

Abstract
The elderly exhibit increased susceptibility to a number of inflammatory or degenerative pathologies. Aging is similarly thought to be associated with increased prevalence and severity of periodontitis, although the underlying causes are poorly understood. Among the plausible mechanisms whereby aging could contribute to increased susceptibility to periodontitis are age-dependent alterations in the innate immune and inflammatory status of the host. This hypothesis is supported by studies in humans and animal models outlined in this Review. Indeed, innate immune cells isolated from elderly subjects exhibit age-related cell-intrinsic defects that could predispose the elderly to deregulated immune and inflammatory responses. Moreover, the investigation of age-related alterations in the tissue environment where recruited inflammatory cells ultimately function could provide complementary, if not better, insights into the impact of aging on periodontitis. Integrative approaches combining in vitro and in vivo mechanistic models are underway and can potentially contribute to targeted molecular therapies that can reverse or mitigate the effects of aging on periodontitis and other inflammatory diseases.
1. Introduction
The prevalence of aging-associated conditions (neurodegenerative disorders; autoimmune, inflammatory, and cardiovascular diseases; cancer; and increased susceptibility to infections) has increased as a result of longer life expectancy in developed countries over the past 170 years, in turn reflecting an improvement of socio-economic conditions [1]. The prevalence of periodontitis also seems to have risen. The most recent epidemiologic data in the adult U.S. population revealed that the prevalence of total periodontitis (i.e., the sum of mild, moderate, and severe forms of the disease) is higher than earlier estimates, affecting almost 50% of adults [2]. However, this increase may be due, at least in part, to improved examination methodology [3]. Periodontitis arises from complex interactions between the host and the subgingival microbiota that lead to the inflammatory destruction of the tooth-supporting connective tissue and alveolar bone [4–6]. The periodontal inflammatory response is modulated by a number of factors, including the genetic background and immuno-inflammatory status of the individuals, and the presence of environmental stressors and/or systemic disease [7–10].
The elderly exhibit increased susceptibility to a number of autoimmune, inflammatory, or infectious diseases, and aging is similarly thought to be associated with increased prevalence and severity of periodontitis [11–17]. Aging is associated with increased severity of naturally occurring periodontitis also in non-human primates and rodents, which are thus useful models for investigating relevant age-related alterations [18, 19].
The impact of aging on periodontitis is poorly understood at the mechanistic level. It has been suggested that aging by itself does not necessarily cause periodontitis [20], which is consistent with the notion that aging per se is not a disease [21]. Aging by itself leads to physiological loss of periodontal attachment and alveolar bone but these changes are quite modest and have little clinical significance, unless in the presence of concomitant periodontal inflammation seen in a significant portion of the elderly population [13, 22]. Briefly stated, periodontitis in the elderly could be a consequence of inflammation independent of the aging process per se. It could thus be argued that the increased prevalence and severity of periodontitis in old age simply reflects the cumulative effect of prolonged exposure to the periodontal microbial challenge (The cumulative effect hypothesis; Fig. 1A).
Figure 1. Impact of aging on periodontitis: competing hypotheses.
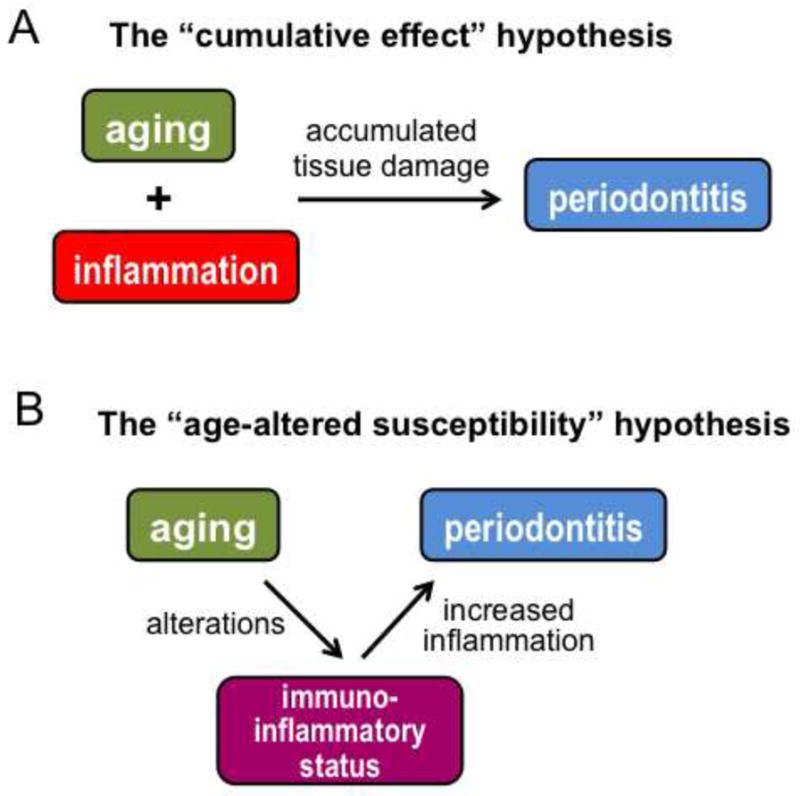
(A) According to the “cumulative effect” hypothesis, aging and inflammation function independently in periodontal pathogenesis. Accordingly, the reason that the elderly have increased prevalence and severity of periodontitis is related to prolonged exposure and thus accumulated tissue damage. (B) According to the “age-altered susceptibility”, aging contributes to periodontal pathogenesis through age-dependent alterations in the immuno-inflammatory status of the host. This is a productive hypothesis that seeks to identify changes in the immunology and/or physiology of the aging periodontal tissue that render the elderly more susceptible to periodontitis than the young.
Alternatively, it could be argued through testable hypotheses that aging causes alterations to the immuno-inflammatory status of the periodontal tissue, which in turn contributes to increased susceptibility to periodontitis (The age-altered susceptibility hypothesis; Fig. 1B). Limited evidence supports a possible role for age-related alterations in the periodontal immune response. In an experimental gingivitis study involving young (20–25 years of age) and elderly (≥ 65 years) individuals, all subjects received professional dental care to establish healthy gingival starting conditions and, subsequently, abstained from oral hygiene for three weeks. Although young and old subjects had comparable dental plaque biofilm accumulation, the latter group developed more severe gingivitis associated with increased accumulation of inflammatory cells [23]. In a similar experimental gingivitis study with young and elderly subjects, the latter group responded with significantly higher levels of IL-1β in the gingival crevicular fluid [24]. Although the mechanistic basis of these observations is uncertain, a likely interpretation is that the regulation of inflammatory cell recruitment to the periodontium becomes defective with aging. Additionally, the possibility that recruited inflammatory cells may exhibit an age-associated hyperactive phenotype cannot be ruled out. To date, only a few studies have attempted to understand the mechanisms, if any, by which aging can affect periodontal inflammation and hence susceptibility to periodontitis. At least in part, this may be due to formidable challenges inherently associated with research on aging (discussed below). Most of the studies on aging have investigated in vitro functional differences between cell types isolated from young and old subjects. Although findings from such studies may not always be relevant to the in vivo setting, the most important findings will be discussed and summarized as they do provide potential insights into how aging may influence the immune response. Overall, the objective of this review was to synthesize available literature into useful concepts for better understanding of the complex interplay between aging and inflammatory diseases such as periodontitis, and to provide stimuli for future research.
2. Challenges in aging research
The genetic heterogeneity among human subjects is a challenge for any type of biomedical research. However, aging research is affected by additional sources of heterogeneity that accrue over time, including heterogeneity in exposure to infectious agents, use of antibiotics, anti-inflammatory agents and other medications, and comorbid medical conditions. To resolve or mitigate the effects of such confounding factors and facilitate the study of the impact of aging per se on the immune system, Ligthart and colleagues developed the SENIEUR protocol. This protocol established rigorous enrollment criteria for immunogerontological studies, on the basis of clinical and laboratory data, and set limits to pharmacological interference [25]. However, at least in one study, only 30% of male and female subjects who considered themselves healthy actually fulfilled the criteria [26] raising questions that such a strict protocol may select for an uncommonly healthy subset of aged individuals [27]. Such studies therefore may yield findings that may not be extrapolatable to the general elderly population, although they could be complementary to the results of studies examining old individuals with comorbid conditions. Therefore, at least in principle, the findings from studies that follow the SENIEUR protocol or recruit subjects with comorbid conditions could collectively contribute to determining the influence of disease versus aging on the immune system. However, the interplay of aging and inflammatory disease is a highly convoluted process without obvious causality or directionality. For instance, it would be very challenging to dissect whether age-related processes have caused or resulted from inflammation, or whether there is bidirectionality. Longitudinal studies can generate much more useful and relevant information than cross-sectional studies. However, longitudinal studies are quite rare in aging research, as the logistical and financial constraints, normally encountered in any human study, become even more challenging in aging studies due their very nature [28].
3. Age-related alterations in innate immune cells
It is thought that many diseases of aging (e.g., autoimmune disorders, cardiovascular diseases, cancer, and increased susceptibility to infections) are linked to immunosenescence, i.e., age-related alterations in the immune system that impair its proper function. Age-related alterations affect both the adaptive and innate arms of immunity, although the impact of aging on the latter is less well understood [12, 29]. It has been firmly established that adaptive immunity declines with age due to decreased production of naïve T cells, decreased diversity of antigen-recognition repertoire, signal transduction defects in T cells with altered cytokine induction patterns, and reduced clonal expansion and function of antigen-specific T and B cells [12, 30]. The decreased production of naïve T cells is in part linked to the involution of the thymus with age. Moreover, the frequency of committed B lymphoid progenitor cells declines with aging, as hematopoietic stem cells undergo a skewing to myeloid-lineage commitment at the expense of lymphoid progenitors [31].
Age-related alterations in innate immunity do not necessarily cause immunodeficiency but rather dysregulation of the host response, leading to either loss or gain of immune activity [14, 29, 32, 33]. A great emphasis has been placed on the study of neutrophils, monocytes/macrophages, and dendritic cells due to their central role in innate immunity. As alluded to above, to better study intrinsic effects of aging on innate immune cells, cells are often isolated from “healthy elderly subjects”, although this may not always be easy to determine. Therefore, studies examining the impact of aging on these cell types have often led to inconsistent results and the discussion below summarizes consensus findings (Table 1) unless otherwise stated.
Table 1.
Effects of aging on phagocytes and antigen-presenting cells
| Functional activity | Cell type | ||
|---|---|---|---|
| Neutrophils | Monocytes/Macrophages | Dendritic cells | |
| Reduced | Signal transduction (Ca2+ influx, phosphorylation of ERK, p38, Akt, PLC-γ) Receptor recruitment to lipid rafts (TLR4, TREM-1) Chemotaxis (fMLP, GM-CSF) CD16 expression Phagocytosis Microbicidal activity Reactive oxygen species (LPS, fMLP, GM-CSF, opsonized bacteria) |
Signal transduction (e.g., total levels and/or activation of STAT-1α, p38 & JNK MAPKs, MyD88, NF-κB) Cytokine production (IL-12, IL-6, TNF, MIP-1α, MIP-1β, MIP-2) Chemotaxis Phagocytosis Reactive oxygen species Reactive nitrogen species Intracellular killing Expression of costimulatory molecules (CD80 and CD86), MHC Class II, Expression of CD14, TLR1, and TLR3* |
Antigen presentation Chemotaxis Endocytosis Production of IFN type I and III (pDC) IL-12 PI3K activity and Akt phosphorylation Expression of TLR-1,-3,-5, -7, and -8 |
| Maintained | Total number of circulating neutrophils Basal levels of receptor expression (GM-CSFR, TLR2, TLR4, CD14, CD11b) Expression of adhesion molecules and adhesion to endothelial cells Apoptosis (spontaneous) |
Expression of IFN-γ receptor and TLR2 Expression of TLR negative regulators (e.g., SOCS-1, IRAK-M, A20, PPAR-γ)** |
Expression of TLR2 (mDC) and TLR9 (pDC) |
| Increased | Apoptosis (under priming conditions; e.g., impaired anti-apoptotic signals after exposure to GM-CSF) Activity of cytokine-signaling inhibitory molecules (SHP-1, SOCS) |
PGE2 production TLR5 expression |
TNF, IL-6, IL-23 Basal expression of CD80, CD83, CD86*** Basal NF-κB activity |
Basal levels only
In mice, not yet reported for humans
DC isolated directly from peripheral blood
3.1. Neutrophils
The total number of circulating neutrophils does not change in old age [14]. Moreover, neutrophils from young or old individuals display comparable expression of adhesion molecules and capacity for adhesion to endothelial cells. On the other hand, neutrophil chemotaxis in response to in vitro stimulation with granulocyte-macrophage colony-stimulating factor (GM-CSF) or the N-formyl-Met-Leu-Phe (FMLP) peptide is significantly reduced in old age, even when the neutrophils are obtained from healthy elderly individuals [34–36]. However, it is uncertain whether impaired chemotaxis is stimulus-specific or applies to also other chemotactic simuli such as chemokine C-X-C receptor 2 ligands. In vitro studies also suggest that neutrophils exhibit a number of age-dependent functional defects in their effector functions (Table 1). These include impaired reactive oxygen species generation, phagocytosis, and microbicidal activity [14, 33, 37]. These changes cannot be readily attributed to reduced expression of receptors involved in microbe recognition and/or neutrophil activation. For example, the expressions of Toll-like receptor (TLR)-2 and TLR4 (which respond to bacterial lipopeptides and LPS, respectively) and GM-CSF receptor (GM-CSFR) are not affected as a function of age [38, 39]. However, the signaling pathways downstream of these receptors may be affected due to age-associated alterations in the membrane lipid rafts that serve as cellular signaling platforms. For instance, altered TLR4 signaling in aged human neutrophils was attributed to changes in the recruitability of this receptor to lipid rafts which experience age-related physicochemical changes [38]. Specifically, the cholesterol content of lipid rafts appears to increase with aging resulting in reduced membrane fluidity [40]. Altered signaling (manifested as reduced phosphorylation of Akt and phospholipase C-γ; PLC-γ) by the triggering receptor expressed on myeloid cells (TREM)-1 in aged neutrophils was similarly attributed to defective recruitment of TREM-1 to lipid rafts [41]. As TREM-1 enhances neutrophil phagocytosis and degranulation, age-related impaired TREM-1 signaling could compromise neutrophil-mediated defenses in old age, thereby contributing to the documented increased susceptibility of the elderly to microbial infections [39]. Neutrophils from elderly individuals additionally show increased activity of cytokine-signaling inhibitory molecules, such as Src homology domain-containing protein tyrosine phosphatase-1 (SHP-1) and suppressors of cytokine signaling (SOCS), which may explain the reduced potency of GM-CSF to prime and to activate the oxidative burst in neutrophils from old subjects [42].
Although neutrophil phagocytosis declines with aging, no significant age-dependent differences were observed in the expression of major phagocytic receptors such as CD11b and CD14 [34]. This suggests that the defect could likewise lie in signal transduction, or that the expression of other important phagocytic receptors is affected. In this regard, CD16 expression is significantly reduced in neutrophils from elderly subjects [35]. Additional age-associated defects in signal transduction, such as decreased FMLP-induced Ca2+ responses and GM-CSF- or FMLP-induced activation of extracellular receptor-activated kinase (ERK) or p38 mitogen-activated protein kinase (MAPK) have also been documented and could similarly contribute to impaired neutrophil function in elderly individuals [29].
No significant differences were noted in the spontaneous apoptosis of neutrophils isolated from young and old subjects; however, the ability of priming agents, such as GM-CSF, complement fragment C5a, or bacterial LPS to induce anti-apoptotic signals is reduced in neutrophils from the elderly [34]. The diminished GM-CSF-induced delayed apoptosis in the neutrophils of the elderly was linked to alterations in the p42/p44 MAPK pathway, leading to overexpression of pro-apoptotic Bcl-2 family members and the activation of caspase-3 [43]. If this observation is relevant in vivo (e.g., during infections of the elderly) neutrophils should die off more rapidly than in young individuals, potentially compromising infection control in old individuals. Moreover, it has been proposed that the clearance of apoptotic cells may be impaired in old age, partly due to reduced expression of macrophage phagocytic receptors such as CD14 [44]. Other possible contributory mechanisms, which would be important to investigate, include reduced expression of phagocytic receptors dedicated to apoptotic cell clearance (e.g., c-mer proto-oncogene tyrosine kinase) or opsonins that bridge these receptors to phosphatidylserine on the apoptotic cell surface. If apoptotic cell phagocytosis is indeed impaired in old age, such defect, in combination with the accelerated neutrophil apoptosis during infection or inflammation, could greatly contribute to inflammatory tissue damage from eventual necrosis of non-cleared apoptotic neutrophils and release of toxic substances.
3.2. Monocytes/macrophages
Unlike neutrophils which can be readily obtained in great numbers resulting in a plethora of aging studies focusing on this cell type, relatively few studies have been performed to investigate monocyte/macrophage age-related functional changes. Studies in humans have shown that several functions of monocytes or macrophages become compromised in old age, including chemotaxis, phagocytosis, production of certain cytokines/chemokines and reactive oxygen or nitrogen species, and expression of MHC Class II and costimulatory molecules [14, 33, 45] (Table 1). Interestingly, the production of prostaglandin E2 is increased in activated macrophages from elderly individuals compared to those from younger subjects, which may explain the age-associated inhibition of MHC Class II expression and IL-12 production [14].
Age-related altered expression and function of monocytic TLRs has also been proposed, but more studies are required to draw firm conclusions. Although the expression of TLR2 is comparable in monocytes from young and elderly individuals, the expression of monocytic TLR1 was reported to decline in old age [46]. This is expected to impair a subset of TLR2-dependent responses since TLR2 signals in obligatory cooperation with either TLR1 or TLR6 [47]. Importantly, reduced TLR1 surface expression on monocytes correlates with increased risk of reactivation of Mycobacterium tuberculosis infection [48]. Macrophages from elderly subjects have lower basal levels of TLR3 compared to macrophages from young individuals. However, whereas the young downregulate TLR3 expression in response to West Nile virus (WNV) infection, the elderly fail to do so [49]. This defect was attributed to impaired signaling in the transducer and activator of transcription 1 (STAT-1)-mediated pathway, and may lead to sustained proinflammatory cytokine production. This in turn could contribute to increased permeability of the blood-brain barrier, thereby suggesting a plausible mechanism for the increased severity of WNV infection in the elderly [49]. In contrast, the expression of TLR5 mRNA and protein in monocytes is increased in the elderly compared to young subjects, leading to increased phosphorylation of MAPK p38 and ERK and higher production of IL-8 [50].
Studies in mice may offer additional insights into TLR expression in old age. An early study showed that macrophages from old mice have reduced mRNA expression of TLR1-9, although at the protein level the results were confirmed only for TLR4 [51]. Accordingly, LPS-induced cytokine responses were found to decline with age [51]. Although this finding was confirmed by an independent study, the age-associated decrease in cytokine responses was attributed to decreased expression of MAPK, rather than decreased TLR4 expression that actually remained unaffected [52].
A study examining age-dependent alterations in key intracellular or transmembrane negative regulators of TLR signaling showed that most of these molecules had comparable expression in macrophages from young and old mice (e.g., suppressor of cytokine signaling-1 [SOCS-1], interleukin-1 receptor-associated kinase M [IRAK-M], the ubiquitin-editing enzyme A20, and peroxisome proliferative activated receptor-γ [PPAR-γ]) [53]. With regard to signaling intermediates that positively regulate macrophage activation, studies in mouse macrophages have shown an age-dependent decrease in the total levels and phosphorylation capacity of STAT-1α and the MAPKs (p38 and Jun N-terminal kinase [JNK]), and the levels of MyD88 and NF-κB [29, 33]. The age-dependent reduced activity of STAT-1α is likely responsible for the suppression of IFN-γ-mediated priming or activation of macrophages, even though the expression of the IFN-γ receptor is preserved in old age [54]. The age-associated reduction in the levels of MAPK and NF-κB, as well as of MyD88, a major TLR signaling adaptor, similarly suggest that the capacity of macrophages for activation may decline with aging. This notion is consistent with the decreased induction of certain cytokine responses in vitro (e.g., IL-6, TNF, and MIP-1α) [29, 54] (Table 1).
3.3 Dendritic cells
Studies in humans have shown that myeloid dendritic cells (mDC) display age-related decline in several functions such as chemotaxis, endocytosis, production of IL-12 and antigen-presentation, resulting in suppressed activation of naïve CD4+ T cells [14, 33] (Table 1). However, mDC from old subjects produce higher levels of certain cytokines such as TNF, IL-6, and IL-23. Some of the above observations may be explained, at least in part, by findings that mDC from old individuals have reduced PI3K activity, which positively regulates phagocytosis and chemotactic migration but negatively regulates TLR signaling [55]. In terms of TLR expression by mDC, old age is associated with reduced expression of TLR-1, -3, -5, and -8 but not of TLR2 [56]. Interestingly, mDC isolated directly from the peripheral blood of elderly subjects display increased basal expression of the DC activation marker CD83 and the co-stimulatory molecules CD80 and CD86, but these differences disappear when the mDC are generated in vitro using peripheral blood mononuclear cells from young and old subjects [33].
In old age, plasmacytoid dendritic cells (pDC), which are important in host defense against viruses, show decreased production of type I and type III interferon, partly attributed to impaired phosphorylation of a crucial transcription factor (IRF-7) [57]. Decreased production of IFN-α by pDC from old individuals, as compared to young controls, was also observed in another study, which attributed the effect to reduced expression of TLR7 [58]. In contrast, TLR9 expression is not reduced in pDC with aging [56]. These changes were accompanied by defects in cytokine production that were in turn associated with poor antibody responses to influenza immunization [56]. Although there is no general consensus as to the effect of aging on the numbers of mDC and pDC, it appears that DC from the elderly exhibit elevated basal NF-κB activity [33]. This primed state of DC from elderly subjects may contribute to their increased reactivity to self-antigen such as DNA, which in turn may contribute to age-associated autoimmune inflammation [59]
4. Inflamm-aging and periodontitis
The capacity to elicit robust immune and inflammatory responses may have been selected during evolution to allow survival to reproductive age, although– ironically– such inclination may be a contributory factor to the development of chronic inflammatory diseases now that humans routinely survive to old age. The term “inflamm-aging” has been aptly coined to describe the heightened chronic inflammatory status often associated with old age in humans [32]. Periodontitis was proposed to be a contributory factor to inflamm-aging [60] since periodontitis in the elderly is associated with elevated systemic inflammatory markers (e.g., C-reactive protein, IL-6, and TNF) [61]. In general, however, it is uncertain whether inflamm-aging contributes to the development of inflammatory or degenerative chronic diseases, or whether it is the chronic pathologies that cause inflamm-aging, or even whether inflamm-aging is both a cause and a consequence in a vicious cycle where inflammation and chronic disease reinforce each other.
Nevertheless, even in the apparent absence of an immune stimulus, elderly individuals without underlying disease may still exhibit significantly higher circulating levels of cytokines, such as IL-6, IL-1β, and TNF, and acute-phase proteins, such as C-reactive protein and fibrinogen [60, 62]. Such low-grade chronic inflammation is associated with poor prognosis following systemic insults (burn, trauma, sepsis) and is a predictor of frailty and mortality in the elderly [60, 62, 63], although its impact on periodontitis has not yet been addressed. The elevated baseline production of inflammatory cytokines in inflamm-aging can co-exist with reduced capacity to produce cytokines upon stimulation, and this combination correlates with increased risk of sepsis following community-acquired pneumonia in the elderly [64]. On the other hand, the systemic cytokine profile of long-lived humans or mice appears to be normal and quite similar to that of their young counterparts [62, 65, 66]. This lends further support to the concept that inflamm-aging contributes to frailty and mortality in old age.
From a mechanistic standpoint, inflamm-aging may not necessarily or exclusively be related to age-related dysregulation of basal cytokine production but may result from a variety of other factors including increased amount of fat tissue (a significant source of inflammatory cytokines), decreased production of sex steroid hormones (many of which have anti-inflammatory action), and lack of physical activity [60, 67, 68]. Conversely, caloric restriction appears to protect against chronic inflammation and recent studies in non-human primates demonstrated that a caloric-restricted diet inhibits periodontal inflammation and clinical attachment loss [69]. Incessant oxidative stress may also contribute to inflamm-aging [70]. Age-associated increases in reactive oxygen species (in part due to a decline in anti-oxidative defense systems) can induce inflammation either directly or through post-translational modifications (e.g., oxidized lipoproteins) by activating redox-sensitive transcription factors associated with TLR pathways or the inflammasome [70, 71]. In this regard, peripheral blood neutrophils from periodontitis patients exhibit a hyper-responsive phenotype in terms of reactive oxygen species production, even in the absence of exogenous stimulation [72]. Therefore, neutrophils recruited to the periodontium can contribute to periodontal pathogenesis, at least in part, by causing oxidative tissue damage [73]. However, whether aging is positively correlated with increased capacity of neutrophils to mediate oxidative damage in the periodontium has not been addressed yet.
To directly determine the importance of oxidative stress in periodontal pathogenesis, a recent study investigated the effects of an anti-oxidant compound, the reduced form of coenzyme Q10 (rCoQ10), in an aging model of rat periodontitis. Long-term topical application of rCoQ10 onto the gingiva of aging rats inhibited oxidative DNA damage, expression of genes encoding inflammatory mediators including components of the Nod-like receptor protein (NLRP)3 inflammasome, and osteoclastogenesis, in contrast to control treatment with vehicle only [74]. Vitamin D is a powerful natural antioxidant and a recent study showed a correlation between vitamin D intake (≥ 800 IU) and protection against periodontal disease progression in relatively old age (men with an average age of 63) [75]. However, it should be noted that vitamin D has pleiotropic effects in immune regulation and inflammation [76] including regulation of local immunity at the periodontal tissue [77].
5. Importance of the local microenvironment: age-associated alterations in the periodontium
The above-discussed age-dependent alterations in innate immune cell function could, at least in principle, contribute to enhanced susceptibility to periodontitis. For instance, the age-associated impairment of phagocytic and microbicidal activities in neutrophils and macrophages could result in uncontrolled growth of periodontal bacteria into dysbiotic communities. Additionally, the age-related increased macrophage production of PGE2 could contribute to inflammatory bone loss. Elevated PGE2 production was also seen in fibroblasts from old rats (as compared to fibroblasts of young rats) as well as in in vitro-aged human gingival fibroblasts and periodontal ligament fibroblasts [78]. Moreover, the age-associated elevated basal NF-κB activity of DC, together with their increased numbers in the periodontium of the elderly [79], might contribute to enhanced local inflammation.
In general, however, results from in vitro studies need to be interpreted with caution. First, cells in their in vivo environment may not simply be affected by age-dependent, cell-intrinsic changes but also by age-dependent alterations to their tissue environment. For instance, the function of macrophages is largely dictated by the type and condition of the tissue they reside [45]. At least some of the innate immune dysfunction seen in old age may be due to inefficient communication between macrophages (or other innate immune cells) and the tissues, which in old age appear to express lower levels of adhesion molecules and display reduced responsiveness to growth factors [45]. In a similar context, aging causes increased basal expression of costimulatory molecules in mDC isolated directly from the peripheral blood but not in mDC generated in vitro [33]. In general, cells from elderly donors may still exhibit normal differentiation when exposed to appropriate growth and other factors in vitro but their cellular phenotype may be altered in their aging microenvironment. Moreover, although induction of TNF production by human dermal macrophages is decreased in old individuals compared to young controls, there are no differences in TNF production upon ex vivo stimulation of such macrophages, further suggesting the importance of the microenvironment in modulating the function of innate immune cells [80]. Therefore, tissues may have greater control, than traditionally appreciated, over the host inflammatory response. This should not be surprising given that tissues can communicate with and regulate the immune system via tissue-derived signals (e.g., cytokines, chemokines, antimicrobial peptides, and growth factors) [45, 81].
Although the in vitro induction of anti-apoptotic signals is reduced in neutrophils from the elderly [34], a study in non-human primates showed that aged animals had higher expression of anti-apoptotic and lower expression of pro-apoptotic genes in their gingival tissue as compared to gingival tissue from young animals [82]. This suggests that the frequency of apoptotic events could be reduced with aging [82]. It should be noted, however, that this study examined apoptotic pathways in the whole gingival tissue and was not specific for neutrophils.
The notion that tissues may have a “regulatory say” over the host inflammatory response is supported by the recent identification of locally produced endogenous modulators of the leukocyte recruitment cascade [83]. One such endogenous inhibitor, which is produced in the periodontal tissue, is a 52-kDa endothelial cell-secreted protein termed developmental endothelial locus-1 (Del-1), also known as EGF-like repeats and discoidin I-like domains 3 (EDIL3) [83]. Del-1 competes with intercellular adhesion molecule-1 (ICAM-1) for binding to the LFA-1 integrin on neutrophils and thereby inhibits their firm adhesion to the endothelium and subsequent transmigration [84]. Consistent with a homeostatic role for Del-1, Del-1–deficient mice develop spontaneous periodontitis featuring excessive neutrophil infiltration and IL-17–dependent inflammatory bone loss [85]. Strikingly, the expression of Del-1 is diminished in the murine gingival tissue in old age, correlating with the development of periodontitis that shares similar features with periodontitis due to genetic Del-1 deficiency. Del-1 and IL-17 are reciprocally regulated and an inverse expression of Del-1 and IL-17 is observed in human gingival biopsy samples, with Del-1 dominating in healthy gingiva and IL-17 in inflamed gingiva [85].
These findings indicate that Del-1 serves as a mechanism whereby a tissue can locally self-regulate persistent inflammation associated with chronic recruitment of neutrophils. This regulatory mechanism breaks down in old age due to an age-related decline in Del-1 mRNA and protein expression (Fig. 2). It is uncertain, however, whether the age-associated downregulation of Del-1 expression is a direct effect of aging or whether the primary defect involves an upregulation of IL-17 with advancing age. Regardless of the precise mechanism, local periodontal treatment with soluble Del-1 in old mice inhibits IL-17 production, LFA-1-dependent neutrophil infiltration, and bone loss [85]. Intriguingly, immune-privileged tissues/organs, such as the eye and the brain, have significantly higher expression of Del-1 than peripheral tissues, such as the lung or the gingiva [84, 85]. Whether these tissues similarly display age-dependent decline in Del-1 expression that could contribute to inflammatory or degenerative diseases (e.g., multiple sclerosis or age-related macular degeneration) is yet to be investigated.
Figure 2. Del-1 vs. IL-17 in aging and periodontitis.
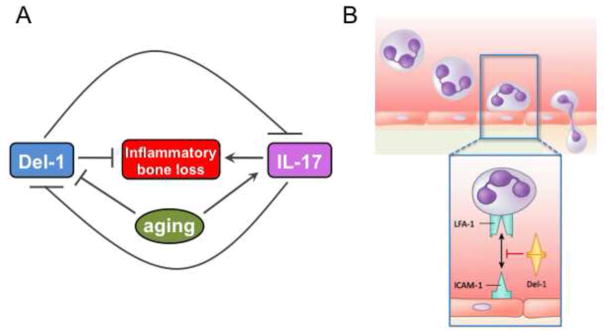
(A) Del-1 and IL-17 exert opposing effects on periodontal pathogenesis, with the former acting homeostatically to block IL-17–mediated inflammatory bone loss. However, Del-1 expression declines with aging resulting in increased production of IL-17, which in turn leads to increased inflammation and periodontal bone loss in old age. As Del-1 and IL-17 are reciprocally regulated, it is uncertain whether the age-associated reduction in Del-1 expression is the cause or the consequence of increased IL-17 expression. (B) Unlike IL-17 which is known to orchestrate neutrophil mobilization and transmigration, Del-1 acts as a local gatekeeper of normal neutrophil recruitment and hence of inflammation. Specifically, Del-1 blocks the interaction of the neutrophil LFA-1 integrin with the endothelial ICAM-1, which is required for firm neutrophil adhesion to the endothelium and subsequent transmigration.
6. Summary and future perspective
A number of studies have shown that aging causes functional alterations to innate immune cells (Table 1). These findings may offer potential insights into how aging could influence immune and inflammatory responses; these alterations could in turn increase susceptibility to chronic diseases such as periodontitis However, most of this work involves in vitro studies in which the cells are isolated from their native tissue environment. On the other hand, tissues are not passive recipients of immune surveillance, as they can regulate the recruitment and activation of leukocytes via tissue-derived signals and thereby can provide homeostatic protection against inflammatory diseases (Fig. 2). In other words, age-related altered function in immune cells in vivo may not necessarily reflect cell-intrinsic genetically-encoded developmental programs, but may alternatively or additionally involve age-dependent changes in the tissue environment where the cells act. Novel integrative approaches are warranted to better understand how aging can affect the immuno-inflammatory status of the elderly and consequently their risk for periodontitis and other age-related diseases. The elucidation of causal mechanisms of innate immune dysfunction in old age may answer current puzzling questions, such as the precise relationships between inflamm-aging and inflammatory/degenerative diseases or the exact role of aging in periodontitis (Fig. 1). Most importantly, such integrative approaches have the potential to facilitate the development of targeted molecular therapies that can benefit the elderly.
Acknowledgments
Studies performed in the author’s laboratory and cited in this review were supported by U.S. Public Health Service Grants DE021580, DE017138, DE021685, and DE015254, from the NIH/NIDCR.
Footnotes
Conflicts of interest
The author declares that there is no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Christensen K, Doblhammer G, Rau R, Vaupel JW. Ageing populations: the challenges ahead. Lancet. 2009;374:1196–208. doi: 10.1016/S0140-6736(09)61460-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eke PI, Dye BA, Wei L, Thornton-Evans GO, Genco RJ. Prevalence of periodontitis in adults in the United States: 2009 and 2010. J Dent Res. 2012;91:914–20. doi: 10.1177/0022034512457373. [DOI] [PubMed] [Google Scholar]
- 3.Papapanou PN. The prevalence of periodontitis in the US: forget what you were told. J Dent Res. 2012;91:907–8. doi: 10.1177/0022034512458692. [DOI] [PubMed] [Google Scholar]
- 4.Hajishengallis G, Lamont RJ. Beyond the red complex and into more complexity: The Polymicrobial Synergy and Dysbiosis (PSD) model of periodontal disease etiology. Mol Oral Microbiol. 2012;27:409–19. doi: 10.1111/j.2041-1014.2012.00663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Darveau RP. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol. 2010;8:481–90. doi: 10.1038/nrmicro2337. [DOI] [PubMed] [Google Scholar]
- 6.Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, McIntosh ML, Alsam A, Kirkwood KL, Lambris JD, Darveau RP, Curtis MA. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe. 2011;10:497–506. doi: 10.1016/j.chom.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stabholz A, Soskolne WA, Shapira L. Genetic and environmental risk factors for chronic periodontitis and aggressive periodontitis. Periodontol 2000. 2010;53:138–53. doi: 10.1111/j.1600-0757.2010.00340.x. [DOI] [PubMed] [Google Scholar]
- 8.Kinane DF, Demuth DR, Gorr SU, Hajishengallis GN, Martin MH. Human variability in innate immunity. Periodontol 2000. 2007;45:14–34. doi: 10.1111/j.1600-0757.2007.00220.x. [DOI] [PubMed] [Google Scholar]
- 9.Graves DT, Liu R, Alikhani M, Al-Mashat H, Trackman PC. Diabetes-enhanced inflammation and apoptosis--impact on periodontal pathology. J Dent Res. 2006;85:15–21. doi: 10.1177/154405910608500103. [DOI] [PubMed] [Google Scholar]
- 10.Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nat Rev Microbiol. 2012;10:717–25. doi: 10.1038/nrmicro2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van der Velden U. The onset age of periodontal destruction. J Clin Periodontol. 1991;18:380–3. doi: 10.1111/j.1600-051x.1991.tb02304.x. [DOI] [PubMed] [Google Scholar]
- 12.Miller RA. The aging immune system: primer and prospectus. Science. 1996;273:70–4. doi: 10.1126/science.273.5271.70. [DOI] [PubMed] [Google Scholar]
- 13.Huttner EA, Machado DC, de Oliveira RB, Antunes AG, Hebling E. Effects of human aging on periodontal tissues. Spec Care Dentist. 2009;29:149–55. doi: 10.1111/j.1754-4505.2009.00082.x. [DOI] [PubMed] [Google Scholar]
- 14.Solana R, Tarazona R, Gayoso I, Lesur O, Dupuis G, Fulop T. Innate immunosenescence: effect of aging on cells and receptors of the innate immune system in humans. Semin Immunol. 2012;24:331–41. doi: 10.1016/j.smim.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 15.Hajishengallis G. Too old to fight? Aging and its toll on innate immunity. Mol Oral Microbiol. 2010;25:25–37. doi: 10.1111/j.2041-1014.2009.00562.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dorshkind K, Montecino-Rodriguez E, Signer RA. The ageing immune system: is it ever too old to become young again? Nat Rev Immunol. 2009;9:57–62. doi: 10.1038/nri2471. [DOI] [PubMed] [Google Scholar]
- 17.Willershausen-Zonnchen B, Gleissner C. Periodontal disease in elderly patients. Eur J Med Res. 1998;3:55–64. [PubMed] [Google Scholar]
- 18.Ebersole JL, Steffen MJ, Gonzalez-Martinez J, Novak MJ. Age and oral disease effects on systemic inflammatory and immune parameters in nonhuman primates. Clin Vaccine Immunol. 2008;15:1067–75. doi: 10.1128/CVI.00258-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang S, Hosur KB, Domon H, Hajishengallis G. Periodontal inflammation and bone loss in aged mice. J Periodontal Res. 2010;45:574–8. doi: 10.1111/j.1600-0765.2009.01245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burt BA. Periodontitis and aging: reviewing recent evidence. J Am Dent Assoc. 1994;125:273–9. doi: 10.14219/jada.archive.1994.0034. [DOI] [PubMed] [Google Scholar]
- 21.Hayflick L. The future of ageing. Nature. 2000;408:267–9. doi: 10.1038/35041709. [DOI] [PubMed] [Google Scholar]
- 22.Kornman KS. Interleukin 1 genetics, inflammatory mechanisms, and nutrigenetic opportunities to modulate diseases of aging. Am J Clin Nutr. 2006;83:475S–83S. doi: 10.1093/ajcn/83.2.475S. [DOI] [PubMed] [Google Scholar]
- 23.Fransson C, Berglundh T, Lindhe J. The effect of age on the development of gingivitis. Clinical, microbiological and histological findings. J Clin Periodontol. 1996;23:379–85. doi: 10.1111/j.1600-051x.1996.tb00561.x. [DOI] [PubMed] [Google Scholar]
- 24.Tsalikis L, Parapanisiou E, Bata-Kyrkou A, Polymenides Z, Konstantinidis A. Crevicular fluid levels of interleukin-1alpha and interleukin-1beta during experimental gingivitis in young and old adults. J Int Acad Periodontol. 2002;4:5–11. [PubMed] [Google Scholar]
- 25.Ligthart GJ, Corberand JX, Fournier C, Galanaud P, Hijmans W, Kennes B, Muller-Hermelink HK, Steinmann GG. Admission criteria for immunogerontological studies in man: the SENIEUR protocol. Mech Ageing Dev. 1984;28:47–55. doi: 10.1016/0047-6374(84)90152-0. [DOI] [PubMed] [Google Scholar]
- 26.Wick G, Grubeck-Loebenstein B. The aging immune system: primary and secondary alterations of immune reactivity in the elderly. Exp Gerontol. 1997;32:401–13. doi: 10.1016/s0531-5565(96)00152-0. [DOI] [PubMed] [Google Scholar]
- 27.Shaw AC, Panda A, Joshi SR, Qian F, Allore HG, Montgomery RR. Dysregulation of human Toll-like receptor function in aging. Ageing Res Rev. 2011;10:346–53. doi: 10.1016/j.arr.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pawelec G, Mu ller L. Introduction to Ageing of the Adaptive Immune System. In: Bosch JA, editor. Immunosenescence. New York: Springer; 2013. pp. 17–33. [Google Scholar]
- 29.Gomez CR, Nomellini V, Faunce DE, Kovacs EJ. Innate immunity and aging. Exp Gerontol. 2008;43:718–28. doi: 10.1016/j.exger.2008.05.0168.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weng NP. Aging of the immune system: how much can the adaptive immune system adapt? Immunity. 2006;24:495–9. doi: 10.1016/j.immuni.2006.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Geiger H, de Haan G, Florian MC. The ageing haematopoietic stem cell compartment. Nat Rev Immunol. 2013;13:376–89. doi: 10.1038/nri3433. [DOI] [PubMed] [Google Scholar]
- 32.Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244–54. doi: 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- 33.Mahbub S, Brubaker AL, Kovacs EJ. Aging of the Innate Immune System: An Update. Curr Immunol Rev. 2011;7:104–15. doi: 10.2174/157339511794474181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Butcher S, Chahel H, Lord JM. Ageing and the neutrophil: no appetite for killing? Immunology. 2000;100:411–6. doi: 10.1046/j.1365-2567.2000.00079.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Butcher SK, Chahal H, Nayak L, Sinclair A, Henriquez NV, Sapey E, O’Mahony D, Lord JM. Senescence in innate immune responses: reduced neutrophil phagocytic capacity and CD16 expression in elderly humans. J Leukoc Biol. 2001;70:881–6. [PubMed] [Google Scholar]
- 36.Biasi D, Carletto A, Dell’Agnola C, Caramaschi P, Montesanti F, Zavateri G, Zeminian S, Bellavite P, Bambara LM. Neutrophil migration, oxidative metabolism, and adhesion in elderly and young subjects. Inflammation. 1996;20:673–81. doi: 10.1007/BF01488803. [DOI] [PubMed] [Google Scholar]
- 37.Wenisch C, Patruta S, Daxbock F, Krause R, Horl W. Effect of age on human neutrophil function. J Leukoc Biol. 2000;67:40–5. doi: 10.1002/jlb.67.1.40. [DOI] [PubMed] [Google Scholar]
- 38.Fulop T, Larbi A, Douziech N, Fortin C, Guerard KP, Lesur O, Khalil A, Dupuis G. Signal transduction and functional changes in neutrophils with aging. Aging Cell. 2004;3:217–26. doi: 10.1111/j.1474-9728.2004.00110.x. [DOI] [PubMed] [Google Scholar]
- 39.Solana R, Pawelec G, Tarazona R. Aging and innate immunity. Immunity. 2006;24:491–4. doi: 10.1016/j.immuni.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 40.Larbi A, Douziech N, Dupuis G, Khalil A, Pelletier H, Guerard KP, Fulop T., Jr Age-associated alterations in the recruitment of signal-transduction proteins to lipid rafts in human T lymphocytes. J Leukoc Biol. 2004;75:373–81. doi: 10.1189/jlb.0703319. [DOI] [PubMed] [Google Scholar]
- 41.Fortin CF, Lesur O, Fulop T., Jr Effects of aging on triggering receptor expressed on myeloid cells (TREM)-1-induced PMN functions. FEBS Lett. 2007;581:1173–8. doi: 10.1016/j.febslet.2007.02.029. [DOI] [PubMed] [Google Scholar]
- 42.Tortorella C, Simone O, Piazzolla G, Stella I, Antonaci S. Age-related impairment of GM-CSF-induced signalling in neutrophils: role of SHP-1 and SOCS proteins. Ageing Res Rev. 2007;6:81–93. doi: 10.1016/j.arr.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 43.Larbi A, Douziech N, Fortin C, Linteau A, Dupuis G, Fulop T., Jr The role of the MAPK pathway alterations in GM-CSF modulated human neutrophil apoptosis with aging. Immun Ageing. 2005;2:6. doi: 10.1186/1742-4933-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Devitt A, Marshall LJ. The innate immune system and the clearance of apoptotic cells. J Leukoc Biol. 2011;90:447–57. doi: 10.1189/jlb.0211095. [DOI] [PubMed] [Google Scholar]
- 45.Stout RD, Suttles J. Immunosenescence and macrophage functional plasticity: dysregulation of macrophage function by age-associated microenvironmental changes. Immunol Rev. 2005;205:60–71. doi: 10.1111/j.0105-2896.2005.00260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Duin D, Mohanty S, Thomas V, Ginter S, Montgomery RR, Fikrig E, Allore HG, Medzhitov R, Shaw AC. Age-associated defect in human TLR-1/2 function. J Immunol. 2007;178:970–5. doi: 10.4049/jimmunol.178.2.970. [DOI] [PubMed] [Google Scholar]
- 47.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–26. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 48.Uciechowski P, Imhoff H, Lange C, Meyer CG, Browne EN, Kirsten DK, Schroder AK, Schaaf B, Al-Lahham A, Reinert RR, Reiling N, Haase H, Hatzmann A, Fleischer D, Heussen N, Kleines M, Rink L. Susceptibility to tuberculosis is associated with TLR1 polymorphisms resulting in a lack of TLR1 cell surface expression. J Leukoc Biol. 2011;90:377–88. doi: 10.1189/jlb.0409233. [DOI] [PubMed] [Google Scholar]
- 49.Kong KF, Delroux K, Wang X, Qian F, Arjona A, Malawista SE, Fikrig E, Montgomery RR. Dysregulation of TLR3 impairs the innate immune response to West Nile virus in the elderly. J Virol. 2008;82:7613–23. doi: 10.1128/JVI.00618-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qian F, Wang X, Zhang L, Chen S, Piecychna M, Allore H, Bockenstedt L, Malawista S, Bucala R, Shaw AC, Fikrig E, Montgomery RR. Age-associated elevation in TLR5 leads to increased inflammatory responses in the elderly. Aging Cell. 2012;11:104–10. doi: 10.1111/j.1474-9726.2011.00759.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Renshaw M, Rockwell J, Engleman C, Gewirtz A, Katz J, Sambhara S. Cutting edge: impaired Toll-like receptor expression and function in aging. J Immunol. 2002;169:4697–701. doi: 10.4049/jimmunol.169.9.4697. [DOI] [PubMed] [Google Scholar]
- 52.Boehmer ED, Goral J, Faunce DE, Kovacs EJ. Age-dependent decrease in Toll-like receptor 4-mediated proinflammatory cytokine production and mitogen-activated protein kinase expression. J Leukoc Biol. 2004;75:342–9. doi: 10.1189/jlb.0803389. [DOI] [PubMed] [Google Scholar]
- 53.Liang S, Domon H, Hosur KB, Wang M, Hajishengallis G. Age-related alterations in innate immune receptor expression and ability of macrophages to respond to pathogen challenge in vitro. Mech Ageing Dev. 2009;130:538–46. doi: 10.1016/j.mad.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kovacs EJ, Palmer JL, Fortin CF, Fulop T, Jr, Goldstein DR, Linton PJ. Aging and innate immunity in the mouse: impact of intrinsic and extrinsic factors. Trends Immunol. 2009;30:319–24. doi: 10.1016/j.it.2009.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Agrawal A, Agrawal S, Cao JN, Su H, Osann K, Gupta S. Altered innate immune functioning of dendritic cells in elderly humans: a role of phosphoinositide 3-kinase-signaling pathway. J Immunol. 2007;178:6912–22. doi: 10.4049/jimmunol.178.11.6912. [DOI] [PubMed] [Google Scholar]
- 56.Panda A, Qian F, Mohanty S, van Duin D, Newman FK, Zhang L, Chen S, Towle V, Belshe RB, Fikrig E, Allore HG, Montgomery RR, Shaw AC. Age-associated decrease in TLR function in primary human dendritic cells predicts influenza vaccine response. J Immunol. 2010;184:2518–27. doi: 10.4049/jimmunol.0901022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sridharan A, Esposo M, Kaushal K, Tay J, Osann K, Agrawal S, Gupta S, Agrawal A. Age-associated impaired plasmacytoid dendritic cell functions lead to decreased CD4 and CD8 T cell immunity. Age (Dordr) 2011;33:363–76. doi: 10.1007/s11357-010-9191-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jing Y, Shaheen E, Drake RR, Chen N, Gravenstein S, Deng Y. Aging is associated with a numerical and functional decline in plasmacytoid dendritic cells, whereas myeloid dendritic cells are relatively unaltered in human peripheral blood. Hum Immunol. 2009;70:777–84. doi: 10.1016/j.humimm.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Agrawal A, Tay J, Ton S, Agrawal S, Gupta S. Increased reactivity of dendritic cells from aged subjects to self-antigen, the human DNA. J Immunol. 2009;182:1138–45. doi: 10.4049/jimmunol.182.2.1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Singh T, Newman AB. Inflammatory markers in population studies of aging. Ageing Res Rev. 2011;10:319–29. doi: 10.1016/j.arr.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bretz WA, Weyant RJ, Corby PM, Ren D, Weissfeld L, Kritchevsky SB, Harris T, Kurella M, Satterfield S, Visser M, Newman AB. Systemic inflammatory markers, periodontal diseases, and periodontal infections in an elderly population. J Am Geriatr Soc. 2005;53:1532–7. doi: 10.1111/j.1532-5415.2005.53468.x. [DOI] [PubMed] [Google Scholar]
- 62.Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, Panourgia MP, Invidia L, Celani L, Scurti M, Cevenini E, Castellani GC, Salvioli S. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. 2007;128:92–105. doi: 10.1016/j.mad.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 63.Nomellini V, Gomez CR, Gamelli RL, Kovacs EJ. Aging and animal models of systemic insult: trauma, burn, and sepsis. Shock. 2009;31:11–20. doi: 10.1097/SHK.0b013e318180f508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kollmann TR, Levy O, Montgomery RR, Goriely S. Innate immune function by Toll-like receptors: distinct responses in newborns and the elderly. Immunity. 2012;37:771–83. doi: 10.1016/j.immuni.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Arranz L, Lord JM, De la Fuente M. Preserved ex vivo inflammatory status and cytokine responses in naturally long-lived mice. Age (Dordr) 2010;32:451–66. doi: 10.1007/s11357-010-9151-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Di Bona D, Vasto S, Capurso C, Christiansen L, Deiana L, Franceschi C, Hurme M, Mocchegiani E, Rea M, Lio D, Candore G, Caruso C. Effect of interleukin-6 polymorphisms on human longevity: a systematic review and meta-analysis. Ageing Res Rev. 2009;8:36–42. doi: 10.1016/j.arr.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 67.Krabbe KS, Pedersen M, Bruunsgaard H. Inflammatory mediators in the elderly. Exp Gerontol. 2004;39:687–99. doi: 10.1016/j.exger.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 68.Lavoie ME, Rabasa-Lhoret R, Doucet E, Mignault D, Messier L, Bastard JP, Faraj M. Association between physical activity energy expenditure and inflammatory markers in sedentary overweight and obese women. Int J Obes (Lond) 2010;34:1387–95. doi: 10.1038/ijo.2010.55. [DOI] [PubMed] [Google Scholar]
- 69.Gonzalez O, Tobia C, Ebersole J, Novak MJ. Caloric restriction and chronic inflammatory diseases. Oral Dis. 2012;18:16–31. doi: 10.1111/j.1601-0825.2011.01830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cannizzo ES, Clement CC, Sahu R, Follo C, Santambrogio L. Oxidative stress, inflamm-aging and immunosenescence. J Proteomics. 2011;74:2313–23. doi: 10.1016/j.jprot.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 71.Chung HY, Lee EK, Choi YJ, Kim JM, Kim DH, Zou Y, Kim CH, Lee J, Kim HS, Kim ND, Jung JH, Yu BP. Molecular inflammation as an underlying mechanism of the aging process and age-related diseases. J Dent Res. 2011;90:830–40. doi: 10.1177/0022034510387794. [DOI] [PubMed] [Google Scholar]
- 72.Matthews JB, Wright HJ, Roberts A, Ling-Mountford N, Cooper PR, Chapple IL. Neutrophil hyper-responsiveness in periodontitis. J Dent Res. 2007;86:718–22. doi: 10.1177/154405910708600806. [DOI] [PubMed] [Google Scholar]
- 73.Chapple IL, Matthews JB. The role of reactive oxygen and antioxidant species in periodontal tissue destruction. Periodontol 2000. 2007;43:160–232. doi: 10.1111/j.1600-0757.2006.00178.x. [DOI] [PubMed] [Google Scholar]
- 74.Yoneda T, Tomofuji T, Ekuni D, Azuma T, Endo Y, Kasuyama K, Machida T, Morita M. Anti-aging Effects of Co-enzyme Q10 on Periodontal Tissues. J Dent Res. 2013;92:735–9. doi: 10.1177/0022034513490959. [DOI] [PubMed] [Google Scholar]
- 75.Alshouibi EN, Kaye EK, Cabral HJ, Leone CW, Garcia RI. Vitamin D and Periodontal Health in Older Men. J Dent Res. 2013;92:689–93. doi: 10.1177/0022034513495239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mora JR, Iwata M, von Andrian UH. Vitamin effects on the immune system: vitamins A and D take centre stage. Nat Rev Immunol. 2008;8:685–98. doi: 10.1038/nri2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McMahon L, Schwartz K, Yilmaz O, Brown E, Ryan LK, Diamond G. Vitamin D-mediated induction of innate immunity in gingival epithelial cells. Infect Immun. 2011;79:2250–6. doi: 10.1128/IAI.00099-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Abiko Y, Shimizu N, Yamaguchi M, Suzuki H, Takiguchi H. Effect of aging on functional changes of periodontal tissue cells. Ann Periodontol. 1998;3:350–69. doi: 10.1902/annals.1998.3.1.350. [DOI] [PubMed] [Google Scholar]
- 79.Bodineau A, Coulomb B, Tedesco AC, Seguier S. Increase of gingival matured dendritic cells number in elderly patients with chronic periodontitis. Arch Oral Biol. 2009;54:12–6. doi: 10.1016/j.archoralbio.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 80.Agius E, Lacy KE, Vukmanovic-Stejic M, Jagger AL, Papageorgiou AP, Hall S, Reed JR, Curnow SJ, Fuentes-Duculan J, Buckley CD, Salmon M, Taams LS, Krueger J, Greenwood J, Klein N, Rustin MH, Akbar AN. Decreased TNF-alpha synthesis by macrophages restricts cutaneous immunosurveillance by memory CD4+ T cells during aging. J Exp Med. 2009;206:1929–40. doi: 10.1084/jem.20090896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Matzinger P, Kamala T. Tissue-based class control: the other side of tolerance. Nat Rev Immunol. 2011;11:221–30. doi: 10.1038/nri2940. [DOI] [PubMed] [Google Scholar]
- 82.Gonzalez OA, Stromberg AJ, Huggins PM, Gonzalez-Martinez J, Novak MJ, Ebersole JL. Apoptotic genes are differentially expressed in aged gingival tissue. J Dent Res. 2011;90:880–6. doi: 10.1177/0022034511403744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hajishengallis G, Chavakis T. Endogenous modulators of inflammatory cell recruitment. Trends Immunol. 2013;34:1–6. doi: 10.1016/j.it.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Choi EY, Chavakis E, Czabanka MA, Langer HF, Fraemohs L, Economopoulou M, Kundu RK, Orlandi A, Zheng YY, Prieto DA, Ballantyne CM, Constant SL, Aird WC, Papayannopoulou T, Gahmberg CG, Udey MC, Vajkoczy P, Quertermous T, Dimmeler S, Weber C, Chavakis T. Del-1, an endogenous leukocyte-endothelial adhesion inhibitor, limits inflammatory cell recruitment. Science. 2008;322:1101–4. doi: 10.1126/science.1165218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Eskan MA, Jotwani R, Abe T, Chmelar J, Lim JH, Liang S, Ciero PA, Krauss JL, Li F, Rauner M, Hofbauer LC, Choi EY, Chung KJ, Hashim A, Curtis MA, Chavakis T, Hajishengallis G. The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nat Immunol. 2012;13:465–73. doi: 10.1038/ni.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]