

Abstract
Caloric restriction (CR), the consumption of fewer calories while avoiding malnutrition, decelerates the rate of aging and the development of age-related diseases. CR has been viewed as less effective in older animals and as acting incrementally to slow or prevent age-related changes in gene expression. Here we demonstrate that CR initiated in 19-month-old mice begins within 2 months to increase the mean time to death by 42% and increase mean and maximum lifespans by 4.7 (P = 0.000017) and 6.0 months (P = 0.000056), respectively. The rate of age-associated mortality was decreased 3.1-fold. Between the first and second breakpoints in the CR survival curve (between 21 and 31 months of age), tumors as a cause of death decreased from 80% to 67% (P = 0.012). Genome-wide microarray analysis of hepatic RNA from old control mice switched to CR for 2, 4, and 8 weeks showed a rapid and progressive shift toward the gene expression profile produced by long-term CR. This shift took place in the time frame required to induce the health and longevity effects of CR. Shifting from long-term CR to a control diet, which returns animals to the control rate of aging, reversed 90% of the gene expression effects of long-term CR within 8 weeks. These results suggest a cause-and-effect relationship between the rate of aging and the CR-associated gene expression biomarkers. Therefore, therapeutics mimicking the gene-expression biomarkers of CR may reproduce its physiological effects.
Caloric restriction (CR), the consumption of fewer calories while avoiding malnutrition, is a robust method of decelerating aging and the development of age-related diseases (1). The effects of CR are conserved in nearly every species tested, perhaps including humans (2). CR delays the onset and reduces the incidence and severity of age-related diseases, including cancer (2, 3).
Quantitative changes in the activity of genes can control the rate of aging and the development of age-related diseases in invertebrates and mammals (4, 5). Numerous cross-sectional studies of the relationship between CR and gene expression have been published (6). Almost without exception, they have been interpreted as though they were performed longitudinally. This has led to the widespread view that the major effect of CR is to prevent age-related changes in gene expression. Funding and publication bias has reinforced this notion (6).
We previously found that in old mice, a 4-week shift from long-term control (LT-CON) to short-term CR (ST-CR) reproduced 55% of the gene-expression changes induced by long-term CR (LT-CR; ref. 7). ST-CR reversed ≈70% of the age-related changes in gene expression seemingly prevented by LT-CR (7). These results suggested for the first time that many of the gene expression changes produced by LT-CR respond rapidly to diet.
The studies described above left several important questions unanswered. First, which of the changes in gene expression are related to the health and longevity effects of CR? Further, what are the kinetics of the changes in gene expression induced by CR? Here we report the use of lifespan and genome-wide microarray studies to answer these questions. Survival studies demonstrated that CR begins to act within 2 months in older mice to reduce cancer incidence and increase time to death. These results are in contrast to the widely held belief that CR is less effective in older mice (8). Further, during this 2-month interval, short-term CR reproduced 72% of the genomic effects of LT-CR. The results closely link the lifespan and genomic effects of CR, suggesting a cause-and-effect relationship.
Materials and Methods
Study Design. Male mice of the long-lived F1 hybrid strain B6C3F1 were purchased (Harlan Breeders, Indianapolis). On arrival, mice were housed four per cage and fed ad libitum a nonpurified diet (no. 5001, Purina). For the survival study, 19-month-old mice were randomly assigned to control and CR groups of 60 each. Control mice were fed 93 kcal per week of chemically defined control diet (AIN-93M, Diet No. F05312, Bioserv, Frenchtown, NJ). This provides ≈10% fewer calories than normally are assumed to be required by a typical mouse (9). This allows comparison of nonobese control with CR mice. CR was introduced by reducing calories to 77 kcal per week of chemically defined CR diet for 2 weeks, followed by 52.2 kcal per week of CR diet thereafter (AIN-93M 40% Restricted, Diet No. F05314, Bioserv). Defined diets were cold packed into 1-g pellets. Mice were checked daily and weighed monthly. Necropsies were performed to determine the cause of death and incidence of tumors. For gene expression studies, the mice were ≈34 months of age when killed. Beginning at 7 months of age, the mice were randomly assigned to LT-CON or LT-CR groups (Fig. 3, which is published as supporting information on the PNAS web site). LT-CON mice were fed 93 kcal per week of control diet. LT-CR mice were reduced to 52.2 kcal per week of CR diet as described above. The CR2 group were LT-CON mice fed 77 kcal of CR diet for 1 week and 52.2 kcal for another week. The CR4 and CR8 groups were LT-CON mice switched to 77 kcal of CR diet per week for 2 weeks and 52.2 kcal per week for 2 and 6 weeks, respectively. The CON8 group was LT-CR mice fed 93 kcal of control diet per week for 8 weeks. For the LT-CON, CON8, and CR8 groups, n = 4. For the LT-CR, CR2, and CR4 groups, n = 3. Mice were fasted 24 h, killed, and their livers excised as described (7). No signs of pathology were detected in any of the animals used. At 9:00 a.m., mice were fed two-sevenths of the weekly allotment of food on Monday and Wednesday and three-sevenths on Friday. Mice were given acidified water ad libitum and maintained at 20–24°C and 50–60% humidity with lights on from 6:00 a.m. to 6:00 p.m. All animal use protocols were approved by the Institutional Animal Use Committee of the University of California, Riverside.
Statistical Analysis of Survival Data. Parametric survival analyses were performed assuming the data followed a normal survival distribution. The observed data were used to estimate the survival function. A change-point regression analysis was performed to find the breakpoints. A failure-time distribution analysis was used to calculate mean and maximum (average age of the last decile of survivors) lifespans. Mortality, tumors, and percent of liver tumors were analyzed by using a two-sample Student t test.
Measurement of Specific mRNA Levels. Liver total RNA was isolated and gene expression quantified by using Affymetrix Mu11K sets A and B oligonucleotide arrays according to standard Affymetrix protocols (Affymetrix, Santa Clara, CA; ref. 7).
Affymetrix Data Reduction and Analysis. Analyses of the array images and data quantification were performed with Affymetrix microarray suite 5.0 by using default parameters (10). Only transcripts “Present” with a signal intensity above the median in at least 75% of the arrays per experimental group were considered for further analysis. Differentially expressed genes were identified using pair-wise comparison between the LT-CON and the other groups. Genes were further considered if 75% of the change calls were the same. An average change of 1.5-fold or greater was considered significant. These criteria reliably identified changed genes that were verified by quantitative PCR (qPCR). Gene names were from the LocusLink and UniGene databases as of April 20, 2003.
Microarray Data Validation by qPCR. The expression of nine genes was examined by qPCR (11). Real-time two-step RT-PCR was performed with a QuantiTect SYBR Green PCR kit (Qiagen, Hilden, Germany) and an ABI PRISM 7700 Sequence Detection System (Applied Biosystems). Primers were designed by using the Netaffx analysis center (Affymetrix) and PCR products sequenced and verified against the public databases (Table 4, which is published as supporting information on the PNAS web site). Primers for transcription elongation factor A (SII) 1 were amplified in parallel with the gene of interest as a control. This mRNA is unaffected by CR (12). Amplification specificity was confirmed by melting curve analysis and agarose gel electrophoresis.
Results
Rapid Effects of CR on Survival. We investigated the hypothesis that the health and lifespan effects of CR are induced rapidly after the initiation of the diet. Control mice were shifted to 40% CR at 19 months of age, a few months before the onset of age-accelerated mortality (Fig. 1). Linear regression and breakpoint analysis showed that between 21 and 31 months of age, newly initiated CR decreased the rate of age-associated mortality 3.1-fold (P < 0.001). Beginning at the first breakpoint, ≈2 months after initiation of the CR diet, the mean time to death increased from 11.8 ± 0.7 (SE) to 16.8 ± 1.2 months (SE; P = 0.004), a 42% increase. LT-CR initiated after weaning extends the lifespan of this mouse genotype by ≈40% (13). After the second breakpoint in the CR survival curve (31 months of age), the mortality rate approximated that of the control mice, but the survival curve was shifted to the right by 4.7 months. CR extended mean lifespan from 30.7 ± 0.7 (SE) to 35.4 ± 0.8 months (P = 0.000017) and extended maximum lifespan from 37.6 to 43.6 months (P = 0.000056). Because CR was initiated ≈2 months before the age-associated mortality breakpoint in the survival curve, 2 months is the minimum time that could have been detected for development of the health and longevity effects of CR.
Fig. 1.

Longevity of mice subjected to CR from 19 months of age. The CR group is represented by the open circles, and the control group is represented by filled circles. The percentage of mice remaining alive at the end of each month is plotted.
Rapid Effects of CR on Carcinogenesis. Both CR and control mice died primarily of tumors, mainly adenomas or carcinomas of the liver or lung. From 21 to 31 months of age, between the first and second breakpoints, 33 control and 10 CR mice died (P = 0.003). Eighty percent of the control and 67% of the CR that died during this time had large tumors (P = 0.012). Sixty-five percent of the control mice and 44% of the CR mice had liver tumors (P = 0.006). However, the total number of mice that died with tumors by the end of the study was approximately the same for the control and CR groups (89.3% and 90.6%, respectively). Thus, CR initiated late in life had no effect on overall tumor incidence, but it delayed the onset and/or growth of tumors and extended average and maximum lifespan.
As shown above, the liver is a major target of the anticancer effects of CR (14, 15). Breakpoint analysis indicated that the beneficial health and lifespan effects of CR developed within 2 months of initiating the diet (Fig. 1). For these reasons, we focused genome-wide microarray studies on liver gene expression during this interval. Affymetrix microarrays containing probe sets for 12,422 mouse transcription units were used to interrogate hepatic RNA purified from old mice shifted from LT-CON to CR for 2, 4, and 8 weeks (CR2, CR4, and CR8, respectively) and from LT-CR to control feeding for 8 weeks (CON8; Figs. 2 and 3).
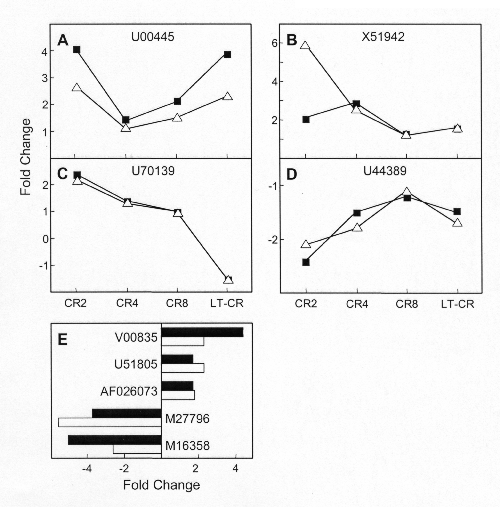
Fig. 2.

Dynamics of the gene expression response in CR2, CR4, CR8, LT-CR, and CON8 mice. (A) Stable genes. (B) Oscillators.
Rapid Genomic Effects of CR. Reliable signals for 2,194 transcripts were obtained after data reduction. LT-CR altered the expression of 123 of these transcripts (6% of the reliably reporting probe sets; Tables 1, 2, 3). The kinetics with which the LT-CR gene expression profile developed are shown in Fig. 2. The CR responsive genes were classified into three categories based on their kinetic behavior. The early genes changed expression after 4 weeks of CR (71 genes, 58% of the LT-CR-responsive genes). Most of the early genes responded within 2 weeks (61 genes; 50% of the LT-CR responsive genes). A group of 17 intermediate genes (14%) changed expression after 4–8 weeks of CR (Fig. 2A). A group of late genes required >8 weeks of CR to change expression (35 genes, 28% of the LT-CR responsive genes).
Table 1. Late genes.
| Category/GenBank | Gene | LT-CR |
|---|---|---|
| Extracellular matrix/cytoskeleton | ||
| AA139495 | Cltc | -1.5 |
| Metabolism | ||
| W81884 | Acat2 (Provisional) | -1.5 |
| M74570 | Aldh1a1 | -1.8 |
| V00719 | Amy1 | 1.5 |
| AA114811 | Atp5c1 | 1.5 |
| L07645 | Hal | 1.8 |
| U58988 | Hgd | 1.5 |
| X95685 | Hsd17b2 | -1.5 |
| W36875 | Siat9 | 1.5 |
| U24493 | Tdo2 | 1.5 |
| Signal transducers, growth factors | ||
| C78610 | Anxa7 | -1.5 |
| AA059550 | Enpp2 | -1.6 |
| M69293 | Idb2 | -1.5 |
| U04268 | Ly6e | -1.7 |
| M16355 | Mup1 | -2.6 |
| Immune response, inflammation | ||
| M17122 | C4bp | -1.8 |
| Stress response, xenobiotic metabolism | ||
| X00479 | Cyp1a2 | -2.2 |
| M77497 | Cyp2f2 | -1.8 |
| D30687 | Gstp2 | -1.7 |
| U96116 | Hadh2 | 2.0 |
| M28723 | Prdx3 | 1.5 |
| X70303 | Psma2 | -1.5 |
| AA008321 | Psma4 | -1.6 |
| Miscellaneous | ||
| U89889 | Hpxn | 1.5 |
| D84391 | L1Md-Tf14 | 1.6 |
| X74504 | T10 | 1.5 |
| AA048018 | EST | -1.6 |
| AA096813 | EST | 2.2 |
| AA213083 | EST | -1.7 |
| AA238331 | EST | -1.9 |
| AA690887 | EST | -1.6 |
| C76068 | EST | 1.6 |
| C77421 | EST | 1.6 |
| C77864 | EST | -1.8 |
| W34969 | EST | -1.5 |
Gene information is listed in this order: GenBank number, gene symbol, fold change.
Table 2. Stables.
| Category/GenBank | Gene | CR2 | CR4 | CR8 | LT-CR | CON8 |
|---|---|---|---|---|---|---|
| Metabolism | ||||||
| AA710204 | Acat1 | -2.0 | -2.5 | -1.5 | -1.5 | NC |
| AA521794 | Cox7b | 4.3 | 3.4 | 1.5 | 1.6 | NC |
| L32836 | Ahcy | NC | NC | 1.5 | 1.9 | NC |
| AA120586 | Apo B-100 | 2.9 | 3.5 | 1.5 | 1.7 | 1.8 |
| U51805 | Arg1 | 3.0 | 2.3 | 1.5 | 2.3 | NC |
| AA237297 | As1 | 2.3 | 1.5 | 1.7 | 2.6 | NC |
| M31690 | Ass1 | NC | NC | 2.1 | 2.5 | NC |
| M27796 | Car3 | -8.2 | -7.5 | -1.8 | -5.5 | NC |
| U31966 | Cbr1 | NC | NC | 1.7 | 1.7 | NC |
| AA521793 | Decr1 | -2.4 | -1.9 | -1.6 | -1.5 | NC |
| AA591003 | Fabp1 | -5.6 | -5.3 | -1.7 | -1.9 | NC |
| U15977 | Facl2 | NC | -3.4 | -2.0 | -1.7 | NC |
| W14826 | Gnmt | NC | NC | 1.8 | 2.0 | NC |
| AA117646 | Slc10a1 | 1.5 | 1.5 | 1.8 | 1.6 | NC |
| AF026073 | Sultn | 5.0 | 3.4 | 1.6 | 1.8 | NC |
| M55154 | Tgm2 | NC | -2.4 | -1.5 | -1.5 | NC |
| Signal transducers, growth factors | ||||||
| Z49085 | Ephb4 | NC | NC | -1.5 | -1.5 | NC |
| AA529583 | Morf4l2 | NC | NC | -1.6 | -1.7 | NC |
| M16358 | Mup4 | -3.8 | -2.5 | -1.6 | -2.7 | NC |
| M16360 | Mup5 | -2.4 | -1.5 | -1.7 | -2.2 | NC |
| W13191 | Thra1 | -3.6 | -2.1 | -1.5 | -2.1 | -1.6 |
| AA106783 | Pabpc1 | 3.8 | 2.4 | 1.6 | 1.7 | NC |
| U92437 | Pten | 2.5 | 1.9 | 1.5 | 1.6 | NC |
| L29441 | Tde1 | 2.4 | 1.8 | 1.5 | 1.8 | NC |
| D89076 | Ttr | -2.3 | -2.2 | -1.5 | -1.8 | NC |
| AF027963 | Xbp1 | NC | NC | -1.5 | -1.7 | NC |
| Immune response, inflammation | ||||||
| X05475 | C9 | NC | NC | -1.5 | -2.0 | NC |
| M27008 | Orm1 | 3.3 | 1.5 | 2.0 | 2.0 | 1.5 |
| U51167 | Idh2 | NC | NC | 1.5 | 1.5 | NC |
| AA415898 | Iigp-pending | NC | NC | -1.5 | -1.9 | NC |
| Stress response | ||||||
| X56603 | Calr | NC | NC | -1.7 | -1.7 | NC |
| AA163552 | Canx | NC | NC | -1.6 | -1.7 | NC |
| AA204094 | Dnajb11 | NC | NC | -1.5 | -1.5 | NC |
| M73329 | Grp58 | NC | -3.0 | -1.7 | -1.7 | NC |
| D78645 | Hspa5 | NC | -2.5 | -1.9 | -2.0 | -1.5 |
| W55140 | Grp94 | -3.1 | -2.6 | -1.7 | -1.5 | NC |
| Xenobiotic metabolism | ||||||
| M60358 | Cyp2b13 | 1.6 | 1.8 | 1.8 | 2.0 | NC |
| J04696 | Gstm2 | 1.6 | 1.8 | 2.4 | 1.9 | NC |
| M21855 | Cyp2b9 | NC | 1.6 | 2.5 | 2.3 | NC |
| V00835 | Mt1 | 4.3 | 2.9 | 1.6 | 2.3 | NC |
| D17571 | Por | 2.8 | 1.7 | 1.5 | 2.1 | 1.6 |
| Miscellaneous | ||||||
| AA408789 | Armet | -3.0 | -1.8 | -1.9 | -2.2 | -1.7 |
| AA120109 | EST | NC | -2.8 | -1.6 | -2.2 | NC |
| AA120387 | EST | -2.1 | -1.5 | -1.6 | -1.7 | NC |
| AA217076 | EST | -4.4 | -3.2 | -1.7 | -1.5 | NC |
| AA537958 | EST | -1.9 | -1.9 | -1.5 | -1.6 | NC |
| AA711625 | EST | -2.1 | -1.8 | -1.6 | -2.2 | NC |
Gene information is listed in this order: GenBank number, gene symbol, fold change. NC, not changed.
Table 3. Oscillators.
| Category/GenBank | Gene | CR2 | CR4 | CR8 | LT-CR | CON8 |
|---|---|---|---|---|---|---|
| Metabolism | ||||||
| D45850 | Akr1c6 | -1.6 | -1.7 | NC | -1.8 | NC |
| X13752 | Alad | -2.7 | -2.3 | NC | -1.6 | NC |
| X06086 | Cts1 | 3.0 | 1.6 | NC | 2.3 | NC |
| U00445 | G6pc | 2.7 | NC | 1.5 | 2.3 | 1.8 |
| U44389 | Hpgd | -2.1 | -1.8 | NC | -1.7 | NC |
| X58426 | Lipc | -2.8 | -2.4 | NC | -1.5 | NC |
| X64837 | Oat | 1.6 | NC | 1.5 | 2.0 | 1.5 |
| X51942 | Pah | 6.0 | 2.5 | NC | 1.6 | NC |
| AA110781 | Pck1 | 1.6 | -2.3 | NC | 1.5 | NC |
| M21285 | Scd1 | 2.7 | NC | 6.5 | 7.3 | 4.0 |
| M88694 | Temt | -2.6 | -2.8 | NC | -1.6 | NC |
| Signal transducers, growth factors | ||||||
| AA240968 | Arhu | -1.9 | -2.4 | NC | -1.6 | NC |
| M81445 | Gjb2 | -2.6 | -2.9 | NC | -1.6 | NC |
| X70533 | Serpina6 | -2.9 | -6.4 | NC | -2.2 | NC |
| Immune response, inflammation | ||||||
| U70139 | Ccrn4l | 2.2 | NC | NC | -1.5 | NC |
| U36220 | Fkbp5 | 2.3 | 1.6 | NC | 1.5 | NC |
| U09010 | Mbll | -2.3 | -1.8 | NC | -1.7 | NC |
| Stress response, xenobiotic metabolism | ||||||
| AA592828 | Akr1c13 | NC | -1.8 | NC | -1.5 | NC |
| U62294 | Cyp2j5 | -1.5 | -1.8 | NC | -1.7 | NC |
| X60452 | Cyp3a11 | NC | -3.2 | NC | 1.9 | 1.5 |
| X63023 | Cyp3a13 | 4.1 | NC | 1.6 | 2.1 | NC |
| D26137 | Cyp3a16 | NC | -1.9 | NC | 1.7 | NC |
| U36993 | Cyp7b1 | -1.5 | -1.9 | NC | -2.3 | NC |
| L11333 | Es31 | -2.4 | -2.2 | NC | -2.2 | NC |
| U90535 | Fmo5 | 2.1 | NC | 1.5 | 1.5 | NC |
| AA254963 | Herpud1 | NC | -1.8 | NC | -1.6 | NC |
| U38652 | Slc22a1 | -2.6 | -2.2 | NC | -1.6 | NC |
| Miscellaneous | ||||||
| W12913 | Hamp | -1.5 | -1.7 | NC | -2.4 | NC |
| AA097626 | EST | 5.8 | 2.0 | NC | 1.7 | NC |
Gene information is listed in this order: GenBank number, gene symbol, fold change. NC, not changed.
Most early genes, termed stables, sustained their initial CR-induced expression levels at all subsequent time points (89 genes; 72% of the total LT-CR-responsive genes; Fig. 2A; Table 2). But others, termed oscillators, returned to control levels transiently before reassuming their LT-CR expression levels (34 genes; 28% of the LT-CR-responsive genes; Fig. 2B and Table 3). It seemed possible that the kinetic behavior of the oscillators arose because some genes did not pass the criteria used for data reduction at one or more time points, even though their gene expression levels were actually altered relative to control. To investigate this possibility, the expression data and data reduction were examined for each oscillator at every time point. In no case did the return to control expression levels appear equivocal. In addition, the expression of four oscillators was examined by using qPCR at the CR2, CR4, CR8, and LT-CR timepoints. In every case, the relative gene expression levels and the kinetic behavior were replicated (Fig. 4 A–D, which is published as supporting information on the PNAS web site). These data indicate that the gene expression patterns found represent the true responses of the genes to CR.
To further ensure that data reduction had produced a low false discovery rate, the expression level of five additional randomly chosen genes was determined by using qPCR. Expression measurements using the same RNA from control and LT-CR mice produced equivalent results with both measurement techniques (Fig. 4E). Therefore, the microarray quantification of nine tested genes was verified as to direction and fold change using qPCR for 21 independent measurements of gene expression. These results suggest that data reduction was reliable and resulted in a false discovery rate of <5%.
It is widely held that the effects of CR dissipate after a return to control caloric intake (16). However, we know of no studies of the transition from the CR to the control state. In the CON8 mice, 110 of the 123 LT-CR responsive transcripts returned to control expression levels in 8 weeks (90%; Fig. 2, CON8). All of the late genes shifted back to control expression in 8 weeks, indicating that they respond readily to caloric intake. These results demonstrate that the expression of all of the LT-CR responsive genes was readily responsive to shifts in dietary state.
Discussion
Here we investigated the widespread belief that a major mechanism of action of CR is to resist incremental age-related changes in gene expression that are deleterious to health and lifespan. Survival and high-density microarray studies revealed that CR acts rapidly after it is initiated to establish a pattern of hepatic gene expression associated in long-term CR mice with enhanced lifespan and reduced tumor incidence. The close temporal linkage between the initiation of the beneficial physiological effects of CR and its genomic effects suggests a cause-and-effect relationship between them.
CR Initiated in Older Animals. The effects of CR on lifespan appeared to be biphasic. Between the first and second breakpoints (from 21 to 31 months of age), CR decelerated the mortality rate and the incidence of cancer as a cause of death. Most notably, breakpoint analysis indicated that this deceleration began after only 2 months of CR. After the second breakpoint in the CR survival curve (after 31 months of age), the mortality rate and the incidence of cancer as a cause of death approximated that of control mice, although mean and maximum lifespans were significantly increased. These results suggest that the major effect of CR initiated later in life is to slow the rate of aging and delay the onset and/or progression of cancer. The kinetics of the survival curves are most consistent with a reduction in the rate of tumor growth. A similar effect of CR on lifespan was recently reported in Drosophila, where mortality rate was rapidly reduced by CR introduced at any age (17).
Our results are in contrast to those of Forster et al. (8), who found that late-life CR had no effect on mouse lifespan (8). However, the abrupt initiation of CR in their study and possible nutritional inadequacy in some CR groups, as indicated by the weights of the mice, may account for their results (1). Others did not observe lifespan extension in older rats (18). However, there was no positive control group in this study. Therefore, there was no evidence that the CR protocol and animal husbandry used were effective at extending lifespan.
Kinetic Behavior of the Genes. The CR-responsive genes were grouped into functional classes (Tables 1, 2, 3). Functional gene categories did not appear to be differentially represented among the early, intermediate, and late genes or among the kinetic categories of oscillators and stables. These results are consistent with the idea that both kinetic classes and every functional category of genes contribute to the longevity and health effects of CR.
Function of Genes Temporally Associated with Onset of the Antineoplastic and Lifespan Effects of CR. The first breakpoint in the survival curve occurs 2 months after initiation of CR. The genes that change expression in this time frame are the ones most closely associated with the initiation of the life-span and antineoplastic effects of CR. These early and intermediate genes were functionally associated with metabolism; signal transduction, growth factors, immune response, inflammation, stress response, and xenobiotic metabolism. These genes and their classes are consistent with those identified previously (7, 19–21).
The metabolic genes included three urea cycle enzymes, Arg1 and As1, which are early genes, and Ass1, an intermediate gene. In combination with our previous studies, these results suggest that the enzymatic capacity for the disposal of nitrogen derived from increased protein degradation for energy generation in the liver and extrahepatic tissues is among the earliest responses to CR (7, 19, 20, 22). Consistent with this idea, Gnmt and Ahcy, which catalyze key steps in methionine degradation, were intermediate stables. The key gating enzymes of gluconeogenesis, Pck1 and G6pc, were early oscillators. In addition, several late genes are involved in amino acid degradation to provide substrates for gluconeogenesis (Ctsl, Pah, Oat, Hpgd, and Hal). Together, the results are consistent with other data suggesting that CR induces a rapid increase in the enzymatic capacity for mobilization of hepatic and extrahepatic protein for energy generation and the transfer of carbon and nitrogen to the liver for gluconeogenesis and disposal, respectively (19, 20, 22–25).
Lipid metabolism was also rapidly altered by CR. Key genes required for lipid biosynthesis and metabolism were down-regulated early after the initiation of CR (Acat1, Facl2, Fabp1, Lipc, and Scd1). CR increased the expression of the early stable Apo B-100, a major component of low-density lipoprotein and very low-density lipoproteins. This change is consistent with its role in the distribution of hepatic lipid to other tissues for use as fuel. Together, these metabolic effects are consistent with the decreased serum triglycerides in CR rodents (26).
CR rapidly altered the expression of genes associated with cellular growth and proliferation. Ttr, the major thyroid hormone carrier protein in rodents, and Thra1, a widely distributed T3 receptor important for maintenance of heart rate and body temperature, were early changers (27). In other studies, we found decreased expression of iodothyronine deiodinase type I in CR4 and LT-CR mice (7). CR reduces serum thyroid hormone levels (28). Reduced thyroid hormone signaling is also a feature of dwarfism in mice, which extends lifespan and reduces the onset of age-related diseases (5, 29, 30).
The expression of two genes associated with angiogenesis, Ephb4, an intermediate stable, and Enpp2, a late gene, was reduced by CR (31). Moreover, CR induced the expression of the early stable Pten, a tumor suppressor gene (32). These changes suggest that CR may enhance antiproliferative growth control soon after its initiation and increase the extent of this control with time.
The down-regulation of endoplasmic chaperone expression was another effect of CR (Table 2). A number of major molecular chaperones of the endoplasmic reticulum were early and intermediate responsive stables (Hspa5, Grp58, DnaJB11, Calr, Canx, and Grp94). This result is in agreement with our previous results showing that the mRNA and protein levels of most hepatic endoplasmic-reticulum chaperones increase with age and decrease with CR (7, 33–35). The linkage between caloric intake and chaperone expression may match the protein processing capacity of liver cells to the level of protein biosynthetic activity (35). However, elevated chaperones also decrease apoptotic responsiveness to genotoxic stress through both the endoplasmic stress and the mitochondrial apoptosis signaling pathways (36, 37). CR and fasting reduce endoplasmic reticulum chaperone levels and enhance apoptosis in liver and other cycling cell types, perhaps accounting for their anticancer benefits (21, 35, 38, 39). Importantly, CR appears to induce chaperone expression in nondividing cell types such as neurons, apparently enhancing their survival after stress (40).
CR rapidly enhanced the expression of Cyp2b9 and Cyp2b13, cytochromes involved in oxidation of fatty acids, xenobiotics, and steroids, including testosterone. Testosterone negatively regulates their expression (41). The decreased testosterone levels in CR mice may account for their induction, which may further decrease the level of the active steroid. CR induced Gstm2 mRNA, which codes for one of two subunits of an enzyme that detoxifies compounds such as the breakdown products of lipid peroxidation, DNA hydroperoxides, electrophilic xenobiotics, and reactive intermediates formed during their biotransformation (42).
Conclusion
We have presented three previously undescribed results. First, CR begun relatively late in the lifespan of mice was as effective as CR begun early in life at decelerating mortality rate, extending remaining lifespan, and delaying the onset and/or progression of cancer as a cause of death. Second, CR appeared to act rapidly to induce these benefits. The deceleration of aging and reduction in cancer mortality began after 2 months of CR. These results suggest that CR may be more proactive in its effects than previously thought. Finally, the initiation of the health and longevity effects of CR coincided with the induction of 72% of the LT-CR-related genomic profile. Ninety percent of the effects of LT-CR were rapidly reversed by control feeding. Thus, the rapid genomic effects of CR may be causally linked to its beneficial physiological effects. These results increase the likelihood that therapies mimicking the rapid gene expression biomarkers of CR will be effective at inducing its beneficial physiological effects.
Supplementary Material
Acknowledgments
We thank BioMarker Pharmaceuticals and the Life Extension Foundation for unrestricted gifts in support of this research.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: CR, caloric restriction; LT-CON, long-term control; LT-CR, long-term CR; qPCR, quantitative PCR.
References
- 1.Weindruch, R. & Walford, R. L. (1988) The Retardation of Aging and Disease by Dietary Restriction (Thomas, Springfield, IL).
- 2.Spindler, S. R. (2003) in Anti-Aging Therapy for Plastic Surgery, eds. Kinney, B. & Carraway, J. (Quality Medical, St. Louis), in press.
- 3.Weindruch, R., Walford, R. L., Fligiel, S. & Guthrie, D. (1986) J. Nutr. 116, 641–654. [DOI] [PubMed] [Google Scholar]
- 4.Guarente, L. & Kenyon, C. (2000) Nature 408, 255–262. [DOI] [PubMed] [Google Scholar]
- 5.Brown-Borg, H. M., Borg, K. E., Meliska, C. J. & Bartke, A. (1996) Nature 384, 33 (lett.). [DOI] [PubMed] [Google Scholar]
- 6.Van Remmen, H., Ward, W. F., Sabia, R. V. & Richardson, A. (1995) in Handbook of Physiology, ed. Masoro, E. J. (Oxford Univ. Press, New York), pp. 171–234.
- 7.Cao, S. X., Dhahbi, J. M., Mote, P. L. & Spindler, S. R. (2001) Proc. Natl. Acad. Sci. USA 98, 10630–10635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Forster, M. J., Morris, P. & Sohal, R. S. (2003) FASEB J. 17, 690–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Subcommittee on Laboratory Animal Nutrition and Committee on Animal Nutrition (1978) in Nutrient Requirements of Laboratory Animals: Rat, Mouse, Gerbil, Guinea Pig, Hamster, Vole, Fish (Natl. Acad. Press, Washington, DC), pp. 38–50.
- 10.Affymetrix (2001) Technical Notes 1, Part No. 701097 Rev. 1 (Affymetrix, Santa Clara, CA).
- 11.Rajeevan, M. S., Vernon, S. D., Taysavang, N. & Unger, E. R. (2001) J. Mol. Diagn. 3, 26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tillman, J. B., Mote, P. L., Dhahbi, J. M., Walford, R. L. & Spindler, S. R. (1996) J. Nutr. 126, 416–423. [DOI] [PubMed] [Google Scholar]
- 13.Cheney, K. E., Liu, R. K., Smith, G. S., Leung, R. E., Mickey, M. R. & Walford, R. L. (1980) Exp. Gerontol. 15, 237–258. [DOI] [PubMed] [Google Scholar]
- 14.Tatar, M., Kopelman, A., Epstein, D., Tu, M. P., Yin, C. M. & Garofalo, R. S. (2001) Science 292, 107–110. [DOI] [PubMed] [Google Scholar]
- 15.Coschigano, K. T., Clemmons, D., Bellush, L. L. & Kopchick, J. J. (2000) Endocrinology 141, 2608–2613. [DOI] [PubMed] [Google Scholar]
- 16.Merry, B. J. (2002) Int. J. Biochem. Cell Biol. 34, 1340–1354. [DOI] [PubMed] [Google Scholar]
- 17.Mair, W., Goymer, P., Pletcher, S. D. & Partridge, L. (2003) Science 301, 1731–1733. [DOI] [PubMed] [Google Scholar]
- 18.Lipman, R. D., Smith, D. E., Blumberg, J. B. & Bronson, R. T. (1998) Aging 10, 463–470. [DOI] [PubMed] [Google Scholar]
- 19.Tillman, J. B., Dhahbi, J. M., Mote, P. L., Walford, R. L. & Spindler, S. R. (1996) J. Biol. Chem. 271, 3500–3506. [DOI] [PubMed] [Google Scholar]
- 20.Dhahbi, J. M., Mote, P. L., Wingo, J., Rowley, B. C., Cao, S. X., Walford, R. & Spindler, S. R. (2001) Mech. Ageing Dev. 122, 35–50. [DOI] [PubMed] [Google Scholar]
- 21.Dhahbi, J. M., Cao, S. X., Mote, P. L., Rowley, B. C., Wingo, J. E. & Spindler, S. R. (2002) J. Nutr. 132, 31–37. [DOI] [PubMed] [Google Scholar]
- 22.Dhahbi, J. M., Mote, P. L., Wingo, J., Tillman, J. B., Walford, R. L. & Spindler, S. R. (1999) Am. J. Physiol. 277, E352–E360. [DOI] [PubMed] [Google Scholar]
- 23.Feuers, R. J., Duffy, P. H., Leakey, J. A., Turturro, A., Mittelstaedt, R. A. & Hart, R. W. (1989) Mech. Ageing Dev. 48, 179–189. [DOI] [PubMed] [Google Scholar]
- 24.Spindler, S. R. (2001) Ann. N.Y. Acad. Sci. 928, 296–304. [DOI] [PubMed] [Google Scholar]
- 25.Spindler, S. R. & Dhahbi, J. M. (2003) in Energy Metabolism and Lifespan Determination: Advances in Cell Aging and Gerontology, ed. Mattson, M. P. (Elsevier, Amsterdam), Vol. 14, pp. 69–86. [Google Scholar]
- 26.Stokkan, K. A., Reiter, R. J., Vaughan, M. K., Nonaka, K. O. & Lerchl, A. (1991) Acta Endocrinol. 125, 93–100. [DOI] [PubMed] [Google Scholar]
- 27.Wikstrom, L., Johansson, C., Salto, C., Barlow, C., Campos, B. A., Baas, F., Forrest, D., Thoren, P. & Vennstrom, B. (1998) EMBO J. 17, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herlihy, J. T., Stacy, C. & Bertrand, H. A. (1990) Mech. Ageing Dev. 53, 9–16. [DOI] [PubMed] [Google Scholar]
- 29.Flurkey, K., Papaconstantinou, J. & Harrison, D. E. (2002) Mech. Ageing Dev. 123, 121–130. [DOI] [PubMed] [Google Scholar]
- 30.Hauck, S. J., Hunter, W. S., Danilovich, N., Kopchick, J. J. & Bartke, A. (2001) Exp. Biol. Med. 226, 552–558. [DOI] [PubMed] [Google Scholar]
- 31.Yang, S. Y., Lee, J., Park, C. G., Kim, S., Hong, S., Chung, H. C., Min, S. K., Han, J. W., Lee, H. W. & Lee, H. Y. (2002) Clin. Exp. Metastasis 19, 603–608. [DOI] [PubMed] [Google Scholar]
- 32.Steck, P. A., Pershouse, M. A., Jasser, S. A., Yung, W. K., Lin, H., Ligon, A. H., Langford, L. A., Baumgard, M. L., Hattier, T., Davis, T., et al. (1997) Nat. Genet. 15, 356–362. [DOI] [PubMed] [Google Scholar]
- 33.Spindler, S. R., Crew, M. D., Mote, P. L., Grizzle, J. M. & Walford, R. L. (1990) J. Nutr. 120, 1412–1417. [DOI] [PubMed] [Google Scholar]
- 34.Dhahbi, J. M., Mote, P. L., Tillman, J. B., Walford, R. L. & Spindler, S. R. (1997) J. Nutr. 127, 1758–1764. [DOI] [PubMed] [Google Scholar]
- 35.Dhahbi, J. M., Cao, S. X., Tillman, J. B., Mote, P. L., Madore, M., Walford, R. L. & Spindler, S. R. (2001) Biochem. Biophys. Res. Commun. 284, 335–339. [DOI] [PubMed] [Google Scholar]
- 36.Suh, Y., Lee, K. A., Kim, W. H., Han, B. G., Vijg, J. & Park, S. C. (2002) Nat. Med. 8, 3–4. [DOI] [PubMed] [Google Scholar]
- 37.Rao, R. V., Peel, A., Logvinova, A., del Rio, G., Hermel, E., Yokota, T., Goldsmith, P. C., Ellerby, L. M., Ellerby, H. M. & Bredesen, D. E. (2002) FEBS Lett. 514, 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grasl-Kraupp, B., Bursch, W., Ruttkay-Nedecky, B., Wagner, A., Lauer, B. & Schulte-Hermann, R. (1994) Proc. Natl. Acad. Sci. USA 91, 9995–9999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jamora, C., Dennert, G. & Lee, A. S. (1996) Proc. Natl. Acad. Sci. USA 93, 7690–7694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu, Z., Luo, H., Fu, W. & Mattson, M. P. (1999) Exp. Neurol. 155, 302–314. [DOI] [PubMed] [Google Scholar]
- 41.Noshiro, M., Lakso, M., Kawajiri, K. & Negishi, M. (1988) Biochemistry 27, 6434–6443. [DOI] [PubMed] [Google Scholar]
- 42.Cnubben, N. H. P., Rietjens, I. M. C. M., Wortelboer, H., van Zanden, J. & van Bladeren, P. J. (2001) Environ. Toxicol. Pharmacol. 10, 141–152. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}