = 1.7–2.3). The chain lengths were lower than expected from the monomer/initiator ratios, and the amounts of incorporated 3DPBPIC in the copolymers were in all cases higher than expected from the monomer ratios. These findings suggested a preferred uptake of the soldiers 16 during the polymerization process. Nevertheless, the distribution of the monomers within the polymer chain was almost statistical. This was shown by NMR-spectroscopic analysis of the relative amounts of the four possible trimers after complete basic degradation of the copolymer (see supporting information). The absolute values of the specific optical rotations of all copolymers (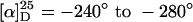 ) were significantly lower than those of the homopolymer poly(DMHIC) (p17) (ref. 1:
) were significantly lower than those of the homopolymer poly(DMHIC) (p17) (ref. 1: 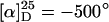 ), even with a sergeant-to-soldier ratio of 40:60, indicating that the chiral bias of the sergeants in these polymers is obviously less pronounced than in p17.
), even with a sergeant-to-soldier ratio of 40:60, indicating that the chiral bias of the sergeants in these polymers is obviously less pronounced than in p17.
Nevertheless, we studied the suitability of these chiral polymeric phosphorus derivatives as ligands in the rhodium-catalyzed asymmetric hydrogenation of N-acetamidocinnamic acid (19) (Scheme 7). After deprotection of p(16-co-17) ([x]:[y] = 60:40) with DABCO and substoichiometric amounts of HCl, p(18-co-17) was obtained in 92% yield with 78 mol % free phosphane groups (determined by 31P-NMR spectroscopy) (Scheme 6). The copolymer p(18-co-17) was successfully complexed by using [Rh(COD)2]OTf as metal source and benzene as solvent. To our delight, after replacement of benzene by CH2Cl2/MeOH this in situ generated rhodium complex was catalytically very active albeit hardly enantioselective (Scheme 7). When the hydrogenation was carried out with only 1 mol % rhodium at room temperature and under H2 at atmospheric pressure, (S)-20 was obtained quantitatively with 14.5% ee.
Scheme 7.

Rhodium-catalyzed asymmetric hydrogenation of 19 with a Rh-p(18-co-17) complex generated in situ as catalyst. COD, cyclooctadiene; OTf, trifluoromethanesulfonate.
In addition to these experiments with P-containing PICs we prepared a PIC containing phenolic side groups. From Monte Carlo simulations we expected this polymer to behave like a multiple-site analogue of 2,2′-dihydroxy-1,1′-binaphthyl (BINOL; see supporting information). Unfortunately, its limited solubility in noncoordinating solvents prevents the application of this polymer as a ligand in Lewis-acid catalysis.
Although these experiments demonstrate the principal suitability of PIC-based ligands for asymmetric transformations, the observed stereoselectivity is clearly dissatisfactory. The main problems with the PICs studied are the limited solubility in noncoordinating solvents and/or the difficulty in controlling their helicity. Moreover, the notoriously low enantioselectivities observed in different stereogenic reactions point to an inefficient transfer of chirality from the helical backbone to the active site of the catalyst. This problem seems to be much less pronounced in the reasonably successful polymethacrylates; we interpret this success to be a consequence of a chiral propeller conformation of the triarylmethyl unit induced by the helically chiral backbone. Similar reasoning has been put forward by Reetz and Neugebauer (32) to explain a local chirality close to the catalytically active center in metal complexes from conformationally labile compounds attached to a chiral backbone. In the light of this perception it looks promising to attach, for example, donor substituted configurationally labile biphenyl units to the polymer to obtain rapidly interconverting, conformationally diastereomeric catalysts with the metal determining the stereochemical outcome of the reaction. This can be provoked either by locking an induced preferred conformation by metal complexation (33, 34) or by preferred reaction of one diastereomorphic conformation (32).
Supplementary Material
Acknowledgments
We thank the Fonds der Chemischen Industrie (Kekulé Fellowship for S.D.) and the Deutsche Forschungsgemeinschaft (DFG Re 1007/3-1) for financial support and the Degussa AG for generous gifts of transition metal salts.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: PIC, polyisocyanates; PB2PyMA, phenyl[bis(2-pyridyl)]methyl methacrylate; DPEDA, N,N′-diphenylethylenediamine; (+)-PMP, (+)-(S)-1-(2-pyrrolidinomethyl)pyrrolidine; DMHIC, 2,6-dimethylheptyl isocyanate; THF, tetrahydrofuran; TrMA, triphenylmethyl methacrylate; D4[DPB]MA, diphenyl-4-[(diphenylphosphanylborane)phenyl]methyl methacrylate; DABCO, diazabicyclo[2.2.2]octane; 3DPBPIC, 3-(diphenylphosphanylborane)propylisocyanate; ee, enantiomeric excess.
References
- 1.Green, M. M., Park, J.-W., Sato, T., Teramoto, A., Lifson, S., Selinger, R. L. B. & Selinger, J. V. (1999) Angew. Chem. Int. Ed. 38, 3139–3154. [DOI] [PubMed] [Google Scholar]
- 2.Bolm, C. & Gerlach, A. (1997) Angew. Chem. Int. Ed. 36, 741–743. [Google Scholar]
- 3.Han, H. S. & Janda, K. D. (1997) Tetrahedron Lett. 38, 1527–1530. [Google Scholar]
- 4.Pu, L. (1998) Chem. Rev. 98, 2405–2494. [DOI] [PubMed] [Google Scholar]
- 5.Gilbertson, S. R. & Wang, X. (1996) Tetrahedron Lett. 37, 6475–6478. [Google Scholar]
- 6.Okamoto, Y. & Nakano, T. (1994) Chem. Rev. 94, 349–372. [Google Scholar]
- 7.Nakano, T. & Okamoto, Y. (2001) Chem. Rev. 101, 4013–4038. [DOI] [PubMed] [Google Scholar]
- 8.Ito, Y., Miyake, T., Hatano, S., Shima, R., Ohara, T. & Suginome, M. (1998) J. Am. Chem. Soc. 120, 11880–11893. [Google Scholar]
- 9.Suginome, M., Collet, S. & Ito, Y. (2002) Org. Lett. 4, 351–354. [DOI] [PubMed] [Google Scholar]
- 10.Bur, A. J. & Fetters, L. J. (1976) Chem. Rev. 76, 727–746. [Google Scholar]
- 11.Yashima, E., Maeda, Y., Matsushima, T. & Okamato, Y. (1997) Chirality 9, 593–600. [Google Scholar]
- 12.Mayer, S., Maxein, G. & Zentel, R. (1999) Macromol. Symp. 137, 67–73. [Google Scholar]
- 13.Green, M. M., Reidy, M. P., Johnson, R. J., Darling, G., O'Leary, D. J. & Willson, G. (1989) J. Am. Chem. Soc. 111, 6452–6454. [Google Scholar]
- 14.Gu, H., Nakamura, Y., Sato, T., Teramoto, A., Green, M. M., Jha, S. K., Andreola, C. & Reidy, M. P. (1998) Macromolecules 31, 6362–6368. [Google Scholar]
- 15.Reggelin, M., Schultz, M. & Holbach, M. (2002) Angew. Chem. Int. Ed. 41, 1614–1617. [DOI] [PubMed] [Google Scholar]
- 16.Helmchen, G. & Pfaltz, A. (2000) Acc. Chem. Res. 33, 336–345. [DOI] [PubMed] [Google Scholar]
- 17.Reif, B., Steinhagen, H., Junker, B., Reggelin, M. & Griesinger, C. (1998) Angew. Chem. Int. Ed. 37, 1903–1906. [Google Scholar]
- 18.Steinhagen, H., Reggelin, M. & Helmchen, G. (1997) Angew. Chem. Int. Ed. 36, 2108–2110. [Google Scholar]
- 19.Trost, B. M. & Vranken, D. L. v. (1996) Chem. Rev. 96, 395–422. [DOI] [PubMed] [Google Scholar]
- 20.Auburn, P. R., Mackenzie, P. B. & Bosnich, B. (1985) J. Am. Chem. Soc. 107, 2033–2046. [Google Scholar]
- 21.Ren, C., Chen, C., Xi, F., Nakano, T. & Okamoto, Y. (1993) J. Polym. Sci. Part A: Polym. Chem. 31, 2721–2728. [Google Scholar]
- 22.Schouten, A. E. (1937) Recl. Trav. Chim. Pays-Bas 56, 541–561. [Google Scholar]
- 23.Corey, E. J., Shibata, S. & Bakshi, R. K. (1988) J. Org. Chem. 53, 2861–2863. [Google Scholar]
- 24.Asami, M. (1990) Bull. Chem. Soc. Jpn. 63, 721–727. [Google Scholar]
- 25.Ren, C., Chen, C. & Xi, F. (1998) J. Polym. Sci. Part A: Polym. Chem. 36, 2127–2133. [Google Scholar]
- 26.Mueller, M. & Zentel, R. (1993) Makromol. Chem. 194, 101–116. [Google Scholar]
- 27.Riegel, N., Darcel, C., Stephan, O. & Juge, S. (1998) J. Organomet. Chem. 567, 219–233. [Google Scholar]
- 28.Sprinz, J. & Helmchen, G. (1993) Tetrahedron Lett. 34, 1769–1772. [Google Scholar]
- 29.Jha, S. K., Cheon, K.-S., Green, M. M. & Selinger, J. V. (1999) J. Am. Chem. Soc. 121, 1665–1673. [Google Scholar]
- 30.Li, J., Schuster, G. B., Cheon, K.-S., Green, M. M. & Selinger, J. V. (2000) J. Am. Chem. Soc. 122, 2603–2612. [Google Scholar]
- 31.Shin, Y.-D., Kim, S.-Y., Ahn, J.-H. & Lee, J.-S. (2001) Macromolecules 34, 2408–2410. [Google Scholar]
- 32.Reetz, M. T. & Neugebauer, T. (1999) Angew. Chem. Int. Ed. 38, 179–181. [DOI] [PubMed] [Google Scholar]
- 33.Luo, Z., Liu, Q., Gong, L., Cui, X., Mi, A. & Jiang, Y. (2002) Angew. Chem. Int. Ed. 41, 4532–4535. [DOI] [PubMed] [Google Scholar]
- 34.Becker, J. J., White, P. S. & Gagné, M. R. (2001) J. Am. Chem. Soc. 123, 9478–9479. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.