

Abstract
Parkinson's disease (PD) is a neurodegenerative disease characterized by Lewy body formation and death of dopaminergic neurons. Mutations in α-synuclein and parkin cause familial forms of PD. Synphilin-1 was shown to interact with α-synuclein and to promote the formation of cytosolic inclusions. We now report that synphilin-1 interacts with the E3 ubiquitin-ligases SIAH-1 and SIAH-2. SIAH proteins ubiquitylate synphilin-1 both in vitro and in vivo, promoting its degradation by the ubiquitin–proteasome system. Inability of the proteasome to degrade synphilin-1/SIAH complex leads to a robust formation of ubiquitylated cytosolic inclusions. Ubiquitylation is required for inclusion formation, because a catalytically inactive mutant of SIAH-1, which still binds to synphilin-1, fails to promote inclusions. Like synphilin-1, α-synuclein associates with SIAH in intact cells, but the interaction with SIAH-2 was much stronger that with SIAH-1. In vitro experiments show that SIAH-2 monoubiquitylates α-synuclein. Further evidence that SIAH proteins may play a role in inclusion formation comes from the demonstration of SIAH immunoreactivity in Lewy bodies of PD patients.
Keywords: α-synuclein-interacting protein, ubiquitin, inclusion bodies
Parkinson's disease (PD) is one of the most common neurodegenerative diseases. It is characterized by loss of dopaminergic neurons in the substantia nigra and the presence of cytoplasmic inclusions called Lewy bodies in surviving neurons (1). Hereditary PD can be caused by mutations in components of the ubiquitin–proteasome system, such as the E3 ubiquitin-ligase parkin (2). Recently, a decrease of proteasomal activity was found in the substantia nigra of PD patients, suggesting that failure of the ubiquitin–proteasome system is involved in the formation of Lewy bodies and death of dopaminergic neurons in PD (3).
α-Synuclein is mutated in families with autosomal dominant PD (4, 5) and is a major component of Lewy bodies (6). Although it is still uncertain whether α-synuclein is degraded by the ubiquitin–proteasome system (7, 8), it was found to be ubiquitylated in purified Lewy bodies (9). The nature of α-synuclein ubiquitylation and the E3 ubiquitin-ligase(s) responsible for its ubiquitylation are still not clear.
We have previously characterized synphilin-1 as a protein that associates with α-synuclein and leads to the formation of inclusion bodies when cotransfected with the non-Aβ component (NAC) portion of α-synuclein in cultured cells (10). The interaction between synphilin-1 and α-synuclein has been confirmed by several groups (11–13). Like α-synuclein, synphilin-1 is a presynaptic protein that associates with synaptic vesicles (14). Additionally, synphilin-1 was shown to be present in many types of cytoplasmic inclusions, where it colocalizes with α-synuclein (15–19), supporting the idea that accumulation of synphilin-1 and its interaction with α-synuclein may be relevant for Lewy body formation and PD. In accordance, synphilin-1 is an intrinsic component of Lewy bodies in PD (20). Further evidence that synphilin-1 may be involved in PD comes from the finding of a mutation in the gene of synphilin-1 in two patients with PD (13).
In an attempt to better understand synphilin-1 function, we sought to look for additional synphilin-1 protein partners. We now report that the E3 ubiquitin-ligases SIAH-1 and SIAH-2 (seven in absentia homologues) interact with and ubiquitylate synphilin-1, promoting its degradation through the ubiquitin–proteasome system. SIAH-2 binds and monoubiquitylates α-synuclein. In addition, SIAH is present in Lewy bodies. Our data indicates a role of ubiquitylation in inclusion body formation with implications for Lewy body generation in PD.
Materials and Methods
Further experimental details are provided as Supporting Text, which is published as supporting information on the PNAS web site.
Coimmunoprecipitation Assays. Transfected HEK 293 cells were incubated with 10 μM lactacystin for 12 h and lysed in buffer containing 50 mM Tris (pH 7.4), 140 mM NaCl, 1% Triton X-100, 30 μM MG132, and protease inhibitors. Cell extracts were clarified and supernatant was incubated with anti-hemagglutinin (HA) or anti-myc as described (10). Immunoprecipitates were washed with lysis buffer containing 500 mM NaCl and detected by Western blot (21). For α-synuclein coimmunoprecipitation with SIAH, 1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate was substituted for 1% Triton X-100 in the buffer. This condition gave a much stronger coimmunoprecipitation, indicating an inhibitory effect of Triton X-100 on SIAH-α-synuclein interaction.
In Vitro Ubiquitylation Assays. Synphilin-1 and α-synuclein were translated by using TnT wheat germ in vitro translation kit from Promega using [35S]methionine (Amersham Pharmacia). In vitro translated proteins were incubated in reaction medium containing 40 mM Tris (pH 7.6), 5 mM MgCl2, 2 mM DTT, 1 mM ATP, 10 mM phosphocreatine, 0.1 mg/ml creatine kinase, 7.5 μg of ubiquitin, 1 μg of ubiquitin aldehyde, 100 ng of E1, and 200 ng of UbcH5b. Reactions were incubated at 37°C for 1 h and resolved on SDS/8% or 12% PAGE gels. 35S-labeled-synphilin-1 and α-synuclein were determined by PhosphorImager analysis.
Immunohistochemistry of PD Tissues. Paraffin-embedded human postmortem brain tissues were cut (7 μm thick), immunostained by the avidin–biotin–peroxidase complex method (Vector Laboratories) and developed by using 4% 3-amino-9-ethylcarbazole as chromogen (Biogenex). This substance gives a purple staining that is easily discernible from the brownish neuromelanin. Antibodies against SIAH were used in a 1:10 (H-18; Santa Cruz Biotechnology) and 1:75 dilution (SIAH 990). The specificity of the immunostainings was checked by incubating adjacent sections with anti-SIAH antibody preabsorbed with H-18 peptide or GST-SIAH-1 (amino acids 77–282).
Results
In yeast two-hybrid experiments using the first 227 N-terminal amino acids of synphilin-1, we identified several clones of different sizes that encode for SIAH-1 and SIAH-2 proteins. Due to the high homology of SIAH-1 and SIAH-2, we first characterized the interaction of SIAH-1 with synphilin-1.
We incubated GST-synphilin-1 (amino acids 1–349) fusion protein with extract of HEK 293 cells transfected with HA-tagged full-length SIAH-1 construct (Fig. 1A Left). HA-SIAH-1 specifically interacted with GST-synphilin-1 but not with the control protein GST (Fig. 1 A Left). Conversely, full-length HA-synphilin-1 expressed in HEK 293 cells specifically interacted with GST-SIAH-1 (amino acids 77–282) but not with control proteins, GST or GST-FKBP12 (FK506-binding protein 12) (Fig. 1 A Right). To test whether endogenous synphilin-1 interacts with SIAH-1, we carried out a GST “pull down” experiment in which full-length GST-SIAH-1 was incubated with adult rat brain homogenate. Indeed, GST-SIAH-1 bound endogenous synphilin-1 (Fig. 1B). To determine the specificity of the binding, we used full-length GST-α-synuclein as a positive control and GST-FKBP12 as a negative control. Endogenous synphilin-1 bound to GST-α-synuclein, but not to GST-FKBP12 (Fig. 1B). myc-SIAH-1 coimmunoprecipitates with HA-synphilin-1 from HEK 293 transfected cells, but not the unrelated protein myc-FKBP12 (Fig. 1C). When anti-myc was used to immunoprecipitate myc-FKBP12 or myc-SIAH-1, H A-synphilin-1 was found to be associated only to myc-SIAH-1 (Fig. 1D). In addition, synphilin-1 specifically coimmunoprecipitated with SIAH-1 from rat brain tissue (Fig. 1E) but not with control beads, indicating that these two protein interact in vivo. Parkin, an E3 ubiquitin-ligase previously shown to interact with synphilin-1 (16), also coimmunoprecipitated with synphilin-1 under our experimental conditions (Fig. 1C).
Fig. 1.

Interaction of synphilin-1 and SIAH-1. (A) (Left) Extract of HEK 293 cells transfected with HA-SIAH-1 was incubated with indicated GST-fusion proteins. Binding was analyzed by using an anti-HA antibody. (Right) Extract of HEK 293 cells transfected with HA-synphilin-1 was incubated with indicated GST-fusion proteins. Binding was analyzed by using anti-HA antibody. (B) Pull down of endogenous synphilin-1 from adult rat brain homogenates by GST-SIAH-1. Brain homogenates were incubated with indicated GST-fusion proteins. Binding of endogenous synphilin-1 was analyzed by using anti-synphilin-1 antibody. (C) SIAH coimmunoprecipitates with synphilin-1 from cotransfected HEK 293 cells. HA-synphilin-1 was immunoprecipitated from extracts of HEK 293 cells by using an anti-HA antibody. Coimmunoprecipitation was determined by Western blot using an anti-myc antibody. (D) The converse coimmunoprecipitation experiment was carried out by immunoprecipitation of myc-SIAH-1 from extracts of cotransfected HEK 293 cells using an anti-myc antibody. Coimmunoprecipitation was checked by using an anti-HA antibody. (E) SIAH-1 coimmunoprecipitates with endogenous synphilin-1. Synphilin-1 was immunoprecipitated from rat brain homogenate by using affinity-purified anti-synphilin-1 antibody, and detection of coimmunoprecipitation was carried out by using anti-SIAH-1 antibody (990).
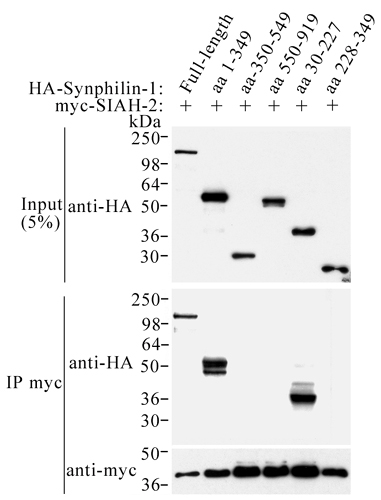
Like SIAH-1, myc-SIAH-2 also coimmunoprecipitated with full-length HA-synphilin-1 in transfected HEK 293 cells (Fig. 7, which is published as supporting information on the PNAS web site). To map the SIAH-binding domain of synphilin-1, we cotransfected different HA-tagged synphilin-1 truncations into HEK 293 cells with myc-SIAH-2 and carried out coimmunoprecipitation experiments. Only truncations retaining the N-terminal region of synphilin-1 coimmunoprecipitated with SIAH-2 (Fig. 7). The minimal region in synphilin-1 responsible for binding SIAH-2 is located on the first N-terminal 227 aa (Fig. 7). Identical binding pattern was observed with SIAH-1 (data not shown).
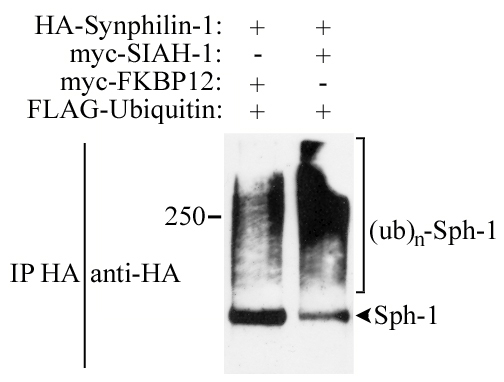
In an attempt to determine whether SIAH proteins ubiquitylate synphilin-1, we carried out in vivo ubiquitylation experiments. We cotransfected HEK 293 cells with HA-synphilin-1, myc-SIAH and FLAG-ubiquitin, and immunoprecipitated HA-synphilin-1 with anti-HA antibody. We found that synphilin-1 is equally ubiquitylated by SIAH-1 and SIAH-2, as shown by the significant anti-FLAG immunoreactivity in the form of smear, which is characteristic of polyubiquitylated proteins (Fig. 2A). Much less ubiquitylation of synphilin-1 was observed when cells were transfected with the unrelated control protein FKBP12, suggesting that SIAH ubiquitylates synphilin-1 in vivo. Detection of immunoprecipitated HA-synphilin-1 with anti-HA antibody also provided a signal in the form of smear, indicating that synphilin-1 itself is polyubiquitylated (Fig. 8, which is published as supporting information on the PNAS web site).
Fig. 2.

SIAH-1 and SIAH-2 ubiquitylate synphilin-1. (A) In vivo ubiquitylation of synphilin-1 by SIAH. Transfected HEK 293 cells were treated for 12 h with 10 μM lactacystin. HA-synphilin-1 was immunoprecipitated by using an anti-HA antibody. Immunoprecipitates were analyzed for ubiquitylation by using anti-FLAG antibody. (B) SIAH ubiquitylates synphilin-1 in vitro. Synphilin-1 was in vitro translated and incubated with the indicated components of the ubiquitin system, in the presence of 0.25 and 1.0 μg GST-SIAH-1, GST-SIAH-2, or GST alone.
To determine the specificity of synphilin-1 ubiquitylation, we carried out in vitro ubiquitylation experiments in which in vitro translated synphilin-1 was incubated with ubiquitin system components (22). We found that synphilin-1 is significantly ubiquitylated in vitro only in the presence of SIAH-1 or SIAH-2, but not with the control protein GST (Fig. 2B).
We determined whether SIAH-1 and SIAH-2 promote synphilin-1 degradation by transfecting HEK 293 cells and analyzing the steady-state level of synphilin-1. A significant decrease in the levels of synphilin-1 was observed when synphilin-1 was cotransfected with SIAH-1 or SIAH-2 (Fig. 3A). When the proteasome inhibitor lactacystin was added to cells cotransfected with synphilin-1 and SIAH, the amount of synphilin-1 returned to levels similar to that observed in the absence of SIAH (Fig. 3A). Furthermore, the steady-state level of synphilin-1 was not significantly decreased by SIAH-1 constructs mutated at the RING-finger (C72S and C55A, C59H, C72S) or lacking the RING-finger domain (amino acids 77–282) (Fig. 9, which is published as supporting information on the PNAS web site). We next carried pulse–chase experiments to determine whether synphilin-1 half-life is altered by SIAH. We found that the degradation rate of 35S-labeled synphilin-1 was significantly accelerated in the presence of SIAH-1 (Fig. 3B). Thus, reduction of synphilin-1 levels by SIAH is due to increased degradation and not to changes in transcription. Taken together, the data indicate that SIAH proteins promote the degradation of synphilin-1 through the ubiquitin–proteasome pathway.
Fig. 3.

SIAH proteins target synphilin-1 for proteasomal degradation. (A) Synphilin-1 and SIAH cotransfected HEK 293 cells were treated with DMSO vehicle or 10 μM lactacystin before harvesting. Extracts HEK 293 cells (25 μg) were analyzed by using an anti-HA antibody. (B) SIAH decreases the half-life of synphilin-1. (Left) Transfected HEK 293 cells were chased for the indicated time points, and HA-synphilin-1 was immunoprecipitated by using an anti-HA antibody. Immunoprecipitates were analyzed by SDS/PAGE and autoradiography. (Right) Quantification of remaining synphilin-1 at different time points.
To determine the cellular effects of ubiquitylated-synphilin-1, we first cotransfected HEK 293 cells with full-length HA-synphilin-1 and myc-SIAH. Immunocytochemistry of cotransfected HEK 293 cells confirmed that SIAH-1 and SIAH-2 promote the degradation of synphilin-1, as observed by the lack of synphilin-1 immunoreactivity in the presence of SIAH proteins (Fig. 4A D–I). To investigate the effects of proteasomal inhibition, we added lactacystin to cells cotransfected with synphilin-1 and SIAH proteins. We found an extensive accumulation of cytosolic inclusions in lactacystin-treated cells cotransfected with synphilin-1 and SIAH proteins (Fig. 4A M–R). These inclusions were found in ≈80% of transfected cells (Fig. 4B) and were positive for both synphilin-1 and SIAH-1/SIAH-2. The inclusions were round, had a very bright halo, and frequently appeared as more than one per cell (Fig. 4A M–R and Fig. 4C). Less than 20% of cytosolic inclusions were observed in lactacystin-treated cells transfected with synphilin-1 alone (Fig. 4B). When present, these inclusions were usually single per cell and had a much fainter halo (Fig. 4C and data not shown). Practically no inclusions were observed in the absence of lactacystin in cells transfected with synphilin-1 alone (<3%, data not shown). Instead, synphilin-1 was homogeneously distributed throughout the cytoplasm of HEK 293 cells (Fig. 4A A).
Fig. 4.

Formation of cytoplasmic inclusions by cotransfection of synphilin-1 and SIAH. (A) Transfected HEK 293 cells were incubated for 12 h with DMSO vehicle (A–I) or 10 μM lactacystin (J–U). Immunocytochemistry was carried out with anti-HA (green) and anti-myc antibody (red). Nuclei were stained with TOTO-3. (Scale bar, 25 μm.) (B) Quantification of inclusion body formation in HEK 293 cells cotransfected with HA-synphilin-1 (WT or V79N, P81N) with vector, myc-SIAH or mutant myc-SIAH-1 constructs in the presence of 10 μM lactacystin. The number of cells containing inclusion bodies is relative to the number of transfected cells. Error bars represent standard deviation of three to eight independent experiments. (C) Quantification of inclusion bodies per cell. HEK 293 cells were cotransfected with HA-synphilin-1 with vector or myc-SIAH-1 in the presence of 10 μM lactacystin. (D) Colocalization of ubiquitin with synphilin-1/SIAH inclusions. HEK 293 cells were transfected with myc-synphilin-1, untagged SIAH-1, and FLAG-ubiquitin or HA-FKBP12 in the presence of 10 μM lactacystin. Immunocytochemistry was carried out with anti-myc (green) and anti-FLAG or anti-HA antibody (red). Nuclei were stained with TOTO-3. (Scale bar, 25 μm.)
To investigate the role of ubiquitylation in inclusion formation, we made a catalytically inactive mutant of SIAH-1 by deleting the first 76 aa encompassing the RING-finger domain. This construct, however, retained full ability to bind to synphilin-1 (Fig. 1 A Right). Lactacystin-treated cells cotransfected with synphilin-1 and inactive SIAH-1 truncation (amino acids 77–282) had practically no inclusion bodies (Fig. 4A S–U and Fig. 4B), indicating that specific ubiquitylation of synphilin-1 is required for inclusion body formation. This finding is in agreement to the fact that the majority of synphilin-1/SIAH inclusions were positive for cotransfected FLAG-tagged ubiquitin (Fig. 4D). Also, endogenous ubiquitin was found to colocalize with synphilin-1/SIAH inclusions (data not shown). By contrast, we did not observe colocalization of the control protein myc-FKB12 in synphilin-1/SIAH inclusions (Fig. 4D). We also found that lactacystin-treated cells cotransfected with synphilin-1 and catalytically deficient triple point mutant SIAH-1 (C55A, H59A, C72S) had much less inclusion body formation, supporting the idea that ubiquitylated-synphilin-1 is important for inclusion body formation (Fig. 4B). The point mutant of SIAH-1 (C72S) still induced significant inclusion body formation, which is consistent to its residual E3 ubiquitin-ligase activity verified by in vitro ubiquitylation assays (data not shown).
It was recently reported that the peptide motif RPVAxVx-PxxR mediate the interaction of SIAH-1 with a range of protein partners (23). Synphilin-1 also displays the consensus for binding SIAH-1. We now found that mutation in the SIAH-binding domain of synphilin-1 (V79N, P81N) resulted in significant decrease in the interaction with SIAH-1 (data not shown). Also, the levels of round cytosolic inclusions in cells cotransfected with synphilin-1 (V79N, P81N) and SIAH-1 was below the basal with synphilin-1 WT alone (Fig. 4B). Thus, endogenous SIAH may be responsible for promoting the basal inclusion body formation of synphilin-1 WT in the absence of transfected SIAH-1 (Fig. 4B).
In triple transfected experiment, HA-α-synuclein was shown to colocalize with synphilin-1/SIAH inclusions (Fig. 10A, which is published as supporting information on the PNAS web site). Because parkin ubiquitylates synphilin-1 (16), we examined whether parkin is able to elicit synphilin-1 inclusion bodies. We found no increase in inclusion body formation in cells cotransfected with HA-synphilin-1 and myc-parkin in the presence of lactacystin (data not shown), suggesting that not all forms of ubiquitylated synphilin-1 are able to promote inclusion body formation.
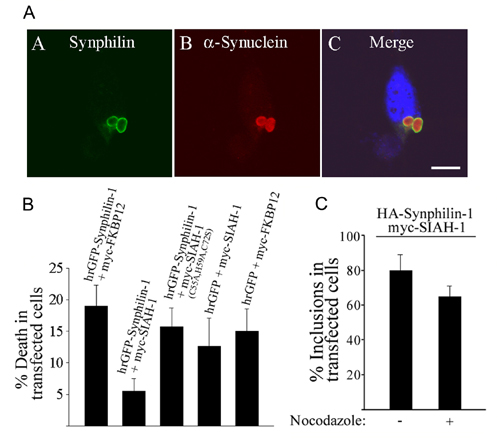
To determine whether synphilin-1/SIAH inclusions are toxic to cells, we incubated cells cotransfected with humanized recombinant GFP (hrGFP)-synphilin-1 and myc-SIAH-1 in the presence of lactacystin, and measured cell death by propidium iodide staining. Most hrGFPsynphilin-1/myc-SIAH-1 transfected cells had cytosolic inclusions with a very bright halo. These inclusions appear to be cytoprotective (Fig. 10B), suggesting that aggregation of ubiquitylated-synphilin-1 may be beneficial to cells.
We next investigated whether synphilin-1/SIAH inclusions display aggresome properties (24). We found by immunocytochemistry that synphilin-1/SIAH inclusions are negative for the centrosome-related protein γ-tubulin, suggesting that these inclusions are not aggresomes (data not shown). In addition, no significant change in synphilin-1/SIAH inclusion formation was observed in the presence of the microtubule-destabilizing drug, nocodazole (Fig. 10C).
We investigated whether α-synuclein itself is a target of SIAH. We cotransfected myc-SIAH-1 and SIAH-2 with HA-α-synuclein into HEK 293 cells. We found that α-synuclein strongly coimmunoprecipitates with SIAH-2 (Fig. 5A). This coimmunoprecipitation was specific because it was not observed with the control protein FKBP12 (Fig. 5A). By contrast, coimmunoprecipitation of α-synuclein with SIAH-1 was faint and only observed after prolonged exposure (Fig. 5A Right, asterisk). To map the SIAH-binding domain of α-synuclein, a series of truncated α-synuclein constructs were generated. Coimmunoprecipitation experiments of HEK 293 cells cotransfected with HA-α-synculein constructs and myc-SIAH-2 indicate that SIAH-2 interacts stronger with the second half of α-synuclein (Fig. 11, which is published as supporting information on the PNAS web site). Cotransfection of HA-α-synuclein with myc-synphilin-1 and untagged SIAH into HEK 293 cells in the presence of lactacystin did not alter the characteristics and number of inclusion bodies, suggesting that α-synuclein neither competes with synphilin-1 for the binding with SIAH-2, nor interferes with synphilin-1 ubiquitylation by SIAH (data not shown).
Fig. 5.

SIAH-2 binds and ubiquitylates α-synuclein. (A) Coimmunoprecipitation of SIAH-2 with α-synuclein from cotransfected HEK 293 cells. HA-α-synuclein was immunoprecipitated by using an anti-HA antibody. Coimmunoprecipitation of myc-SIAH-1 and myc-SIAH-2 was checked by using an anti-myc antibody. The asterisk depicts the faint SIAH-1 coimmunoprecipitation observed after prolonged exposure of the membrane. (B) Monoubiquitylation of α-synuclein was observed in the presence of GST-SIAH-2. Synphilin-1 was in vitro translated and incubated with the indicated components of the ubiquitin system, in the presence of 1 μg of GST-SIAH-2 or GST alone.
We next carried out in vitro ubiquitylation experiments in which in vitro translated α-synuclein was incubated with recombinant SIAH-2 and purified components of the ubiquitin system. We found that SIAH-2 promotes monoubiquitylation of α-synuclein in a specific manner (Fig. 5B). We did not observe significant degradation of α-synuclein when cotransfected with SIAH-2 in HEK 293 cells (data not shown), consistent with the nondegradative role of monoubiquitylation.
To determine whether SIAH is present in Lewy bodies, we first examined the specificity of anti-SIAH antibody (H-18), which recognizes the C terminus of both SIAH-1 and SIAH-2 (Fig. 6A). H-18 antibody detects SIAH from transfected cells, but does not detect the low levels of endogenous SIAH in Western blot. Nevertheless, H-18 robustly immunoprecipitates SIAH, suggesting that it is much more efficient in the recognition of native instead of denatured protein (Fig. 6A). Immunohistochemical experiments using H-18 antibody revealed that ≈30% of the Lewy bodies from substantia nigra of PD patients were SIAH-positive (Fig. 6B). SIAH immunoreactivity in Lewy bodies (purple staining, using 3-amino-9-ethylcarbazole chromogen) is specific because no signal was observed when H-18 antibody was preincubated with antigen (Fig. 6B Lower). Purified polyclonal anti-SIAH-1 antibody 990 also recognized SIAH present in Lewy bodies (data not shown).
Fig. 6.

SIAH is present in Lewy bodies of PD patients. (A)(Left) Equal amounts of protein extracts (50 μg) were analyzed by Western blot using H-18 antibody. (Right) H-18 antibody immunoprecipitates endogenous SIAH. SIAH was immunoprecipitated from rat brain homogenate by using H-18 antibody, and detection of immunoprecipitation was carried out by using anti-SIAH-1 antibody (990). (B) SIAH immunoreactivity in substantia nigra of PD patients. Immunohistochemistry experiments were carried out in substantia nigra sections of PD patients by using H-18 antibody. The presence of SIAH in Lewy bodies is shown by a purple staining corresponding to 3-amino-9-ethylcarbazole chromogen. Staining of Lewy bodies is specific because no immunolabeling was observed in the control by using preabsorbed antibody (Lower). Arrows indicate the limits of Lewy bodies. Sections were counterstained with hematoxylin.
Discussion
Ubiquitin ligases (E3) are responsible for the formation of polyubiquitin chains in substrate proteins, which in general work as a tag for proteasomal degradation (22). Our data demonstrate that SIAH interacts in vivo with synphilin-1 and promotes its polyubiquitylation and proteasomal degradation by working as an E3 ubiquitin ligase. Ubiquitylation of synphilin-1 by SIAH-1 and SIAH-2 is required for the formation of Lewy body-like inclusions in the presence of proteasome inhibitor. We also found that SIAH-2 interacts with and monoubiquitylates α-synuclein. The presence of SIAH in Lewy bodies suggests a role of SIAH in the pathogenesis of the disease.
SIAH proteins have been implicated in a variety of cellular processes, such as apoptosis and tumor suppression (25). Although the N-terminal region of SIAH encodes for a RING-finger domain that confers its ability to work as an E3 ubiquitin-ligase, its C-terminal region encodes for a domain implicated in the binding of different substrate proteins (26), among them, the presynaptic protein synaptophysin (27).
Synphilin-1 and α-synuclein were previously shown to be localized to presynaptic terminals (14, 28). Thus, a physiological role of SIAH may be to modulate the turnover and function of presynaptic proteins, such as synphilin-1 and α-synuclein.
Dysfunction of the ubiquitin–proteasome system in PD (3) may lead to accumulation of synphilin-1. We found that synphilin-1/SIAH inclusions are not toxic to HEK 293 cells, suggesting that accumulation of synphilin-1 into inclusions could be cytoprotective. Other E3 ubiquitin ligases, such as parkin and dorfin, were reported to ubiquitylate synphilin-1 but, unlike SIAH, they were not shown to target synphilin-1 for degradation (16, 29). It is still not clear whether these ligases would be synergic to SIAH in ubiquitylating synphilin-1.
It is notable that inclusions are formed in the presence of the proteasome inhibitor in cells cotransfected with synphilin-1 and wild-type SIAH, but much less in those transfected with synphilin-1 alone or with catalytically inactive SIAH (Fig. 4). This observation indicates that the accumulation of nonubiquitylated synphilin-1, in complex with SIAH, is not sufficient for inclusion body formation, and that ubiquitylation of synphilin-1 has to take place. It is possible that ubiquitylated synphilin-1 has a greater tendency to form aggregates with other proteins than nonubiquitylated synphilin-1. This notion is supported by the observation that proteins linked to polyubiquitin chain are generally more hydrophobic than the same proteins (A. Hershko, personal communication).
Monoubiquitylated proteins are not targeted for degradation by the proteasome system (30). In agreement with this, the monoubiquitylation of α-synuclein by SIAH-2 observed in the present study was not accompanied by degradation. SIAH-1 and SIAH-2 proteins are highly homologous but significantly differ in their N-terminal region before their RING-finger domain (31). The stronger binding of SIAH-2 to α-synuclein (Fig. 5A) suggests that the N-terminal region of SIAH-2 is important for the interaction. Additionally, α-synuclein seems to bind to SIAH-2 more strongly through its C-terminal region.
UCH-L1 was shown to be able to ubiquitylate α-synuclein in vitro (32). Parkin was also shown to ubiquitylate a glycosylated form of α-synuclein, but does not act on unmodified α-synuclein (33). Although the relative contribution of the different types of α-synuclein ubiquitylation still needs to be addressed, the ubiquitylation of α-synuclein promoted by SIAH-2 is similar to the endogenous ubiquitylation pattern observed in biochemically purified Lewy bodies (9). It is conceivable that synphilin-1/SIAH inclusions may be relevant for Lewy body formation. SIAH represents a component of the ubiquitin–proteasome system linked to the pathogenesis of the disease.
Recently, Nagano et al. (34) reported that SIAH-1 interacts with and ubiquitylates synphilin-1, which confirms our present results. In addition, we found that both synphilin-1 and α-synuclein interact with, and are ubiquitylated by, SIAH-2. Also, we now show that inability of the proteasome to degrade ubiquitylated synphilin-1 leads to robust formation of cytosolic inclusions. Finally, we report SIAH immunoreactivity in Lewy bodies of PD patients.
Supplementary Material
Acknowledgments
We are grateful to Prof. A. Ciechanover for helpful suggestions regarding the disruption of the RING-finger domain of SIAH-1 and for supplying ubiquitin components for the in vitro ubiquitylation experiments. We thank B. Bercovich and H. Gonen for valuable advice. This work was supported by Israel Academy of Sciences, a National Institutes of Health–Fogarty International Collaboration Award, and by Technion Research funds (to S.E.). C.A.R. acknowledges the Parkinson's Disease Center Grant NS38377.
Abbreviations: PD, Parkinson's disease; HA, hemagglutinin.
References
- 1.Dunnett, S. B. & Bjorklund, A. (1999) Nature 399, A32–A39. [DOI] [PubMed] [Google Scholar]
- 2.Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., Yokochi, M., Mizuno, Y. & Shimizu, N. (1998) Nature 392, 605–608. [DOI] [PubMed] [Google Scholar]
- 3.McNaught, K. S. & Jenner, P. (2001) Neurosci. Lett. 297, 191–194. [DOI] [PubMed] [Google Scholar]
- 4.Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., Pike, B., Root, H., Rubenstein, J., Boyer, R., et al. (1997) Science 276, 2045–2047. [DOI] [PubMed] [Google Scholar]
- 5.Krüger, R., Kuhn, W., Muller, T., Woitalla, D., Graeber, M., Kosel, S., Przuntek, H., Epplen, J. T., Schols, L. & Riess, O. (1998) Nat. Genet. 18, 106–108. [DOI] [PubMed] [Google Scholar]
- 6.Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R. & Goedert, M. (1997) Nature 388, 839–840. [DOI] [PubMed] [Google Scholar]
- 7.Bennett, M. C., Bishop, J. F., Leng, Y., Chock, P. B., Chase, T. N. & Mouradian, M. M. (1999) J. Biol. Chem. 274, 33855–33858. [DOI] [PubMed] [Google Scholar]
- 8.Ancolio, K., Alves da Costa, C., Ueda, K. & Checler, F. (2000) Neurosci. Lett. 285, 79–82. [DOI] [PubMed] [Google Scholar]
- 9.Hasegawa, M., Fujiwara, H., Nonaka, T., Wakabayashi, K., Takahashi, H., Lee, V. M., Trojanowski, J. Q., Mann, D. & Iwatsubo, T. (2002) J. Biol. Chem. 277, 49071–49076. [DOI] [PubMed] [Google Scholar]
- 10.Engelender, S., Kaminsky, Z., Guo, X., Sharp, A. H., Amaravi, R. K., Kleiderlein, J. J., Margolis, R. L., Troncoso, J. C., Lanahan, A. A., Worley, P. F., et al. (1999) Nat. Genet. 22, 110–114. [DOI] [PubMed] [Google Scholar]
- 11.Kawamata, H., McLean, P. J., Sharma, N. & Hyman, B. T. (2001) J. Neurochem. 77, 929–934. [DOI] [PubMed] [Google Scholar]
- 12.Neystat, M., Rzhetskaya, M., Kholodilov, N. & Burke, R. E. (2002) Neurosci. Lett. 325, 119–123. [DOI] [PubMed] [Google Scholar]
- 13.Marx, F. P., Holzmann, C., Strauss, K. M., Li, L., Eberhardt, O., Gerhardt, E., Cookson, M. R., Hernandez, D., Farrer, M. J., Kachergus, J., et al. (2003) Hum. Mol. Genet. 12, 1223–1231. [DOI] [PubMed] [Google Scholar]
- 14.Ribeiro, C. S., Carneiro, K., Ross, C. A., Menezes, J. R. & Engelender, S. (2002) J. Biol. Chem. 277, 23927–23933. [DOI] [PubMed] [Google Scholar]
- 15.McLean, P. J., Kawamata, H. & Hyman, B. T. (2001) Neuroscience 104, 901–912. [DOI] [PubMed] [Google Scholar]
- 16.Chung, K. K., Zhang, Y., Lim, K. L., Tanaka, Y., Huang, H., Gao, J., Ross, C. A., Dawson, V. L. & Dawson, T. M. (2001) Nat. Med. 7, 1144–1150. [DOI] [PubMed] [Google Scholar]
- 17.O'Farrell, C., Murphy, D. D., Petrucelli, L., Singleton, A. B., Hussey, J., Farrer, M., Hardy, J., Dickson, D. W. & Cookson, M. R. (2001) Brain Res. Mol. Brain Res. 97, 94–102. [DOI] [PubMed] [Google Scholar]
- 18.Lee, G., Junn, E., Tanaka, M., Kim, Y. M. & Mouradian, M. M. (2002) J. Neurochem. 83, 346–352. [DOI] [PubMed] [Google Scholar]
- 19.Ihara, M., Tomimoto, H., Kitayama, H., Morioka, Y., Akiguchi, I., Shibasaki, H., Noda, M. & Kinoshita, M. (2003) J. Biol. Chem. 278, 24095–24102. [DOI] [PubMed] [Google Scholar]
- 20.Wakabayashi, K., Engelender, S., Yoshimoto, M., Tsuji, S., Ross, C. A. & Takahashi, H. (2000) Ann. Neurol. 47, 521–523. [PubMed] [Google Scholar]
- 21.Engelender, S., Sharp, A. H., Colomer V., Tokito, M. K., Lanahan, A., Worley, P., Holzbaur, E. L. F. & Ross, C. A. (1997) Hum. Mol. Genet. 6, 2205–2212. [DOI] [PubMed] [Google Scholar]
- 22.Hershko, A. & Ciechanover, A. (1998) Annu. Rev. Biochem. 67, 425–479. [DOI] [PubMed] [Google Scholar]
- 23.House, C. M., Frew, I. J., Huang, H. L., Wiche, G., Traficante, N., Nice, E., Catimel, B. & Bowtell, D. D. (2003) Proc. Natl. Acad. Sci. USA 100, 3101–3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kopito, R. R. (2000) Trends Cell Biol. 10, 524–530. [DOI] [PubMed] [Google Scholar]
- 25.Roperch, J. P., Lethrone, F., Prieur, S., Piouffre, L., Israeli, D., Tuynder, M., Nemani, M., Pasturaud, P., Gendron, M. C., Dausset, J., et al. (1999) Proc. Natl. Acad. Sci. USA 96, 8070–8073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu, G. & Fearon, E. R. (1999) Mol. Cell. Biol. 19, 724–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wheeler, T. C., Chin, L. S., Li, Y., Roudabush, F. L. & Li, L. (2002) J. Biol. Chem. 277, 10273–10282. [DOI] [PubMed] [Google Scholar]
- 28.Iwai, A., Masliah, E., Yoshimoto, M., Ge, N., Flanagan, L., de Silva, H. A., Kittel, A. & Saitoh, T. (1995) Neuron 4, 467–475. [DOI] [PubMed] [Google Scholar]
- 29.Ito, T., Niwa, J., Hishikawa, N., Ishigaki, S., Doyu, M. & Sobue, G. (2003) J. Biol. Chem. 278, 29106–29114. [DOI] [PubMed] [Google Scholar]
- 30.Hicke, L. (2001) Nat. Rev. Mol. Cell. Biol. 2, 195–201. [DOI] [PubMed] [Google Scholar]
- 31.Hu, G., Chung, Y.-L., Glover, T., Valentine, V., Look, A. T. & Fearon, E. R. (1997) Genomics 46, 103–111. [DOI] [PubMed] [Google Scholar]
- 32.Liu, Y., Fallon, L., Lashuel, H. A., Liu, Z. & Lansbury, P. T., Jr. (2002) Cell 111, 209–218. [DOI] [PubMed] [Google Scholar]
- 33.Shimura, H., Schlossmacher, M. G., Hattori, N., Frosch, M. P., Trockenbacher, A., Schneider, R., Mizuno, Y., Kosik, K. S. & Selkoe, D. J. (2001) Science 293, 263–269. [DOI] [PubMed] [Google Scholar]
- 34.Nagano, Y., Yamashita, H., Takahashi, T., Kishida, S., Nakamura, T., Iseki, E., Hattori, N., Mizuno, Y., Kikuchi, A. & Matsumoto, M. (2003) J. Biol. Chem. 278, 51504–51514. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}