

Abstract
Exposure to arsenic affects large human populations worldwide, and has been associated with a long list of human diseases, including skin, bladder, lung, and liver cancers, diabetes, and cardiovascular disorders. In addition, there are large individual differences in susceptibility to arsenic-induced diseases, which are frequently associated with different patterns of arsenic metabolism. Several underlying mechanisms, such as genetic polymorphisms and epigenetics, have been proposed, as these factors closely impact the individuals' capacity to metabolize arsenic. In this context, the role of the gut microbiome in directly metabolizing arsenic and triggering systemic responses in diverse organs raises the possibility that perturbations of the gut microbial communities affect the spectrum of metabolized arsenic species and subsequent toxicological effects. In this study, we used an animal model with altered gut microbiome induced by bacterial infection, 16S rRNA gene sequencing and inductively coupled plasma mass spectrometry (ICP-MS)-based arsenic speciation to examine the effect of gut microbiome perturbations on the biotransformation of arsenic. Metagenomics sequencing revealed that bacterial infection significantly perturbed the gut microbiome composition in C57BL/6 mice, which in turn resulted in altered spectra of arsenic metabolites in urine, with inorganic arsenic species, methylated and thiolated arsenic being perturbed. These data clearly illustrated that gut microbiome phenotypes significantly affected arsenic metabolic reactions, including reduction, methylation and thiolation. These findings improve our understanding of how infectious diseases and environmental exposure interact, and may also provide novel insight regarding the gut microbiome composition as a new risk factor of individual susceptibility to environmental chemicals.
1. Introduction
Exposure to arsenic affects large human populations worldwide through the contamination of drinking water by geological sources of inorganic arsenic. Hundreds of millions of people around the world, especially in South and East Asia, drink water with inorganic arsenic levels that far exceed the 10 μg/L guideline, established or accepted by WHO and US EPA.1 In the United States, as many as 25 million people are estimated to drink water with an arsenic level above 10 μg/L, as private wells are not regulated by EPA and other agencies.2 Arsenic exposure has been associated with a number of diseases, such as skin, bladder, lung, and liver cancers, diabetes, as well as cardiovascular disorders and reproductive defects.1,3-5 More recently, arsenic exposure has been linked to an increased incidence of diabetes in animal models and human population studies.6,7 Numerous mechanisms, including the interaction with sulfur, oxidative stress, genotoxicity, altered DNA repair and signal transduction, cell proliferation and epigenetics, have been proposed for arsenic-induced diseases.1,3,8-10 In addition, there are large differences in susceptibility to arsenic-induced diseases among individuals,11,12 with several underlying mechanisms, such as genetic polymorphisms, epigenetics and nutrition homeostasis, being proposed. Individual susceptibility is frequently associated with different spectra of arsenic metabolism. Accumulating evidence indicates that perturbations of the gut microbiome and functions may play an important role in the development of human diseases. The essential role of the gut microbiome in a variety of aspects of human health and metabolic processing of xenobiotics raise the possibility that gut microbiome phenotypes affect the biotransformation of arsenic. This hypothesis is supported by several previous studies that have clearly demonstrated the involvement of the gut microbiome in reduction, methylation and thiolation of arsenic species.13,14
The human body harbors 100 trillion gut microbes, ∼10-times more than all human cells.15 It is estimated that the ∼500-1000 species residing in the human gut encode 100-fold more unique genes than our human genome. The gut microbiota has important functions in metabolic processing, energy production, immune cell development, food digestion, epithelial homeostasis, and so forth.16,17 Mounting evidence indicates that dysregulated gut microflora contributes to a variety of diseases, including diabetes, obesity, cardiovascular diseases, allergies, inflammatory bowel disease and others.18-21 The composition of the gut microbiome is highly diverse, and this diversity can be readily affected by external factors, such as environment, diet, bacterial/viral infection and antibiotics. Furthermore, previous studies have shown that altered gut microflora dramatically changed activities of diverse enzymes in the liver, including P450s and phase II enzymes responsible for the metabolic activation of xenobiotics.22 This raises the possibility that gut microbiome phenotypes play a role in defining individual response via altering metabolic capacity in the host when exposed to environmental chemicals.
In particular, inorganic arsenic is metabolized via a series of methylation and reduction reactions, with S-adenosylmethionine (SAM) as the main methyl donor.23,24 The widely-accepted metabolic pathway of inorganic arsenic consists of alternating reduction from pentavalent arsenic to trivalent and oxidative methylation of the trivalent arsenic metabolites. The six arsenic species involved in this pathway in humans are inorganic arsenic (iAsV and iAsIII), monomethylarsonic acid (MMAsV), monomethylarsonous acid (MMAsIII), dimethylarsinic acid (DMAsV) and dimethylarsinous acid (DMAsIII). In addition to arsenic metabolism catalyzed by specific enzymes, such as arsenic (+3 oxidation state) methyltransferase (As3mt), in diverse tissues,25 a few experiments have demonstrated that the gut microbiome participates in the metabolism of arsenic species.13,14,26,27 For example, Rowland and Davies reported the reduction of iAsV to iAsIII, as well as the formation of MMAs and DMAs, by rat cecal bacteria.28 Likewise, the thiolation of methylated As oxides in the cecal contents of a mouse 13 and the in vivo thiolation of arsenic have been reported.29-31 More recently, Van de Wiele, et al., found a high degree of methylation of iAs and As-contaminated soils by human gut microbiota.14 Besides the formation of MMAsV, they also detected the formation of highly toxic MMAsIII, and microbial thiolated monomethylmonothioarsonic acid (MMMTAsV).14
Given the role of the gut microbiome in direct metabolic reactions of arsenic and triggered systemic responses in other organs, we thus hypothesize that perturbations of the gut microbial community affect the spectrum of metabolized arsenic species and subsequent toxicological responses. In the present study, we used an animal model with altered gut microbiome, 16S rRNA gene sequencing and ICP-MS-based arsenic speciation to examine the effect of gut microbiome perturbations on the biotransformation of arsenic. Metagenomics sequencing revealed that bacterial infection significantly perturbed the gut microbiome composition in C57BL/6 mice, which in turn resulted in altered spectra of arsenic metabolites, with inorganic arsenic species and methylated arsenic being up-regulated and down-regulated, respectively. These data clearly illustrated that gut microbiome perturbations arising from bacterial infection significantly affect the metabolism of arsenic. These findings may provide novel insight regarding the gut microbiome composition as a new risk factor of individual susceptibility to environmental chemicals.
2. Materials and Methods
Chemicals
Sodium arsenite and arsenobetaine (AsB) were obtained from Fisher Scientific (Pittsburgh, PA) and Sigma-Aldrich (Milwaukee, WI), respectively. Dimethylarsinic acid (DMAsV) and disodium monomethylarsonate (MMAsV) were obtained from Chem Service (West Chester, PA). MMAsIII (oxomethylarsine), DMAsIII (iododimethylarsine), dimethylmonothioarsinic acid (DMTAs) and trimethyl arsine oxide (TMAsO) were provided by Professor William Cullen, University of British Columbia (Canada). Other reagents used for the HPLC mobile phase and diluent were Puratonic® 99.999% purity grade ammonium carbonate (Alfa Aesar, Ward Hill, MA), ammonium sulfate (Mallinckrodt, Hazelwood, MO), and ammonium acetate (ICN Biochemicals, Aurora, OH).
Animal infection and exposure
Helicobacter-free C57BL/6 (∼8 weeks old) mice were purchased from Jackson Laboratories (Bar Harbor, ME). Mice were provided pelleted rodent diet (ProLab 3000; Purina Mills, St. Louis, MO) and filtered water ad libitum and were maintained in AAALAC accredited facilities in microisolator caging under standard environmental conditions. All experiments were approved by the MIT Committee on Animal Care. Each grouping comprised 10 mice unless otherwise stated. Mice (Groups B and C only, Figure 1) were dosed at 9 to 10 weeks of age with 2 × 107 H. trogontum three times on alternate days by oral gavage. Inorganic arsenic (10 ppm) was then administered to mice (Groups B and D only, Figure 1) through drinking water for 4 weeks. The helicobacter infection status of these separately housed colonies was confirmed by fecal PCR at the end of the study. Urine samples were collected using a metabolic cage with dry ice placed around the urine collection vessel to prevent oxidation or degradation of metabolites during the collection period (∼16 h). Fecal pellets were also collected from individual animals. Plasma samples and tissues were collected during necropsy at the end of the study.
Figure 1.
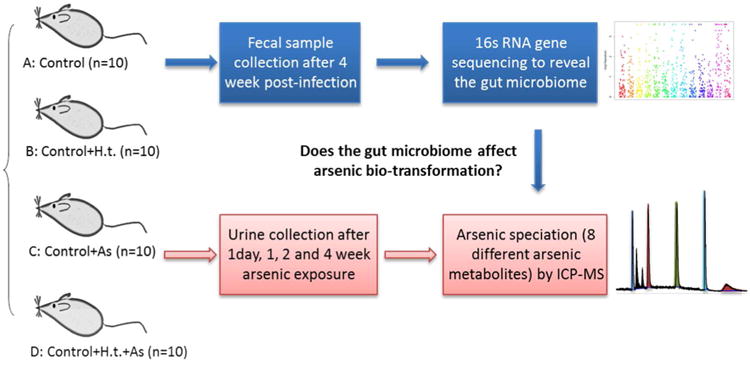
The integration approach combining a bacterial-infection animal model, 16S rRNA gene sequencing and ICP-MS-based arsenic speciation to explore the impact of gut microbiome changes on the biotransformation of arsenic. (Helicobacter troguntum: H.t.)
Animal monitoring and histological analysis
Throughout the experiments, mice were assessed for evidence of diarrhea, dehydration, and deteriorating body condition. Mice were euthanized with CO2 and necropsied after 4 weeks of arsenic consumption. Formalin-fixed tissues were routinely processed, embedded in paraffin, sectioned at 4 μm, stained with hematoxylin and eosin, and evaluated by a board-certified veterinary pathologist blinded to the sample identity. Inflammation, edema, epithelial defects, hyperplasia, and dysplasia of multiple regions of liver and colon were scored on an ascending scale (0 to 4, with 0 being normal) of severity and invasiveness of the lesion if any.
16S rRNA metagenomics sequencing
DNA was isolated from fecal pellets using a PowerSoil® DNA Isolation Kit as instructed by the manufacturer (MO BIO Laboratories, CA). The resultant DNA was quantified by a UV spectroscopy and stored at -80°C for further analysis. DNA was amplified using universal primers of U515 (GTGCCAGCMGCCGCGGTAA) and E786 (GGACTACHVGGGTWTCTAAT) to target the V4 regions of 16S rRNA of bacteria. Individual samples were barcoded, pooled to construct the sequencing library, followed by sequencing with an Illumina Miseq to generate pair-ended 150 × 150 reads.
Data analysis of 16s rRNA sequencing data
The raw mate-paired fastq files were quality-filtered, demultiplexed and analyzed using Quantitative Insights into Microbial Ecology (QIIME).32 For quality filtering, the default parameters of QIIME were maintained in which reads with a minimum Phred quality score less than 20, containing ambiguous base calls and containing fewer than 113bp of consecutive high quality base calls, were discarded.
Additionally, reads with three consecutive low quality bases were truncated. The samples sequenced were demultiplexed using 8bp barcodes, allowing 1.5 errors in the barcode. UCLUST was used to choose the Operational Taxonomic Units (OTUs) with a threshold of 97% sequence similarity.33 A representative set of sequences from each OTU was selected for taxonomic identification of each OTU using the Ribosomal Database Project (RDP) classifier.34 The Greengenes OTUs (4feb2011 build) reference sequences (97% sequence similarity) were used as the training sequences for RDP. A 0.80 confidence threshold was used for taxonomic assignment.
Arsenic speciation
Arsenic species were measured using an Agilent 7500 ICP-MS (Santa Clara, CA), as described previously.35 The ICP-MS was interfaced with an Agilent 1260 HPLC, and a Hamilton PRP-X100 column was used to separate arsenic species. The mobile phase A was 10 mM ammonium carbonate and 10 mM TRIS (pH=8.7), and the mobile phase B included 10 mM ammonium carbonate, 10 mM TRIS and 15 mM ammonium sulfate (pH 8.0). The following gradient was run: 0 – 5 min 0% B to 100% B, 5–11 min 100% B, 11-16 min 100% A. Sample preparation was conducted as follows: 10 μL of urine sample or arsenic reference standards was diluted with 0.1 M ammonium acetate, pH=5, to make a total volume of 40 μL in a polypropylene microcentrifuge tube. Microcentrifuge tubes containing samples were centrifuged for 5 min at 12,000 rpm at 4 °C in a refrigerated centrifuge. Following centrifugation, 35 μL of supernatant was transferred to an HPLC autosampler vial, without disturbing the solid pellet at the bottom of the tube, followed by injection of 5-10 μL into the mass spectrometer.
Statistical analysis
Data analysis was conducted with multivariate statistical methods to compare gut microbiome communities between groups. Principal coordinate analysis (PCoA) was performed to examine intrinsic clusters within the observations. In addition, Jackknifed beta diversity and hierarchical clustering analysis via Unweighted Pair Group Method with Arithmetic mean (UPGMAD) was used to differentiate the gut microbiome profiles of the controls and infected samples. The difference in the individual gut microbiome composition between control and treatment groups was assessed using a non-parametric test with Metastats as described previously.36 The abundance of arsenic species in the control and treatment groups were compared by Student's t test, and results were considered significant when p < 0.05. The correlation between different arsenic metabolites was generated using Pearson's correlation coefficient. One-way ANOVA with repeated measures was used to assess the time effect of gut microbiome changes on arsenic metabolites. A linear mixed statistical model was also applied to examine the impact of gut microbiome alterations on the formation of arsenic species. All data handling and statistical analyses were performed using the statistical R package or SAS.
3. Results
3.1 Experimental workflow
Figure 1 shows the experimental workflow for the 16S rRNA sequencing and arsenic metabolite profiling. First, two groups of mice (B and D groups, 10/each group) were infected with H. troguntum with three doses in a week, as described previously.37 Next, arsenic was administrated to two groups of mice (one control, and another consisting of mice with altered gut microbiome arising from bacterial infection, i.e. C and D groups in Figure 1). For the gut microbiome profiling, DNA was isolated from fecal pellets, amplified by PCR using 16S rRNA specific primers, and followed by 150 × 150 bp paired-end sequencing using an Illumina Miseq platform. The resultant sequencing reads were processed by the QIIME and Metastats software packages to reveal infection-induced gut microbiome changes. For arsenic speciation analysis, urine was collected and analyzed by HPLC-ICP-MS using a Hamilton PRP-X100 column. The resultant intensity counts (m/z = 75) of each arsenic metabolite were further normalized to reduce the impact of individual variations of animals on uptaking arsenic during exposure, followed by statistical analysis to reveal any altered metabolites between the control and treatment group. Urine samples were collected at multiple time points including 1 day, 1, week, 2 and 4 weeks to examine potential dynamic effects of gut microbiome changes on the metabolism of arsenic.
3.2 Arsenic speciation by ICP-MS
Figure 2A illustrates an arsenic metabolism pathway that involves reduction, methylation and thiolation, with 8 major arsenic metabolites being measured. The formation of DMTAsV is unique, as gut bacteria are important in the generation of this thiolated arsenic metabolite. Figure 2B shows a HPLC-ICP-MS chromatogram from the urine sample of a mouse exposed to iAsIII for 1 week. AsB and TMAsO eluted first but overlapped, followed by iAsIII, MMAsIII, DMAsV, MMAsV, iAsV and DMTAsV. Among these arsenic species, DMAsV is predominant (∼90% counts), followed by iAsV (∼5% counts), and the remaining arsenic metabolites account for a total of ∼5% counts, as illustrated in Figure 2C. DMAsIII could not be resolved on the column as it coeluted with the large peak of DMAsV.
Figure 2.
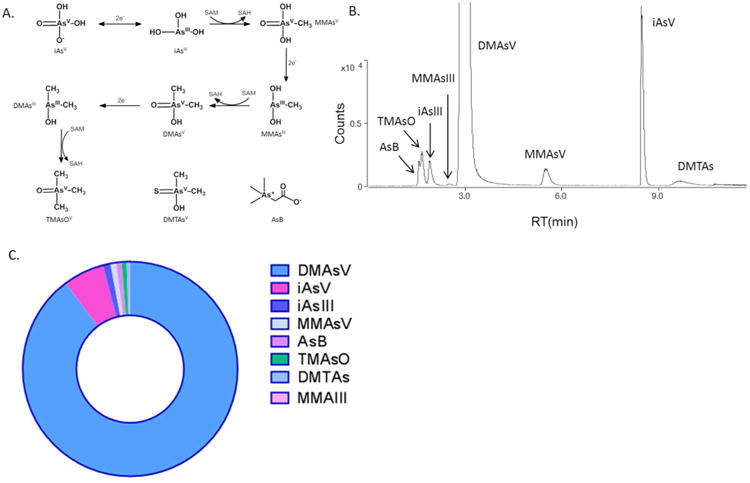
The metabolism pathway and major arsenic species (A), the ICP-MS chromatogram of urine sample from a mouse exposed to 10 ppm arsenic (B), and the relative abundance of different arsenic species after normalization using the total counts of arsenic at m/z = 75, with DMAsV and iAsV being the top 2 arsenic metabolites (C).
3.3 Gut microbiome changes induced by bacteria
As described in the method section, H. trogontum was used to disturb the gut microbiome to construct an animal model with bacterial-induced gut microbiome alterations and also to serve as a model to understand how infection influences individual's response to environmental chemicals.
To probe gut microbiome changes arising from bacterial infection, 16S rRNA gene sequencing was conducted. Figure 3A shows the identified gut bacteria assigned at family level from 16S rRNA sequencing reads, with each color representing an individual bacterial family. In terms of the assignment at phylum level, Fimicutes (53.7%) and Bacteroidetes (41.1%) are predominant in the gut bacteria of mice, followed by Tenericutes (2.7%), Actinobacteria (0.2%), Cyanobacteria (0.05%) and Proteobacteria (0.02%), with 2.2% sequences unmatched with the database (Figure S1). The observations and assignments of gut bacteria at phylum level are consistent with previous reports that the gut microbiome consists of only several phyla.38 Figure 3B further illustrates all the gut bacteria families in control and infection groups, as revealed by 16S sequencing at the family level. The forward read and reverse read identify similar numbers and consistent types of gut bacteria at the family level, with 42 and 46 for the forward and reverse read, respectively. Significantly changed gut bacteria families (those above the dotted line, p<0.05) are illustrated in the Figure 3B and their family level taxa assignments and fold changes are illustrated in Figure 3C. In addition to helicobacter detected in the infection group only, two Bacteroidetes families increased ∼2000 and 15 fold compared to control (p_Bacteroidetes;c_Bacteroidia;o_Bacteroidales;f_Porphyromonadaceae and p_Bacteroidetes;c_Bacteroidia;o_Bacteroidales;Other). Five bacterial families belonging to Firmicutes were all down-regulated in infected animals, with fold changes from -2 to -11.
Figure 3.
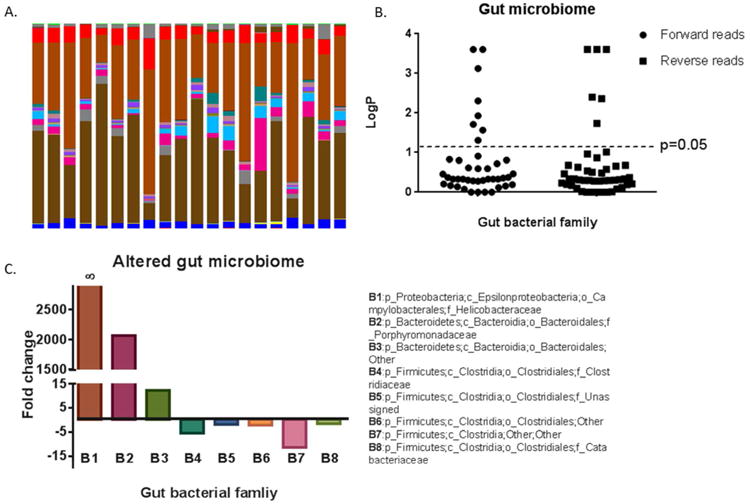
Major gut bacteria at family level in individual animals, as revealed by 16S rRNA sequencing, with each color being one bacterial family (A); Statistically significantly altered gut bacterial families (those above the dotted line, p<0.05) by 16S rRNA gene sequencing forward and reverse reads (B); The fold changes and taxa assignments of perturbed gut bacteria at family level in infected mice compared to controls (C). (p: phylum; c: class; o: order; f: family, ∞: helicobacter was detected only in the infection group).
The difference in the gut microbiome patterns arising from H. trogontum infection could be readily differentiated when comparing beta diversity UniFrac distances,39,40 as shown by the PCoA plot in Figure 4A. The control (Red) and treated animals (Blue) are well separated, with 22.8%, 12.1% and 8.6% variation explained by PC1, PC2 and PC3, respectively. Consistent with the PCoA plot, the Jackknifed beta diversity and hierarchical clustering analysis via UPGMAD demonstrate that infected animals and controls typically cluster in their own groups, but with one treated mouse being grouped with the control group, as shown in Figure 4B.
Figure 4.
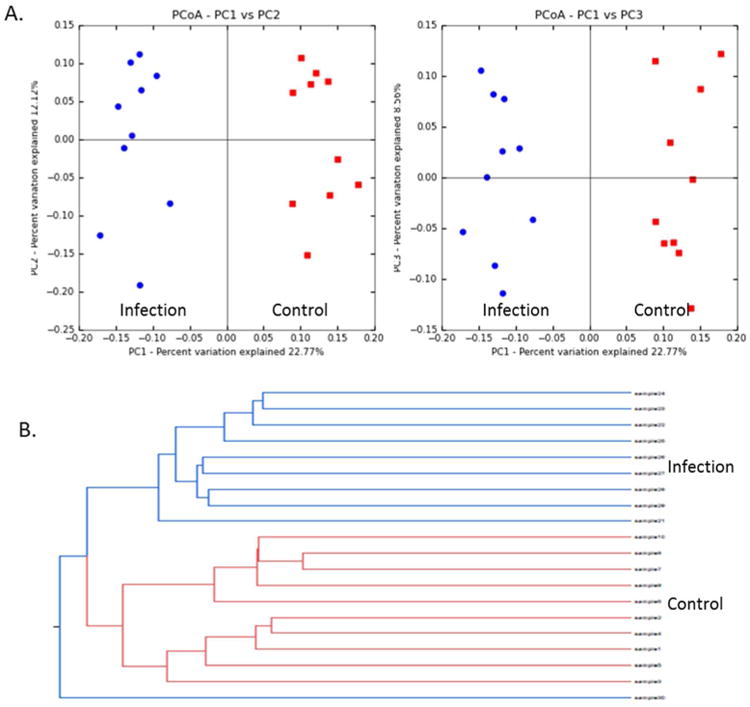
The gut microbiome patterns of control samples (Red) and infected (Blue) mice are readily differentiated by Principal Coordinate Analysis (PCoA), with 22.8%, 12.1% and 8.6% variation explained by PC1, PC2 and PC3, respectively (A); Hierarchical Clustering analysis by Unweighted Pair Group Method with Arithmetic mean indicates that controls and infected mice cluster in their own groups (Red for control, Blue for treatment group) (B).
3.4 Histological analysis
Histology analysis was also conducted to examine whether H. trogontum infection caused detectable tissue pathological changes. Histological scoring revealed no statistically significant differences between the controls and infected animals in terms of inflammation, edema, epithelial defects, crypt atrophy, hyperplasia and dysplasia in multiple regions of liver and colon (Figure S2). These histological results are consistent with previous studies, which reveal that C57BL/6 mice are not susceptible to lower bowel inflammation due to Helicobacter spp. infection, despite persistent infection.37
3.5 Gut microbiome affects the metabolism of arsenic
As shown in Figure 3 and 4, the gut microbiome was significantly perturbed by H. trogontum infection. We next compared the relative abundance of measured arsenic species to examine whether the gut microbiome alterations impact the biotransformation of arsenic, as shown in Figure 5 A to F. DMTAsV, a gut-flora-generated thiolated arsenic metabolite, DMAsV and MMAsIII were significantly decreased after 1 day post arsenic exposure, while iAsV was increased in the infection group, suggesting that gut microbiome changes resulting from bacterial infection inhibit the detoxification of arsenic. Moreover, inorganic arsenic (iAsV) in urine from 1 day-exposed mice is highly negatively correlated with methylated and thiolated arsenic metabolites, including DMAsV and DMTAsV, as shown in Figure S3. Similar to the case of 1 day exposure, iAsV was increased, while methylated arsenic metabolites, such as DMAsV, were significantly down-regulated at other time points, including 1 week or 2 weeks post arsenic exposure. These findings clearly indicate that bacterial infection-induced gut microbiome changes alter the reduction, methylation and thiolation of arsenic, and consequently inhibit its detoxification.
Figure 5.
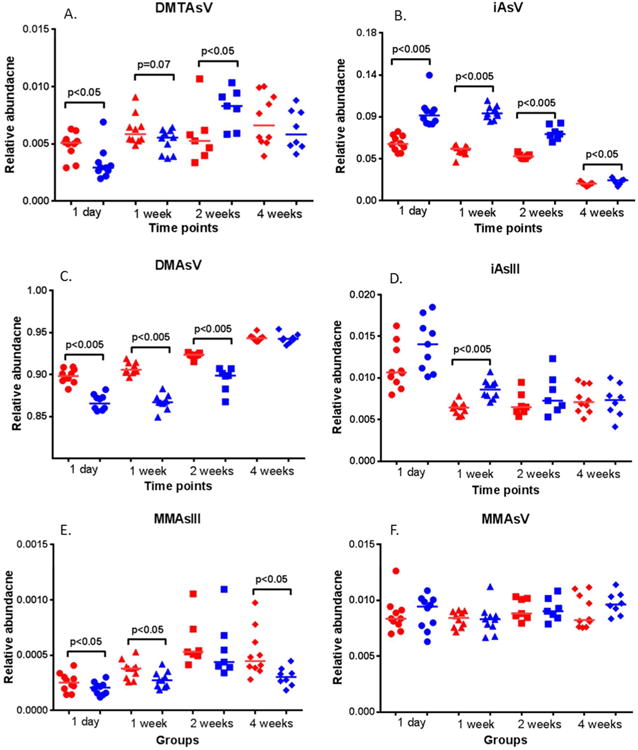
Arsenic metabolites were statistically significantly disturbed in infected animals compared to the controls (p<0.05), with inorganic arsenic species, methylated and thiolated arsenic metabolites being perturbed at multiple time points: DMTAsV (A), iAsV (B), DMAsV (C), iAsIII (D), MMAsIII (E) and MMAsV (F). (Red: Controls; Blue: infection groups).
It is well documented that the establishment or recovery of the gut microbiome is a temporal process.41-43 Thus, the interaction between the gut microbiome and arsenic metabolism could be dynamic. Figure S4 A and B illustrate the time effects of gut microbiome on the formation of DMTAsV. Although there is no time dependence for DMTAsV in the control groups, a statistically significant time-dependent effect was observed in infection groups (p<0.005), highlighting a potential dynamic impact of gut microbiome changes on the biotransformation of arsenic. Time-dependent effects were observed for the formation of DMAsV in both controls and treated animals, as shown in Figure S4 C and D. However, a linear mixed statistical model further identified the influence of gut microbiome perturbations on the methylation of arsenic, with the intercepts of 0.0017 and 0.0032 being determined for the control and treatment groups, respectively.
4. Discussion
In this study, we used a bacterial-infection animal model, high-throughput 16S rRNA gene sequencing and ICP-MS based arsenic speciation to study the impact of bacterial-induced gut microbiome perturbations on the biotransformation of arsenic. The data clearly demonstrated that bacterial infection caused significant changes in the gut microbiome of mice, which in turn affected the spectrum of metabolized arsenic species in mice. These findings may provide novel mechanistic insights regarding the gut microbiome as a new risk factor of individual responses when exposed to diverse environmental chemicals.
We used bacterium-infected mice as an animal model to probe the impact of gut microbiome changes on arsenic metabolism. This selection was grounded in the understanding that bacterial infections are naturally-occurring and universal. More importantly, infectious diseases have been intertwined with environmental exposure in complex human exposure scenarios, but the role of infection is not often well-considered in the common toxicological paradigm.44 Our results are a beginning insight into the relationships between bacterial infections and environmental exposures, and their consequent impacts on the toxicity of toxicants. Specifically, we discovered that several gut bacteria families changed arising from H. trogontum infection, as shown in Figure 3. In addition to the H. trogontum, another two Bacteroidetes families were significantly up-regulated. They increased ∼2000 and 15 fold compared to control, respectively (p_Bacteroidetes;c_Bacteroidia;o_Bacteroidales;f_Porphyromonadaceae and p_Bacteroidetes;c_Bacteroidia;o_Bacteroidales;Other). Porphyromonadaceae is a family of pathogenic bacteria generally composed of two genera of environmental bacteria, g_Porphyromonas and g_Dysgonomonas.45 Thus, it appears that H. trogontum infection increases the expansion of opportunistic pathogens. Five bacterial families belonging to Firmicutes were all down-regulated in infected animals, with fold changes ranging from -2 to -11. These data clearly demonstrated that bacterial infection significantly altered the gut microbiome. It should be noted that our analyses were conducted at the family level due to higher confidence in the assignment of taxa based on the sequencing reads, therefore, a significant change at the family level may reflect changes of multiple gut bacteria at genus and species levels.
To examine whether infection and perturbed gut microbiome impact the biotransformation of arsenic, we measured arsenic species in urine and found a significant change in the spectrum of arsenic metabolites. Among of these disturbed arsenic metabolites, DMTAsV is of particular interest, since previous studies have documented that thiolation of arsenic could be carried out by the gut microflora.13,14 For example, the capacity of the anaerobic microflora from a mouse cecum to metabolize dimethylarsinic acid was examined, and DMAsV was thiolated to DMTAsV and dimethyldithioarsinic acid (DMDTAs) after incubated under anaerobic conditions at 37°C for up to 24 hours.13 Another study, taking advantage of a dynamic model of the human gut, discovered that microbial thiolation led to an additional thiolated arsenic metabolite, MMMTAsV, after incubating the colon microbiata with iAs and As-contaminated urban soils.14 Moreover, urine analysis after iAs exposure has already revealed the formation of both DMTAsV and MMMTAsV in animals and humans.46,47 In this study, we measured the abundance of DMTAsV, which was significantly different between the control and treatment group at 1 day and 2 week post-exposure to arsenic. For example, DMTAsV was decreased in urine (fold change=∼-1.5, p<0.05) after a 1-day exposure in mice with disturbed gut microbiome, highlighting the role of the gut flora on generating thiolated arsenic species.
Methylation has long been considered a detoxification pathway, with As3mt being the primary enzyme for the methylation of arsenic. As3mt is widely distributed in tissues, such as liver, kidney, testis, and lung,25 and plays a key role in arsenic metabolism and toxicity, as revealed by a few studies using As3mt knockout mice.48-51 In addition to the methylation catalyzed by As3mt, a high degree of methylation of iAs (10 μg methylarsenical/g biomass/hr) and As-contaminated soils (up to 28 μg/g biomass/hr) by the gut microbiota was also reported.14 DMAsV is the primary metabolite during arsenic detoxification and is readily excreted in urine. In this study, we discovered that DMAsV excretion in urine was lower in the treatment group and typically accompanied by increased inorganic arsenic species, indicating the detoxification was impaired by the gut microbiome perturbations. Since both tissue enzymes and gut microflora are responsible for the methylation of arsenic, any measurement on methylated arsenic species reflects the superimposed effects of both factors. Significantly modulated DMAsV may therefore result from perturbed gut microbiome communities or changes in As metabolic capacity of tissues during infection. Previous studies, however, have revealed that gut microbiome changes can also induce different expressions of liver metabolizing enzymes and functions of other organs distant from the gut.22,43 For instance, colonization of gut bacteria was associated with modifications of hepatic Cyp8b1 expression and the subsequent alteration of bile acid metabolites, including taurocholate and tauromuricholate.22 Expression and activity of major drug-metabolizing enzymes, such as Cyp3A11 and Cyp2C29, were also significantly stimulated by gut microbiome changes.22 The systemic effects of the gut microbiome can even reach the brain, and microbial colonization initiates signaling mechanisms that affect neuronal circuits involved in motor control and anxiety behavior.43 It is therefore also likely that enzyme systems that regulate the metabolism of arsenic could be modulated by gut microbiome perturbations, which in turn contribute to reduced methylation capacity in the treatment group. Answers to this intriguing question await future experiments.
There is considerable individual variation in arsenic metabolism and toxicity,52-55 and some individuals methylate and excrete arsenic less efficiently than others, which could place them at higher risks at developing arsenic-induced diseases. Different mechanisms for these variations e.g., genetic variants, epigenetic regulation, and nutrition homeostasis, have been proposed.56-58 Genetic susceptibility factors for arsenic-induced diseases have been extensively studied.59-61 For example, single nucleotide polymorphisms of four genes, including purine nucleoside phosphorylase (PNP), As3mt, glutathione S-transferase omega 1 and omega 2, which mediate multiple-step metabolism of arsenic through sequential reduction and oxidative methylation, were examined and three exonic polymorphisms, His20His, Gly51Ser, and Pro57Pro of PNP, were recently found to be associated with arsenicism.60 The abundance and patterns of arsenic species have long been closely associated with toxicity in individuals exposed to arsenic, with the MMAsV/DMAsV ratio showing a positive association with arsenic toxicity in several studies.61,62 In particular, large individual variations in the gut microbiome and metagenomic genotypes have been reported by others.63-65 In this complementary and specific study, we showed that infection-induced gut microbiome perturbations affect the spectra of arsenic metabolites, thus implicating the gut microbiome as a potential risk factor associated with individual susceptibility to arsenic exposure.
We have also demonstrated that the infection and/or gut microbiome has a dynamic effect on the spectra of arsenic metabolites (Figure S4), which may reflect the temporal establishment of the gut microbiome communities following infection. It is well documented that infants acquire their gut microbiome from the environment through the interaction with their mothers,66 and that the establishment of the gut microflora is a temporal process, with bacterial abundances increasing ∼ 6 orders of magnitude within weeks of life and becoming more adult-like within the first year of life.41 Similarly, animal studies also show that reconstruction of the gut microbiota ecosystem in germ-free mice through colonization of gut bacteria is a dynamic event, and gut microbiome compositions in colonized mice are highly time-dependent.22 In particular, we discovered that the formation of DMTAsV, a gut-flora-generated arsenic metabolite, is time-dependent, highlighting the dynamic effects of gut microbiome changes on the biotransformation of specific arsenic metabolites.
Measuring arsenic species in urine only represents a limitation of the study, since these data do not reveal the retention/redistribution of As species in tissues. Future studies are needed to pinpoint the effect of bacterial infection on the tissue arsenic burden. In addition, we took advantage of an animal model with perturbed gut microbiome from bacterial infections to emulate naturally-occurring and universal events in human exposure scenarios. Although it has been demonstrated that C57BL/6 mice are not susceptible to Helicobacter spp. and infection-induced immune response is limited, it would be challenging to completely rule out the role of immune response in regulating arsenic biotransformation, since the immune system is intrinsically intertwined with the gut microbiome. Animal models established via other approaches, such as transplantation of the caecal microbiota, may serve as better tools to further elucidate the role of the gut microbiome in mediating arsenic metabolism in future studies.
In summary, we have combined an animal model with altered gut microbiome, 16S rRNA gene sequencing and arsenic speciation to analyze the impact of gut microbiome changes on the biotransformation of arsenic. Several arsenic species were significantly modulated by gut microbiome perturbations resulting from bacterial infection, and a dynamic effect of gut microbiome on the metabolism of arsenic was also revealed. Taken together, these data show that gut microbiome compositions play a role in defining the abundance of specific toxic species, supporting the hypothesis that gut microbiome phenotypes affect the spectra of arsenic metabolites. The finding that bacterial infection impacts the biotransformation of arsenic may improve our understanding how infectious diseases and environmental exposure interact. Moreover, this study highlights the gut microbiome phenotype as a potential novel risk factor associated with individual susceptibility to diverse environmental chemicals.
Supplementary Material
Acknowledgments
We are grateful to the MIT Division of Comparative Medicine and associated professionals for their timely and indispensable assistance during this study. We thank Agilent Technologies for graciously providing access to liquid chromatography and mass spectrometer systems.
Funding Support: The authors thank the MIT Center for Environmental Health Sciences for financial support through a pilot project under NIEHS grant (P30 ES002109). The author also thanks the UNC Center for Environmental Health and Susceptibility for partial financial support by NIEHS grant (P30 ES010126).
Abbreviations
- ICP-MS
Inductively coupled plasma mass spectrometry
- H.
Helicobacter
- SAM
S-adenosylmethionine
- iAs
Inorganic arsenic
- MMAsV
monomethylarsonic acid
- MMAsIII
monomethylarsonous acid
- DMAsV
dimethylarsinic acid
- DMAsIII
dimethylarsinous acid
- MMAsV
monomethylarsonic acid
- DMTAsV
dimethylmonothioarsinic acid
- MMMTAsV
monomethylmonothioarsonic acid
- AsB
arsenobetaine
- TMAsO
trimethyl arsine oxide
- QIIME
Quantitative Insights into Microbial Ecology
- OTUs
Operational Taxonomic Units
- RDP
Ribosomal Database Project
- PCoA
Principal coordinate analysis
- UPGMAD
Unweighted Pair Group Method with Arithmetic mean
- DMDTAs
dimethyldithioarsinic acid
- As3mt
arsenic (+3 oxidation state) methyltransferase
- DMDTAs
dimethyldithioarsinic acid
Footnotes
Supporting Information: Gut bacteria abundance at phylum level of each sample revealed by 16S rRNA sequencing; Histological analysis for the controls and infected animals, with inflammation being scored for multiple regions of the colon and liver; Correlation analysis between iAsV with methylated (DMAsV) and thiloated arsenic species (DMTAs) in 1 day-exposed samples; the dynamic interaction between the gut microbiome perturbations and arsenic species. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hughes MF, Beck BD, Chen Y, Lewis AS, Thomas DJ. Arsenic exposure and toxicology: a historical perspective. Toxicol Sci. 2011;123:305–332. doi: 10.1093/toxsci/kfr184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kozul CD, Ely KH, Enelow RI, Hamilton JW. Low-dose arsenic compromises the immune response to influenza A infection in vivo. Environ Health Perspect. 2009;117:1441–1447. doi: 10.1289/ehp.0900911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fry RC, Navasumrit P, Valiathan C, Svensson JP, Hogan BJ, Luo M, Bhattacharya S, Kandjanapa K, Soontararuks S, Nookabkaew S, Mahidol C, Ruchirawat M, Samson LD. Activation of inflammation/NF-kappaB signaling in infants born to arsenic-exposed mothers. PLoS Genet. 2007;3:e207. doi: 10.1371/journal.pgen.0030207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gribble MO, Howard BV, Umans JG, Shara NM, Francesconi KA, Goessler W, Crainiceanu CM, Silbergeld EK, Guallar E, Navas-Acien A. Arsenic exposure, diabetes prevalence, and diabetes control in the strong heart study. Am J Epidemiol. 2012;176:865–874. doi: 10.1093/aje/kws153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hou Y, Xue P, Woods CG, Wang X, Fu J, Yarborough K, Qu W, Zhang Q, Andersen ME, Pi J. Association between Arsenic Suppression of Adipogenesis and Induction of CHOP10 via the Endoplasmic Reticulum Stress Response. Environ Health Perspect. 2012;121:237–243. doi: 10.1289/ehp.1205731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paul DS, Walton FS, Saunders RJ, Styblo M. Characterization of the impaired glucose homeostasis produced in C57BL/6 mice by chronic exposure to arsenic and high-fat diet. Environ Health Perspect. 2011;119:1104–1109. doi: 10.1289/ehp.1003324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maull EA, Ahsan H, Edwards J, Longnecker MP, Navas-Acien A, Pi J, Silbergeld EK, Styblo M, Tseng CH, Thayer KA, Loomis D. Evaluation of the association between arsenic and diabetes: a National Toxicology Program workshop review. Environ Health Perspect. 2012;120:1658–1670. doi: 10.1289/ehp.1104579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smeester L, Rager JE, Bailey KA, Guan X, Smith N, Garcia-Vargas G, Del Razo LM, Drobna Z, Kelkar H, Styblo M, Fry RC. Epigenetic changes in individuals with arsenicosis. Chem Res Toxicol. 2011;24:165–167. doi: 10.1021/tx1004419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang XJ, Sun Z, Chen W, Eblin KE, Gandolfi JA, Zhang DD. Nrf2 protects human bladder urothelial cells from arsenite and monomethylarsonous acid toxicity. Toxicol Appl Pharmacol. 2007;225:206–213. doi: 10.1016/j.taap.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiong L, Wang Y. Quantitative proteomic analysis reveals the perturbation of multiple cellular pathways in HL-60 cells induced by arsenite treatment. J Proteome Res. 2010;9:1129–1137. doi: 10.1021/pr9011359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hernandez A, Marcos R. Genetic variations associated with interindividual sensitivity in the response to arsenic exposure. Pharmacogenomics. 2008;9:1113–1132. doi: 10.2217/14622416.9.8.1113. [DOI] [PubMed] [Google Scholar]
- 12.Steinmaus C, Yuan Y, Kalman D, Rey OA, Skibola CF, Dauphine D, Basu A, Porter KE, Hubbard A, Bates MN, Smith MT, Smith AH. Individual differences in arsenic metabolism and lung cancer in a case-control study in Cordoba, Argentina. Toxicol Appl Pharmacol. 2010;247:138–145. doi: 10.1016/j.taap.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kubachka KM, Kohan MC, Herbin-Davis K, Creed JT, Thomas DJ. Exploring the in vitro formation of trimethylarsine sulfide from dimethylthioarsinic acid in anaerobic microflora of mouse cecum using HPLC-ICP-MS and HPLC-ESI-MS. Toxicol Appl Pharmacol. 2009;239:137–143. doi: 10.1016/j.taap.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 14.Van de Wiele T, Gallawa CM, Kubachka KM, Creed JT, Basta N, Dayton EA, Whitacre S, Du LG, Bradham K. Arsenic metabolism by human gut microbiota upon in vitro digestion of contaminated soils. Environ Health Perspect. 2010;118:1004–1009. doi: 10.1289/ehp.0901794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124:837–848. doi: 10.1016/j.cell.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 16.Holmes E, Li JV, Athanasiou T, Ashrafian H, Nicholson JK. Understanding the role of gut microbiome-host metabolic signal disruption in health and disease. Trends Microbiol. 2011;19:349–359. doi: 10.1016/j.tim.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 17.Tremaroli V, Backhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489:242–249. doi: 10.1038/nature11552. [DOI] [PubMed] [Google Scholar]
- 18.Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, Wu Y, Schauer P, Smith JD, Allayee H, Tang WH, DiDonato JA, Lusis AJ, Hazen SL. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Graessler J, Qin Y, Zhong H, Zhang J, Licinio J, Wong ML, Xu A, Chavakis T, Bornstein AB, Ehrhart-Bornstein M, Lamounier-Zepter V, Lohmann T, Wolf T, Bornstein SR. Metagenomic sequencing of the human gut microbiome before and after bariatric surgery in obese patients with type 2 diabetes: correlation with inflammatory and metabolic parameters. Pharmacogenomics J. 2012 doi: 10.1038/tpj.2012.43. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 21.Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, Peng Y, Zhang D, Jie Z, Wu W, Qin Y, Xue W, Li J, Han L, Lu D, Wu P, Dai Y, Sun X, Li Z, Tang A, Zhong S, Li X, Chen W, Xu R, Wang M, Feng Q, Gong M, Yu J, Zhang Y, Zhang M, Hansen T, Sanchez G, Raes J, Falony G, Okuda S, Almeida M, LeChatelier E, Renault P, Pons N, Batto JM, Zhang Z, Chen H, Yang R, Zheng W, Li S, Yang H, Wang J, Ehrlich SD, Nielsen R, Pedersen O, Kristiansen K, Wang J. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 22.Claus SP, Ellero SL, Berger B, Krause L, Bruttin A, Molina J, Paris A, Want EJ, de WI, Cloarec O, Richards SE, Wang Y, Dumas ME, Ross A, Rezzi S, Kochhar S, Van BP, Lindon JC, Holmes E, Nicholson JK. Colonization-induced host-gut microbial metabolic interaction. MBio. 2011;2:e00271–10. doi: 10.1128/mBio.00271-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watanabe T, Hirano S. Metabolism of arsenic and its toxicological relevance. Arch Toxicol. 2013;87:969–979. doi: 10.1007/s00204-012-0904-5. [DOI] [PubMed] [Google Scholar]
- 24.Healy SM, Wildfang E, Zakharyan RA, Aposhian HV. Diversity of inorganic arsenite biotransformation. Biol Trace Elem Res. 1999;68:249–266. doi: 10.1007/BF02783907. [DOI] [PubMed] [Google Scholar]
- 25.Healy SM, Casarez EA, yala-Fierro F, Aposhian H. Enzymatic methylation of arsenic compounds. V. Arsenite methyltransferase activity in tissues of mice. Toxicol Appl Pharmacol. 1998;148:65–70. doi: 10.1006/taap.1997.8306. [DOI] [PubMed] [Google Scholar]
- 26.Conklin SD, Ackerman AH, Fricke MW, Creed PA, Creed JT, Kohan MC, Herbin-Davis K, Thomas DJ. In vitro biotransformation of an arsenosugar by mouse anaerobic cecal microflora and cecal tissue as examined using IC-ICP-MS and LC-ESI-MS/MS. Analyst. 2006;131:648–655. doi: 10.1039/b516275k. [DOI] [PubMed] [Google Scholar]
- 27.Alava P, Tack F, Laing GD, Van de Wiele T. Arsenic undergoes significant speciation changes upon incubation of contaminated rice with human colon micro biota. J Hazard Mater. 2012 doi: 10.1016/j.jhazmat.2012.05.042. dx.doi.org/10.1016/j.jhazmat.2012.05.042. in press. [DOI] [PubMed] [Google Scholar]
- 28.Rowland IR, Davies MJ. In vitro metabolism of inorganic arsenic by the gastrointestinal microflora of the rat. J Appl Toxicol. 1981;1:278–283. doi: 10.1002/jat.2550010508. [DOI] [PubMed] [Google Scholar]
- 29.Kuroda K, Yoshida K, Yoshimura M, Endo Y, Wanibuchi H, Fukushima S, Endo G. Microbial metabolite of dimethylarsinic acid is highly toxic and genotoxic. Toxicol Appl Pharmacol. 2004;198:345–353. doi: 10.1016/j.taap.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 30.Naranmandura H, Suzuki KT. Formation of dimethylthioarsenicals in red blood cells. Toxicol Appl Pharmacol. 2008;227:390–399. doi: 10.1016/j.taap.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 31.Naranmandura H, Suzuki N, Suzuki KT. Reaction mechanism underlying the in vitro transformation of thioarsenicals. Toxicol Appl Pharmacol. 2008;231:328–335. doi: 10.1016/j.taap.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 32.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 34.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Verdon CP, Caldwell KL, Fresquez MR, Jones RL. Determination of seven arsenic compounds in urine by HPLC-ICP-DRC-MS: a CDC population biomonitoring method. Anal Bioanal Chem. 2009;393:939–947. doi: 10.1007/s00216-008-2537-3. [DOI] [PubMed] [Google Scholar]
- 36.White JR, Nagarajan N, Pop M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput Biol. 2009;5:e1000352. doi: 10.1371/journal.pcbi.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whary MT, Danon SJ, Feng Y, Ge Z, Sundina N, Ng V, Taylor NS, Rogers AB, Fox JG. Rapid onset of ulcerative typhlocolitis in B6.129P2-IL10tm1Cgn (IL-10-/-) mice infected with Helicobacter trogontum is associated with decreased colonization by altered Schaedler's flora. Infect Immun. 2006;74:6615–6623. doi: 10.1128/IAI.01091-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 39.Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. Development of the human infant intestinal microbiota. PLoS Biol. 2007;5:e177. doi: 10.1371/journal.pbio.0050177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ge Z, Feng Y, Taylor NS, Ohtani M, Polz MF, Schauer DB, Fox JG. Colonization dynamics of altered Schaedler flora is influenced by gender, aging, and Helicobacter hepaticus infection in the intestines of Swiss Webster mice. Appl Environ Microbiol. 2006;72:5100–5103. doi: 10.1128/AEM.01934-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Diaz HR, Wang S, Anuar F, Qian Y, Bjorkholm B, Samuelsson A, Hibberd ML, Forssberg H, Pettersson S. Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci U S A. 2011;108:3047–3052. doi: 10.1073/pnas.1010529108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feingold BJ, Vegosen L, Davis M, Leibler J, Peterson A, Silbergeld EK. A niche for infectious disease in environmental health: rethinking the toxicological paradigm. Environ Health Perspect. 2010;118:1165–1172. doi: 10.1289/ehp.0901866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bajaj JS, Heuman DM, Sanyal AJ, Hylemon PB, Sterling RK, Stravitz RT, Fuchs M, Ridlon JM, Daita K, Monteith P, Noble NA, White MB, Fisher A, Sikaroodi M, Rangwala H, Gillevet PM. Modulation of the metabiome by rifaximin in patients with cirrhosis and minimal hepatic encephalopathy. PLoS One. 2013;8:e60042. doi: 10.1371/journal.pone.0060042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raml R, Rumpler A, Goessler W, Vahter M, Li L, Ochi T, Francesconi KA. Thio-dimethylarsinate is a common metabolite in urine samples from arsenic-exposed women in Bangladesh. Toxicol Appl Pharmacol. 2007;222:374–380. doi: 10.1016/j.taap.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 47.Naranmandura H, Suzuki N, Iwata K, Hirano S, Suzuki KT. Arsenic metabolism and thioarsenicals in hamsters and rats. Chem Res Toxicol. 2007;20:616–624. doi: 10.1021/tx700038x. [DOI] [PubMed] [Google Scholar]
- 48.Drobna Z, Naranmandura H, Kubachka KM, Edwards BC, Herbin-Davis K, Styblo M, Le XC, Creed JT, Maeda N, Hughes MF, Thomas DJ. Disruption of the arsenic (+3 oxidation state) methyltransferase gene in the mouse alters the phenotype for methylation of arsenic and affects distribution and retention of orally administered arsenate. Chem Res Toxicol. 2009;22:1713–1720. doi: 10.1021/tx900179r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen B, Arnold LL, Cohen SM, Thomas DJ, Le XC. Mouse arsenic (+3 oxidation state) methyltransferase genotype affects metabolism and tissue dosimetry of arsenicals after arsenite administration in drinking water. Toxicol Sci. 2011;124:320–326. doi: 10.1093/toxsci/kfr246. [DOI] [PubMed] [Google Scholar]
- 50.Dodmane PR, Arnold LL, Pennington KL, Thomas DJ, Cohen SM. Effect of dietary treatment with dimethylarsinous acid (DMA(III)) on the urinary bladder epithelium of arsenic (+3 oxidation state) methyltransferase (As3mt) knockout and C57BL/6 wild type female mice. Toxicology. 2013;305:130–135. doi: 10.1016/j.tox.2013.01.015. [DOI] [PubMed] [Google Scholar]
- 51.Naranmandura H, Rehman K, Le XC, Thomas DJ. Formation of methylated oxyarsenicals and thioarsenicals in wild-type and arsenic (+3 oxidation state) methyltransferase knockout mice exposed to arsenate. Anal Bioanal Chem. 2013;405:1885–1891. doi: 10.1007/s00216-012-6207-0. [DOI] [PubMed] [Google Scholar]
- 52.Drobna Z, Waters SB, Walton FS, LeCluyse EL, Thomas DJ, Styblo M. Interindividual variation in the metabolism of arsenic in cultured primary human hepatocytes. Toxicol Appl Pharmacol. 2004;201:166–177. doi: 10.1016/j.taap.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 53.Chung JS, Kalman DA, Moore LE, Kosnett MJ, Arroyo AP, Beeris M, Mazumder DN, Hernandez AL, Smith AH. Family correlations of arsenic methylation patterns in children and parents exposed to high concentrations of arsenic in drinking water. Environ Health Perspect. 2002;110:729–733. doi: 10.1289/ehp.02110729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lesseur C, Gilbert-Diamond D, Andrew AS, Ekstrom RM, Li Z, Kelsey KT, Marsit CJ, Karagas MR. A case-control study of polymorphisms in xenobiotic and arsenic metabolism genes and arsenic-related bladder cancer in New Hampshire. Toxicol Lett. 2012;210:100–106. doi: 10.1016/j.toxlet.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hernandez A, Xamena N, Sekaran C, Tokunaga H, Sampayo-Reyes A, Quinteros D, Creus A, Marcos R. High arsenic metabolic efficiency in AS3MT287Thr allele carriers. Pharmacogenet Genomics. 2008;18:349–355. doi: 10.1097/FPC.0b013e3282f7f46b. [DOI] [PubMed] [Google Scholar]
- 56.Hernandez A, Marcos R. Genetic variations associated with interindividual sensitivity in the response to arsenic exposure. Pharmacogenomics. 2008;9:1113–1132. doi: 10.2217/14622416.9.8.1113. [DOI] [PubMed] [Google Scholar]
- 57.Bailey KA, Wu MC, Ward WO, Smeester L, Rager JE, Garcia-Vargas G, Del Razo LM, Drobna Z, Styblo M, Fry RC. Arsenic and the epigenome: interindividual differences in arsenic metabolism related to distinct patterns of DNA methylation. J Biochem Mol Toxicol. 2013;27:106–115. doi: 10.1002/jbt.21462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsang V, Fry RC, Niculescu MD, Rager JE, Saunders J, Paul DS, Zeisel SH, Waalkes MP, Styblo M, Drobna Z. The epigenetic effects of a high prenatal folate intake in male mouse fetuses exposed in utero to arsenic. Toxicol Appl Pharmacol. 2012;264:439–450. doi: 10.1016/j.taap.2012.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rodrigues EG, Kile M, Hoffman E, Quamruzzaman Q, Rahman M, Mahiuddin G, Hsueh Y, Christiani DC. GSTO and AS3MT genetic polymorphisms and differences in urinary arsenic concentrations among residents in Bangladesh. Biomarkers. 2012;17:240–247. doi: 10.3109/1354750X.2012.658863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.De CS, Ghosh P, Sarma N, Majumdar P, Sau TJ, Basu S, Roychoudhury S, Ray K, Giri AK. Genetic variants associated with arsenic susceptibility: study of purine nucleoside phosphorylase, arsenic (+3) methyltransferase, and glutathione S-transferase omega genes. Environ Health Perspect. 2008;116:501–505. doi: 10.1289/ehp.10581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ahsan H, Chen Y, Kibriya MG, Slavkovich V, Parvez F, Jasmine F, Gamble MV, Graziano JH. Arsenic metabolism, genetic susceptibility, and risk of premalignant skin lesions in Bangladesh. Cancer Epidemiol Biomarkers Prev. 2007;16:1270–1278. doi: 10.1158/1055-9965.EPI-06-0676. [DOI] [PubMed] [Google Scholar]
- 62.Kile ML, Hoffman E, Rodrigues EG, Breton CV, Quamruzzaman Q, Rahman M, Mahiuddin G, Hsueh YM, Christiani DC. A pathway-based analysis of urinary arsenic metabolites and skin lesions. Am J Epidemiol. 2011;173:778–786. doi: 10.1093/aje/kwq427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schloissnig S, Arumugam M, Sunagawa S, Mitreva M, Tap J, Zhu A, Waller A, Mende DR, Kultima JR, Martin J, Kota K, Sunyaev SR, Weinstock GM, Bork P. Genomic variation landscape of the human gut microbiome. Nature. 2013;493:45–50. doi: 10.1038/nature11711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li K, Bihan M, Yooseph S, Methe BA. Analyses of the microbial diversity across the human microbiome. PLoS One. 2012;7:e32118. doi: 10.1371/journal.pone.0032118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Arumugam M, Raes J, Pelletier E, Le PD, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, Bertalan M, Borruel N, Casellas F, Fernandez L, Gautier L, Hansen T, Hattori M, Hayashi T, Kleerebezem M, Kurokawa K, Leclerc M, Levenez F, Manichanh C, Nielsen HB, Nielsen T, Pons N, Poulain J, Qin J, Sicheritz-Ponten T, Tims S, Torrents D, Ugarte E, Zoetendal EG, Wang J, Guarner F, Pedersen O, de Vos WM, Brunak S, Dore J, Antolin M, Artiguenave F, Blottiere HM, Almeida M, Brechot C, Cara C, Chervaux C, Cultrone A, Delorme C, Denariaz G, Dervyn R, Foerstner KU, Friss C, van de GM, Guedon E, Haimet F, Huber W, van Hylckama-Vlieg J, Jamet A, Juste C, Kaci G, Knol J, Lakhdari O, Layec S, Le RK, Maguin E, Merieux A, Melo MR, M'rini C, Muller J, Oozeer R, Parkhill J, Renault P, Rescigno M, Sanchez N, Sunagawa S, Torrejon A, Turner K, Vandemeulebrouck G, Varela E, Winogradsky Y, Zeller G, Weissenbach J, Ehrlich SD, Bork P. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, Knight R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A. 2010;107:11971–11975. doi: 10.1073/pnas.1002601107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.