

Abstract
Janus kinase 3 (JAK3) is a non-receptor tyrosine kinases family of protein which is comprised of JAK1, JAK2, JAK3 and TYK2. It plays an important role in immune function and lymphoid development and it only resides in the hematopoietic system. Therefore, selective targeting JAK3 is a rational approach in developing new therapeutic molecule. In this study, about 116 JAK3 inhibitors were collected from the literature and were used to build four-point pharmacophore model using Phase (Schrodinger module). The statistically significant pharmacophore hypothesis of AAHR.92 with r2 value of 0.942 was used as 3D query to search against 3D database namely Zincpharmer. A total of 2, 27,483 compounds obtained as hit were subjected to high throughput virtual screening (HTVS module of Schrodinger). Among the hits, ten compounds with good G-score ranging from -12.96 to -11.18 with good binding energy to JAK3 were identified.
Keywords: JAK3, 3DQSAR, Phase, Pharmacophore modelling, Inhibitor
Background
Janus Kinases (JAKs) belongs to the family of non receptor tyrosine kinase consisting of JAK1, JAK2, JAK3 and TYK2 which are activated after cytokine receptor activation. JAK activation results in phosphorylation of the STAT transcription factor members and translocated into a nucleus, binds DNA and promotes transcription [1– 3]. The mutation or increased localized concentration of cytokines causes over activation of JAK-STAT signalling, which leads to various inflammatory diseases [4, 5], autoimmune disease [6], cancer [7, 8] and graft rejection [9]. Among the JAK family, JAK3 is abundantly expressed in hemopoietic cells and plays an important role in normal lymphocyte development and function, whereas JAK1, JAK2 and TYK2 are ubiquitously expressed in vertebrates [10, 11].
The drug that is under the clinical trial for JAK3 inhibitor is tofacitinib for rheumatoid arthritis [12, 13] which was found to have adverse reactions due to less JAK3 selectivity [14, 15]. R348, another potent JAK3 inhibitor for inflammatory skin disease such as psoriasis, but there was no reported clinical trials [16].Therefore selective targeting of JAK3 may have a therapeutic benefit over broader JAK signaling inhibition for the treatment in various areas like oncology, organ transplantation and autoimmune diseases [17, 18].
In the present study, pharmacophore model was generated for Human JAK3 inhibitors using Phase module 3.5 (Schrödinger module). Subsequently an atom based 3D-QSAR model was obtained and database screening was done in search of novel lead compounds. The lead compounds were then docked with JAK3 to study the interaction of inhibitors with the protein.
Methodology
Dataset:
A total of 116 JAK3 inhibitors were collected from the literature [1, 11, 19–22] and the pIC50 (pIC50 = -logIC50) values were calculated. The dataset contains different chemical classes, namely phenyl aminoprymidines, Nphenylmethanesulfonamide, nitrile carboxamide, Ncyanomethylbenzamide, 2-aminoethylketone, 2- Benzimidazolyl-9-(chroman-4-yl)-purinone, di-substituted pyrimidine, tri-substituted pyrimidine and 5H-pyrrolo(2,3-b) pyrazine-2-phenyl ethers. The structures were drawn using Build panel of Maestro version 9.4 and prepared using LigPrep 2.6 modules. Different conformer was generated using rapid torsion search of Macromodel. Energy minimization was done using OPLS 2005 with an implicit distance-dependent dielectric solvation treatment.
The Generation of Pharmacophore hypothesis and 3D-QSAR model building:
The pharmacophore and 3D-QSAR model was generated using Phase version 3.5, Schrödinger suite 9.4 [23]. There are six builtin pharmacological features in Phase, namely hydrogen bond receptor (A), hydrogen bond donor (D), hydrophobic group (H), negatively ionisable (N), positively ionisable (P) and aromatic ring (R). The pharmacophore model was developed using a set of pharmacophore features to generate sites for all the compounds. The alignment was measured using survival score [24] and the default values have been used for the hypothesis generation.
A total of 116 molecules were ranked based on pIC50 values. Every 5th compound was chosen as a test set, so 19 were selected as a test set and remaining were used as training set to generate atom-based QSAR models. This type of test set selection procedure was employed to represent the range of biological activities similar to the training set molecule. To encompass the space occupied by the aligned training set molecules the rectangular grid was generated with the spacing of 1.2Å. Each model contains five or more partial least square (PLS) factors tend to fit the pIC50 values beyond their experimental uncertainty. The statistical parameters R2 (coefficient of determination) and SD (standard deviation of regression) were calculated to evaluate the overall significance of the model.
Virtual screening:
Virtual screening was carried out using ZincPharmer (zincpharmer.csb.pitt.edu ) which uses the pharmacophore to efficiently search the ZINC database of fixed conformers [25]. We also used constraints that included maximum of 0.7 Root Mean Square Deviation (RMSD), 10 rotatable bond cut-off and molecular weight range of 180–500 Dalton to get the best similarity hits from ZINC database. The hit compounds were subsequently subjected to the addition of hydrogen, removal of salt ionization and generation of low energy ring conformation using LigPrep.
Protein Preparation:
The X-ray crystallographic structure of JAK3 complexes with inhibitor was downloaded from the PDB database (PDB ID: 4HVD [26]) and prepared using the protein preparation wizard of Schrödinger Module by retaining the water molecule within 5 Å of ligand. OPLS-2005 force field was used for energy minimization. Hydrogen atoms were added to the protein to correct ionization and tautomeric states of amino acid residues. The receptor grid was generated using Receptor Grid Generation Panel.
Docking study:
The Virtual Screening Workflow in Maestro was used to dock and to score the lead-like compounds to identify potential ligands. It provides the different level of docking precision, namely High Throughput Virtual Screening (HTVS), Standard Precision (SP) and Extra Precision (XP) [27–29]. We first carried out HTVS calculation, and then SP and finally XP mode for further refinement of good ligand pose. The screened compounds were also filtered by Lipinski׳s rule of five (QikProp version 3.6 [30]). And also XP docking was carried out for 116 ligands which were used to build the 3D-QSAR model.
Post Docking Analysis:
The free energy of binding was calculated for the best scoring pose of the Glide-XP docking results using Multi-ligand bimolecular association with energetic (eMBrAcE) of the Macromodel module. It uses physics-based rescoring procedure to calculate binding energies between ligand and proteins using OPLS force field for docked conformation. For each ligand, the protein–ligand complex (Elig–prot), the free protein (Eprot), and the free ligand (Elig) were subjected to energy minimization in implicit solvent (water) using the OPLS_2001 force field with a constant dielectric electrostatic treatment of 1.0 Å. A conjugate gradient energy minimization protocol was performed. The energy difference mode was used for the calculation of Glide output and it was calculated using the following equation.
▲E= Ecomplex − Eligand − Eprotein
All computational and molecular modelling were done on the centOS Linux platform in HCL Intel Xeon Server with 4GB RAM and using the Maestro window of Schrödinger version 9.4, LLC, New York 2012 and 2013.
Results & Discussion
Structural Analysis of JAK3:
The available 3D structure of JAK3 in the PDB databank (www.rcsb.org) was analyzed to see the critical amino acid for JAK3 inhibitor. All the ligands in complex structures were found to interact with Glu903, Leu956 and Leu905 either by hydrogen bonding or hydrophobic interactions Table 1 (see supplementary material). For the present study, based on the resolution 4HVD (1.85 Å) was selected as target protein.
JAK3 exhibits bilobed structure which is conserved among the catalytic domain of all protein kinases. JAK3 is having N and C lobe, C helix, glycine rich loop or P-loop and hinge regions. It also has additional helix between the residues 1029 to 1038 named as a FG helix. The residues in the active site region are Leu828, Phe833, Val836, Ala853, Lys855, Met902, Glu903, Cys909, Arg953, Leu956, Ala966 and Asp967. JAK3 shares the high sequence similarity with JAK2 and JAK1. The sequence difference between JAK3 and JAK2 that are proximal to the catalytic cleft are Ser828, Leu838, Arg 916, Gln 988 Cys909 and Ala966 in JAK3 which is substituted by Gln, Met, Lys, Glu, Ser and Gly respectively in JAK2. The two important differences are Cys909 and Ala966 which are in the ATP binding site of JAK3 [31].
Validation of docking:
Water molecules are considered as an important factor in the docking process, i.e. the binding affinity of the ligand molecule is improved and stabilized by hydrogen-bonded network of water molecules with protein [32– 35]. We have carried out two methods of protein preparation for the target protein 4HVD: one by removing all water molecules and other by retaining water molecules within 5 Å of ligand. Further, the results of XP docking was validated by re-docking the co-crystallized ligand (2-Cyclopropyl-5H-pyrrolo [2, 3-b] pyrazine-7-carboxylic acid ((S)-1, 2, 2-trimethyl-propyl)-amide) to the binding site of JAK3. The protein structure prepared by retaining water molecules within 5 Å of ligand has generated lowest RMSD (0.296 Å) than without water molecules (1.68 Å) when superimposed with experimental structure (Figure 1). Hence, the target protein JAK3 was prepared with water molecules to find the best pharmacophore hits.
Figure 1.
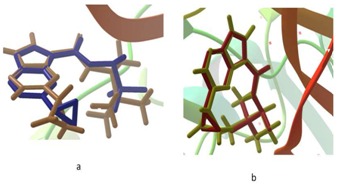
Superposition of co-crystallized ligand 2-Cyclopropyl- 5H-pyrrolo [2, 3-b] pyrazine-7-carboxylic acid ((S)-1, 2, 2- trimethyl-propyl)-amide in experimental binding mode (in maroon) with redocked pose a) without water molecules (RMSD 1.68 Å) and b) with water molecules (RMSD 0.296 Å).
Determination of Common Pharmacophore:
In order to identify common pharmacophores, the collected dataset was divided into active (pIC50>7.2), inactive (pIC50<5) and moderatively active (pIC50 between 5 and 7). In Phase, common pharmacophores are identified from a set of variants. For defining variants, the maximum number of sites to be included in the hypothesis was selected as 5 and a minimum number of sites to be 4. With the above said criteria, 28 compounds were matched out of 30 active compounds. Among the 12 variants listed out, seven was found to have common pharmacophore (ADHR 3, DHRR 3, AHRR 18, ADRR 6, AAHR 73, AADH 4 and AADR 8). Then 115 pharmacophores were subsequently scored by scoring with respect to active and inactive using default score. The scoring procedure provides a ranking of the different hypothesis, allowing us to make rational choice. The scoring algorithm includes the alignment of site points and vectors, volume overlap, number of ligands matched, selectivity, relative conformational energy and activity. A total of 14 hypotheses survived the scoring processes which were used to build an atom based QSAR model.
Building and Validation of 3D-QSAR:
An atom based 3D-QSAR model was generated for the pharmacophore hypothesis. The best QSAR model was identified and validated by predicting activities of 19 test set compounds. Regression was constructed for a series of model with the increasing number of Partial Least-Square (PLS factor). As the number of PLS factors increased, the statistical significance and predictive ability of the model was also incrementally increased up to 7. It was found that AAHR.92 has higher R2 value (0.9422), predictive power q2 value of 0.6023, Pearson R value of 0.8178 and lower Standard Deviation (SD) of 0.2666 Table 2 (see supplementary material). Larger the value of F (186) with the smaller value of p (8.8e-47) indicates a statistically significant regression model with high degree of confidence. The small value of SD of 0.2666 and Root-Mean- Square-Error value (RMSE) of 0.6472 indicates that the data used for the analysis of the QSAR model was good even though QSAR model was generated using the different set of chemical class compounds. Besides all these, the best QSAR model should have high predictive ability, so the best model should have a high cross validated correlation coefficient. The q2 value is more reliable since it is obtained by external validation of the test set model. AAHR.92 has q2 value of 0.60 indicates the goodness of the model. A graph of actual versus predicted pIC50 value of the test and training set is shown in Figure 2.
Figure 2.
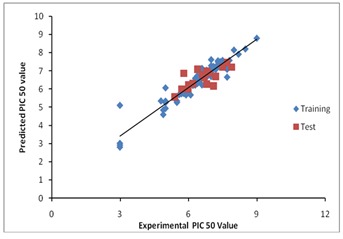
A graph of actual vs predicted pIC50 value of the test and training set of AAHR.92
The selected pharmacophore hypothesis included the following features (Figure 3): the hydrogen bond acceptor (sphere with arrow A2 and A3), hydrophobic group (H9 green sphere) and the aromatic ring (R15 circle). The alignment generated by the best pharmacophore model for active and inactive compound is shown in Figure 4. The key pharmacophore element of the AAHR.92 hypothesis: the aromatic ring (R15), which is mapped into the benzene ring of benzimidazole or azobenzimidazole of all active compounds, the hydrogen bond acceptor (A2 and A3) features are in N of prymidine, N and O of Purinone and O of the chroman ring and the hydrophobic groups are featured on alkyl substituent.
Figure 3.
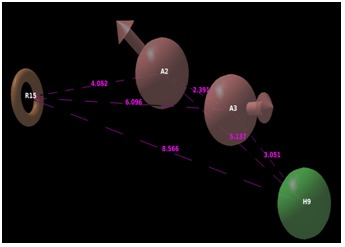
Pharmacophoric features of active ligands
Figure 4.
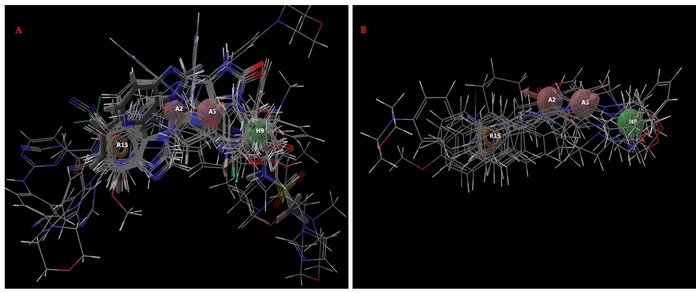
Alignment of active a) and inactive ligands; b) to the pharmacophore model AAHR.92
Virtual Screening:
AAHR.92 was used to search 3D database for structures that match the pharmacophoric features of the model. A total 2,26,248 compounds were retrieved as hits from ZincPharmer and were placed through virtual screening workflow. A total of 107 compounds with XP docking score ranged between -12.96 and -9.00 were obtained. Finally, we selected 10 ligands which have good interaction with active site of JAK3 and are shown in Table 3 (see supplementary material) and Figure 5.
Figure 5.
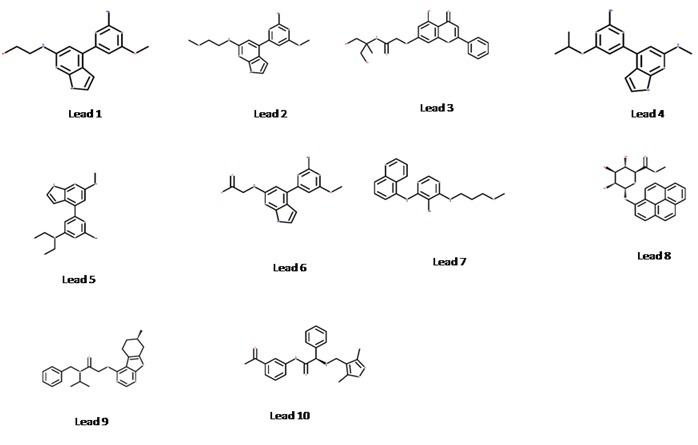
Structure of lead compounds
The pharmacophore model was developed using pyrimidine, pyrrolo pyrimdine, pyrolopyrazine, purinone with chromane substitution and naphyl ketone. The best ten lead compounds were found to have these groups. CP-690550 (Tofacitinib) a potent JAK3 inhibitor was also included as a reference compound in the docking protocol. The docking score and binding energy of CP-690550 was found to be -9.86 and -150.29 respectively.
The Chemical name of the leads are, lead 1: 2-{[4-(2-amino-6- methoxypyrimidin-4-yl)-1H-pyrrolo[2,3-b]pyridin-6- yl]amino}ethanol, lead 2: 4-(2-amino-6-methoxypyrimidin-4- yl)-N-(2-methoxyethyl)-1H-pyrrolo[2,3-b]pyridin-6-amine, lead 3: N-(1,3-dihydroxy-2-methylpropan-2-yl)-2-[(5-hydroxy-4- oxo-2-phenyl-4H-chromen-7-yl)oxy]acetamide, lead 4: 4-(2- amino-6-isopropoxypyrimidin-4-yl)-N-methyl-1H-pyrrolo[2,3- b]pyridin-6-amine, lead 5: N~4~,N~4~-diethyl-6-[6- (methylamino)-1H-pyrrolo[2,3-b]pyridin-4-yl]pyrimidine-2,4- diamine, lead 6: N-[4-(2-amino-6-methoxypyrimidin-4-yl)- 1H-pyrrolo[2,3-b]pyridin-6-yl]glycine, lead 7: N4-(3- methoxypropyl)-N6-(5-quinolyl)pyrimidine-4,5,6-triamine, lead 8: 1-Hydroxypyrene beta-D-Glucuronide Methyl Ester, lead 9: N-benzyl-N-isopropyl-2-[[(7S)-7-methyl-5,6,7,8- tetrahydrobenzothiopheno[2,3-d]pyrimidin-4-yl]oxy]ace and lead 10: (2S)-N-(3-acetylphenyl)-2-[(3,5-dimethylisoxazol-4- yl)methylamino]-2-phenyl-acetamide.
Post Docking Scoring:
In the present work, the post docked scoring approach, namely eMBrAcE was used to evaluate the molecular docking of JAK3 with inhibitors Table 4 (available with authors). The Multiligand Bimolecular Association with Energetics (MBAE) of the best active compounds 64 and 105 was found to be -145.34 and - 172.38 respectively. The binding energy of the least active compound 46 was found to be -67.43. The MBAE energy of the lead compounds ranged from -80.78 to -157.68 (Table 3).
ADME Analysis:
Lipinski׳s rule of five is based on the physicochemical properties of drugs and candidate drugs in clinical trials to evaluate drug likeness. The rule includes molecular weight, logP, Hydrogen bond donor and acceptor. The molecules that pass through the Lipinski׳s rule of five are expected to be active after oral administration in human. The percentage of the human oral absorption of selected lead compounds was found to be 52 to 100%. For selected lead compounds, the partition coefficient (QPlogPo/w) and water solubility (QPlogS) was within the permissible range of -2.0 to 6.5 and -6.5 to 0.5 respectively. All the pharmacokinetic parameters analysed were found to be within the permissible range Table 5 (see supplementary material).
Conclusion
JAK3 is an important component associated with sensitivity towards cytokine signalling and its deficiency is associated with severe combined immunodeficiency phenotype. JAK3 inhibitors are immunomodulatory agents with immunosuppressive, anti-inflammatory, anti-allergic, antithrombotic and anti-leukemic properties. The preclinical trial drug tofacitinib found to have an inhibitory effect also on JAK1 and JAK2. The identification of selective JAK3 inhibitor may effectively act as immunomodulatory agent. In this study, pharmacophore based virtual screening and molecular docking approach was carried out to identify JAK3 inhibitor. A series of phenylaminopyrmidines, napthylketones, di and tri-substituted pyrmidines and pyrole pyrazines compounds were used for 3D-QSAR analysis. A four-point pharmacophore model AAHR.92 was developed which comprises of two hydrogen bond acceptor, one hydrophobic group and aromatic ring. AAHR.92 was used as 3D query and a total of 2,26,248 compounds were obtained as hit. These compounds were taken for high throughput virtual screening. Finally, ten lead compounds with good docking score (-12.96 to -11.18) and ADME properties were identified. All the lead compounds were within the acceptable range defined for human use, thereby indicating their potential as drug-like molecules.
Supplementary material
Footnotes
Citation:Rajeswari et al, Bioinformation 10(3): 157-163 (2014)
References
- 1.Burns CJ, et al. Bioorg Med Chem Lett. 2009;19:5887. doi: 10.1016/j.bmcl.2009.08.071. [DOI] [PubMed] [Google Scholar]
- 2.Chem E, et al. Immunity. 2012;36:529. doi: 10.1016/j.immuni.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ghoreschi K, et al. Immunological Rev. 2009;228:273. doi: 10.1111/j.1600-065X.2008.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malaviya R, et al. Int Immunopharmacol. 2012;10:829. doi: 10.1016/j.intimp.2010.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O׳shea JJ, et al. Mol Immuno. 2004;41:727. [Google Scholar]
- 6.Cetkovic-Cvrlje M, et al. Clin Immunol. 2003;106:213. doi: 10.1016/s1521-6616(02)00049-9. [DOI] [PubMed] [Google Scholar]
- 7.Vainchenker W, et al. Semin Cell Dev Biol. 2008;19:385. doi: 10.1016/j.semcdb.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 8.Chen E, et al. Immunity. 2012;36:529. doi: 10.1016/j.immuni.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cetkovic-Cvrlje M, et al. Blood. 2001;98:1607. doi: 10.1182/blood.v98.5.1607. [DOI] [PubMed] [Google Scholar]
- 10.O׳Shea JJ, et al. Nat Rev Drug Discov. 2004;3:555. doi: 10.1038/nrd1441. [DOI] [PubMed] [Google Scholar]
- 11.Jaime-Figueroa S, et al. Bioorg Med Chem Lett. 2013;23:2522. doi: 10.1016/j.bmcl.2013.03.015. [DOI] [PubMed] [Google Scholar]
- 12.Kudlacz E, et al. Am J Transplant. 2004;4:51. doi: 10.1046/j.1600-6143.2003.00281.x. [DOI] [PubMed] [Google Scholar]
- 13.Tanaka Y, et al. Arthritis Care Res. 2011;63:1150. doi: 10.1002/acr.20494. [DOI] [PubMed] [Google Scholar]
- 14.Riese RJ, et al. Best Pract Res Clin Rheumatol. 2010;4:513. doi: 10.1016/j.berh.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 15.Kremer JM, et al. Arthritis Rheum. 2009;60:1895. doi: 10.1002/art.24567. [DOI] [PubMed] [Google Scholar]
- 16.Chang YB, et al. J Immunol. 2009;189:2183. doi: 10.4049/jimmunol.0804063. [DOI] [PubMed] [Google Scholar]
- 17.Trieu VN, et al. Biochem Biophys Res Commun. 2000;267:22. doi: 10.1006/bbrc.1999.1905. [DOI] [PubMed] [Google Scholar]
- 18.Cox L, Cools J. Chem Biol. 2011;18:277. doi: 10.1016/j.chembiol.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 19.Brown GR, et al. Bioorg Med Chem Lett. 2000;10:575. doi: 10.1016/s0960-894x(00)00051-2. [DOI] [PubMed] [Google Scholar]
- 20.Cole AG, et al. Bioorg Med Chem Lett. 2009;19:6788. doi: 10.1016/j.bmcl.2009.09.080. [DOI] [PubMed] [Google Scholar]
- 21.Chen JJ, et al. Bioorg Med Chem Lett. 2006;16:5633. doi: 10.1016/j.bmcl.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 22.Clark MP, et al. Bioorg Med Chem Lett. 2007;17:1250. doi: 10.1016/j.bmcl.2006.12.018. [DOI] [PubMed] [Google Scholar]
- 23.Phase 3.5. Schrodinger. New York, NY: LLC; 2013. [Google Scholar]
- 24.Dixon SL, et al. J Comput Aided Mol Des. 2006;20:647. doi: 10.1007/s10822-006-9087-6. [DOI] [PubMed] [Google Scholar]
- 25.Koes RD, Camacho JC. Nucl Acids Res. 2012;40:W409. doi: 10.1093/nar/gks378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soth M, et al. J Med Chem. 2013;56:345. doi: 10.1021/jm301646k. [DOI] [PubMed] [Google Scholar]
- 27.Halgren TA, et al. J Med Chem. 2004;47:1750. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 28.Friesner RA, et al. J Med Chem. 2006;49:6177. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 29.Small-Molecule Drug Discovery Suite 2013-1: Glide. version 5.9, Schrödinger. New York: LLC; 2013. [Google Scholar]
- 30.QikProp Version 3.6, Schrodinger. New York, NY: LLC; 2013. [Google Scholar]
- 31.Boggon TJ, et al. Blood. 2005;106:996. doi: 10.1182/blood-2005-02-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sekhar YN, et al. J Mol Graph Model. 2008;26:1338. doi: 10.1016/j.jmgm.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 33.Verdonk ML, et al. J Med Chem. 2005;48:6504. doi: 10.1021/jm050543p. [DOI] [PubMed] [Google Scholar]
- 34.de Graaf C, et al. J Med Chem. 2005;48:2308. doi: 10.1021/jm049650u. [DOI] [PubMed] [Google Scholar]
- 35.Moitessier N, et al. J Med Chem. 2006;49:1023. doi: 10.1021/jm0508437. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.