

Abstract
Myosin V, a double-headed molecular motor, transports organelles within cells by walking processively along actin, a process that requires coordination between the heads. To understand the mechanism underlying this coordination, processive runs of single myosin V molecules were perturbed by varying nucleotide content. Contrary to current views, our results show that the two heads of a myosin V molecule communicate, not through any one mechanism but through an elaborate system of cooperative mechanisms involving multiple kinetic pathways. These mechanisms introduce redundancy and safeguards that ensure robust processivity under differing physiologic demands.
As a processive motor, a myosin V molecule carries its intracellular cargo for long distances along an actin track, taking multiple steps before detaching and deriving the energy for each step from the hydrolysis of ATP (1–3). Determining the mechanism of this processive movement is essential for explaining diseases such as Griscelli syndrome in which a mutation to human myosin Va leads to hypopigmentation and severe neurological impairment (4, 5). Each of the two heads of myosin V catalyzes the hydrolysis of ATP and after release of hydrolysis products (Pi and ADP) generates motion with a rotation of its long lever arm (6–8). When both heads function together, the myosin V molecule walks along actin in a hand-over-hand fashion, taking 36-nm strides (7, 9–12). One of the most critical and intriguing questions, however, remains unanswered. How do the two heads coordinate their biochemical and mechanical cycles to maintain processive movement? Solution kinetic studies to date have focused only on single-headed myosin V constructs, from which coordination between the heads can only be inferred (13–15). Therefore, we expressed double-headed, heavy meromyosin V molecules with a C-terminal yellow fluorescent protein (YFP-HMMM5) such that processive movement of individual myosin V molecules could be visualized by total internal reflectance fluorescence (TIRF) microscopy (16) and then described in terms of run length and velocity (17). By perturbing the biochemical cycle through addition of ATP, ADP, or Pi, we assessed the impact of these ligands on myosin V processivity, identified the myosin state from which a processive run most likely terminates, and showed that the two heads of myosin V are coordinated to generate processive movement through an elaborate system of cooperative mechanisms.
Methods
YFP-HMMM5 Expression. YFP was cloned onto the C terminus of HMMM5. The final construct contains the first 1,098 amino acids (G1098) of murine myosin V HMM, followed by a linker region coding for the amino acids VTGS, followed by YFP and a FLAG epitope for purification. Sf9 cells were coinfected with two recombinant viruses, one encoding for the myosin V heavy chain and one for calmodulin. The calmodulin was a mutant deficient in calcium binding (CaMΔall) to ensure complete occupancy of all the IQ motifs (18). After 72 h of infection, cells were pelleted and then lysed with buffer that contained 10 mM NaPI (pH 7.2), 0.6 M NaCl, 5 mM MgCl2, 3 mM NaN3, 7% sucrose, 2 mM EGTA, 1% Nonidet P-40, 2 mM DTT, 0.5 mM 4-(2-aminoethyl)benzenesulfonyl fluoride, 5 μg/ml leupeptin, and 0.78 mg/ml benzamidine, to which 25 μg/ml of CaMΔall was added. The lysate was fractionated with 25% and then 70% ammonium sulfate. CaMΔall (25 μg/ml) was added to the resuspended 70% ammonium sulfate pellet, which was dialyzed overnight versus 10 mM imidazole (pH 7.4)/0.3 M NaCl/1 mM EGTA/1 mM DTT/1 mM NaN3/5 μg/ml leupeptin. The dialyzed protein was clarified at 50,000 rpm for 45 min (Beckman, rotor Ti60) and applied to a FLAG column equilibrated in 10 mM imidazole (pH 7.4)/0.3 M NaCl/1 mM EGTA/1 mM NaN3. The protein was eluted with FLAG peptide and dialyzed versus 50% glycerol/10 mM imidazole (pH 7.4)/0.3 M NaCl/1 mM EGTA/1 mM DTT/1 mM NaN3/5 μg/ml leupeptin.
Motility Buffers. Myosin buffer contained 0.3 M KCl, 25 mM imidazole, 1 mM EGTA, 4 mM MgCl2, and 10 mM DTT, adjusted to pH 7.4. Actin buffer (AB) contained 25 or 100 mM KCl, 25 mM imidazole, 1 mM EGTA, 4 mM MgCl2, 10 mM DTT, and oxygen scavengers (0.1 mg·ml–1 glucose oxidase, 0.018 mg·ml–1 catalase, and 2.3 mg·ml–1 glucose), adjusted to pH 7.4. Ligands (1 μM to 1 mM MgATP, 0–5 mM MgADP, and 0–40 mM Pi) were added to AB (AB+ATP), varying KCl and MgCl2 concentrations to maintain a constant ionic strength and a 3 mM free Mg+2 (19). To visualize actin filaments at a wavelength that did not overlap with YFP-HMMM5 emission and to fix the filaments to the experimental chamber surface through a neutravidin–biotin linkage, 1 μM filamentous actin prepared from chicken pectoralis (20) was incubated in AB containing 0.5 μM Alexa 660-phalloidin (Molecular Probes, A-22285), 0.5 μM biotin-XX phalloidin (Molecular Probes, B-7474), and 10 μM DTT for 18 h at 4°C. For ensemble measurements of YFP-HMMM5, tetramethylrhodamine B isothiocyanate-phalloidin-labeled actin filaments (tetramethylrhodamine B isothiocyanate-actin) were prepared as described (20).
Single YFP-HMMM5 Motility Measured with TIRF Microscopy. A 30-μl experimental flow-cell chamber was constructed by placing a glass coverslip (no. 1 Fisher premium, 24 × 30 mm) supported by two 125-μm Mylar shim strips along its long edges onto a larger glass coverslip (no. 1 Fisher premium, 24 × 60 mm). The shims were dipped in optical adhesive (Norland no. 61, Cranbury, NJ) such that the chamber could be glued and cured under UV light. Solutions in 30-μl volumes then were added to the flow cell with the following series of incubations: (i)1mg·ml–1 neutravidin (Molecular Probes, A-2666) in AB for 2 min; (ii) 2× AB wash; (iii) 2× 0.1% Triton X-100 (Sigma, T9284) in AB, incubated for 2 min on the final wash; (iv) 4× AB wash; (v) Alexa 660/biotin-phalloidin-actin in AB for 3min; (vi)4× AB+ATP wash; (vii) YFP-HMMM5 in myosin buffer + BSA for 1 min; and (viii) YFP-HMMM5 in AB+ATP. YFP-HMMM5 concentrations were adjusted between 0.5 and 2.0 nM such that the density of YFP-HMMM5 undergoing processive runs on a given actin filament were frequent yet distinguishable as individual runs. Experiments were performed at room temperature, which averaged 25 ± 1°C.
The flow cell with added proteins was placed on the stage of a Nikon TE2000-U microscope equipped with a PlanApo objective lens (×100, numerical aperture 1.45) for through-the-objective TIRF (16). The Alexa 660/biotin-actin filaments were visualized in epifluorescence, excited with an HBO 100-W lamp (Zeiss), by using an HQ Cy5.5 Chroma (Chroma Technology, Brattleboro, VT) filter set. Once a field of actin that had sufficient filament density but little filament overlap was identified, the fluorescence excitation was switched to an argon laser (488 nm, Spectra-Physics model 163) so that the YFP-HMMM5 could be visualized in TIRF mode through a Chroma HQ560/80 filter. Both the fluorescent actin filaments and individual YFP-HMMM5 molecules were detected at a magnification of ×200 by using an image-intensified digital charge-coupled device camera (DVC Intensicam IV S, Austin, TX), resulting in a final pixel resolution of 22 nm. Digital images were acquired with an integration time of 110 msec but were stored at a rate of 3.4 frames per sec by using image-capturing software (QED Imaging, Pittsburgh). The image-acquisition rate was chosen to provide sufficient time to improve the signal-to-noise ratio as well as to resolve the movement of YFP-HMMM5 at its fastest velocities (i.e. ≈0.6 μm/sec) with more than four frames. Image files then were analyzed by using scion image (Scion Corporation) to manually track the movement of a single YFP-HMMM5 between frames. The total distance traveled from initial appearance to disappearance of a YFP-HMMM5 defined its run length, L, whereas the number of frames for a run divided by the frame-storage rate defined its run time, τr. These two parameters were used to calculate the velocity (V = L/τr) for each YFP-HMMM5 processive run. More than 50 runs were recorded under each condition from which an average velocity, V, was calculated. A run-length constant, λ, was determined by fitting the distribution of measured run lengths, L, to a single exponential, Ae–L/λ. Data for YFP-HMMM5 runs that terminated at the end of an actin filament were discarded, because the run length would be limited by the actin length and not factors associated with the YFP-HMMM5 itself. In addition, processive runs lasting <1 sec were discarded from our analysis, because short, unidirectional movement of YFP-HMMM5 (<1 sec) was observed in the absence of actin because of the Brownian motion of molecules within the evanescent field.
Ensemble Motility Using Epifluorescence. Movement of tetramethylrhodamine B isothiocyanate-actin filaments over a YFP-HMMM5-coated surface was visualized as described (20), and actin filament velocities, Vactin, were determined from video recordings of filament movement by using a custom filament-tracking program (21). Solutions were added to flow cells, previously coated with nitrocellulose (20), with the following series of incubations: (i) 20 μl of 50 μg·ml–1 anti-AFP monoclonal antibody 3E6 (AFP5002, Qbiogene, Carlsbad, CA) in AB for 1 min; (ii) 2 × 20 μl of 0.5 mg·ml–1 BSA in AB wash; (iii) 20 μl of 17.5 μg·ml–1 YFP-HMMM5 for 1 min; (iv)2 × 20 μl 100 nM tetramethylrhodamine B isothiocyanate-actin for 1 min; (v) 3 × 20 μl AB wash; and (vi) 3 × 20 μl of AB-ATP with desired ligands. Experiments were performed at 30°C.
Kinetic Modeling. The model in Fig. 4 links myosin V kinetics and movement through three different pathways (A–C). It is widely assumed that, although challenges to this assumption exist (22), myosin V movement and kinetics are tightly coupled. According to this view, the velocity, V, is related to the ATPase rate per head, v, as
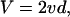 |
[1] |
where 2d = 72 nm is the net displacement of the molecule after each head takes its turn hydrolyzing an ATP.
Fig. 4.

A simple six-state model showing the multiple pathways that the two heads of a myosin V molecule might follow during their individual ATPase cycles as they process along an actin filament (green helical structure). The scheme represents only half of a cycle for a given head (the second half would have the heads swapping roles as lead and trail heads, states 6 → 1′) and is arranged such that transitions from top to bottom are the trail head's (light blue) biochemical transitions (ADP release and ATP binding), with transitions from left to right being the lead head's (dark blue) working step. The working step involves a relatively discrete rotation of the light-chain domain of myosin (six small ovals) relative to its catalytic domain (large oval) and is closely associated with Pi release and strong binding to actin (illustrated as the catalytic domain making contact with the actin filament). Weak binding is indicated by the +++ signs and is assumed to be a rapid, nonspecific attachment and detachment of the myosin head to actin. The working step can be partitioned between movement (illustrated as translocation along a scale) and the generation of intramolecular strain (illustrated as the bending of the myosin light-chain domain). An additional light-chain domain rotation associated with ADP release (26, 27) results in additional movement (state 1 → 2) and an energetically favorable relaxation of any intramolecular strain (state 4 → 5). The complete relaxation of intramolecular strain after ATP binding may result in additional movement (state 5 → 6). Finally, termination of a processive run, at a rate of kterm 1, is assumed to occur from a state at which only one ADP-bound head is attached to actin. Although myosin V might terminate from state 6, kinetic studies (13) indicate that the lifetime of this state (<5 msec) is short relative to the lifetime of state 1, making the probability of detachment from this state relatively low.
The per-head ATPase rate, v, through paths A and B (the pathways suggested by our results) is the sum of the flux through each pathway, or
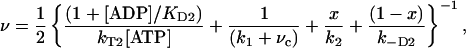 |
[2] |
where x = vc/(vc + k1) is the probability that myosin V takes path B over all possible paths (i.e., A and B) and vc is described below. The factor of ½ is needed because the reaction in Fig. 4 represents only half of the ATPase cycle for a given head.
A simple expression for the average length of a myosin V processive run is
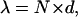 |
[3] |
where n = 1/P is the number of steps taken by the molecule and P is the probability that myosin V terminates a processive run during a given ATPase cycle. A processive run through a combination of paths A and B is most likely to terminate from state 1 with a probability, P = kterm 1/(kterm 1 + k1 + vc), where vc = 1/(1/k–D1 + [ADP]/KD1k2) is the flux through path B, which we assume is limited by ADP release. Substituting P into Eq. 3:
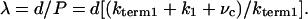 |
[4] |
Results and Discussion
The processive movement of individual YFP-HMMM5 molecules was characterized (see Table 1) by a myosin V run-length constant, λ, of 0.8 μm and an average velocity, V, of 0.55 μm/sec under near-physiological conditions (1 mM ATP/100 mM KCl at 25°C). These processive runs were not limited by the actin filament length (average = 7.2 ± 2.4 μm), “road blocks” caused by inaccessible regions along the actin filament, or premature termination caused by YFP photobleaching kinetics (see Fig. 1). Assuming a 36-nm stride length (1, 2, 7, 12, 22) these run lengths suggest that YFP-HMMM5 takes ≈23 steps before detaching from actin.
Table 1. YFP-HMMM5 results summary.
| V, μm·sec-1 | λ, μm | KM, μM | |
|---|---|---|---|
| Single molecule (25 mM KCl) | 0.38 ± 0.04 (n=4) | 1.3 ± 0.2 | 6.1 ± 1.1 |
| Ensemble (25 mM KCl) | 0.59 ± 0.03 | NA | 17 ± 2 |
| Single molecule (100 mM KCl) | 0.55 ± 0.04 (n=3) | 0.8 ± 0.2 | 163 ± 21 |
| Ensemble (100 mM KCl) | 0.86 ± 0.04 | NA | 114 ± 12 |
| Single molecule + 40 mM Pi | 0.44 ± 0.03 (n=4) | 0.5 ± 0.1 | NA |
Single-molecule and ensemble data were gathered at 25°C and 30°C, respectively. Velocities, V, and run lengths were obtained at 1 mM ATP. Velocities are the mean ± SD (n), where n is the number of experiments, each containing > 18 processive runs for the single-molecule data and > 40 filaments for the ensemble data. KM is [ATP] at half-maximal velocity ± SD determined from a linear regression to the double-reciprocal plot (Fig. 2, solid line). Run-length constants, λ, were obtained from the best fit of the equation, Ae-L/λ, (Figs. 2a and 3a, solid lines) to run-length, L, histograms. Forty millimolar Pi is the ionic equivalent of ≈92 mM KCl. NA, not applicable.
Fig. 1.

(a) A series of images showing a single YFP-HMMM5 molecule moving along an actin filament. The Alexa 660/biotin-actin filament (red) was first imaged in epifluorescence. At the same location, a series of YFP-HMMM5 images (green) were subsequently obtained at 0.3-sec intervals in TIRF mode. The stacked red and green images show regions of overlap (colocalization) as yellow, thus highlighting the position and movement of YFP-HMMM5 on actin. YFP-HMMM5 movement was always unidirectional, suggesting that movement occurred on single actin filaments. (b) YFP-HMMM5 molecules travel the full length of the actin filament. A 120-frame average (≈36 sec) of ≈40 YFP-HMMM5 processive runs (Top, green, imaged with TIRF) along an actin filament (Middle, red, imaged with epifluorescence). The stacked images (Bottom), with regions of overlap shown in yellow, demonstrate our observation that as the YFP-HMMM5 travels along the actin filament, it had access to the entire length of actin, suggesting that few inaccessible regions exist. The green spots in the Top and Bottom images are YFP-HMMM5 molecules that are bound nonspecifically to the surface at which actin was not present. (c) Time course of fluorescence intensity [arbitrary units (a.u.)] showing photobleaching of a single YFP-HMMM5 molecule. Most (>95%) YFP-HMMM5 molecules photobleached with either one or two steps, the latter (shown here) corresponding to the photobleaching of two YFP molecules, one bound to the C terminus of each of the two heavy chains of HMMM5. The absence of photobleaching events that occur in more than two steps indicates that the fluorescent images originated from a single myosin V molecule and not from an aggregate. (d) Time course of fluorescence intensity (arbitrary units) showing photobleaching of many YFP-HMMM5 molecules. A single exponential fitofthe fluorescence decay (red line) of many YFP-HMMM5 molecules bound to actin filaments in rigor gave a photobleaching rate of ≈0.1 sec–1, which is significantly slower than the range of inverse run times reported in this article (0.17–0.7 sec–1), indicating that photobleaching kinetics do not influence our run-time estimates significantly. Indeed, including the photobleaching rates in our analysis did not change our conclusions.
At both 25 and 100 mM KCl, decreasing [ATP] and increasing [ADP] or [Pi] resulted in a decreased velocity for both single-molecule and ensemble motility (Figs. 2 and 3 and Table 1), which according to Eq. 1 corresponds to a decrease in the YFP-HMMM5 ATPase rate. These qualitative observations are expected for an ATPase that binds ATP and reversibly releases ADP and Pi. However, to understand how head-to-head interactions affect YFP-HMMM5 kinetics, we must develop a more quantitative analysis of these data, using models that explicitly link myosin V biochemistry and mechanics. Fig. 4 illustrates such a model in which the double-headed myosin V ATPase is described by a six-state model with three separate kinetic pathways (A–C).
Fig. 2.

Velocities and run lengths versus [ATP]. (a) Run lengths, L, observed at four different ATP concentrations (10, 25, 50, and 500 μm) are plotted as histograms. Solid lines are Ae–L/λ, where λ is the run-length constant. Inverse velocity (1/V, ○) and run lengths constant (▵) are plotted versus 1/[ATP] for a single YFP-HMMM5 at 100 mM KCl (b) and 25 mM KCl (c). The solid and dashed lines are linear regressions of the velocity and run-length data, respectively.
Fig. 3.

Velocities and run lengths versus [ADP] at 1 mM ATP and 100 mM KCl. (a) Run lengths, L, observed at three different ADP concentrations (75, 150, and 1,000 μM) are plotted as histograms. Solid lines are Ae–L/λ, where λ is the run-length constant. (b) Velocities are plotted versus [ADP] for a single YFP-HMMM5. The solid line is the best fit of Eq. 1 (substituting in Eq. 2) to the velocity data. (c) Run-length constants are plotted versus [ADP]. The solid line is the best fitofEq. 4 to the run-length data.
Which pathway(s) myosin V takes during a processive run is currently debated (23). Neither velocity data alone nor existing data in the literature can resolve this issue; however, the observed effects of ligands on the run length of YFP-HMMM5 do place clear constraints on the kinetic scheme and provide information about the dissociation kinetics that terminate a processive run. We observed that decreasing [ATP] had little effect on the YFP-HMMM5 run length (Fig. 2), whereas increasing [ADP] resulted in a decrease in run length that saturates at a nonzero value (Fig. 3). With the addition of ADP, the decrease in run length implies that myosin V either spontaneously dissociates from actin in an ADP-bound state (Fig. 4, state 1) or undergoes futile cycling from this state. The nonzero saturation of this decline indicates that myosin V travels down multiple pathways. With changes in [ATP], high [ATP] would tend to draw myosin V down path C, risking termination at state 3, whereas low [ATP] favors safe passage through path B (24). Thus, the relatively small decline in run length observed with a 20-fold increase in [ATP] (Fig. 2) suggests that ATP concentrations much greater than 1 mM are needed to significantly populate path C. Based on this simple model in which paths A and B are traveled most frequently, we developed analytical expressions (see Methods and Eqs. 1–4) for the [ATP] and [ADP] dependences of run length and velocity. By assuming a value for the ADP-binding constant (KD1 = 1 μM) that is consistent with single-headed myosin V kinetic studies (13–15), these equations were fit (R2 > 0.92) to the data in Figs. 2 and 3 (solid lines) by using a least-squares fitting routine that provided estimates for transition rates through key steps in the cycle (see Table 2).
Table 2. Rate constants obtained from best-fit to six-state kinetic model.
| kT2, 106 M-1·sec-1 | k-D1, sec-1 | k-D2, sec-1 | k1, sec-1 | k2, sec-1 | kterm 1, sec-1 | KD1, μM | KD2, μM |
|---|---|---|---|---|---|---|---|
| 0.050 ± 0.01 | 7.5 ± 2 | >25 | 5 ± 2 | 572 ± 228 | 1.1 ± 0.1 | 1 | >60 |
A value for KD1 of 1 μM was assumed based on solution studies (13-15). Values for k1, k2, and kterm 1 were obtained from the best fit of Eq. 4 to the run-length data in Fig. 3. A value for k-D1 was obtained from Eq. 1 and the maximum velocity. kT2 was obtained from the best fit of Eq. 1 to velocity data in Fig. 2a. KD2 was obtained from the best fit of Eq. 1 to velocity data in Fig. 3. The errors of the estimates were obtained based on a nonlinear regression analysis.
Our data and analysis provide insight into the critical transitions that govern myosin V processivity and its termination. Starting in state 1 (Fig. 4), the molecule begins its processive run by first choosing between two paths, A and B (k–D1 = 7.5 sec–1 versus k1 = 5 sec–1), with nearly equal probability. Regardless of which path it takes, the transition out of state 1 is rate-limiting for the entire cycle, with a rate similar to that determined both in solution and optical-trap studies (2, 13–15). Thus, the predominant state during processive movement is one having a single strongly attached head (state 1) (24). When the motor travels down path A, the leading head attaches to actin, releases Pi, and undergoes its working step (state 1 → 4) at an effective rate, k1, of 5 sec–1 before the trailing head releases ADP. Our estimate for k1 is significantly slower than values obtained in solution studies (13), indicating that the attached trail head mechanically inhibits attachment by the lead head, making this transition rate-limiting for path A. This finding is in contrast to previous studies (2, 7) that suggest that ADP release from the trailing head in state 4 is rate-limiting. In fact, ADP release from state 4 is significantly faster in the two-headed molecule (k–D2 > 25 sec–1) than measured in single-headed solution studies, indicating that attachment of the lead head (state 1 → 4) mechanically accelerates ADP release from the trail head (state 4 → 5), which biases the motor toward forward motion. In addition, the ADP affinity for state 4 is relatively low (KD2 > 60 μM), suggesting that the strain generated by the leading head on the trail head reduces the free energy for ADP release from the trail head (25). When the motor travels along path B, the effective rate of attachment, Pi release, and working step of the lead head (state 2 → 5, k2 = 572 sec–1) is nearly 2 orders of magnitude greater than the equivalent step through path A (state 1 → 4, k1 = 5 sec–1), indicating that this transition is made more favorable by the ADP-induced rotation of the lever (26–29) of the trailing head moving the leading head closer to its actin-binding site. Thus, depending on its chemical state, the trailing head has the capacity to modulate the rate at which the lead head attaches to actin. This cooperative mechanism may be critical for myosin V processivity.
What terminates a processive run? Because both reduced [ATP] and increased [ADP] should increase the probability that at least one head of a myosin V molecule is bound strongly to actin, the observations that the run length decreased only slightly with increasing [ATP] and decreased dramatically with increasing [ADP] were both quite surprising (Figs. 2 and 3). A processive run is at risk of being terminated when the motor runs a gauntlet through a state that is vulnerable to detachment, presumably a state with only one head strongly bound to actin or both heads weakly bound. Our data imply that state 1 is the vulnerable state (as well as the predominant state; see above) and that spontaneous detachment from this state occurs at a relatively low rate (kterm 1 = 1.1 sec–1), as expected for a singly attached, strongly bound head. This ensures a low probability of run termination (≈0.04) and an average processive run of ≈23 steps. The fact that kterm 1 (1.1 sec–1) is significantly larger than the value of 0.032 sec–1 obtained in solutions studies of single-headed myosin V (13) indicates that either there are differences between single- and double-headed myosin V constructs or that all the factors that contribute to termination are not fully understood yet. Any perturbation that increases the lifetime of the vulnerable state will necessarily decrease run length. With the motor traveling down both paths A and B, the rapid decline in run length with ADP (Fig. 3) is caused by ADP binding with a high affinity (KD1 = 1 μM) to a singly attached head (state 2), backing myosin V into the vulnerable state (i.e., state 1). At ADP concentrations that saturate the singly attached head, myosin V is forced down path A, at which further addition of ADP backs myosin V into a state that is not vulnerable to detachment (state 4, with two strongly bound heads), consistent with little additional effect on run length from reversal of this path (see Fig. 3). Decreasing [ATP] below 1 mM has little effect on run length, because the time spent waiting in state 5 for ATP to bind does not change the fact that myosin must run a gauntlet through state 1 to take a step forward. We suggest that only at ATP concentrations much greater than 1 mM will path C be significantly populated and run lengths be dramatically diminished.
It seems that myosin V has evolved an elaborate system of cooperative mechanisms to help maintain its processive movement. Depending on the kinetic pathway traveled, the two heads vary their communication scheme in an effort to keep their biochemical cycles out of phase such that at least one head is always attached. When traveling down path A, the heads behave like opposing magnets, where the attached trail head inhibits attachment of the lead head (state 1 → 4), but once attached the lead head accelerates the detachment of the trail head (state 4 → 5). Whereas if the molecule chooses path B, the attached trail head is now one step closer to a highly vulnerable state (state 3) and thus accelerates the attachment of the lead head (state 2 → 5) to ensure that the lead head is attached before the trail head detaches. With multiple pathways to choose from, a level of flexibility may be built into the myosin V molecule so that the heads can adopt different mechanisms of coordination to maintain processivity under physiological conditions that may vary within a cell. For example, the intracellular milieu in which the myosin V must drag its cargo may result in loads that alter the kinetics for processive movement (2), favoring one path over the other, as proposed for kinesin, a microtubular-based processive motor (30, 31).
Acknowledgments
We thank A. Federico for assistance with experiments; J. Moore, N. Kad, and N. Debold for helpful discussions; and S. Work for software development. We want to especially thank Dr. Andrej Vilfan for his insightful review and suggestions to our kinetic model. This study was funded by National Institutes of Health Grants AR47906 (to D.M.W. and K.M.T.) and HL38113 (to K.M.T.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: YFP, yellow fluorescent protein; HMMM5, heavy meromyosin V; TIRF, total internal reflectance fluorescence; AB, actin buffer.
References
- 1.Mehta, A. D., Rock, R. S., Rief, M., Spudich, J. A., Mooseker, M. S. & Cheney, R. E. (1999) Nature 400, 590–593. [DOI] [PubMed] [Google Scholar]
- 2.Rief, M., Rock, R. S., Mehta, A. D., Mooseker, M. S., Cheney, R. E. & Spudich, J. A. (2000) Proc. Natl. Acad. Sci. USA 97, 9482–9486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sakamoto, T., Amitani, I., Yokota, E. & Ando, T. (2000) Biochem. Biophys. Res. Commun. 272, 586–590. [DOI] [PubMed] [Google Scholar]
- 4.Pastural, E., Barrat, F. J., Dufourcq-Lagelouse, R., Certain, S., Sanal, O., Jabado, N., Seger, R., Griscelli, C., Fischer, A. & de Saint, B. G. (1997) Nat. Genet. 16, 289–292. [DOI] [PubMed] [Google Scholar]
- 5.Menasche, G., Ho, C. H., Sanal, O., Feldmann, J., Tezcan, I., Ersoy, F., Houdusse, A., Fischer, A. & de Saint, B. G. (2003) J. Clin. Invest. 112, 450–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moore, J. R., Krementsova, E. B., Trybus, K. M. & Warshaw, D. M. (2001) J. Cell Biol. 155, 625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Veigel, C., Wang, F., Bartoo, M. L., Sellers, J. R. & Molloy, J. E. (2002) Nat. Cell Biol. 4, 59–65. [DOI] [PubMed] [Google Scholar]
- 8.Purcell, T. J., Morris, C., Spudich, J. A. & Sweeney, H. L. (2002) Proc. Natl. Acad. Sci. USA 99, 14159–14164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walker, M. L., Burgess, S. A., Sellers, J. R., Wang, F., Hammer, J. A., III, Trinick, J. & Knight, P. J. (2000) Nature 405, 804–807. [DOI] [PubMed] [Google Scholar]
- 10.Ali, M. Y., Uemura, S., Adachi, K., Itoh, H., Kinosita, K., Jr., & Ishiwata, S. (2002) Nat. Struct. Biol. 9, 464–467. [DOI] [PubMed] [Google Scholar]
- 11.Yildiz, A., Forkey, J. N., McKinney, S. A., Ha, T., Goldman, Y. E. & Selvin, P. R. (2003) Science 300, 2061–2065. [DOI] [PubMed] [Google Scholar]
- 12.Forkey, J. N., Quinlan, M. E., Shaw, M. A., Corrie, J. E. & Goldman, Y. E. (2003) Nature 422, 399–404. [DOI] [PubMed] [Google Scholar]
- 13.De la Cruz, E. M., Wells, A. L., Rosenfeld, S. S., Ostap, E. M. & Sweeney, H. L. (1999) Proc. Natl. Acad. Sci. USA 96, 13726–13731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trybus, K. M., Krementsova, E. & Freyzon, Y. (1999) J. Biol. Chem. 274, 27448–27456. [DOI] [PubMed] [Google Scholar]
- 15.Wang, F., Chen, L., Arcucci, O., Harvey, E. V., Bowers, B., Xu, Y., Hammer, J. A., III, & Sellers, J. R. (2000) J. Biol. Chem. 275, 4329–4335. [DOI] [PubMed] [Google Scholar]
- 16.Axelrod, D. (1989) Methods Cell Biol. 30, 245–270. [DOI] [PubMed] [Google Scholar]
- 17.Sakamoto, T., Wang, F., Schmitz, S., Xu, Y., Xu, Q., Molloy, J. E., Veigel, C. & Sellers, J. R. (2003) J. Biol. Chem. 278, 29201–29207. [DOI] [PubMed] [Google Scholar]
- 18.Krementsov, D. N., Krementsova, E. B. & Trybus, K. M. (2004) J. Cell. Biol. 164, 877–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fabiato, A. & Fabiato, F. (1979) J. Physiol. (Paris) 75, 463–505. [PubMed] [Google Scholar]
- 20.Warshaw, D. M., Desrosiers, J. M., Work, S. S. & Trybus, K. M. (1990) J. Cell Biol. 111, 453–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Work, S. S. & Warshaw, D. M. (1992) Anal. Biochem. 202, 275–285. [DOI] [PubMed] [Google Scholar]
- 22.Tanaka, H., Homma, K., Iwane, A. H., Katayama, E., Ikebe, R., Saito, J., Yanagida, T. & Ikebe, M. (2002) Nature 415, 192–195. [DOI] [PubMed] [Google Scholar]
- 23.Mehta, A. (2001) J. Cell Sci. 114, 1981–1998. [DOI] [PubMed] [Google Scholar]
- 24.De la Cruz, E. M., Ostap, E. M. & Sweeney, H. L. (2001) J. Biol. Chem. 276, 32373–32381. [DOI] [PubMed] [Google Scholar]
- 25.Baker, J. E., Brosseau, C., Fagnant, P. & Warshaw, D. M. (2003) J. Biol. Chem. 278, 28533–28539. [DOI] [PubMed] [Google Scholar]
- 26.Whittaker, M., Wilson-Kubalek, E. M., Smith, J. E., Faust, L., Milligan, R. A. & Sweeney, H. L. (1995) Nature 378, 748–751. [DOI] [PubMed] [Google Scholar]
- 27.Gollub, J., Cremo, C. R. & Cooke, R. (1996) Nat. Struct. Biol. 3, 796–802. [DOI] [PubMed] [Google Scholar]
- 28.Cremo, C. R. & Geeves, M. A. (1998) Biochemistry 37, 1969–1978. [DOI] [PubMed] [Google Scholar]
- 29.Veigel, C., Molloy, J. E., Schmitz, S. & Kendrick-Jones, J. (2003) Nat. Cell Biol. 5, 980–986. [DOI] [PubMed] [Google Scholar]
- 30.Schnitzer, M. J., Visscher, K. & Block, S. M. (2000) Nat. Cell Biol. 2, 718–723. [DOI] [PubMed] [Google Scholar]
- 31.Yajima, J., Alonso, M. C., Cross, R. A. & Toyoshima, Y. Y. (2002) Curr. Biol. 12, 301–306. [DOI] [PubMed] [Google Scholar]