

Abstract
Oxidative stress leads to the up-regulation of many antioxidant enzymes including Cu,Zn superoxide dismutase (SOD1) via transcriptional mechanisms; however, few examples of posttranslational regulation are known. The copper chaperone for SOD1 (CCS) is involved in physiological SOD1 activation, and its primary function is thought to be delivery of copper to the enzyme. Data presented here are consistent with a previously uncharacterized function for CCS in the SOD1 pathway, namely mediating enzyme activation in response to increases in oxygen tension. Activity assays with pure proteins and cell extracts reveal that O2 (or superoxide) is required for activation of SOD1 by CCS. Dose–response studies with a translational blocking agent demonstrate that the cellular oxidative response to O2 is multitiered: existing apo-pools of SOD1 are activated by CCS in the early response, followed by increasing expression of SOD1 protein with persistent oxidative stress. This CCS function provides oxidant-responsive posttranslational regulation of SOD1 activity and may be relevant to a wide array of physiological stresses that involve a sudden elevation of oxygen availability.
The production of reactive oxygen species (ROS) such as superoxide (O–2) and hydroxyl radicals occurs during cellular respiration and is a consequence of aerobic life. Cells have evolved a variety of inducible responses to attenuate oxidative damage, including superoxide dismutase enzymes that catalyze the disproportionation O–2 to H2O2 and O2 (1–3). Early studies by Fridovich and coworkers (2–4) showed that Cu,Zn superoxide dismutase (SOD1) is predominately found in the cytosol, with a smaller fraction in the inner membrane space of the mitochondria, whereas Mn superoxide dismutase (SOD2) is found in the mitochondrial matrix (2). Further, they demonstrated that superoxide dismutase activity in the yeast Saccharomyces cerevisiae could be enhanced by increases in oxygen tension, and survival of cells to hyperbaric O2 is increased by pretreatment of cells with 100% O2 (5). Consistent with this, respiring yeast have higher SOD1 protein and activity levels than anaerobic or fermenting cells (6, 7). Treatment of anaerobically grown cells with copper reportedly results in an increase in SOD1 activity, and maximal activation occurs in the presence of both copper and oxygen (7). These studies led to the prediction that activation of apo-SOD1 in yeast depends on oxygen metabolism (7). Investigations in animal models mirror the results in yeast. Repeated exposure to oxidative stress in the form of endurance training enhanced the levels of antioxidant enzymes and the resistance to ROS produced during acute bouts of exercise (8–10). Thus, while oxidative stress leads to increased transcription and/or translation of the SOD1 gene (5–11), such stresses may lead to activation of SOD1 at the posttranslation level, although the molecular mechanisms remain unknown.
Most structurally characterized forms of active eukaryotic SOD1 are dimeric, contain a single copper and zinc ion, although lower metal stoichiometries have been reported, and one disulfide bond per monomer (12). The redox active copper is essential for dismutase activity (13), and although CCS independent activation has been reported in some mammalian proteins (14), in both yeast and human cells physiological activation of most SOD1 involves the copper chaperone for SOD1 (CCS) (15, 16). Purified CCS protein from various species has been characterized as binding several equivalents of copper (17–19); however, neither the minimal stoichiometry nor the chemical basis of the metal transfer to SOD1 have been established. Mechanistic studies indicate that CCS binds Cu(I) tightly but nonetheless transfers it into apo-SOD1, even in the presence of stringent copper chelators (17, 20, 21). These results suggest that free copper ion is not available in the cytosol to apo-SOD1 and are consistent with direct insertion of copper by CCS in vivo (17). Recent biochemical and structural studies indicate that direct copper transfer is most likely accomplished within a heterodimeric complex of SOD1 and CCS (17, 20–23).
In the studies presented here we demonstrate an essential role for O2 or O–2 in the posttranslational activation of SOD1 by CCS. Activation of SOD1 in vitro requires both Cu–CCS and O2 exposure, and studies using translational blocking agents show that the active enzyme is undetectable in S. cerevisiae cells deprived of O2. Transition of anaerobic cultures to aerobic conditions results in the rapid appearance of SOD1 activity, even in the absence of new protein synthesis. The results are consistent with a model in which CCS mediates the posttranslational regulation of superoxide dismutase activity in response to increases in cellular oxygen tension by modification of a preexisting pool of the immature form of SOD1 protein. Thus, oxidants like O2 not only induce transcription and/or translation of SOD1 but can also increase the ratio of active to inactive SOD1 in a manner that may be relevant to mammalian physiology and disease.
Materials and Methods
Purification and Preparation of Proteins. Proteins were purified according to published protocols (17, 21). The Cu(I) form of CCS and the apo, reduced, and denatured (ARD) forms of yeast (ySOD1) and human (hSOD1) SOD1 were also prepared as described (17, 21). All of the forms used for in vitro assays were prepared under N2 in a Vacuum Atmospheres (Hawthorne, CA) chamber.
EPR Spectroscopy of in Vitro Assay Mixtures. EPR samples were made in 20% glycerol. Mixtures containing 100 μM ARD hSOD1, 100 μM Cu(I)–hCCS (human CCS), and 50 μM ZnSO4 were incubated for 60 min at 37°C either under N2 or in air. Then 100 μl of the reaction mixtures was frozen under N2 or in air. Additionally, 100 μM holo hSOD1, 100 μM CuSO4, and 100 μM Cu(I)–hCCS (under N2 and after 60 min of air exposure at 37°C) were analyzed individually. EPR was performed by using a Varian EPR at 100 K under flowing N2 at 9.1 GHz microwave frequency. Instrument settings were as follows: modulation amplitude, 5 G; microwave power, 20 mW; time constant, 125 ms; scan time, 8 min.
In Vitro Anaerobic SOD1 Activation. For in vitro assay of CCS activity, 2 μM ARD hSOD1 was mixed with 200 μM bathocuproinedisulfonic acid in one tube and 20 μM ZnSO4, 1 mM GSH, and either 10 μMCu–hCCS or 10 μM Cu(I) (CH3CN)4 PF6 was mixed in another tube. Reactants were incubated anaerobically for 1 h at 37°C. Equal volumes of reaction mixture and native gel sample buffer containing 1 mM EDTA were mixed and run on a nondenaturing 12% Tris·HCl gel. To separate CCS and SOD1 under anaerobic conditions, native gels were run inside the anaerobic chamber, and the gels removed from the chamber for activity staining by standard p-nitroblue tetrazolium chloride (NBT) assay (24). “aerobic” samples were prepared in anaerobic conditions, then removed from the chamber after 30 min and incubated in air at 37°C for an additional 30 min. Samples were returned to the glove box, and gel electrophoresis was run anaerobically as above. For the time course, 10-μl aliquots of the anaerobic reactions were removed from the N2 atmosphere for 5, 30, and 60 min then brought back into the anaerobic chamber for separation on native gel.
Anaerobic KO2 assays were performed by adding 2.5 mg of KO2 to 100 μl of anhydrous DMSO under N2. The dissolved KO2 was then added to a final concentration of 700 μM to each reaction mixture and the reaction immediately loaded onto a native gel.
Strains and Culture Conditions. S. cerevisiae strain SY1699 (α, leu2, ura3, ade1, his3) was used for all in vivo studies. Strains lacking yeast CCS (yCCS), 1699Δlys7 (α, leu2, ura3, ade2, his3, lys7::LEU2), and ySOD1, KS107 (α, leu2-3, 112, his3Δ1, GAL+, trp1-289a, ura3-52, sod1Δ::TRP1) were used as controls (25). Strains were maintained aerobically on yeast extract/peptone/dextrose (YPD) medium. Anaerobic growth of yeast requires 0.2% Tween-80 and 20 μg/ml ergosterol; therefore, all YPD cultures were supplemented as a control.
Anaerobic Growth and Extraction. Anaerobic cultures were grown in a Vacuum Atmospheres glove box maintained at 28–30°C. All media and extraction buffers were maintained under anaerobic conditions throughout the experiments. Single colonies of SY1699, 1699Δlys7, and KS107 were brought into the glove box and used to inoculate 3-ml precultures. To reduce the amount of active ySOD1 that was generated during aerobic culture to background levels, strains were maintained anaerobically for at least 5 days before beginning experiments and diluted 30-fold into fresh media daily. The cultures were grown to an A600 of 0.6–0.9, and cytoplasmic extracts were made by means of glass bead lysis as described (26), with the exception that all lysis steps were carried out anaerobically.
In Vivo SOD1 Activation Time Course. Aliquots of anaerobic cultures were spun down, and the cell pellet was frozen. The remaining culture was removed from the chamber and maintained in a 30°C water bath with 21% O2 bubbling through it. At the indicated times, aliquots were removed, and cells were spun down and immediately frozen in liquid N2. All pellets were brought in to the anaerobic chamber, and extracts were made. Where indicated, anaerobic cultures were split and a portion incubated with 100 μg/ml cycloheximide for 30 min before exposure to air. In these studies a portion of each extract was concentrated three to five times to enhance the detection of SOD1 activity by NBT assay. All NBT assays were performed under N2 as described above.
Western Blot of SOD1. Antibodies against ySOD1 and yCCS were prepared as described (23). Total protein concentrations of lysates were determined by published procedures (27). Polyclonal antibodies for either ySOD1 or yCCS were used as the primary antibodies (1:1,250 dilution), and goat anti-rabbit IgG HRP conjugate (Bio-Rad) was used as the secondary antibody (1:6,500 dilution). Blots were developed by using the ECL Plus Western Blotting Detection System (Amersham Biosciences). Estimates of protein concentration from Western blots were calculated by using densitometry (kodak 1d image analysis software, Version 3.6).
Inductively Coupled Plasma (ICP)–MS. Cell cultures were grown to early–mid exponential phase anaerobically or aerobically as above. To eliminate extracellular background, each cell pellet was washed extensively in 1 mM EDTA made with Milli-Q-treated water. The cell pellets were dried overnight at 80°C in Teflon tubes, then dissolved completely in concentrated nitric acid. The metal content was determined by ICP-MS (PQ Excell, TJA Solutions, Franklin, MA).
Results
O2 Is Required for the Formation of Cu(II)–hSOD1 via Cu(I)–hCCS. To test whether oxidation of Cu(I) occurs in CCS before activation of SOD1, the reactions of the pure human forms of the proteins were examined by EPR spectroscopy. Cu(I)–hCCS samples frozen anaerobically under N2 (Fig. 1A) or after a 60-min exposure to air (Fig. 1B) were EPR silent, indicating that the Cu(I) form of the chaperone is not readily oxidized in air to an EPR-detectable Cu(II) state. Nor was a Cu(II) EPR signal detected in a reaction mixture of Cu(I)–hCCS and ARD SOD1 incubated at 37°C in the absence of O2 before freezing (Fig. 1C). In the presence of O2, however, a characteristic Cu(II)–SOD1 EPR signal (12, 13) was detected for the same reaction mixture after 60 min at 37°C (Fig. 1D). Based on comparisons of the signal in Fig. 1D with a holo-hSOD1 control measured under the same conditions, we estimate that about three-quarters of the copper in hCCS was transferred to apo-hSOD1 (data not shown). These spectroscopic results suggest that the formation of active SOD1 requires both oxygen and the copper chaperone but do not reveal whether other forms of SOD1 [i.e., Cu(I)] can form in the absence of O2. To test this directly, an anaerobic assay for SOD1 was developed in which metal transfer and protein separation could be conducted under anaerobic conditions, while the SOD1 enzyme activity assays, which require O2, could be conducted aerobically after separation of the enzyme from copper donors.
Fig. 1.

EPR spectra of purified Cu(I)–hCCS and hSOD1. Each reaction mixture was incubated under aerobic or anaerobic conditions for 60 min at 37°C before EPR analysis. EPR spectra of 100 μM Cu(I)–hCCS alone, anaerobic (A) or aerobic (B), and 100 μM Cu(I)–hCCS + 100 μM apo hSOD1, anaerobic (C) or aerobic (D) are shown.
Oxygen and Cu(I)–CCS Are Required for Apo-SOD1 Activation in Vitro. When assayed in the presence of air, the copper-loaded form of human CCS alone was sufficient to activate the apo form of human SOD1 (Fig. 2A) as reported (21). To determine whether hSOD1 activation by hCCS can also occur in the absence of oxygen, the assay was performed anaerobically in a Vacuum Atmospheres chamber. After the designated time, hSOD1 in the reaction mixture was separated from copper donors (i.e., low-molecular-weight copper or Cu–hCCS) by native gel electrophoresis in the anaerobic chamber using Ar purged buffers. No SOD1 activity was observed by NBT assay for any fully anaerobic reaction mixture, regardless of whether copper was provided as a Cu(I)–hCCS complex or in the form of Cu(I)–glutathione complex (Fig. 2B). None of the anaerobic procedures per se altered holo-SOD1, which retained full activity throughout these procedures (lane 1 of each gel).
Fig. 2.

Aerobic and anaerobic activation of purified SOD1. Reaction mixtures with 100 ng of total hSOD1 were analyzed by NBT assays as described under aerobic (A) or anaerobic (B) conditions. (C) Time course samples were prepared anaerobically, then exposed to air for the indicated times before determining hSOD1 activity.
To determine whether ARD hSOD1 activation by Cu(I)–hCCS required the presence of O2, the above reaction mixture was removed from the anaerobic chamber and exposed to air for various times before being returned to the chamber and loaded on the native gel. In the absence of air exposure, no activity was observed from a mixture of Cu(I)–hCCS and ARD hSOD1 (Fig. 2C). Significant SOD1 activity, however, was observed after as little as 5 min of air exposure, and maximal activity was apparent within 30 min, indicating rapid metal transfer and activation of the apo-SOD1 by CCS and oxygen exposure. Anaerobic addition of potassium superoxide to ARD hSOD1/Cu(I)–hCCS mixtures was also able to stimulate activation of ARD hSOD1 by Cu(I)–hCCS in a dose-dependent manner (Fig. 2B). In this case, some spontaneous disproportionation could have provided a small amount of O2; thus, a direct oxygen role cannot be ruled out. These results demonstrate an absolute requirement for an oxidant such as O2 and possibly O–2 itself in the activation of SOD1 by CCS in vitro. This finding led to the hypothesis that oxygen availability or reactive oxygen species (ROS) levels can posttranslationally regulate SOD1 activity in vivo. To test this, the extent of activation of SOD1 in yeast undergoing an anaerobic to aerobic shift was examined.
SOD1 Expression and Activity in Vivo Is Regulated in an O2 Dose-Responsive Manner. Wild type and cells lacking CCS, lys7Δ mutants, of S. cerevisiae were grown to early–mid exponential phase in a Vacuum Atmosphere glove box heated to 28–30°C. Both strains were grown aerobically in a 30°C water bath as well, sparged with either 21% or 100% O2. Consistent with earlier studies, we find no SOD1 activity from lys7Δ mutants (15), regardless of the O2 tension during growth and despite the increase in ySOD1 expression (Fig. 3). In agreement with in vitro results using hSOD1, no ySOD1 activity is detected in extracts from anaerobic cells. An additional band is present above the SOD1 band in activity gels with extracts, including those from the sod1Δ mutant strain. Although the identity of this band remains unknown, it is routinely seen by others (12). Their presence in extracts from yeast strains lacking SOD1 or CCS indicate they are not due to SOD1 and are most likely an additional form of SOD2.
Fig. 3.

SOD1 activity in S. cerevisiae cultured with increasing O2 tension. (A) NBT assay using 5 μg of total cell lysate from anaerobically cultured yeast or from cells cultured in 21% or 100% O2. Fifty micrograms of total protein was used for extracts from sod1Δ cells. (B) Western blot of 300 ng of total protein from the same lysates as in A.
An increase in oxygenation produces a significant increase in ySOD1 activity in the wild-type strain but not the lys7Δ mutants, indicating dose-responsive activation that depends on yCCS. Western blots revealed that ySOD1 protein was between 1 and 2.5 ng SOD1 protein per 300 ng of total protein loaded in extracts from anaerobically grown cells but more abundant in extracts from cells grown aerobically (Fig. 3B). The total protein loaded in the Western gel was low to prevent the saturation of the 21% and 100% O2 samples. Higher concentrations of total extract were used in subsequent Western blots to bring the SOD1 protein levels in anaerobic samples farther above the detection limit. Expression of the ySOD1 protein increased with increasing oxygen tension and the lys7Δ mutants mirrored the quantitative increase of ySOD1 protein with increasing oxygen tension seen in wild-type cells. This result indicates that the O2 regulation of SOD1 protein expression is CCS-independent, in accord with previous studies (15). The level of the yCCS protein, as determined by Western blot, remained constant under all of the oxygenation conditions studied indicating that CCS expression is not regulated in an O2-dependent manner (data not shown).
Cellular Response to O2 Is Rapid and Prolonged. To determine whether the in vivo response to oxygen occurred on the same time frame as that seen in vitro, an anaerobic to aerobic time course study was conducted by using cell cultures. Given that SOD1 protein in anaerobic samples was difficult to detect, we increased the total protein loaded in each lane. We can now observe ≈2.5 ng of SOD1 protein per 1.5 μg of total loaded protein. Western blot analysis of these extracts indicated that total ySOD1 protein increased dramatically over the time course (Fig. 4A). The ySOD activity in these extracts also increased parallel to protein expression, with the highest activity observed after a 60-min air exposure (Fig. 4B). No SOD1 activity was detected in the lys7Δ mutants in a parallel time course (only the anaerobic and 60-min time points are shown; Fig. 4B). Additional controls indicate that this SOD1 activation phenomenon is not the result of O2 metabolism causing an increase in copper accumulation by the cells. Inductively coupled plasma (ICP)–MS analysis reveals that anaerobic cells accumulate 2-fold higher concentrations of copper than do aerobically grown cells (6.3 × 10–3 mg of copper per g of dry weight of anaerobic cells versus 3.2 × 10–3 mg of copper per g of dry weight of aerobic cells). Despite this increase in intracellular copper, no activity is detected in anaerobic extracts from either wild type or lys7Δ mutants. Taken together, these results strongly support the proposal that the O2-mediated activation of SOD1 in vivo strictly depends on CCS.
Fig. 4.

Time course of SOD1 air activation in S. cerevisiae. Yeast extract was prepared from anaerobic cultures exposed to air for the indicated times. (A) Western blot of 1.5 μg of total protein. (B) Active SOD1 protein was detected in 80 μg of total protein via NBT assay from same extracts as in A.
In agreement with earlier studies (5–7), the increase in SOD1 expression with increasing and/or prolonged O2 exposure demonstrates that new protein synthesis contributes to the oxidative stress response (Fig. 4). However, the oxygen-dependent activation of purified protein (Fig. 2) suggests a posttranslational component to SOD1 regulation as well. To examine whether oxygen mediates the activation of a preexisting pool of apo-SOD1 or solely nascent protein, cells were treated with inhibitors of protein synthesis and tested as above.
Cells Exhibit  and CCS-Dependent Posttranslational Activation of Apo-SOD1. Anaerobic cultures were grown and treated with cycloheximide for 30 min before air exposure. Western analysis demonstrates that wild-type yeast treated with cycloheximide have a constant level of ySOD1 protein (≈5 ng of SOD1 protein per 3 μg of total protein loaded) despite prolonged aeration under normoxic conditions, whereas controls without cycloheximide show increased ySOD1 expression, as expected (Fig. 5A). Anaerobic controls from cultures treated with and without cycloheximide showed no SOD1 activity (Fig. 5B), and no SOD1 activity was detected in extracts from strains lacking either CCS and SOD1 (Fig. 5 B and C). In the absence of cycloheximide, cultures that were aerated for 60 min show very strong SOD1 activity (Fig. 5B). Cultures treated with cycloheximide demonstrated a significant increase in activity once cells were transferred to an aerobic environment (Fig. 5B). Taken together, these results suggest that the initial cellular response to a sudden increase in oxidative stress is the rapid activation of a preexisting pool of SOD1 protein, while continued exposure to oxidative stress leads to an increase in the synthesis and activation of nascent protein.
and CCS-Dependent Posttranslational Activation of Apo-SOD1. Anaerobic cultures were grown and treated with cycloheximide for 30 min before air exposure. Western analysis demonstrates that wild-type yeast treated with cycloheximide have a constant level of ySOD1 protein (≈5 ng of SOD1 protein per 3 μg of total protein loaded) despite prolonged aeration under normoxic conditions, whereas controls without cycloheximide show increased ySOD1 expression, as expected (Fig. 5A). Anaerobic controls from cultures treated with and without cycloheximide showed no SOD1 activity (Fig. 5B), and no SOD1 activity was detected in extracts from strains lacking either CCS and SOD1 (Fig. 5 B and C). In the absence of cycloheximide, cultures that were aerated for 60 min show very strong SOD1 activity (Fig. 5B). Cultures treated with cycloheximide demonstrated a significant increase in activity once cells were transferred to an aerobic environment (Fig. 5B). Taken together, these results suggest that the initial cellular response to a sudden increase in oxidative stress is the rapid activation of a preexisting pool of SOD1 protein, while continued exposure to oxidative stress leads to an increase in the synthesis and activation of nascent protein.
Fig. 5.

SOD1 activation after translational block. Anaerobically grown cells were exposed to cycloheximide before exposure to 21% O2 and extracts made. (A) Western blot with 3 μg of total protein from wild-type, lys7Δ, and sod1Δ extracts made at the indicated times after air exposure. (B and C) Extracts used in A were concentrated for use in NBT assays. To each lane was loaded 420 μg of total protein.
Discussion
Studies in S. cerevisiae conducted before the discovery of the copper chaperone for SOD1 showed that O2 alters the transcription and/or translation of the Cu,Zn superoxide dismutase gene (6, 7). Here we show that O2 (or superoxide) can also play a direct posttranslational role in controlling the amount of the active form of enzyme in vitro and in vivo. Furthermore, these results indicate that both yeast and mammalian forms of CCS mediate this O2 or O–2 responsive activation of apo-SOD1. The in vitro hCCS and hSOD1 assays reveal that Cu(I)–CCS cannot activate the apo-form of the enzyme in the absence of oxygen. In the presence of O2, however, Cu(I)–CCS-mediated activation of SOD1 occurs within 5 min. The in vivo data indicate that physiological activation of SOD1 also strictly depends on O2 and CCS. No SOD1 activity was detected in extracts from anaerobic cells or cells lacking CCS or SOD1; however, wild-type cells exhibit SOD1 activity that increases in step with increasing O2 tension (Fig. 3). When translation of new protein is blocked in anaerobic cultures by the addition of cycloheximide, we find a pool of SOD1 that is rapidly activated on shifting the cells to an aerobic environment (Fig. 5). Our studies also suggest that under some conditions cells maintain a pool of the immature form of SOD1 that can be modified into the mature, active form of the protein. This finding is consistent with reports that show increased activation of SOD1 by the aerobic addition of copper to translationally blocked cells (7, 28).
Although a mechanistic role for O2 in the activation of SOD1 is not clear from these data, it is likely that CCS mediates oxidative steps converting Cu(I) to Cu(II) and/or conserved cysteines to the disulfide. The conserved disulfide bond found in all structurally characterized SOD1 has been proposed to play a role in the proper folding and stabilization of the active form of the protein (29, 30). Recent investigations demonstrate that formation of the conserved disulfide bond must occur or the enzyme is inactive (Y. Furukawa, A.S.T., and T.V.O., unpublished work). Furthermore, this disulfide must be reduced before the protein can be translocated into the mitochondrial inner membrane space, where CCS then activates the protein (4). Although the chemical mechanism is not yet certain, the results, taken together, indicate that CCS directly mediates the oxygen-dependent activation of SOD1 and suggest a new physiological function for the copper chaperone, namely the posttranslational regulation of SOD1 activity in response to changes in oxygen tension.
The results presented here appear to be in contrast with an earlier study where SOD1 activity was observed from cells grown under anaerobic conditions (7). In the previous investigations yeast cultures were grown and treated with chloramphenicol and cycloheximide anaerobically; however, cells were exposed to air for ≈80–100 min before cell extracts were prepared (7). This is in contrast to the protocol herein where all steps were carried out in an anaerobic chamber, from growth to protein separation on native gels. Further, we were still able to detect SOD1 activity in anaerobic extracts 30–36 h (≈12–14 doublings) after cells were introduced into the anaerobic chamber (data not shown). This activity was eliminated by both extended growth of the strains in a N2 environment, coupled with a 30-fold dilution of the culture each day. We propose that the differences in anaerobic SOD1 activity is perhaps best explained by both the presence of residual active SOD1 enzyme from aerobic growth and the duration of air exposure before cell lysis in the previous studies.
Taken together the data suggest that one of the earliest cellular responses to oxidative stress is the immediate posttranslational activation of the available apo-SOD1 protein pool. The ability of cells to maintain even a small pool of the immature form of SOD1 is likely to represent an important physiological response to an increase in oxidative stress. It has been shown that cellular SOD1 concentrations as little as 0.01% of total cellular protein can rescue cells from the sod1Δ phenotype, including oxidative stress (14). In Fig. 3 the total SOD1 protein found in anaerobic cells was <2.5 ng but still detectable. In this example, activation of even 1 ng of SOD1 protein (the lower detection limit of Western analysis) would still represent 0.33% of the total protein loaded (300 ng), >30 times greater than the lower limit required for an effective defense against sudden oxidative stress. If the stress persists, SOD1 expression is rapidly increased and the nascent protein is activated to provide adequate protection.
Implication for Mammalian Physiology. The effect of oxygen on SOD1 activity has implications for a variety of physiological conditions and may be relevant when rapid transition from anoxic or hypoxic states to normal oxygenation occurs. In many conditions, including ischemia-reperfusion injury, stroke, myocardial infarction, and exercise, tissues experience an oxidative burst that must be quickly cleared to minimize damage. In studies of exercise physiology, posttranslational processes have been proposed to account for the increased activity of antioxidant enzymes, including SOD1, observed in animals during exercise training (8, 10). Situations involving overexpression of SOD1, such as in Down syndrome patients or cell culture lines, suggest that a regulated balance between radicals and antioxidant enzymes is essential: overexpression of SOD1 corresponded to increases in lipid peroxidation in these studies (31). A posttranslational mechanism of SOD1 activation would not only allow for an immediate response to oxidative stress, but would limit the commitment of cellular resources should the oxidative stress prove to be short-lived. The O2-dependent regulation of SOD1 activity described here expands the complexity of the biochemical pathways that regulate this antioxidant response. These results will also have implications for the study and treatment of one form of familial amyotrophic lateral sclerosis (fALS), a neurodegenerative disease that effects the motor neurons of the brain and spinal cord (32). Changes in O2 tension may alter the distribution of SOD1 between immature, partially modified and fully active pools of the protein, some of which may be more prone to aggregation. In this manner, changes in oxygen levels that effect the SOD1 pool may ultimately influence the pathological mechanisms related to fALS.
Acknowledgments
SOD2 protein and yeast strains were a kind gift from V. Culotta. We thank B. M. Hoffman for EPR facilities and expertise, and V. Culotta, Y. Furukawa, and the O'Halloran group for helpful discussions. This work was supported by National Institutes of Health Grants GM 54111 (to T.V.O.), HL13531 (to B. M. Hoffman), and DK60305-02 (to N.M.B.), the Illinois Minority Graduate Incentive Program (A.S.T.), and the ALS Association (T.V.O.).
Abbreviations: ARD, apo, reduced, and denatured; CCS, copper chaperone for SOD1; hCCS, human CCS; yCCS, yeast CCS; NBT, p-nitroblue tetrazolium chloride; 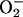 , superoxide radical; SOD1, Cu,Zn superoxide dismutase; hSOD1, human SOD1; ySOD1, yeast SOD1.
, superoxide radical; SOD1, Cu,Zn superoxide dismutase; hSOD1, human SOD1; ySOD1, yeast SOD1.
References
- 1.McCord, J. M. & Fridovich, I. (1969) J. Biol. Chem. 244, 6049–6055. [PubMed] [Google Scholar]
- 2.Weisiger, R. A. & Fridovich, I. (1973) J. Biol. Chem. 248, 4793–4796. [PubMed] [Google Scholar]
- 3.Weisiger, R. A. & Fridovich, I. (1973) J. Biol. Chem. 248, 3582–3592. [PubMed] [Google Scholar]
- 4.Sturtz, L. A., Diekert, K., Jensen, L. T., Lill, R. & Culotta, V. C. (2001) J. Biol. Chem. 276, 38084–38089. [DOI] [PubMed] [Google Scholar]
- 5.Gregory, E. M., Goscin, S. A. & Fridovich, I. (1974) J. Bacteriol. 117, 456–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galiazzo, F. & Labbe-Bois, R. (1993) FEBS Lett. 315, 197–200. [DOI] [PubMed] [Google Scholar]
- 7.Galiazzo, F., Ciriolo, M. R., Carri, M. T., Civitareale, P., Marcocci, L., Marmocchi, F. & Rotilio, G. (1991) Eur. J. Biochem. 196, 545–549. [DOI] [PubMed] [Google Scholar]
- 8.Oh-Ishi, S., Kizaki, T., Ookawara, T., Sakurai, T., Izawa, T., Nagata, N. & Ohno, H. (1997) Am. J. Respir. Crit. Care Med. 156, 1579–1585. [DOI] [PubMed] [Google Scholar]
- 9.Bejma, J. & Ji, L. L. (1999) J. Appl. Phys. 87, 465–470. [DOI] [PubMed] [Google Scholar]
- 10.Ji, L. L. (2002) Ann. N.Y. Acad. Sci. 959, 82–92. [DOI] [PubMed] [Google Scholar]
- 11.Gralla, E. B. (1997) in Oxidative Stress and the Molecular Biology of Antioxidant Defenses, ed. Scandalios, J. G. (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 495–525.
- 12.Valentine, J. S. & Pantoliano, M. W. (1981) in Copper Proteins, ed. Spiro, T. G. (Wiley, New York), Vol. 3, pp. 292–358. [Google Scholar]
- 13.Bertini, I., Mangani, S. & Viezzoli, M. S. (1998) Adv. Inorg. Chem. 45, 127–250. [Google Scholar]
- 14.Corson, L. B., Strain, J. J., Culotta, V. C. & Cleveland, D. W. (1998) Proc. Natl. Acad. Sci. USA 95, 6361–6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Culotta, V. C., Klomp, L. W. J., Strain, J., Casareno, R. L. B., Krems, B. & Gitlin, J. D. (1997) J. Biol. Chem. 272, 23469–23472. [DOI] [PubMed] [Google Scholar]
- 16.Wong, P. C., Waggoner, D., Subramaniam, J. R., Tessarollo, L., Bartnikas, T. B., Culotta, V. C., Price, D. L., Rothstein, J. & Gitlin, J. D. (2000) Proc. Natl. Acad. Sci. USA 97, 2886–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rae, T. D., Schmidt, P. J., Pufahl, R. A., Culotta, V. C. & O'Halloran, T. V. (1999) Science 284, 805–808. [DOI] [PubMed] [Google Scholar]
- 18.Eisses, J. F., Stasser, J. P., Ralle, M., Kaplan, J. H. & Blackburn, N. J. (2000) Biochemistry 39, 7337–7342. [DOI] [PubMed] [Google Scholar]
- 19.Zhu, H., Shipp, E., Sanchez, R. J., Liba, A., Stine, J. E., Hart, P. J., Gralla, E. B., Nersissian, A. M. & Valentine, J. S. (2000) Biochemistry 39, 5413–5421. [DOI] [PubMed] [Google Scholar]
- 20.Schmidt, P. J., Rae, T. D., Pufahl, R. A., Hamma, T., Strain, J., O'Halloran, T. V. & Culotta, V. C. (1999) J. Biol. Chem. 274, 23719–23725. [DOI] [PubMed] [Google Scholar]
- 21.Rae, T. D., Torres, A. S., Pufahl, R. A. & O'Halloran, T. V. (2001) J. Biol. Chem. 276, 5166–5176. [DOI] [PubMed] [Google Scholar]
- 22.Lamb, A. L., Torres, A. S., O'Halloran, T. V. & Rosenzweig, A. C. (2001) Nat. Struct. Biol. 8, 751–755. [DOI] [PubMed] [Google Scholar]
- 23.Torres, A. S., Petri, V., Rae, T. D. & O'Halloran, T. V. (2001) J. Biol. Chem. 276, 38410–38416. [DOI] [PubMed] [Google Scholar]
- 24.Beauchamp, C. & Fridovich, I. (1971) Anal. Biochem. 44, 276–287. [DOI] [PubMed] [Google Scholar]
- 25.Horecka, J., Kinsey, P. T. & Sprague, G. F. (1995) Gene 162, 87–92. [DOI] [PubMed] [Google Scholar]
- 26.Brown, N. M., Anderson, S. A., Steffen, D. W., Carpenter, T. B., Kennedy, M. C., Walden, W. E. & Eisenstein, R. S. (1998) Proc. Natl. Acad. Sci. USA 95, 15235–15240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Groves, W. E., Davis, F. C., Jr., & Sells, B. H. (1968) Anal. Biochem. 22, 195–210. [DOI] [PubMed] [Google Scholar]
- 28.Schmidt, P. J., Kunst, C. & Culotta, V. C. (2000) J. Biol. Chem. 275, 33771–33776. [DOI] [PubMed] [Google Scholar]
- 29.Bordo, D., Djinovic, K. & Bolognesi, M. (1994) J. Mol. Biol. 238, 366–386. [DOI] [PubMed] [Google Scholar]
- 30.Battistoni, A., Mazzetti, A. P. & Rotilio, G. (1999) FEBS Lett. 443, 313–316. [DOI] [PubMed] [Google Scholar]
- 31.McCord, J. M. (2002) Methods Enzymol. 349, 331–341. [DOI] [PubMed] [Google Scholar]
- 32.Cleveland, D. W. & Rothstein, J. D. (2001) Nat. Rev. Neurosci. 2, 806–819. [DOI] [PubMed] [Google Scholar]