

Abstract
Synaptotagmin 1 (Syt1) is a major Ca2+-sensor that evokes neurotransmitter release. Here we used site-specific fluorescence resonance energy transfer (FRET) assay to investigate the effects of Syt1 on SNAREpin assembly. C2AB, a soluble version of Syt1, had virtually no stimulatory effect on the rate of the FRET at N-terminus of SNARE complex both with and without Ca2+, indicating C2AB does not interfere with the initial nucleation of SNARE assembly. However, C2AB-Ca2+ accelerated the FRET rate significantly at membrane proximal region, indicating C2AB-Ca2+ promotes the transition from a partially assembled SNARE complex to the fusion-competent SNAREpin. Similar enhancement was also observed at the end of the transmembrane domain of SNARE proteins. The stimulatory effect disappeared if there was no membrane or only neutral membrane present.
The neurotransmitter release at synapses underlies major brain functions such as cognition, emotion, and memory. The precise temporal control of the release is essential for healthy brain activities. Soluble NSF attachment protein receptors (SNAREs) are known to be the core fusion machinery in neuro-exocytotic pathways1,2,3. SNARE complex formation in the chasm of two membranes is mediated by the cognate coiled-coil motifs on vesicle and target membrane (v- and t-) SNAREs. One such motif from t-SNARE syntaxin 1A, two from t-SNARE SNAP-25, and another from v-SNARE vesicle-associated membrane protein 2 (VAMP2) forms all parallel four-stranded coiled coil, which brings about the apposition of two membranes4,5. The SNARE proteins, however, do not have the required regulatory function that confers the temporal on/off switching capability. A vesicular protein synaptotagmin1 (Syt1) is instead believed to be the key regulator for the temporal control of SNARE-dependent synaptic vesicle fusion6,7,8. Syt1 senses the spike of the Ca2+-level, which resulted from the action potential, and helps trigger fast fusion9,10.
In molecular level, Syt1 is known to bind the membrane11,12,13 and the SNARE complex14,15,16 in the presence of Ca2+. The importance of the membrane binding has been well described in several in vivo studies employing Syt1 mutants17,18. Interestingly, it is recently suggested that Syt1 has an ability to buckle the membrane to create the locally positive curvature19,20, which is believed to help membrane fusion21.
What is then the function of Syt1 binding to the SNARE complex? Clues to this question may be found in its interplay with complexin in SNARE binding22,23. Complexins are a family of small soluble proteins that bind to the SNAREpins24,25. The complexin clamp model proposes that complexin binding to the SNAREpin clamps the fusion26,27,28,29 by preventing the membrane-proximal half from complex formation29. But upon the rise of the Ca2+ level, Syt1 displaces complexin from the SNARE complex26,28, allowing complete SNARE assembly to a fusion competent SNAREpin30,31. In such a case, Syt1 binding to the SNARE complex could be simply the end product of the competitive displacement of complexin from the SNARE complex.
Mechanistically, it is thought that SNARE complex formation proceeds from membrane-distal N-terminal region towards membrane-proximal C-terminal region2,32,33,34. Such sequential zippering could facilitate progressive apposition of two opposing membranes. It has been shown that Syt1 interacts with the C-terminal region of the SNARE complex14,15,35. However, whether Syt1 only specifically acts on C-terminal SNARE assembly to give a final push on SNARE zippering remains unclear.
In this work, we were able to dissect the effects of Syt1 and Syt1-Ca2+ on SNARE zippering by using fluorescence resonance energy transfer (FRET) technology through placing FRET pairs at strategic locations on the t- and v-SNAREs. We concluded that Syt1-Ca2+ not only help vesicle docking, but also play roles on C-terminal SNARE complex formation via crosslinking two membranes, which in turn facilitates the fusion pore opening.
Results
C2AB and Ca2+ had no effect on N-terminal trans SNARE assembly
Syt1 has two soluble Ca2+-binding tandem C2 domains and a single membrane-spanning helix at the C-terminal region that anchors Syt1 to the vesicle membrane6,36. C2AB, a recombinant soluble version of Syt1 lacking the transmembrane part, is often used as an alternative model in a variety of in vitro studies19,20,37. C2AB is shown to recapitulate not all, but some essential features of Syt1 functions such as Ca2+-triggered enhancement of SNARE-driven fusion38. To test the effect of Syt1 on N-terminal initiation of SNARE assembly, the FRET assay was employed with N-terminal labeled v- and t-SNARE pairs. The cysteine free version of full-length syntaxin 1A (SyxF), VAMP2, and SNAP-25 were used in our system. To generate the N-terminally labeled protein, the N-terminal amino acids I187 of SyxF and G18 of VAMP2, which are both located outside but close to the N-terminal ends of SNARE motifs, were changed to cysteines, and the cysteine mutants SyxF I187C and VAMP2 G18C were labeled with fluorescence donor Alexa Fluor 546 (A546) and acceptor Alexa Fluor 647 (A647), respectively. As shown on SDS-PAGE gels, the labeled proteins run slightly slower than wild type proteins, due to the addition of molecular weight of the dye molecules to the proteins (Supplementary Figure S1). The labeled t-SNARE, SyxF I187C-A546 combined with SNAP-25, was reconstituted into a population of vesicles (t-vesicles), while VAMP2 G18C-A647 was reconstituted into a separate population of vesicles (v-vesicles, Figure 1A). Here, both t- and v-vesicles contained 45 mol% POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine), 15 mol% DOPS (1, 2-dioleoyl-sn-glycero-3-phosphatidylserine) and 40 mol% cholesterol, which mimics the lipid composition in synaptic vesicles39. The lipid to protein ratio was kept to be 200:1 for all measurements. The activities of fluorophore-labeled SNARE proteins were checked by the lipid-mixing assay. The results suggested the dyes did not affect the SNARE activities (Supplementary Figure S2A).
Figure 1. The NN-FRET assay with Syt1 and Ca2+.

(A) The schematic diagram for the NN-FRET assay. SynF I187C-A546 (red) and SNAP-25 (green) are reconstituted to t-vesicles and VAMP2 G18C-A647 (blue) is to v-vesicles. C2AB is shown in pink. (B) The increase of FRET signal was recorded. The red line is the control with labeled SNAREs only without C2AB. The green line represents the NN-FRET assay with 1 μM C2AB and 1 mM EDTA, while the blue line represents the reaction with 1 μM C2AB and 100 μM Ca2+. The black line is the negative control with 10 μM soluble VAMP2 (Vps). (C) Relative initial rates of the NN-FRET assays at different Ca2+ concentrations. Error bars, which represent standard deviation, were obtained from 3 independent measurements with 3 different preparations. (D) C2AB and C2AB-Ca2+show similar effects on the initial rates of N-terminal trans SNARE assembly at different lipid concentrations (0.06, 0.10, and 0.20 mM). The initial rate was measured from the slope at time near 0 (*P < 0.05, **P < 0.01, and n.s means not significant).
FRET assay has been widely used to determine whether two fluorophores are within a certain distance of each other40,41,42,43,44,45,46. When t- and v-vesicles dock or fuse with each other due to the interaction between t- and v-SNAREs, the proximity between A546 on SyxF I187C and A647 on VAMP2 G18C leads to FRET (NN-FRET), results in the increase of the acceptor signal (Figure 1B, red line). On the contrast, the 80% FRET signal will be inhibited during the time course in presence of 10 uM Vps (Figure 1B, black line). When 1 μM C2AB was added to the fusion reaction mixture, however, we did not observe any further stimulation, even in the presence of Ca2+ (Figure 1B, green and blue lines). Instead, Ca2+caused slight inhibition of NN-FRET (Figure 1B, blue lines). The inhibition was less than a factor of two in initial rates for all Ca2+ concentrations studied (Figure 1C), which we might attribute to the self-association of v- and t-vesicles induced by cross-binding of C2B domains in the Ca2+ presence47,48. Meanwhile, we also excluded the possibility that the individual factor such as C2AB or Ca2+ did not change the donor or acceptor signal under the same condition (supplementary Figure S3). Thus, the results show that C2AB and C2AB-Ca2+ have no stimulatory effects on NN-FRET or vesicle docking. To test whether different FRET probes will affect the FRET signal, we also observed the similar results by using Cy3-labeled SyxF I187C and Cy5-labeled VAMP2 G18C (Supplementary Figure S4), further confirmed our conclusion. Next, we performed the same experiment at three different lipid concentrations at 0.06, 0.1, and 0.2 mM (Figure 1D). Surprisingly, we observed stronger inhibitory effect by C2AB/Ca2+ at high lipid concentration level. We attribute this inhibition to the non-specific aggregation of vesicles caused by C2AB/Ca2+, which would reduce the available t- and v- vesicles for SNARE nucleation at N-terminus. Here, we note that soluble VAMP2 (amino acids 1–96, Vps in Figure 1B), which is often used as a competitive inhibitor for SNARE-dependent membrane fusion, does not completely inhibit NN-FRET at 10 μM, different from the complete inhibition observed for other C-terminally labeled SNARE FRET pairs (see below). Overall, the results of NN-FRET assay suggest that C2AB did not involve in stimulating the initiation of SNAREpin zippering, indicating it may play roles during SNARE zippering at the later step.
C2AB and Ca2+ facilitates SNARE zippering at the membrane-proximal region
Previous studies have already shown that C2AB binds to the membrane proximal region of SNARE core35,49 and mutations at the C-terminal SNARE motif of SNAP-25 could also impair the Ca2+-triggered neurotransmitter release35,50. Thus, it is anticipated that binding of Syt1 to the C-terminal region of SNARE motif is critical to its function as a Ca2+ sensor. To further address the impact of Syt1 binding to the SNARE core, we generated another site-specifically labeled v- and t-SNARE FRET pairs, SyxF Y257C-A546 and VAMP2 K87C-A647, in which labeled sites are localized near the membrane-proximal end of SNARE motifs (MM-FRET pairs) (Figure 2A). With labeled SNAREs only, but without C2AB and Ca2+, the rate change of MM-FRET in time course was much slower than that of the NN-FRET assay (Figure 1), indicating that SNARE zippering is indeed sequential, consistent with the zipper model2,8,32,51. Moreover, the slower rate for MM-FRET implies that the C-terminal zippering step might have higher activation energy barrier. In contrast to the NN-FRET assay, we observed a clear factor of 10 enhancement of the initial rate in MM-FRET when both 1 μM C2AB and 100 μM Ca2+were added, while there is no any enhancement observed in the absence of Ca2+ (Figure 2B). As expected, Ca2+ alone also did not affect the initial rate of the MM-FRET (Supplementary Figure S5). All these data suggested that role of Syt1 on SNARE core zippering was Ca2+ dependent, which was consistent with early findings. Furthermore, the stimulatory effect of C2AB with Ca2+ on MM-FRET as well as MM-FRET in the absence of C2AB and Ca2+ were completely blocked by 10 μM soluble VAMP2 (Vps) (Figure 2B yellow and black lines), indicating that the stimulation by C2AB and Ca2+ is also SNARE-dependent. As shown above, here the lipid-mixing assay was performed again to confirm that fluorescence probes did not affect the activities of SNARE proteins (Supplementary Figure S2B). In summary, our results show that in the presence of Ca2+, C2AB dramatically promotes the SNARE zippering at the membrane-proximal C-terminal region while does not affect the nucleation of SNARE assembly at the N-terminal region (Figure 1). When higher concentration of C2AB (5 μM) used in the assay, similar results for MM-FRET were observed (Supplementary Figure S6).
Figure 2. Ca2+-dependent stimulation of SNARE zippering by C2AB.

(A)The schematic diagram for the MM-FRET assay. SyxF Y257C-A546 (red) combined with SNAP-25 (green) are reconstituted to t-vesicles, while VAMP2 K87C-A647 (blue) was reconstituted to v-vesicles. C2AB is shown in pink. (B) Changes of the FRET signal in time after mixing t- and v-vesicles are shown. The red line is the control with labeled SNAREs only without C2AB or Ca2+. The green line represents FRET signal with 1 μM C2AB and 1 mM EDTA, while the blue line represents FRET signal with 1 μM C2AB and 100 μM Ca2+. The yellow line represents FRET signal with 1 μM C2AB, 100 μM Ca2+ and 10 μM Vps. The black line is with 10 μM Vps without C2AB and Ca2+. (C) Initial rate of FRET signal in the MM-FRET assays at different Ca2+ concentrations. The error bars, which represent standard deviation, were obtained from 3 independent measurements from 3 different preparations (*P < 0.05, **P < 0.01, and n.s means not significant).
C2AB-Ca2+stimulates cis SNARE assembly assessed by FRET at the ends of transmembrane domains
Next, we attached fluorescence probes at or near the C-terminal end of t- and v-SNAREs (SyxFG288C-A546 and VAMP2 T116C-A647, respectively) to further investigate the effects of C2AB and Ca2+ on cis SNARE assembly, which occurs after fusion pore opening and reflects the coexistence of two transmembrane domains in the same membrane (CC-FRET pairs, Figure 3A). Again, we verified the activities of the C-terminally labeled SNARE pairs by the lipid-mixing assay (Supplementary Figure S2C). In the CC-FRET assay, as expected, we observed clear Ca2+-dependent stimulation of CC-FRET in the presence of 1 μM C2AB and Ca2+ (Figure 3B). At 100 μM Ca2+, we observed again the same factor of 10 enhancements of the initial rates when compared with that in the absence of C2AB. Furthermore, CC-FRET was completely blocked by 10 μM Vps (Figure 3B, black line). Neither did Ca2+ alone in the absence of C2AB, nor did C2AB alone in the absence of Ca2+ give any stimulation of CC-FRET (Supplementary Figure S5C). Thus, our results show that C2AB-Ca2+ enhances the rate of cis SNARE assembly, which may help to enlarge the fusion pore later.
Figure 3. Ca2+-dependent stimulation of cis SNARE assembly by C2AB.

(A) The schematic diagram for the CC-FRET assay. SyxF G288C-A546 (red) combined with SNAP-25 (green) are reconstituted to t-vesicles, while VAMP2 T116C-A647 (blue) was reconstituted to v-vesicles. C2AB is shown in pink. (B) Changes of the FRET signal in time. The red line is the control with labeled SNAREs only without C2AB or Ca2+. The green line represents signal with 1 μM C2AB and 1 mM EDTA, while the blue line represents signal with 1 μM C2AB and 100 μM Ca2+. The black line is the control with 10 μM Vps. (C) Initial rate of FRET signal in the CC-FRET assays at different Ca2+ concentrations. The error bars, which represent standard deviation, were obtained from 3 independent measurements from 3 different preparations (*P < 0.05, **P < 0.01, and n.s means not significant).
Membrane is required for C2AB-Ca2+to stimulate SNAREpin formation
It has been reported that enhancement of SNARE assembly by C2AB-Ca2+ might be contributed by two ways. One is binding of Syt1 to SNARE complex14, while the other is cross-linking two membrane38,48,52. Up to now, however, it remains unclear that which one is dominant in this process. To answer this question, we further studied the effect of C2AB-Ca2+ on SNARE complex assembly in the absence of membrane. Interestingly, we did not observe any stimulation of initial rate in MM-FRET with the presence of C2AB-Ca2+ when there was no membrane (Figure 4A), indicating that the binding of C2AB-Ca2+ to membrane was required. In this study, we also examined whether detergent or lipid affects SNARE-C2AB interaction. Our results clearly showed that both detergent and lipid had little effect on the interaction between SNARE proteins and C2AB (Supplementary Figure S7). Furthermore, when we removed the acidic lipid from the vesicle membrane to block the binding of C2AB-Ca2+ to the membrane, we observed no stimulatory effect on MM-FRET by C2AB-Ca2+ (Figure 4B). All these data suggested the sequential assembly of SNARE core region requires two opposite membranes. Moreover, our dynamics light scattering measurement shows only C2AB and Ca2+ can efficiently enlarge the size of vesicle particle from 100 nm to 700 nm, indicating that the two opposing membrane bridging by C2AB was Ca2+ dependent (Supplementary Figure S8). To further confirm our conclusions, We interfered the formation of SNARE complex by applying the botulium neurotoxin (BoNT) type A and E digested version of SNAP-25: SNAP-25 BoNT/A and SNAP-25 BoNT/E, in which SNAP-25 is cleaved at amino acid Gln197 and Arg180, respectively. As expected, in absence of Ca2+, SNAP25 BoNT/A and BoNT/E both show inhibition in SNARE assembly due to the impairment in SNARE motif of SNAP-25 (green and blue line in Figure 4C). However, we observed that C2AB-Ca2+ could still facilitate the SNARE assembly in the presence of membrane though SNARE complex formation has been partially blocked (Figure 4C). We noticed that due to the severe defect in SNARE motif of SNAP-25 BoNT/E, the SNARE assembly with SNAP25 BoNT/E can not achieve the level of wild type in presence of Ca2+, even though the stimulatory effect by C2AB and Ca2+ is still substantial (dotted blue line in Figure 4C). Thus, we concluded that membrane was required for the SNARE zippering process by Syt1-Ca2+ in the SNARE core region.
Figure 4. C2AB-Ca2+ stimulates SNAREpin formation via crosslink two opposition membranes.

(A) MM-FRET in presence of 0.8% OG. The red line is the control with labeled SNAREs only without C2AB or Ca2+. The green line represents FRET signal with 0.5 μM C2AB and 100 μM Ca2+, while the blue line represents signal with 0.05 μM C2AB and 50 μM Ca2+. (B) The MM-FRET with neutral membrane. The red line is the control with labeled SNAREs only without C2AB or Ca2+. The green line represents signal with 1 μM C2AB and 1 mM EDTA, while the blue line represents signal with 1 μM C2AB and 100 μM Ca2+. (C) MM-FRET with SNAP-25 BoNT/A and BoNT/E. The dotted lines represent MM-FRET with C2AB with Ca2+, while the sold lines represent MM-FRET with C2AB only. And the red line is the control with labeled SNAREs with WT SNAP25. The green line is with SNAP25 BoNT/A, while the blue line is with SNAP25 BoNT/E.
Discussion
We developed the FRET assay employing fluorescently labeled SNAREs to investigate the function of Syt1 in vesicle exocytosis. With a set of labeled SNAREs for which FRET pairs are site-specifically attached at strategic positions, we were able to dissect the impact of C2AB-Ca2+ at three different stages of vesicle fusion: NN-FRET for the nucleation of SNARE assembly, MM-FRET for SNARE zippering at the membrane-proximal region, and CC-FRET for fusion pore opening. When FRET happens between the donor and acceptor dyes, we could observe the enhancement of the acceptor intensity during the time course, while the donor intensity is declining at the same time (Supplementary Figure S9). Our results show that C2AB alone, in the absence of Ca2+, did not influence the kinetics of any of the three steps. However, Syt1 alone can help vesicle docking, indicating that C2AB and Syt1 might have a distinct mechanism to regulate vesicle docking38. Furthermore, the result show that C2AB in the presence of Ca2+ stimulates the kinetics of MM-FRET and CC-FRET with little stimulatory effect on NN-FRET, indicating that C2AB-Ca2+ specifically works on the SNARE zippering and the fusion pore opening steps after docking. As we did not see the stimulation in NN-FRET, it excluded the possibility that the enhancement of NN-FRET caused the increase of MM-FRET. Therefore, the results provide the direct in vitro evidence that Syt1 may stimulate SNARE zippering specifically at the membrane proximal half. Finally, via using detergent, neutral membrane and SNAP25 mutants, we figured out that the membrane binding by C2AB-Ca2+ is required for the enhancement on SNARE complex assembly.
Based on the results of CC-FRET, C2AB-Ca2+ appears to stimulate the rate of the fusion pore opening by 10-folds. But the enhancement factor is virtually the same as that of SNARE zippering at the membrane-proximal region detected in the MM-FRET analysis. Therefore, we speculate the rate enhancement in CC-FRET is just the consequence of the stimulation of MM-FRET by C2AB-Ca2+. If so, the main function of C2AB and Ca2+ might be to stimulate SNARE-zippering at the membrane proximal region, and the observed stimulation of the fusion pore opening may be a collateral consequence of this main thrust at the membrane-proximal region of SNAREs. In our system, we analyzed all the FRET assays and defined the initial rates instead of the rate constants as our data, because the rate constants of the negative control and FRET sample could become the same level when they reach the steady state (Supplementary Figure S10).
Recently, some in vivo study53,54 have suggested that Syt1 is required for vesicle docking to the plasma membrane via the interaction with the binary t-SNARE complex. In this work, we have strategically used C2AB, the recombinant soluble version of Syt1, instead of the full length Syt1 in order to suppress its involvement in vesicle docking and single out its effect on SNARE complex formation. As anticipated, we did not observe any enhancement of the rate of NN-FRET by C2AB both in the presence and absence of Ca2+ (Figure 1). The main reason for this discrepancy between Syt1 and C2AB may be the lack of the transmembrane anchor in C2AB. A recent study with the single vesicle fusion assay demonstrated that transmembrane anchor is essential for the enhancement of docking by Syt19.
Combining the current and previously reported results it appears that Syt1 involves every step along the fusion pathway from docking to fusion pore opening (Figure 5). The docking enhancement is mediated by the interaction of Syt1 to t-SNARE9,54,55, for which anchoring of Syt1 to the vesicle membrane is essential. This interaction appears to be Ca2+-independent. Next, C2AB, the tandem C2 domains of Sty1, was specifically involved in stimulating the C-terminal SNARE zippering in the presence of Ca2+. We speculate that this step involves the interaction of C2AB with acidic membrane. Thus, C2AB-Ca2+ cross-linked two membranes and shorten the distance which is favorable for SNARE zippering.
Figure 5. The model of the stimulatory SNARE zippering by Syt1 and Ca2+.
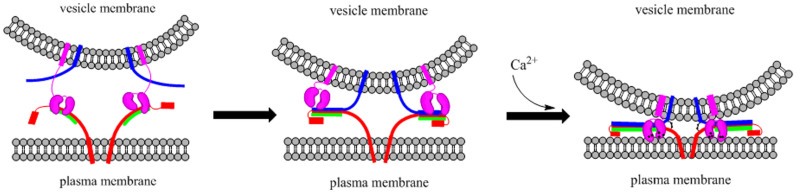
In this model, syntaxin-1A (red) and SNAP-25 (green) pre-assemble into the binary t-SNARE complex at plasma membrane, while VAMP2 (blue) and Syt1 (pink) are anchored on the vesicle membrane. In the stage of vesicle docking, Syt1 binding to the binary t-SNARE complex facilitates the synaptic vesicle docking with the plasma membrane53,54. The N-terminus of the binary t-SNARE complex then start zippering with the N-terminus of VAMP2, and form into a partially zipped trans SNARE complex. Upon the rise of the Ca2+ level Syt1-Ca2+ interacts with the partially zipped complex and facilitates the SNARE zippering at membrane proximal region, which drives the vesicle fusion.
Finally, C2AB stimulate fusion pore opening. This step also requires Ca2+. The molecular interactions underlying this particular action are unclear, and we speculate this could be simply the consequence of the main push on the SNARE zippering at the C-terminal region of Snares. Or, possibly, it does involve further specific interaction between Syt1 and SNAREs, warranting further investigation.
Methods
Plasmid constructs and site-directed mutagenesis
DNA sequences encoding syntaxin 1A (amino acids 1–288 with three cysteines replaced by alanines), VAMP2 (amino acids 1–116 with C103 replaced by alanines), SNAP-25 (amino acids 1–206 with four native cysteines replaced by alanines), soluble VAMP2 (amino acids 1–96) and soluble synaptotagmin-1 C2AB (amino acid 96–421) were inserted into the pGEX-KG vector as N-terminal glutathione S-transferase (GST) fusion proteins. We used the Quick Change site-directed mutagenesis kit (Stratagene) to generate all cysteine mutants, including SyxF I187C, SyxF Y257C, SyxF G288C, VAMP2 G18C, VAMP2 K87C and VAMP2 T116C. Sequencing Facility at Iowa State University confirmed all of the DNA sequences.
Protein expression, purification and labeling
All cysteine mutant recombinant neuronal SNARE proteins, SNAP-25, and C2AB were expressed as N-terminal glutathione-S-transferase fusion proteins. Recombinant proteins were expressed in Escherichia coli Rosetta (DE3) pLysS (Novagene). The cells were grown at 37°C in LB medium with 100 μg ml−1 ampicillin until the absorbance at 600 nm reached 0.6–0.8. The cells were further grown for overnight after adding IPTG (0.5 mM final concentration) at 16°C. We purified the proteins using glutathione-agarose chromatography. Cell pellets were resuspended in 20 ml PBS, pH 7.4, containing 0.2 v/v% Triton X-100, with final concentrations of 1 mM 4-(2-aminoethyl)-benzenesulfonyl fluoride (AEBSF), 2 mM DTT. Cells were broken by sonication in an ice bath and centrifuged at 15,000 g for 30 min at 4°C. The supernatant was mixed with 2 ml glutathione-agarose beads in PBS and nutated in the cold room (4°C) for 2 h. The proteins were then cleaved by thrombin in cleavage buffer (50 mM TrisHCl, 150 mM NaCl, pH 8.0) with 0.8 g per 100 ml n-octyl-D-glucopyranoside (OG) for cysteine mutants and SNAP-25, or cleaved by thrombin in cleavage buffer for soluble VAMP2 and C2AB. Purified proteins were examined with 15% SDS-PAGE, and the purity was at least 85% for all proteins.
SyxF and VAMP2 cysteine mutants were labeled with the fluorescence labels Alexa Fluor 546 and Alexa Fluor 647 (Invitrogen) or Cy3 and Cy5 maleimide (Amersham) (Supplementary Figure S4), respectively. The reaction mixture, with the dye to protein ratio of more than 5:1, was left overnight at 4°C. The free dye was removed using the PD-10 desalting column (Amersham). All labeled proteins were analyzed by the SDS-polyacryamide gel in which the labeled proteins ran a little slower than the wild-type, and the labeling efficiency were over 90% for all proteins (Supplementary Figure S1).
Membrane reconstitution
The mixture of POPC (1-palmitoyl-2-dioleoyl-sn-glycero-3-phosphatidylcholine), DOPS (1,2-dioleoyl-sn-glycero-3-phosphatidylserine) and Cholesterol (molar ratio of 45:15:40) in chloroform was dried in a vacuum and was resuspended in a buffer (25 mM HEPES/KOH, 100 mM KCl, pH 7.4) to make the total lipid concentration about 25 mM. Protein-free large unilamellar vesicles (~100 nm in diameter) were prepared by extrusion through polycarbonate filters (Avanti Polar Lipids). For the lipid mixing assay, the t-vesicles contained 2 mol% DiI in replacement of equimolar POPC, whereas the v-SNARE vesicles contained 2 mol% DiD.
Labeled SyxF and SNAP-25, in a molar ratio of 1:1.5, were premixed, and the mixture was left at room temperature for 1 hour to form the complex before the reconstitution. For membrane reconstitution, proteins were mixed with vesicles at the protein to lipid molar ratio of 1:200 with ~0.8 g per 100 ml OG in the buffer at 4°C for 15 min. The mixture was diluted two times with dialysis buffer (25 mM HEPES, 100 mM KCl, pH 7.4), and this diluted mixture was then dialyzed in 2 L dialysis buffer at 4°C for overnight.
For each reaction, there is 10 nmole lipids for each t- and v- vesicle, accordingly, 0.05 nmole SyxF or VAMP2 was needed. For each preparation, we need to reconstitute each vesicle for 10 ~ 20 reactions.
Fluorescence spectrum scanning
The reconstituted vesicles were diluted with HEPES buffer (25 mM HEPES, pH 7.4, 150 mM KCl) to 0.1 mM using a quartz cell of 100 μL with a 2 mm path length. The fluorescence emission spectrum was detected by a Varian Cary Eclipse spectrophotometer, with the excitation wavelength 530 nm and the emission wavelength 550–700 nm for t-vesicles and with the excitation wavelength 630 nm and the emission wavelength 650–800 nm for v-vesicles, respectively. Each spectrum was averaged from five repeated scans with 1 min interval. 1 μM C2AB was added to vesicles with or without 100 mM Ca2+. All measurements were performed at 35°C.
Dynamic light scattering
DLS experiments were performed on a Protein Solutions DynaPro instrument equipped with a temperature-controlled microsampler, using 10 s acquisition time and 10% laser power. The samples contained 0.1 mM lipid vesicles (POPC/Cholesterol/DOPS = 65:20:15) in 25 mM HEPES (pH 7.4) and 100 mM KCl with various conditions: No addition, 1 μM C2AB and 1 μM C2AB/1 mM Ca2+. Samples were prepared in reconstitution buffer in a total volume of 20 μl or incubated with C2AB for 30 minutes in 20°C, and each measurement was done as an average of 30 data points in 20°C. Intensity data from each sample were collected in three replicates and analyzed by the Precision Deconvolve software.
GST Pull-down assay
To test binding of C2AB to the ternary SNARE complex, purified soluble syntaxin 1A (SH3, 191–266) and soluble VAMP2 (Vps, 1–96) was mixed with His-tagged SNAP-25 at about 1:1:1 ratio before loading to Ni-NTA beads. The ternary complex was purified following the same procedure described for His-tagged SNAP-25 followed by size-exclusion chromatography using Superdex 200 HR 10/30 column on AKTA system (GE Healthcare). Ternary SNARE fractions was concentrated and binding of GST-C2AB was carried out in dialysis buffer, and dialysis buffer containing 0.1 mM lipids (components are same as the vesicles) or 0.1% triton. The GST Pull-down assay samples were resolved on precast 15% SDS-PAGE and visualized by Coomassie blue staining.
FRET assays
Reconstituted t-and v-vesicles were mixed at a ratio of 1:1. The final lipid concentrations in the reaction ranged from 0.06 to 0.2 mM. The fluorescence intensity was monitored in two channels with the excitation wavelength of 556 nm and emission wavelengths of 573 and 665 nm for Alexa Fluorescence dyes, respectively, or with the excitation wavelength of 545 nm and emission wavelengths of 570 and 668 nm for AmershamCy Dye. Fluorescence changes were recorded with the Varian Cary Eclipse model fluorescence spectrophotometer using a quartz cell of 100 μL with a 2 mm path length. All measurements were performed at 35°C. FRET value is calculated by the formula: E = Ia/(Ia + Id), where Ia is the fluorescence intensity of the acceptor A647 and Id is that of the donor A546.
Lipid mixing assay
Reconstituted t vesicle and v-vesicle were mixed at a ratio of 1:1. The total lipid concentration in the reaction is 0.2 mM. The fluorescence intensity was monitored in two channels with the excitation wavelength of 530 nm and emission wavelengths of 570 and 670 nm for DiI and DiD dye pairs, respectively. Fluorescence changes were recorded with the same Varian fluorimeter. All measurements were performed at 35°C.
Statistics analysis
Values are given as mean ± SD. For all the data sets requiring a single pairwise comparison, we used Student's t test to compute significance. Significance was defined by the criterion *P < 0.05, **P < 0.01.
Author Contributions
L.Y. performed most of the experiments. L.X.C. performed additional experiments including the data presented in Supplementary Figure S3, S7, and S8. W.C.Q. and X.T. helped to discuss with the result. L.Y., X.T. and T.J.S. designed and analyzed the data. L.Y. and T.J.S. wrote the manuscript.
Supplementary Material
Supplementary information
Acknowledgments
We greatly thank professor Yeon-Kyun Shin at Iowa State University for his kind assistance. This work was supported by NIH grant GM051290, which was funded to professor Shin, Chinese 1000Plan Professorship for Young Talents and Creative Research Awards from Huazhong University of Science and Technology, which were funded to professor Tian Xia.
References
- Weber T. et al. SNAREpins: Minimal machinery for membrane fusion. Cell 92, 759–772 (1998). [DOI] [PubMed] [Google Scholar]
- Brunger A. T. Structure and function of SNARE and SNARE-interacting proteins. Q Rev Biophys 38, 1–47 (2005). [DOI] [PubMed] [Google Scholar]
- Jahn R. & Scheller R. H. SNAREs--engines for membrane fusion. Nat Rev Mol Cell Biol 7, 631–643 (2006). [DOI] [PubMed] [Google Scholar]
- Poirier M. A. et al. The synaptic SNARE complex is a parallel four-stranded helical bundle. Nat Struct Biol 5, 765–769 (1998). [DOI] [PubMed] [Google Scholar]
- Sutton R. B., Fasshauer D., Jahn R. & Brunger A. T. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature 395, 347–353 (1998). [DOI] [PubMed] [Google Scholar]
- Chapman E. R. Synaptotagmin: a Ca(2+) sensor that triggers exocytosis? Nat Rev Mol Cell Biol 3, 498–508 (2002). [DOI] [PubMed] [Google Scholar]
- Sudhof T. C. The synaptic vesicle cycle. Annu Rev Neurosci 27, 509–547 (2004). [DOI] [PubMed] [Google Scholar]
- Rizo J. & Rosenmund C. Synaptic vesicle fusion. Nat Struct Mol Biol 15, 665–674 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. K. et al. Dynamic Ca2+-dependent stimulation of vesicle fusion by membrane-anchored synaptotagmin 1. Science 328, 760–763 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara M. & Littleton J. T. Synaptotagmin I functions as a calcium sensor to synchronize neurotransmitter release. Neuron 36, 897–908 (2002). [DOI] [PubMed] [Google Scholar]
- Davletov B. A. & Sudhof T. C. A single C2 domain from synaptotagmin I is sufficient for high affinity Ca2+/phospholipid binding. J Biol Chem 268, 26386–26390 (1993). [PubMed] [Google Scholar]
- Chapman E. R. & Davis A. F. Direct interaction of a Ca2+-binding loop of synaptotagmin with lipid bilayers. J Biol Chem 273, 13995–14001 (1998). [DOI] [PubMed] [Google Scholar]
- Fernandez I. et al. Three-dimensional structure of the synaptotagmin 1 C2B-domain: synaptotagmin 1 as a phospholipid binding machine. Neuron 32, 1057–1069 (2001). [DOI] [PubMed] [Google Scholar]
- Choi U. B. et al. Single-molecule FRET-derived model of the synaptotagmin 1-SNARE fusion complex. Nat Struct Mol Biol 17, 318–324 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai H., Shen N., Arac D. & Rizo J. A quaternary SNARE-synaptotagmin-Ca2+-phospholipid complex in neurotransmitter release. J Mol Biol 367, 848–863 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrljic M. et al. Molecular mechanism of the synaptotagmin-SNARE interaction in Ca2+-triggered vesicle fusion. Nat Struct Mol Biol 17, 325–331 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Chacon R. et al. Synaptotagmin I functions as a calcium regulator of release probability. Nature 410, 41–49 (2001). [DOI] [PubMed] [Google Scholar]
- Rhee J. S. et al. Augmenting neurotransmitter release by enhancing the apparent Ca2+ affinity of synaptotagmin 1. Proc Natl Acad Sci USA 102, 18664–18669 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui E., Johnson C. P., Yao J., Dunning F. M. & Chapman E. R. Synaptotagmin-mediated bending of the target membrane is a critical step in Ca(2+)-regulated fusion. Cell 138, 709–721 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens S., Kozlov M. M. & McMahon H. T. How synaptotagmin promotes membrane fusion. Science 316, 1205–1208 (2007). [DOI] [PubMed] [Google Scholar]
- Chernomordik L. V. & Kozlov M. M. Protein-lipid interplay in fusion and fission of biological membranes. Annu Rev Biochem 72, 175–207 (2003). [DOI] [PubMed] [Google Scholar]
- Yoon T. Y. et al. Complexin and Ca(2+) stimulate SNARE-mediated membrane fusion. Nat Struct Mol Biol 15, 707–713 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao J. et al. Complexin-1 enhances the on-rate of vesicle docking via simultaneous SNARE and membrane interactions. J Am Chem Soc 135, 15274–15277 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. et al. Three-dimensional structure of the complexin/SNARE complex. Neuron 33, 397–409 (2002). [DOI] [PubMed] [Google Scholar]
- Pabst S. et al. Rapid and selective binding to the synaptic SNARE complex suggests a modulatory role of complexins in neuroexocytosis. J Biol Chem 277, 7838–7848 (2002). [DOI] [PubMed] [Google Scholar]
- Giraudo C. G., Eng W. S., Melia T. J. & Rothman J. E. A clamping mechanism involved in SNARE-dependent exocytosis. Science 313, 676–680 (2006). [DOI] [PubMed] [Google Scholar]
- Schaub J. R., Lu X., Doneske B., Shin Y. K. & McNew J. A. Hemifusion arrest by complexin is relieved by Ca2+-synaptotagmin I. Nat Struct Mol Biol 13, 748–750 (2006). [DOI] [PubMed] [Google Scholar]
- Tang J. et al. A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell 126, 1175–1187 (2006). [DOI] [PubMed] [Google Scholar]
- Giraudo C. G. et al. Alternative zippering as an on-off switch for SNARE-mediated fusion. Science 323, 512–516 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao J. et al. Synaptic proteins promote calcium-triggered fast transition from point contact to full fusion. eLife 1, 1–21 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y. et al. Fusion pore formation and expansion induced by Ca2+ and synaptotagmin 1. Proc Natl Acad Sci USA 110, 1333–1338 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen J. B. SNARE complexes prepare for membrane fusion. Trends Neurosci 28, 453–455 (2005). [DOI] [PubMed] [Google Scholar]
- Sorensen J. B. et al. Sequential N- to C-terminal SNARE complex assembly drives priming and fusion of secretory vesicles. EMBO J 25, 955–966 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao J. et al. A single-vesicle content mixing assay for SNARE-mediated membrane fusion. Nat Commun 1, 54–65 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerona R. R., Larsen E. C., Kowalchyk J. A. & Martin T. F. The C terminus of SNAP25 is essential for Ca(2+)-dependent binding of synaptotagmin to SNARE complexes. J Biol Chem 275, 6328–6336 (2000). [DOI] [PubMed] [Google Scholar]
- Sudhof T. C. & Rizo J. Synaptotagmins: C2-domain proteins that regulate membrane traffic. Neuron 17, 379–388 (1996). [DOI] [PubMed] [Google Scholar]
- Tucker W. C., Weber T. & Chapman E. R. Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science 304, 435–438 (2004). [DOI] [PubMed] [Google Scholar]
- Wang Z., Liu H., Gu Y. & Chapman E. R. Reconstituted synaptotagmin I mediates vesicle docking, priming, and fusion. J Cell Biol 195, 1159–1170 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S. et al. Molecular anatomy of a trafficking organelle. Cell 127, 831–846 (2006). [DOI] [PubMed] [Google Scholar]
- Diao J. et al. A single vesicle-vesicle fusion assay for in vitro studies of SNAREs and accessory proteins. Nat Protoc 7, 921–934 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzi A. The application of fluorescent probes in membrane studies. Q Rev Biophys 8, 237–316 (1975). [DOI] [PubMed] [Google Scholar]
- Blumenthal R., Gallo S. A., Viard M., Raviv Y. & Puri A. Fluorescent lipid probes in the study of viral membrane fusion. Chem Phys Lipids 116, 39–55 (2002). [DOI] [PubMed] [Google Scholar]
- Ha T. Single-molecule fluorescence resonance energy transfer. Methods 25, 78–86 (2001). [DOI] [PubMed] [Google Scholar]
- Heyduk T. Measuring protein conformational changes by FRET/LRET. Curr Opin Biotech 13, 292–296 (2002). [DOI] [PubMed] [Google Scholar]
- Mendelsohn A. R. & Brent R. Protein interaction methods--toward an endgame. Science 284, 1948–1950 (1999). [DOI] [PubMed] [Google Scholar]
- Tong J., Borbat P. P., Freed J. H. & Shin Y. K. A scissors mechanism for stimulation of SNARE-mediated lipid mixing by cholesterol. Proc Natl Acad Sci USA 106, 5141–5146 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arac D. et al. Close membrane-membrane proximity induced by Ca(2+)-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat Struct Mol Biol 13, 209–217 (2006). [DOI] [PubMed] [Google Scholar]
- Xue M., Ma C., Craig T. K., Rosenmund C. & Rizo J. The Janus-faced nature of the C(2)B domain is fundamental for synaptotagmin-1 function. Nat Struct Mol Biol 15, 1160–1168 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee Y. & Scheller R. H. Localization of synaptotagmin-binding domains on syntaxin. J Neurosci 16, 1975–1981 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. A., Scales S. J., Patel S. M., Doung Y. C. & Scheller R. H. SNARE complex formation is triggered by Ca2+ and drives membrane fusion. Cell 97, 165–174 (1999). [DOI] [PubMed] [Google Scholar]
- Fasshauer D. Structural insights into the SNARE mechanism. Biochim Biophys Acta 1641, 87–97 (2003). [DOI] [PubMed] [Google Scholar]
- Diao J., Yoon T. Y., Su Z. L., Shin Y. K. & Ha T. C2AB: A Molecular Glue for Lipid Vesicles with a Negatively Charged Surface. Langmuir 25, 7177–7180 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewen C. A., Lee S. M., Shin Y. K. & Reist N. E. C2B polylysine motif of synaptotagmin facilitates a Ca2+-independent stage of synaptic vesicle priming in vivo. Mol Biol Cell 17, 5211–5226 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit H. et al. Synaptotagmin-1 Docks Secretory Vesicles to Syntaxin-1/SNAP-25 Acceptor Complexes. Cell 138, 935–946 (2009). [DOI] [PubMed] [Google Scholar]
- Lai Y. & Shin Y. K. The importance of an asymmetric distribution of acidic lipids for synaptotagmin 1 function as a Ca2+ sensor. Biochem J 443, 223–229 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information