

Abstract
in silico modeling, using Psipred and ExPASy servers was employed to determine the structural elements of Bcr-Abl oncoprotein (p210BCR-ABL) isoforms, b2a2 and b3a2, expressed in Chronic Myelogenous Leukemia (CML). Both these proteins are tyrosine kinases having masses of 210-kDa and differing only by 25 amino acids coded by the b3 exonand an amino acidsubstitution (Glu903Asp). The secondary structure elements of the two proteins show differences in five α-helices and nine β-strands which relates to differences in the SH3, SH2, SH1 and DNA-binding domains. These differences can result in different roles played by the two isoforms in mediating signal transduction during the course of CML.
Background
Chronic Myelogenous Leukemia (CML) develops when a single, hematopoietic stem cell acquires a Philadelphia (Ph) chromosome carrying the BCR-ABL fusion oncogene which gives its progeny an advantage for proliferation over normal RBCs and allows the Ph-positive clone gradually to displace normal RBCs during hematopoiesis [1, 2, 3, 4]. The abnormal Ph chromosome is produced by the translocation between chromosome 9 and 22 (Figure 1a).The major consequence of Philadelphia translocation is the fusion of the ABL gene on chromosome 9 with the BCR gene on chromosome 22 [5]. The BCR-ABL fusion oncogene encodes new fusion proteins of 190, 210 and 230 kDa molecular weight [6, 7]. The p210BCR-ABL isoforms have an increased level of tyrosine kinase activity, which is important for the development of the disease [8]. The production of fusion proteins increases the diversity of proteinprotein binding domains associated with tyrosine kinase activity.
Figure 1.
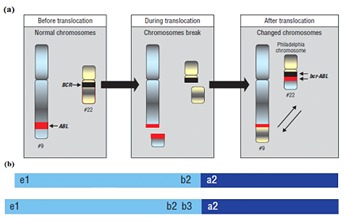
a) Shown is the Philadelphia chromosome (also called derivative 22) produced by reciprocal translocation [4]; b) Schematic showing the different lengths of the two p210 BCRABL protein isoforms (b2a2 and b3a2) expressed by the Philadelphia chromosome.
The normal product of the BCR gene is a 160 kDa (p160BCR) cytosolic phosphoprotein whose physiologic role is not clearly defined. It has been shown to form cytoplasmic complexes with p210BCR-ABL in Ph-positive CML cells, as well as with a 53 kDa protein of unknown function in both Ph-positive and Phnegative cell lines [9, 10]. The sequences encoded by the first exon of BCR gene are responsible for the p160BCR serine/threonine kinase activity [11]. The N-terminus of p160BCR comprises a coiled–coil domain that allows dimer formation in vivo. At the center, are DBL-like [12] and Pleckstrin-homology [13] domains that stimulate the exchange of guanidine triphosphate (GTP) for guanidine diphosphate (GDP) and activate transcription factors such as NF-kB. The Cterminus of p160BCR contains GTPase activity for Rac. In addition, p160BCR can be phosphorylated at several tyrosine residues, such as Tyrosine 177, which binds Grb-2, an adapter protein involved in the activation of the Ras pathway.
The normal product of the ABL gene is a 145-kDa protein (p145ABL) [14]. It is a protein tyrosine kinase that is involved in cell differentiation, cell division, cell adhesion, and stress response. The activity of p145ABL is regulated by its SH3 domain, and deletion of the SH3 domain turns it into an oncoprotein. The Philadelphia translocation results in the headto- tail fusion of p145ABL to p160BCR [15].
The two p210 BCR-ABL onco-protein isoforms, b2a2 and b3a2 (Figure 1b), formed by the head-to-tail fusion of p160BCR and p145ABL proteins, differ by a 25 amino acid insertion coded by the b3 exon and a Glu903Asp substitution between b2a2 and b3a2. Several studies have examined whether the type of fusion transcript has any influence on the clinical outcome [16, 7, 18, 9, 20, 21]. However, the data remains controversial. In fact, several groups did not succeed in demonstrating any such correlation [6, 22, 23]. A study has shown a correlation between the b3a2 transcript and a higher platelet count at diagnosis in a group of CML patients [21].
Recently, the crystal structure of the oligomerization domain at the N-terminus of both b2a2 and b3a2 (residues 1–72) has been published. The investigators have reported a novel mode of oligomer formation which involves dimerization of two monomers by swapping of N-terminus helices and by formation of an antiparallel coiled coil between C-terminus helices. The two dimers then stack onto each other to form a tetramer [24]. In the present study, we performed a comparison of the structural elements of p210BCR-ABL protein isoforms, b2a2 and b3a2. For this purpose, Psipred and ExPASy servers were employed [25, 26].
Methodology
The amino acid sequences of the p210BCR-ABL protein isoforms, b2a2 and b3a2, were subjected to computer predictive analysis in order to reveal possible differences in terms of secondary structure and tertiary structure, Different tools and database were used for molecular modeling of these proteins such as GenBank-NCBI, Protein Data Bank, Psipred [25] and ExPASy servers [26]. The sequence of the proteins was retrieved in FASTA format from NCBI database for homology modeling. The procedure of homology modeling procedure comprised three sequential steps: (i) template selection, (ii) target template alignment, and (iii) model building. The secondary structures of both b2a2 and b3a2 were predicted using Psipred server. The tertiary structures were predicted using the ExPASy server.
Results
The use of Psipred server [25] provided the secondary structure elements of both b2a2 and b3a2 isoforms. The amino acid sequence of b2a2 has 2006 residues and b3a2 has 2031 residues. They differ by 26 amino acids. There is an amino acid substitution (Glu903Asp) and a 25 amino acid insertion just before Ala 904 in the sequence of b3a2. The secondary structure of b2a2 comprises 48 α-helices (α1 to α48) and 37 β-strands (β1 to β37). The b3a2 isoform also contains similar secondary structure elements along with an additional α-helix (α') and two β-strands (β' and β"). A short β-strand, β33 is also missing in b3a2 (Schematic 1, supplementary material). In total, there are differences in five α-helices and nine β-strands of the two proteins which relates to differences in the SH3, SH2, SH1 and DNA-binding domains Table 1 (see supplementary material). The tertiary structures of the two proteins were obtained via the ExPASy server [26]. The tertiary structures show that both the proteins possess a-helical and β-sheet domains (Figures 2a & b). The tertiary structure of b3a2 protein reveals that it has greater amount of α-helical content.
Figure 2.
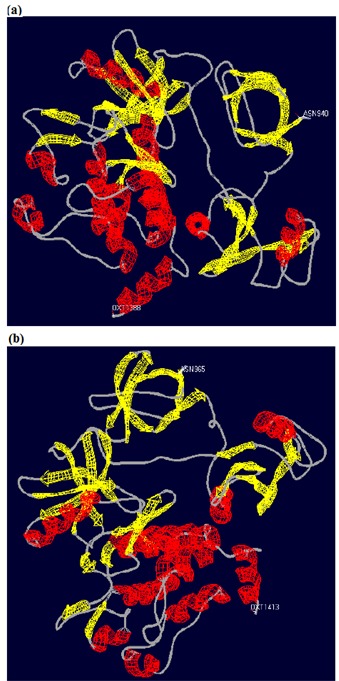
(a, b) Tertiary structures of b2a2 and b3a2 proteins obtained via the ExPASy server. The structures show that both the proteins possess α-helical and β-sheet domains. The b3a2 protein shows a greater amount of α-helical structure.
Discussion
The oncogenic potential of p210BCR-ABL protein isoforms is due to the fact that the normally regulated tyrosine kinase activity of the ABL protein (p145ABL) becomes unregulated in both b2a2 and b3a2 isoforms. ABL proteins are non-receptor tyrosine kinases that have important roles in signal transduction and the regulation of cell growth [27]. At the N-terminus, there are three SRC homology domains (SH3, SH2 and SH1). SH2 and SH3 domains regulate the tyrosine kinase function of ABL protein and SH1 domain contains the tyrosine kinase activity of ABL protein. SH3 has a negative regulatory effect on the tyrosine kinase function. Deletion of SH3 or mutation in SH3 facilitates tyrosine kinase activity of ABL protein [28, 29, 30]. Mutations in SH2 decrease phospho-tyrosine binding activity and reduce transforming capacities of ABL protein [31]. The C-terminus of ABL protein contains a DNA-binding domain, nuclear localization signals, and a binding site for actin [32] (Figure 3).
Figure 3.
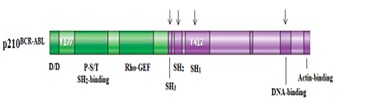
The domain organization of p210BCR-ABL proteins. The part that is derived from BCR gene is shown in green and the other half that is derived from ABL gene is shown in purple. The structural differences found in the SH3, SH2, SH1 and DNAbinding domains of p210BCR-ABL are indicated by arrows. Also shown in the figure are oligomerization domain (D/D), actinbinding domain and the positions of Tyr 177 and Tyr 412 [33].
The structure of the oligomerization domain present at the Nterminus of both the p210BCR-ABL protein isoforms (b2a2 and b3a2) has been reported earlier [24]. It consists of a short Nterminal helix (α1), a flexible loop and a long C-terminal helix (α2). The two a-helices, together form an N-shaped structure, with the loop allowing the two helices to assume a parallel orientation. The monomeric domains associate into a dimer through the formation of an antiparallel coiled coil between the α2 helices and domain swapping of two α1 helices, where one α1 helix swings back and packs against the α2 helix from the second monomer. The two dimers then associate into a tetramer [24]. The oligomerization domain promotes clustering of a C-terminal actin-binding domain that cross-links actin filaments [34, 35]. The two p210BCR-ABL protein isoforms can induce a redistribution of F-actin from the cortical cytoskeleton into aggregates [35]. Oligomerization also plays a role in the activation of the p210BCR-ABL tyrosine kinase activity [34]. A comparison of the secondary structure elements of both b2a2 and b3a2 with the structural elements determined by X-ray crystallography revealed that both similarities and differences. The length of α1 and α2 are longer in the crystal structure. Also, the position of a2 is shifted in the crystal structure. An additional β-strand (β1) is present in the secondary structural elements obtained via Psipred.
The experimental data shows that p210BCR-ABL protein isoforms, b2a2 and b3a2, exhibit differences in their secondary structure elements Table 1 (see supplementary material). A twenty five amino acid insertion just before Ala 904 in the sequence of b3a2 shifts some of the secondary structure elements and also produces some additional ones Schematic 1 (see supplementary material). Both p210BCR-ABL proteins can cause pleiotropic effects on many signal transduction pathways that can affect cell survival, disease progression, and genomic stability. The signals controlled by p210BCR-ABL proteins are important for normal hematopoiesis. An earlier paper using the GOR, NnPredict and PHD methods to predict the structural elements of b2a2 and b3a2 did not find any difference between the two full-length protein types. However, a discrepancy was observed for the 25 amino acids coded by the b3 exon. In one case, it was seen as completely solvent accessible loop region (PHD method) whilst in the other cases (GOR and NnPredict methods) it comprised a short β-strand and a short a-helix [36].
Conclusion
The p210BCR-ABL protein isoforms, b2a2 and b3a2 show differences in their secondary structure elements mainly due to the insertion of a 25 amino acid segment coded by the b3 exon in b3a2. In total, structural differences are found between the two proteins in five α-helices (α25, α', α26, α27 and α29) and nine β-strands (β12, β13, β15, β', β17, β30, β", β34 and β35). These differing structural elements are present in the SH3, SH2, SH1 and DNA-binding domains which can result in different roles played by the two isoforms in mediating signal transduction during the course of Chronic Myelogenous Leukemia.
Supplementary material
Footnotes
Citation:Hai et al, Bioinformation 10(3): 108-114 (2014)
References
- 1.Eaves CJ, Eaves AC. Baillieres Clin Haematol. 1997;10:233. doi: 10.1016/s0950-3536(97)80005-4. [DOI] [PubMed] [Google Scholar]
- 2.Fialkow PJ, et al. Proc Natl Acad Sci USA. 1967;58:1468. doi: 10.1073/pnas.58.4.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nowell P, Hungerford D. Science. 1960;142:1497. [Google Scholar]
- 4.Hazlehurst LA, et al. Cancer Control. 2009;16:100. doi: 10.1177/107327480901600202. [DOI] [PubMed] [Google Scholar]
- 5.Shtivelman E, et al. Nature. 1985;315:550. doi: 10.1038/315550a0. [DOI] [PubMed] [Google Scholar]
- 6.Melo JV. Leukemia. 1996;10:751. [PubMed] [Google Scholar]
- 7.Faderl S, et al. N Engl J Med. 1999;341:164. doi: 10.1056/NEJM199907153410306. [DOI] [PubMed] [Google Scholar]
- 8.Daley GQ, et al. Science. 1990;247:824. doi: 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- 9.Campbell ML, et al. Oncogene. 1990;5:773. [PubMed] [Google Scholar]
- 10.Li W, et al. Oncogene. 1989;4:127. [PubMed] [Google Scholar]
- 11.Maru Y, Witte ON. Cell. 1991;67:459. doi: 10.1016/0092-8674(91)90521-y. [DOI] [PubMed] [Google Scholar]
- 12.Aghazadeh B, et al. Nat Struct Biol. 1998;5:1098. doi: 10.1038/4209. [DOI] [PubMed] [Google Scholar]
- 13.Mayer BJ, et al. Cell. 1993;73:629. doi: 10.1016/0092-8674(93)90244-k. [DOI] [PubMed] [Google Scholar]
- 14.Tani K. J Biol Chem. 2003;278:21685. doi: 10.1074/jbc.M301447200. [DOI] [PubMed] [Google Scholar]
- 15.Shtivelman E, et al. Cell. 1986;47:277. doi: 10.1016/0092-8674(86)90450-2. [DOI] [PubMed] [Google Scholar]
- 16.Mills KI, et al. Blood. 1991;78:1155. [PubMed] [Google Scholar]
- 17.Futaki M, et al. Leukemia Res. 1992;16:1071. doi: 10.1016/0145-2126(92)90045-9. [DOI] [PubMed] [Google Scholar]
- 18.Zaccaria A, et al. Br J Haematol. 1993;84:265. doi: 10.1111/j.1365-2141.1993.tb03062.x. [DOI] [PubMed] [Google Scholar]
- 19.Rozman C, et al. Leukemia. 1995;9:1104. [PubMed] [Google Scholar]
- 20.Elliott SL, et al. Leukemia. 1995;9:946. [Google Scholar]
- 21.Shepherd P, et al. Br J Haematol. 1995;89:546. doi: 10.1111/j.1365-2141.1995.tb08362.x. [DOI] [PubMed] [Google Scholar]
- 22.Shepherd PC, et al. Blood. 1992;80:556. [PubMed] [Google Scholar]
- 23.Opalka B, et al. Blood. 1992;80:1854. [PubMed] [Google Scholar]
- 24.Zhao X, et al. Nat Struct Biol. 2002;9:117. doi: 10.1038/nsb747. [DOI] [PubMed] [Google Scholar]
- 25.McGuffin LJ, et al. Bioinformatics. 2000;16:404. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 26.Gasteiger E, et al. Nucleic Acids Res. 2003;31:3784. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang JY. Curr Opin Genet Dev. 1993;3:35. doi: 10.1016/s0959-437x(05)80338-7. [DOI] [PubMed] [Google Scholar]
- 28.Jackson P, Baltimore D. Embo J. 1989;8:449. doi: 10.1002/j.1460-2075.1989.tb03397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sawyers CL, et al. Cancer Surv. 1992;15:37. [PubMed] [Google Scholar]
- 30.Franz WM, et al. Embo J. 1989;8:137. doi: 10.1002/j.1460-2075.1989.tb03358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gale RP, et al. Leukemia. 1993;7:653. [PubMed] [Google Scholar]
- 32.Chung SW, et al. Crit Rev Oncog. 1996;7:33. doi: 10.1615/critrevoncog.v7.i1-2.30. [DOI] [PubMed] [Google Scholar]
- 33.Goldman JM, Melo JV. New England J Med. 2003;349:1451. doi: 10.1056/NEJMra020777. [DOI] [PubMed] [Google Scholar]
- 34.McWhirter JR, et al. Mol Cell Biol. 1993;13:7587. doi: 10.1128/mcb.13.12.7587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McWhirter JR, Wang JY. EMBO J. 1993;12:1533. doi: 10.1002/j.1460-2075.1993.tb05797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perego RA, et al. Eur J Cancer. 2000;36:1395. doi: 10.1016/s0959-8049(00)00128-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.