

Abstract
Synthetic heme-binding four-α-helix bundles show promise as working model systems, maquettes, for understanding heme cofactor–protein assembly and function in oxidoreductases. Despite successful inclusion of several key functional elements of natural proteins into a family of heme protein maquettes, the lack of 3D structures, due principally to conformational heterogeneity, has prevented them from achieving their full potential. We report here the design and synthesis of HP-1, a disulfide-bridged two-α-helix peptide that self-assembles to form an antiparallel twofold symmetric diheme four-α-helix bundle protein with a stable conformation on the NMR time-scale. The HP-1 design strategy began with the x-ray crystal structure of the apomaquette L31M, an apomaquette derived from the structurally heterogeneous tetraheme-binding H10H24 prototype. L31M was functionally redesigned to accommodate two hemes ligated to histidines and to retain the strong coupling of heme oxidation-reduction to glutamate acid–base transitions and proton exchange that was characterized in molten globule predecessors. Heme insertion was modeled with angular constraints statistically derived from natural proteins, and the pattern of hydrophobic and hydrophilic residues on each helix was then altered to account for this large structural reorganization. The transition to structured holomaquette involved the alteration of 6 of 31 residues in each of the four identical helices and, unlike our earlier efforts, required no design intermediates. Oxidation-reduction of both hemes displays an unusually low midpoint potential (–248 mV vs. normal hydrogen electrode at pH 9.0), which is strongly coupled to proton binding, as designed.
De novo designed proteins show promise as robust, simplified working models, maquettes, with which to determine structure–function relationships that are obscured in their much more complicated natural counterparts (1–5). Maquettes have proven to be ready scaffolds for incorporating single and multiple redox cofactors and metals (6–11). They also provide interiors that support clearly defined functional elements, such as redox-coupled proton exchange (12) or charge-driven conformational switches (13), which are integral components of catalysis and energy conversion in natural proteins (14). It has been known since the earliest heme protein maquette designs that these functional characteristics can be catalyzed by proteins with a distinct secondary structure but without a singular tertiary structure (1). Despite the insight that exquisitely defined singular tertiary structure is not necessarily a prerequisite for some functions, structural visualization of the protein and the atomic-level details of the heme-binding site are essential if we are to appreciate the mechanism or integrate the elements of function to develop structures with designed enzyme activity.
Here we outline the completion of a redesign strategy that began with a conformationally heterogeneous heme protein maquette that displays specific glutamate-mediated proton exchange coupled to heme oxidation-reduction and ends with a singularly structured diheme maquette without loss of the redox proton coupling. The prototype hemoprotein maquette, H10H24, is a de novo-designed four-α-helix bundle derived from both the cytochrome bc1 and the four-helix de novo-designed protein α2B (1, 15, 16). H10H24 and related maquettes are composed of a noncovalent homodimer of disulfide-linked two-α-helix subunits in the form (α-SS-α)2. The maquette and its derivatives are composed of amino acids laid out in an alternating hydrophobic/hydrophilic binary pattern that has been found to promote helical bundle formation (17). The incorporation of histidine residues provides for heme binding, whereas the arginine residue was used because it was considered to be the source of the raised reduction potential of one of the bound hemes in the cytochrome bc1, which corresponds to the H24-binding site in H10H24 (1).
In the first stage of the redesign of H10H24, structural specificity was introduced into the apoform after iterative variation of the residues thought to reside in the protein interior (18). The solution structure of the resultant apoprotein, H10H24-L6I,L13F, was then solved with NMR (19). Although the structure of each two-α-helix monomer was well defined, the intermonomer-packing interface was unobservable. Subsequently, an x-ray crystal structure of the H10H24-L6I,L13F apoprotein was determined with one additional nonperturbative sequence alteration, L31M (hereafter called L31M), introduced to enable the creation of heavy-metal derivatives (20). The monomers in the homodimer in the x-ray structure proved to be arranged in an anti-topology. Not surprisingly, both of these proteins displayed the properties of a molten globule on binding heme. Because maquettes are intended to serve as small models of protein–cofactor complexes, our immediate goal has been the design of a heme-binding maquette that has a singular structure as a holoprotein. As we demonstrate here, apomaquette structures can provide a firm starting platform for the design and synthesis of singularly structured holomaquettes by using the growing body of biochemical and statistical data on factors governing natural protein structure and stability, especially in combination with the collected functional characterizations of molten globular predecessors of the heme maquette family and related de novo-designed peptides.
Materials and Methods
Materials. Peptide synthesis reagents were purchased from PerSeptive Biosystems (Foster City, CA). Iron(III) protoporphyrin IX-chloride (Fe-PPIX) was from Fluka, and zinc(II) protoporphyrin IX-chloride (Zn-PPIX) was from Aldrich. Deuterium oxide and (trimethylsilyl)-propionate were from Cambridge Isotope Laboratories (Andover, MA). All other solvents and reagents were either from Fisher Scientific or Sigma.
Histidine Conformational Statistics and Maquette Modeling. A program that calculates histidine side chain torsion angles and identifies heme ligand histidines based on the distance between heme irons and histidine nitrogens was written by using the ActiveState (Vancouver) perl 5.6.1 on a Hewlett–Packard personal computer. α-Helical histidines were identified by using backbone torsion angles of residues from i – 4 to i + 4, thereby covering two full turns about the central histidine. If seven or more of these nine residues had helical backbone torsion angles, the histidine was taken to be α-helical. This program was run on the August 1994 PDB select nonredundant structure set created by using a 25% sequence identity filter (21). Statistics were improved by adding selected nonhomologous multiheme structures that have been solved after 1994. Source code for this program is available on request from the authors. Maquette modeling was performed with insight (Molecular Simulations, Waltham, MA) or the program pymol (Delano Scientific, San Carlos, CA).
Synthesis and Purification. Solid-phase peptide syntheses were performed on a continuous-flow PerSeptive Biosystems (Framingham, MA) peptide synthesizer as described (12, 18). Crude peptides were purified to homogeneity by reversed-phase C18 HPLC with aqueous-acetonitrile gradients containing 0.1% (vol/vol) trifluoroacetic acid. Peptide homogeneity and composition were assayed by analytical HPLC and laser desorption mass spectrometry. The N-terminal cysteine residues of the purified peptides were air-oxidized to the symmetric disulfides in 100 mM ammonium carbonate buffer, pH 9.5 (2–5 h), and peptide dimerization was followed by analytical C18 HPLC. Peptide sequences were as follows, with changes indicated in boldface and the position of the deletion indicated as a Δ.
 |
[1] |
Heme Incorporation. DMSO solutions of either Fe-PPIX or Zn-PPIX were added in 0.2-eq aliquots to 3-ml peptide solutions (20 mM KH2PO4/100 mM KCl, pH 8.0) with 20-min equilibrations between additions. After the addition of 1.4 eq, holomaquettes were purified from oligomerized free porphyrins by using a PD-10 desalting column (Amersham Pharmacia). NMR samples were concentrated and further purified on a 2.6 × 100 cm Sephadex G-50 size-exclusion chromatography column.
Solution Molecular Weight Determination. The oligomerization states of the holomaqettes were determined by using size exclusion chromatography on a Beckman System Gold HPLC pump equipped with a diode array detector and a Superdex 75 column (Amersham Pharmacia Biotech). Protein was eluted with 20 mM K2HPO4/100 mM KCl (pH 8.0) at a 0.5 ml/min flow rate. The column was standardized as described (18).
UV-Visible Spectroscopy. UV-visible spectra were recorded on a Perkin–Elmer Lambda 35 spectrophotometer with quartz cells of 0.2-, 0.4-, and 1.0-cm path lengths. Peptide concentrations were determined by using ε280 = 5,810 M–1·cm–1 for Trp.
Redox Potentiometry. The pH dependence of the reduction potential of diheme HP-1 was determined as described (12, 22). Em vs. pH curves were analyzed by using the equation of Clark (22, 23):
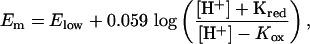 |
[2] |
where Elow is the limiting reduction midpoint potential at low pH, when the coupled protonation site is fully protonated, Kred is the association constant for proton binding to the reduced holomaquette, and Kox is the association constant for proton binding to the oxidized maquette.
Magnetic Resonance Spectroscopy. EPR spectra were collected on 10 μM holoprotein at 20 K and 10 mW power as described (1). For NMR, protein samples were exchanged into deuterium oxide buffer containing 20 mM K2DPO4/50 mM KCl [–log(deuterium ion) (pD) 8.0]. All samples were 0.8–1.2 mM in four-helix bundle concentration except for the Zn-PPIX holomaquette, which was 120 μM. One-dimensional 1H spectra and 2D 1H-13C sensitivity-enhanced constant-time heteronuclear single-quantum coherence spectra were collected as described (18).
Results and Discussion
Rational Redesign of Apo L31M. The recent availability of a full structure of one of our apomaquette scaffolds gives us the opportunity to use the iterative redesign strategy (18) to take us from a structured apomaquette to a structured holomaquette incorporating two hemes. Each of the two sets of four histidines within the antiparallel four-helix bundle structure of apo L31M point into the core of the protein (see Fig. 1A) (20). Bis-histidine ligation of Fe-porphyrins typically entails the coordination of the iron with the N(ε) of the two axial imidazole side chains (24) aligned linearly in a vector through the heme iron and perpendicular to the plane of the porphyrin (see Fig. 1B) (25). Consequently, heme binding by L31M is expected to be accompanied by substantial reorganization of the four-helix bundle structure into a conformation that will both accommodate the relatively large heme macrocycle within the interior and align the histidines in a suitable conformation for heme ligation. This conformational reorganization could take the form of either a rigid-body rotation of the two helices that bind each heme or a local perturbation of the four-helix bundle structure that leaves the rest of the L31M structure intact. Some success in treating helices as rigid bodies in automated structure prediction algorithms has occurred recently (26, 27). Moreover, IR spectroscopy and site-directed sequence alteration experiments performed on the related, but unstructured, holomaquette H10A24 prototype indicate that glutamate residues along the entire length of each helix that are solvent-exposed in the apoprotein become buried in the maquette interior in the holoprotein, supporting a rigid-body helical rotation concomitant with heme binding (12). Such helix rotations would cause the binary patterning used in the original design of the bundle to become misaligned by exposing a number of previously buried hydrophobic residues and burying a number of polar residues. Thus, rational redesign of L31M entails the modification of the original alternating hydrophobic/hydrophilic pattern (16, 17) to accommodate this anticipated rigid-body structural reorientation. To simplify the resultant holomaquette structure and to avoid steric clashes between bound hemes, the second heme-binding site was removed by changing histidine 24 to a phenylalanine. Arginine 27, which was originally intended to interact with the deleted heme-binding site, was itself deleted. Finally, because it was inserted to facilitate x-ray structure determination, the C-terminal methionine M31 was reverted to leucine of the original apostructure.
Fig. 1.

(A) Orientation of histidine residues in the antiparallel (α-ss-α)2 bundle L31M. (B) Section of the transmembrane four-helix cytochrome B subunit of bovine cytochrome bc1. To accommodate a heme cofactor, L31M would be forced to undergo a substantial conformational change, resulting in the reorientation of each α-ss-α monomer into an alignment similar to that depicted in B. The figure was drawn with the program pymol (DeLano Scientific).
To estimate the degree of helix rotation required to place the histidines in an arrangement suitable for heme ligation, we surveyed the Protein Data Bank for the conformational distribution of histidine residues that are ligated to various forms of heme. Because the intended maquette scaffold is a four-α-helix bundle, our program further differentiated between α-helical histidines and histidines in other secondary-structure environments on the basis of the backbone torsion angles of the histidine and all residues within a full turn (four residues) before and after it on a possible helix. To account for some helix distortion, a minimum of seven of these nine residues were required to be in an α-helical backbone conformation for the residue to be considered resident in an α-helix. Although these restrictive criteria will not detect every histidine on a helix in the database, they are sufficient to define the conformational distributions of histidines in relatively unperturbed helices similar to those of L31M.
The overall distribution of histidine conformations we have observed is similar to that observed previously (28), with three major clusters of χ1, near –60°, +60°, and ±180° (hereafter called the m, p, and t torsion-angle populations according to the nomenclature of Lovell et al. (29); see Scheme 1B) and two major populations of χ2, near –80° and +80°, and one minor cluster near 180° (all denoted by their angle). The data for all heme ligand histidines display two significant deviations from this distribution; these deviations are seen as a 68% decrease in the relative p population of χ1 and a narrowing of the distributions of each population of χ2 by at least 10o (see Fig. 2 and Table 1). Statistics for the dataset restricted to α-helical histidines were similar to those observed (30, 31), with the χ1 angles presenting two distinct populations, m and t, and the χ2 again clustered near –80° and +80°. The data for α-helical heme ligand histidines differ from the data of all α-helical histidines in that the χ1 populations show a greater preference for the t conformation and that the χ2 distributions both narrow significantly and show a 3-fold greater preference for the +80° over the –80° conformation. The latter difference may be due in part to the difficulty, at lower resolutions, in distinguishing between the two rotamers crystallographically in the absence of a bound cofactor (29, 32)
Scheme 1.

Fig. 2.

Conformational statistics for histidines derived from the Protein Data Bank select nonredundant protein database (21). Population distributions derived for all histidines, depicted in green, correspond to the left axis and distributions derived for heme ligand histidines, depicted in red, correspond to the right axis.
Table 1. Conformational distribution of histidine side chains in the Protein Data Bank.
| Sample size | χ1 torsion-angle mean (fractional population)* | χ1 torsion angle, σ | χ2 torsion-angle mean (fractional population)* | χ2 torsion angle, σ | |
|---|---|---|---|---|---|
| All histidines | 19,221 | 66 ± 3 (14.2) | 15 ± 3 | 87 ± 2 (36.3) | 25 ± 1 |
| -170 ± 1 (34.2) | 16 ± 1 | 174 ± 6 (19.5) | 39 ± 9 | ||
| -63 ± 1 (51.7) | 13 ± 1 | -76 ± 1 (44.2) | 29 ± 1 | ||
| Heme ligand histidines | 209 | 57 ± 14 (4.5) | 16 ± 14 | 81 ± 1 (41.7) | 15 ± 1 |
| -178 ± 4 (26.7) | 23 ± 4 | 169 ± 9 (16.4) | 29 ± 9 | ||
| -64 ± 1 (68.7) | 10 ± 1 | -74 ± 1 (41.9) | 13 ± 1 | ||
| All α-helical histidines | 3,292 | -175 ± 1 (53.9) | 12 ± 1 | 76 ± 1 (47.8) | 25 ± 1 |
| -67 ± 1 (46.1) | 10 ± 1 | 160 ± 4 (11.9) | 26 ± 8 | ||
| -84 ± 3 (40.3) | 36 ± 3 | ||||
| Heme ligand α-helical histidines | 66 | -179 ± 3 (61.5) | 18 ± 3 | 67 ± 3 (67.5) | 18 ± 2 |
| -67 ± 1 (38.5) | 8 ± 1 | 174 ± 4 (14.1) | 8 ± 4 | ||
| -72 ± 6 (18.4) | 13 ± 6 |
Values in parentheses are percentages.
This database analysis offers a straightforward method for the design of a heme-binding site in L31M. Simply use the most probable (and presumably the lowest energy) rotamer for an α-helical histidine bound to a heme iron to model the expected rigid-body reorientation of the two helices on heme binding. A 30-residue, all-alanine α-helix was generated by using insight. Residue 10 was made into a histidine, with χ1 and χ2 chosen to be –179° and +67°, respectively, based on the mostly likely rotamer as predicted by our database analysis. A molecule of Fe-PPIX was attached to the N(ε) of the histidine (24) with the histidine imidazole moiety perpendicular to the plane of the heme macrocycle and the heme propionates pointing to the outside of the bundle, perpendicular to the bundle axis (25). Another identical helix was attached to the opposite side of the heme, and two of these dihelical monomers were docked by placing them the same distance apart as in the L31M x-ray crystal structure. The sequence of L31M, minus the deleted arginine, was mapped onto this simple model. Inspection (see Fig. 3) provides the prediction that helix rotation causes the exposure of the hydrophobic residues Leu-9 and Leu-23 to solvent and the burial of the charged residues Glu-11 and Glu-18 in the hydrophobic core of the predicted holomaquette structure. To correct for these anticipated problems, a new peptide, denoted HP-1, was designed in which Leu-9 and Leu-23 were converted to glutamine to attenuate the degree of surface hydrophobicity. The glutamate residues were not altered with the intention of retaining the pH-dependent reduction potentials of our previous maquettes (12). Hydrophobic residues occupying interfacial positions, intermediate between solvent-exposed and -buried, have been shown to play an important role in conferring stability and structural specificity in proteins (33, 34). Marshall and Mayo (34) have shown that conformational specificity of natural proteins can be lost when the interfacial side-chain population is made more hydrophobic. In light of this, Phe-13 and Leu-20, both predicted to be interfacial in the model, were each changed to alanine in HP-1.
Fig. 3.

Simple model of the heme-induced helical rotation. A 30-residue, all-alanine α-helix was generated by using insight (Molecular Simulations). Residue 10 was changed into a histidine with χ1 and χ2 chosen to be –179° and +67°, respectively, based on the most likely rotamer as depicted in Table 1. A molecule of Fe-PPIX was added in an orientation perpendicular to the imidazole side chain with the Fe-PPIX iron bound to the histidine N(ε) and the heme propionates pointed to the outside of the bundle (25). An identical helix–Fe-PPIX pair was placed in the position it would occupy in an antiparallel bundle. Mapping the sequence of L31M onto the resultant model indicates that the hydrophobic residues Leu-9 and Leu-23 (β-carbons indicated in green) will become solvent-exposed and that Phe-13 and Leu-20 (indicated in red) are predicted to occupy interfacial positions in the homodimer.
HP-1 was synthesized, and the holoprotein was analyzed by using NMR. The sample showed structural specificity on the NMR timescale, as demonstrated by both narrow proton line widths in 1D spectra and exceptional chemical-shift dispersion in 13C-heteronuclear single-quantum coherence spectra (see Fig. 4). Peak counts were appropriate for a symmetric homodimer with side chain peak doublets caused by heme insertion isomers arising from the two possible orientations of the bound asymmetric Fe-PPIX (35). Complexation with the symmetric heme iron-protoporphyrin III eliminated these peak doublets, confirming that the doublets were a result of heme insertion isomers and not slow structural interconversion between two or more protein conformations (R.L.K., K. M. Smith, P.L.D., and A.J.W., unpublished work).
Fig. 4.

Methyl region expansion of the natural abundance 13C-heteronuclear single-quantum coherence spectra of diheme HP-1. Solution conditions were ≈1 mM protein complex, 25 mM KD2PO4, 50 mM KCl, 100% D2O[–log(deuterium ion) (pD) 8.0], 32°C.
Physical Properties of Diheme HP-1. HP-1 forms a tight complex with ferric Fe-PPIX, binding two hemes per homodimer (i.e., four-helix bundle), both hemes display Kd values of <20 nM (data not shown). The diheme HP-1 complex displays reduced and oxidized absorption spectra (reduced minus oxidized α- and β-band λmax = 563 and 533 nm, respectively; see Fig. 5A) and EPR spectra (highly rhombic gz, gy, and gx values of 2.91, 2.28, and 1.54; see Fig. 5B) consistent with bis-histidine coordination of two identical low-spin ferric hemes. No evidence exists of a high-spin heme, which would signify a population of single histidine-ligated heme or unligated heme. Size exclusion chromatography of the complex proves it to be a single species with a predicted molecular mass of 19.5 ± 0.5 kDa, similar to the calculated molecular mass of 15.5 kDa. The room-temperature reduction potential of the HP-1–Fe-PPIX complex in 20 mM potassium phosphate/100 mM KCl (pH 9.0) is –248 mV vs. the normal hydrogen electrode (NHE) (see Fig. 5A). As with our earlier designs (12), HP-1 displays proton-coupled reduction potentials: 1.0 proton is exchanged per heme oxidation/reduction between a pK value of 6.8 when the heme is reduced and a pK value in the oxidized state well below 5.0, too low to measure because of the instability of the complex at low pH. At all pH values, redox titrations were best fit with a single Nernst equation with n = 1, suggesting that the hemes are not adjacent in the structure and, hence, that the homodimer is in an antiparallel conformation in both oxidation states over this pH range (13).
Fig. 5.

Redox and spectral properties of HP-1. (A) Oxidized minus reduced visible absorption difference spectrum of Fe-PPIX–HP-1. (B) EPR spectrum of Fe-PPIX–HP-1. (C) Nernst plot of the dithionite-induced reduction and ferricyanide-induced oxidation of Fe-PPIX–HP-1 in 20 mM KH2BO4/100 mM KCl (pH 9.0). Line drawn is a best fit with n = 1 and a reduction midpoint potential of –248 mV vs. NHE. (D) pH dependence of the midpoint potential. Each point represents an individual titration as shown in C. The line drawn is a best fit with Eq. 1, assuming the protonation of one acid–base pair in the reduced protein with a pKa of 6.80.
Heme Binding Imparts Structural Specificity in HP-1. Both L31M and its variant H10H24-L6I,L13F are conformationally specific in the apostate but become molten globules on binding ferric heme (19, 20, 36). In direct contrast, HP-1 is a molten globule apoprotein that becomes structurally specific on binding ferric heme (see Fig. 6). As a test of our rigid-body rotation model of the heme-binding induced structural reorganization, Zn-PPIX was bound to apo-HP-1. Zn-PPIX is isosteric with Fe-PPIX but obligately binds only a single histidine, resulting in five-coordinate ligation instead of the six-coordinate bis-histidine ligation routinely observed in ferric diheme maquettes (37, 38). As with earlier maquettes, Zn-PPIX binds with high affinity to apo-HP-1. However, the five-coordinate ligation complex, which would be expected to align only one of the two helices in each dihelix monomer, displays the NMR spectral properties of a molten globule (see Fig. 6B). Clearly, bis-histidine ligation is required for structural specificity in HP-1.
Fig. 6.

Heme-binding-mediated structural reorientation in HP-1. One-dimensional proton NMR spectra of apo-HP-1 (A), the Zn-PPIX–HP-1 complex (B), and the ferric heme–HP-1 complex (C). The structural specificity evident in C demonstrates that both helices must be oriented through the formation of two histidine imidazole–heme bonds for the complex to have native-like properties, as opposed to the orientation of only one (B) or zero (A) helices in this manner.
This behavior has some similarity to that found in the natural heme-binding four-helix bundle protein cytochrome b562. This protein binds a single Fe-PPIX with methionine-histidine ligation originating from the first and second antiparallel helices, respectively. Removal of the Fe-PPIX results in a protein that is partially molten globular as detected by NMR (39). Although helices 2, 3, and 4 are relatively unperturbed, most of helix 1 becomes unstructured. Likewise, removal of one of the axial heme ligands in this protein, Met-7, results in a protein that readily binds heme but still has partial molten globule character (40), analogous to the behavior of Zn-PPIX-bound HP-1.
Concluding Comments. The first stage of the transformation of the molten globular H10H24 prototype (1) into a conformationally specific diheme HP-1 involved the establishment of a structured version of the molten globular apoform of H10H24. Permutations of interior residues of H10H24, achieved with two sequence changes in each identical helix comprising the four-helix bundle, yielded six native-like apomaquettes from 20 variants (20). Similar findings have been reported in more generalized four-helix bundle sequence searches (17, 41). The apomaquette selected for structural determination, L31M, incorporated two sequence changes, L6I and L13F. Despite the unusual non-native-like interface between the two two-helix monomers, L31M has provided enough structural information to enable the second redesign stage, resulting in HP-1. For this redesign simple structural models were generated to predict the structural reorganization produced by insertion of the heme cofactors. These models were fashioned by using a statistical analysis of natural protein structures, the body of biochemical knowledge derived from natural proteins, and the accumulated characterizations of the H10H24 derivatives. Six substitutions in each helix, including a further change at position 13, were chosen based on these models. These changes based on the L31M structure were made in a single step to yield the conformationally specific HP-1 with the concomitant loss of singular structure in the apoform. Note that conformational specificity is achieved by the insertion and ligation of the heme; it is clear too that what is likely to be the internalization of the glutamates (which are solvent-exposed in L31M), their pK alterations, and promotion of proton coupling to the heme oxidation-reduction is also a consequence of the heme binding.
Progress reported here, made in such a straightforward way, suggests that relatively simple “binary patterning” models (17) can be successfully applied to the design of cofactor-binding proteins when the structural rearrangement induced by cofactor incorporation is taken into account. It seems likely that many routes to conformational specificity and to a range of related functions exist.‡ In HP-1, we have created a simple, structurally defined, quantifiable model system to study the assembly and mechanisms of proton-coupled electron transfer processes fundamental to oxidoreductase enzymes and membrane redox proteins at the heart of energy conversion, respiration, and photosynthesis.
Acknowledgments
We thank Dr. Vikas Nanda for help with the program insight and Dr. William Degrado for valuable discussions. This work was supported by National Institutes of Health Grants GM48130 and GM41048 (to P.L.D.), GM35940 (to A.J.W.), and GM64090 (to R.L.K.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: Fe-PPIX, iron(III) protoporphyrin IX-chloride; Zn-PPIX, zinc(II) protoporphyrin IX-chloride; NHE, normal hydrogen electrode.
Footnotes
A conformationally specific diheme-binding four-helix bundle similarly inspired by the natural cytochrome bc1 but designed by computational methods has been synthesized by G. Ghirlanda and W. DeGrado in this department and completed within the same period as this work (G. Ghirlanda, private communication).
References
- 1.Robertson, D. E., Farid, R. S., Moser, C. C., Urbauer, J. L., Mulholland, S. E., Pidikiti, R., Lear, J. D., Wand, A. J., Degrado, W. F. & Dutton, P. L. (1994) Nature 368, 425–431. [DOI] [PubMed] [Google Scholar]
- 2.Choma, C. T., Lear, J. D., Nelson, M. J., Dutton, P. L., Robertson, D. E. & Degrado, W. F. (1994) J. Am. Chem. Soc. 116, 856–865. [Google Scholar]
- 3.Sharp, R. E., Moser, C. C., Rabanal, F. & Dutton, P. L. (1998) Proc. Natl. Acad. Sci. USA 95, 10465–10470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gibney, B. R., Isogai, Y., Rabanal, F., Reddy, K. S., Grosset, A. M., Moser, C. C. & Dutton, P. L. (2000) Biochemistry 39, 11041–11049. [DOI] [PubMed] [Google Scholar]
- 5.Marsh, E. N. G. & DeGrado, W. F. (2002) Proc. Natl. Acad. Sci. USA 99, 5150–5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen, X. X., Discher, B. M., Pilloud, D. L., Gibney, B. R., Moser, C. C. & Dutton, P. L. (2002) J. Phys. Chem. B 106, 617–624. [Google Scholar]
- 7.Gibney, B. R., Huang, S. S., Skalicky, J. J., Fuentes, E. J., Wand, A. J. & Dutton, P. L. (2001) Biochemistry 40, 10550–10561. [DOI] [PubMed] [Google Scholar]
- 8.Shifman, J. M., Gibney, B. R., Sharp, R. E. & Dutton, P. L. (2000) Biochemistry 39, 14813–14821. [DOI] [PubMed] [Google Scholar]
- 9.Gibney, B. R. & Dutton, P. L. (1999) Protein Sci. 8, 1888–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tommos, C., Skalicky, J. J., Pilloud, D. L., Wand, A. J. & Dutton, P. L. (1999) Biochemistry 38, 9495–9507. [DOI] [PubMed] [Google Scholar]
- 11.Lombardi, A., Summa, C. M., Geremia, S., Randaccio, L., Pavone, V. & DeGrado, W. F. (2000) Proc. Natl. Acad. Sci. USA 97, 6298–6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shifman, J. M., Moser, C. C., Kalsbeck, W. A., Bocian, D. F. & Dutton, P. L. (1998) Biochemistry 37, 16815–16827. [DOI] [PubMed] [Google Scholar]
- 13.Grosset, A. M., Gibney, B. R., Rabanal, F., Moser, C. C. & Dutton, P. L. (2001) Biochemistry 40, 5474–5487. [DOI] [PubMed] [Google Scholar]
- 14.Discher, B. M., Koder, R. L., Moser, C. C. & Dutton, P. L. (2003) Curr. Opin. Chem. Biol. 7, 741–748. [DOI] [PubMed] [Google Scholar]
- 15.Degrado, W. F., Wasserman, Z. R. & Lear, J. D. (1989) Science 243, 622–628. [DOI] [PubMed] [Google Scholar]
- 16.Ho, S. P. & Degrado, W. F. (1987) J. Am. Chem. Soc. 109, 6751–6758. [Google Scholar]
- 17.Kamtekar, S., Schiffer, J. M., Xiong, H. Y., Babik, J. M. & Hecht, M. H. (1993) Science 262, 1680–1685. [DOI] [PubMed] [Google Scholar]
- 18.Gibney, B. R., Rabanal, F., Skalicky, J. J., Wand, A. J. & Dutton, P. L. (1999) J. Am. Chem. Soc. 121, 4952–4960. [Google Scholar]
- 19.Skalicky, J. J., Gibney, B. R., Rabanal, F., Urbauer, R. J. B., Dutton, P. L. & Wand, A. J. (1999) J. Am. Chem. Soc. 121, 4941–4951. [Google Scholar]
- 20.Huang, S. S., Gibney, B. R., Stayrook, S. E., Dutton, P. L. & Lewis, M. (2003) J. Mol. Biol. 326, 1219–1225. [DOI] [PubMed] [Google Scholar]
- 21.Hobohm, U. & Sander, C. (1994) Protein Sci. 3, 522–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dutton, P. L. (1978) Methods Enzymol. 54, 411–435. [DOI] [PubMed] [Google Scholar]
- 23.Clarke, W. M. (1960) Oxidation Reduction Potentials of Organic Systems (Williams & Wilkins, Baltimore).
- 24.Chakrabarti, P. (1990) Protein Eng. 4, 57–63. [DOI] [PubMed] [Google Scholar]
- 25.Zaric, S. D., Popovic, D. M. & Knapp, E. W. (2001) Biochemistry 40, 7914–7928. [DOI] [PubMed] [Google Scholar]
- 26.Zhang, C., Hou, J. T. & Kim, S. H. (2002) Proc. Natl. Acad. Sci. USA 99, 3581–3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nanias, M., Chinchio, M., Pillardy, J., Ripoll, D. R. & Scheraga, H. A. (2003) Proc. Natl. Acad. Sci. USA 100, 1706–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ponder, J. W. & Richards, F. M. (1987) J. Mol. Biol. 193, 775–791. [DOI] [PubMed] [Google Scholar]
- 29.Lovell, S. C., Word, J. M., Richardson, J. S. & Richardson, D. C. (2000) Proteins 40, 389–408. [PubMed] [Google Scholar]
- 30.Dunbrack, R. L. & Karplus, M. (1993) J. Mol. Biol. 230, 543–574. [DOI] [PubMed] [Google Scholar]
- 31.McGregor, M. J., Islam, S. A. & Sternberg, M. J. E. (1987) J. Mol. Biol. 198, 295–310. [DOI] [PubMed] [Google Scholar]
- 32.Lovell, S. C., Word, J. M., Richardson, J. S. & Richardson, D. C. (1999) Proc. Natl. Acad. Sci. USA 96, 400–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Street, A. G. & Mayo, S. L. (1998) Folding Des. 3, 253–258. [DOI] [PubMed] [Google Scholar]
- 34.Marshall, S. A. & Mayo, S. L. (2001) J. Mol. Biol. 305, 619–631. [DOI] [PubMed] [Google Scholar]
- 35.Lamar, G. N., Budd, D. L., Viscio, D. B., Smith, K. M. & Langry, K. C. (1978) Proc. Natl. Acad. Sci. USA 75, 5755–5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gibney, B. R., Rabanal, F., Skalicky, J. J., Wand, A. J. & Dutton, P. L. (1997) J. Am. Chem. Soc. 119, 2323–2324. [Google Scholar]
- 37.Feitelson, J. & Spiro, T. G. (1986) Inorg. Chem. 25, 861–865. [Google Scholar]
- 38.Sharp, R. E., Diers, J. R., Bocian, D. F. & Dutton, P. L. (1998) J. Am. Chem. Soc. 120, 7103–7104. [Google Scholar]
- 39.Feng, Y. Q., Sligar, S. G. & Wand, A. J. (1994) Nat. Struct. Biol. 1, 30–35. [DOI] [PubMed] [Google Scholar]
- 40.Kamiya, N., Okimoto, Y., Ding, Z., Ohtomo, H., Shimizu, M., Kitayama, A., Morii, H. & Nagamune, T. (2001) Protein Eng. 14, 415–419. [DOI] [PubMed] [Google Scholar]
- 41.Wei, Y. N., Liu, T., Sazinsky, S. L., Moffet, D. A., Pelczer, I. & Hecht, M. H. (2003) Protein Sci. 12, 92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]