

Abstract
Cysteine dioxygenase (CDO) is a mononuclear non-heme iron(II)-dependent enzyme critical for maintaining appropriate cysteine (Cys) and taurine levels in eukaryotic systems. Since CDO possesses both an unusual 3-His facial ligation sphere to the iron center and a rare Cys-Tyr crosslink near the active site, the mechanism by which it converts Cys and molecular oxygen to cysteine sulfinic acid is of broad interest. However, as of yet direct experimental support for any of the proposed mechanisms is still lacking. In this study, we have used NO as a substrate analogue for O2 to prepare a species that mimics the geometric and electronic structures of an early reaction intermediate. The resultant unusual S=1/2 {FeNO}7 species was characterized by magnetic circular dichroism, electron paramagnetic resonance, and electronic absorption spectroscopies, as well as computational methods including density functional theory and semi-empirical calculations. The NO adducts of Cys- and selenocysteine (Sec)-bound Fe(II)CDO exhibit virtually identical electronic properties; yet, CDO is unable to oxidize Sec. To explore the differences in reactivity between Cys- and Sec-bound CDO, the geometries and energies of viable O2-bound intermediates were evaluated computationally, and it was found that a low-energy quintet-spin intermediate on the Cys reaction pathway adopts a different geometry for the Sec-bound adduct. The absence of a low-energy O2 adduct for Sec-bound CDO is consistent with our experimental data and may explain why Sec does not act as a substrate for CDO.
Cysteine dioxygenase (CDO) is a non-heme Fe(II)-dependent dioxygenase that catalyzes the first step in oxidative cysteine catabolism, using both O atoms from molecular oxygen to convert L-cysteine (Cys) to cysteine sulfinic acid (CSA).1, 2 Cys is a necessary component in protein biosynthesis and also supplies the reactive thiol group for the synthesis of the biologically essential molecules coenzyme A and glutathione.3, 4 Elevated Cys levels have an excitotoxic effect on neurons, and an imbalance in Cys metabolism has been implicated in a variety of neurological diseases (e.g., Parkinson’s, Alzheimer’s, and motor neuron diseases) and autoimmune disorders (e.g., rheumatoid arthritis and systemic lupus erythematosus).5–9 Furthermore, the CDO product, CSA, is an important precursor in the biosynthesis of both taurine and pyruvate.3 Consequently, CDO plays a vital role in both producing important metabolites and maintaining appropriate Cys levels.
X-ray crystal structures of mammalian CDO have revealed that the active site of this enzyme features an Fe(II) ion ligated by three His residues and a solvent molecule arranged in a distorted tetrahedral coordination geometry.10, 11 This facial 3His metal-binding motif, observed in only a few other enzymes to date,12–14 is a fairly unique variation of the more common 2His1Carboxylate motif used by several other Fe(II)-dependent oxygenases.12, 15–17 The active site of CDO also contains a rare covalent crosslink between the phenol ring of Tyr157 and the thiol group of Cys93. Although not itself a ligand to the Fe(II) ion, the second-sphere Tyr residue is close enough to potentially engage in H-bonding interactions with coordinated solvent molecules within the active site of the resting enzyme and/or with substrates during catalysis. A similar posttranslational modification has been observed in galactose oxidase (where it plays a vital part in catalysis)18, 19 and in NirA (where the function of the crosslink remains unknown).20–22 Hence it is not surprising that the C93-Y157 crosslink has been shown to be instrumental for catalytic activity in CDO,23 though its specific role during enzymatic turnover has yet to be established. Intriguingly, however, in prokaryotic forms of CDO, Cys93 is replaced by a Gly residue, and thus the Cys-Tyr crosslink is absent.24 Despite this difference, the prokaryotic enzymes are still capable of oxidizing Cys at a rate similar to that of their mammalian CDO orthologs.24
Following the publication of the X-ray crystal structures of both resting10 and substrate-bound forms of CDO,11 several different reaction mechanisms were proposed for this enzyme.10, 11, 25–27 However, direct experimental evidence supporting any of these mechanisms is still lacking. Tight binding of the Fe(II) center to the 3His protein motif has been demonstrated through the use of Mossbauer spectroscopy.28 Crystallographic studies have revealed bidentate binding of the substrate Cys to the CDO active site through both its thiol and amino groups to yield a trigonal bipyramidal Fe(II) ligand environment.11 Additionally, the ordered binding of substrates (Cys first, then O2) and the formation of a 6-coordinate, Cys- and O2-bound Fe(II) center were proposed on the basis of an electron paramagnetic resonance (EPR) investigation of the NO adduct of Cys-bound Fe(II)CDO.27 However, while several proposed mechanisms invoke subsequent electron transfer from the iron to the O2 moiety, resulting in an iron(III)-superoxo type complex, the proposed mechanisms diverge widely after this point. One mechanism suggests the formation of a four-membered Fe(II)-S-O-O ring, with subsequent O-O bond scission and an oxidation state for iron that never exceeds +3.27 Alternatively, it was proposed elsewhere that an Fe(IV)-oxo species is generated during the transfer of the first oxygen to sulfur, after which a three-membered Fe(III)-S-O ring is formed to facilitate the transfer of the second oxygen to the sulfur.11 A third, purely theoretical investigation by de Visser and coworkers led to the suggestion that the formation of an Fe(IV)-oxo complex is followed by a ‘rebound’ step, where the iron-sulfur bond is broken and an Fe-O-S linkage is formed. Transfer of the second oxygen to sulfur was then proposed to complete the reaction and restore the original Fe(II)-bound CDO resting state.25, 26 Obviously, additional experimental work is needed to distinguish between any of these proposed catalytic mechanisms for CDO.
In a previous spectroscopic and computational study, we obtained detailed insight into the nature of the interaction between substrate Cys and the enzyme active site.29 By utilizing NO to mimic the reactivity of Cys-bound Fe(II)CDO with O2, a rather unusual species with an S=1/2 ground state was obtained,27 as opposed to a coupled S=3/2 spin system (consisting of a high-spin Fe(III) coupled antiferromagnetically to NO−) that is typically observed for NO adducts of non-heme Fe(II) enzymes.30 By using magnetic circular dichroism (MCD) spectroscopy, we subsequently determined that Cys binds to both Fe(II)- and Fe(III)CDO, and that the electronic transitions for the reduced and oxidized species are well separated in energy.29 The substrate analogue selenocysteine (Sec) was also found to bind to both forms of CDO, as evidenced by the appearance of spectral features reminiscent of Cys-CDO, though red-shifted by 1,500–2,000 cm−1. Additionally, resonance Raman spectra obtained upon excitation into the S/Se→Fe(III) charge transfer absorption bands were found to exhibit two features consistent with a direct Fe-S/Se bonding interaction. The MCD spectral signatures of Cys-Fe(II)CDO decayed when this putative reaction intermediate was exposed to oxygen, while those associated with Cys-Fe(III)CDO and all Sec-bound CDO species persisted, suggesting that only the Cys-Fe(II)CDO complex is catalytically active. Thus, although Cys and Sec bind in a similar fashion to the CDO active site, Sec cannot be oxidized by CDO despite the lower reduction potential of Sec vs. Cys at physiological pH (E°(Sec·/Sec−) = +0.43V and E°(Cys·/CysH) = +0.92V vs. NHE).29, 31
In this study, we have utilized Cys and its substrate analogue Sec, in conjunction with the O2 analogue NO, to mimic key steps in the reaction between O2 and substrate-bound Fe(II)CDO. The resulting substrate (analogue)-bound species were characterized using a combination of electronic absorption (Abs), MCD, and EPR spectroscopic techniques along with density functional theory (DFT) and semi-empirical computational methods. Collectively, our spectroscopic and computational data provide new, experimentally supported insights into the CDO mechanism and afford clues as to why Sec is a competitive enzyme inhibitor rather than an alternative substrate for CDO.
Experimental Procedures
CDO Gene Expression and Protein Purification
The cdo gene from Mus musculus was expressed with an N-terminal maltose binding protein (MBP) tag in Escherichia coli BL21(DE3)RIPL cells. Cell growth and induction of gene expression using isopropyl β-D-1-thiogalactopyranoside (IPTG) were performed as described previously.27 The harvested cell paste was resuspended in 25 mM HEPES (pH 7.5), treated with lysozyme for 30 minutes, and pulse sonicated on ice for 10 minutes to disrupt the cells. All subsequent steps in the isolation and purification of CDO were conducted at 4 ° C. This suspension was then centrifuged for 75 minutes at 39,200 ×g. To isolate CDO, the cell-free extract was diluted to a volume of 400 mL and applied to a pre-equilibrated diethylaminoethanol (DEAE) sepharose anion exchange column, which was then washed with two column volumes of buffer, followed by a linear gradient of 0 to 400 mM NaCl in HEPES buffer. Fractions containing the fusion protein were identified by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and further purified by loading them onto an amylose affinity column, to which the fusion protein selectively binds. After washing the non-binding proteins from the column, the target fusion protein was eluted in buffer (25 mM HEPES, 200 mM NaCl, 10 mM maltose, pH 7.5). The MBP tag was cleaved from CDO by incubating the solution containing the fusion protein with tobacco etch virus overnight at 4 °C. To remove the cleaved MBP, the protein solution was applied to an S-100 size exclusion column. Fractions containing CDO were identified by SDS-PAGE, pooled, and concentrated. As has been previously reported for CDO,32 our as-isolated protein consisted of two nearly equal fractions, one with and the other without the C93-Y157 crosslink being present (as revealed by the presence of a double-band pattern on an SDS-PAGE gel, see Figure S1). No efforts were undertaken to separate these two species or to change their relative populations.
Sample Preparation
Approximate protein concentrations were determined spectrophotometrically, using an ε280 value of 25,300 M−1cm−1 (note that the CDO concentration may have been slightly overestimated due to residual MBP in solution).29 The Fe content was determined using a colorimetric assay with bathophenanthrolinedisulfonic acid as described previously,27 and ranged from ~55–75% among all preparations. Samples used for spectroscopy were 2 mM in Fe-bound CDO. Degassed, as-isolated CDO was reduced under an N2 atmosphere to Fe(II)CDO by adding a slight molar excess of sodium dithionite, which led to a complete loss of color. Samples containing substrate (analogue) were prepared anaerobically and contained a 5-fold excess of Cys or Sec. Sec was prepared by anaerobic reduction of selenocystine with tris(2-carboxyethyl)phosphine.33 NO(g) was generated through the reaction of sodium nitrite with Cu(II) chloride and ascorbate under an argon atmosphere.34 The NO produced in this reaction was transferred via gas-tight syringe to the head-space of a small Ar-purged vial containing substrate (analogue)-bound Fe(II)CDO and allowed to react with the protein for up to an hour on ice. Sufficient NO gas was transferred such that additional gas did not result in a further change in color. All samples used for low-temperature (LT) Abs and MCD experiments also contained 55% (v/v) glycerol.
Spectroscopy
Room temperature (RT) Abs spectra were obtained using a Varian Cary 5e spectrophotometer while the sample compartment was purged with N2(g). LT Abs and MCD spectra were collected with a Jasco J-715 spectropolarimeter in conjunction with an Oxford Instruments SM4000-8T magnetocryostat. The MCD spectra presented herein were obtained by taking the difference between spectra obtained with the magnetic field aligned parallel and antiparallel to the light propagation axis to reduce contributions from the natural CD and glass strain. Iterative Gaussian deconvolutions of Abs and MCD spectra were conducted using Igor 6.22a.35
X-band EPR data were collected using a Bruker ESP 300E spectrometer equipped with a Varian EIP model 625A continuous wave frequency counter. The sample temperature was maintained at 20 K by an Oxford ESR 900 continuous flow liquid He cryostat that was regulated by an Oxford ITC4 temperature controller. Spectra were fit using the W95EPR program developed by Dr. F. Neese.36
Computations
Quantum mechanics/molecular mechanics (QM/MM) calculations were performed with Gaussian09.37 For the QM region, density functional theory (DFT) was utilized in conjunction with Becke’s three-parameter hybrid exchange functional with the Lee-Yang-Parr correlation functional (B3LYP),38, 39 as well as the 6-31G basis set40 for all atoms except iron, its immediately ligated atoms, and the NO or O2 ligand, for which the TZVP basis set41 was used. For the MM portion of the calculations, the Amber95 force field42 was utilized. Residues R60, H86, H88, C93, H140, H155, and Y157 were placed in the QM region, as were the iron center, six crystallographically defined water molecules within 10 Å of the Fe atom, and all substrates and substrate analogues. For each of these protein residues, the QM/MM boundary was placed between the alpha and beta carbons, with hydrogen used as the link atom. The alpha carbons of these residues were frozen during the optimization process. As a crystal structure of mouse CDO with only Cys bound at the active site was unavailable at the time this work was initiated, we utilized two separate crystal structures in the generation of our starting coordinates, namely the 1.4 Å resolution crystal structure of persulfenate-bound CDO from rat (PDB file 3ELN43) for all non-hydrogen atoms except those in the QM region, for which the 2.7 Å resolution structure of Cys-bound CDO from human (PDB file 2IC111) was used instead. All H atoms were added using the Reduce 3.14 program.44 Based on an inspection of potential hydrogen bonding interactions, His residues were protonated at Nε, except for H86, H88, H140, and H155, which were protonated at Nδ. For the NO adducts, the N and O atoms were manually added trans to H86 in a manner approximating a bent Fe-N-O net bonding interaction.
For comparative purposes, analogous QM/MM geometry optimizations were also carried out using an alternative functional (BP8645, 46) and basis set (Slater’s single valence polarized, SV/C41). In the case of the Cys adduct of Fe(II)-CDO, these calculations predicted an S=0 ground state, rather than the S=2 ground state we observe experimentally and properly reproduce with the B3LYP functional. Also, while the computations with the BP86 functional correctly predicted an S=1/2 spin ground state for the NO/Cys-Fe(II)CDO complex, the resulting electron configuration for this {FeNO}7 species (a low-spin Fe(III) coupled antiferromagnetically to NO−) is distinctly different from the configuration we determined experimentally (a low-spin Fe(II) ion coupled to an NO·) and obtained using the B3LYP functional. Especially in light of this latter observation, we opted to use the B3LYP functional in this work.
In the generation of small active site models from the QM/MM energy-minimized protein structures, iron, the substrates, H86, H88, and H140 were included. The protein residues were truncated at the alpha carbon and the immediate protein backbone atoms were substituted with hydrogens with a C-H bond distance of 1.1 Å. Spin-unrestricted single-point DFT calculations were performed on these active site models using the Orca 2.9 package developed by Dr. F. Neese,47 with the same functional and basis sets that were utilized for the QM/MM computations (vide supra). Electronic transition energies and absorption intensities for the NO adduct of Cys-Fe(II)CDO were computed by utilizing the INDO/S-CI module as implemented in Orca 2.9. In this program, spin-orbit coupling was introduced by implementing an effective potential/mean-field approach for evaluating one-electron terms and an exact calculation of the Coulomb potential. PyMOL version 1.5.0.448 was employed to generate isosurface plots of molecular orbitals (MOs) and electron density difference maps (EDDMs) using isodensity values of 0.06 au and 0.001 au, respectively. EPR parameters for the active site models were computed with Orca by solving the coupled-perturbed self-consistent field (CP-SCF) equations49, 50 using the B3LYP hybrid functional and the TZVP basis set on all atoms, except iron and the nitrogen of the NO ligand, for which CP(PPP)51, 52 and IGLO-III53 were used, respectively. These calculations included all orbitals within a ±100 hartree window around the HOMO-LUMO gap, where the origin of the g tensor was defined as the center of electronic charge. A high-resolution radial grid with an integration accuracy of 7.0 was used for the Fe atom and the N atom of the NO ligand.
The coordinates from the QM/MM-optimized NO adducts of Cys- and Sec-Fe(II)CDO were used as a starting point for geometry optimizations of the corresponding O2 adducts, where three viable multiplicities for the QM region were considered (singlet, triplet, and quintet). The oxygen adducts were further analyzed computationally by modifying their coordinates to approximate previously proposed reaction intermediates.26 The same QM/MM methodology described above for the NO adduct was then utilized to minimize the energies of complete protein models of these intermediates. The model energies obtained from a single-point calculation of small active site models derived from the QM/MM-optimized coordinates were drastically different from the SCF energies of the converged, whole-protein QM/MM models. In addition, the energetic ordering of the proposed O2-bound species changed if only the contributions from the first coordination sphere were considered. Thus, the energies of all models reported herein are the SCF energies from the QM region of the QM/MM optimizations.
Results
Spectroscopy
The RT Abs and LT MCD spectra exhibited by the species obtained by adding first Cys and then NO to Fe(II)CDO are shown in Figure 2. The temperature-dependent MCD features observed at high energy (>28,000 cm−1) are strikingly similar to those observed in the MCD spectrum of Cys-bound Fe(II)CDO in the absence of NO;29 thus, we assign them as S→Fe(II) charge transfer (CT) transitions arising from a fraction of Cys-bound Fe(II)CDO species that did not react with NO. However, additional temperature-dependent MCD features are observed in the lower-energy region (<28,000 cm−1) of the MCD spectrum obtained in the presence of NO. The appearance of these features indicates that NO likely coordinates directly to the Fe(II) ion. This conclusion is corroborated by a previous EPR study, which revealed that the NO adduct of Cys-Fe(II)CDO is best described as a six-coordinate S = 1/2 {FeNO}7 species (utilizing the nomenclature of Enemark and Feltham, where the superscript denotes the sum of the Fe 3d and NO π* electrons54) consisting of an NO radical (S = 1/2) directly ligated to a low-spin (S = 0) Fe(II) ion.27
Figure 2.

Room temperature Abs (top) and 4.5 K MCD (bottom) spectra of Cys-Fe(II)CDO and Sec-Fe(II)CDO after exposure to NO(g). Features present due to unreacted Sec- or Cys-Fe(II)CDO or glass strain are marked by *.
The Abs and MCD spectra obtained for the NO adduct of Sec-bound Fe(II)CDO are also shown in Figure 2. As in the case of the Cys-bound NO adduct, several new MCD features are observed in the near-IR and visible spectral regions upon addition of NO to Sec-bound Fe(II)CDO, providing compelling evidence that NO also coordinates directly to the Fe(II) center in this species. The MCD features of NO-bound Sec-Fe(II)CDO are red-shifted between 200 and 2,400 cm−1 from those of the Cys-bound analogue, which is reminiscent of the red-shifts displayed by the Abs and MCD features of the Sec- vs. Cys-bound Fe(II)CDO species in the absence of NO.29 These shifts reflect the fact that the occupied frontier molecular orbitals (MOs) of Sec are higher in energy than those of Cys and are thus closer in energy to the Fe 3d-based unoccupied MOs.
The similar natures of the NO adducts of Sec-Fe(II)CDO and Cys-Fe(II)CDO are further highlighted by the similarity between the corresponding EPR spectra (Figure 3). In each case, a rhombic EPR spectrum is observed, with resolvable hyperfine coupling to an I=1 nucleus in the region of g2. Fits of the experimental EPR data yielded the g values and hyperfine coupling constants listed in Table 1. Previously, the small spread in g values and the well-resolved hyperfine structure exhibited by the NO/Cys-Fe(II)CDO species were shown to be characteristic of a low-spin Fe(II) center coordinated to an NO·.27 As anticipated on the basis of their similar EPR spectra, the NO adducts of Cys- and Sec-Fe(II)CDO display nearly identical g values and hyperfine coupling constants, indicating that the two species have very similar electronic, and thus likely geometric, structures.
Figure 3.

20 K EPR spectra of NO/Cys-Fe(II)CDO and NO/Sec-Fe(II)CDO. Note that a minor underlying signal attributed to the in situ formation of a dinitrosyl iron complex is also present (*).
Table 1.
Experimentally determined and computed EPR parameters for the NO adducts of Cys- and Sec-bound Fe(II)-CDO
| Species | Experimental | Computed | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| g values | A values (MHz) | g values | A values (MHz) | |||||||||
| NO/Cys-CDO | 1.979 | 2.028 | 2.071 | <30 | 100 | <30 | 1.962 | 1.990 | 2.016 | 15 | 90 | 11 |
| NO/Sec-CDO | 1.978 | 2.028 | 2.077 | <30 | 55 | <30 | 1.975 | 1.991 | 2.007 | 15 | 92 | 12 |
Computations
To further characterize the NO adducts of Cys- and Sec-bound Fe(II)CDO, QM/MM geometry optimizations were performed on complete protein models. The corresponding active site structures were used in subsequent single-point DFT and semi-empirical calculations to allow for a direct comparison with the experimental data. In the QM/MM-optimized structures, the iron center adopts a distorted octahedral coordination environment, with the substrate cysteine coordinating in a bidentate fashion through both its thiol and amino groups, as observed in the X-ray crystal structure of Cys-bound CDO.11 The relevant bond distances and angles of the QM/MM-optimized active site models are in reasonable agreement with those determined by X-ray crystallography for the Cys-bound CDO crystal structure, as well as for synthetic S=1/2 {FeNO}7 complexes (see Table 2).55–57
Table 2.
Comparison of relevant bond lengths (in Å) and angles (in degrees) of the QM/MM optimized active site models, a crystal structure of Cys-bound CDO, and two crystallographically characterized synthetic S=1/2 {FeNO}7 complexes
The EPR parameters computed for these active site models reproduce both the g spread and the magnitude of the 14N hyperfine coupling constant obtained from fits of our experimental spectra quite well. Although the actual g values are underestimated by the calculation, similar discrepancies are observed between the experimental and computed EPR parameters for synthetic, structurally characterized S=1/2 {FeNO}7 species (see Supporting Information, Table S1 for more details). While similar optimized geometries and computed EPR parameters were obtained with both functionals (B3LYP and BP86) and basis sets (6-31G/TZVP and SV/C/TZVP) we tested, the results of the INDO calculations described below exhibited much better agreement with experimental data when the B3LYP and 6-31G/TZVP QM/MM-optimized models were used (see Figure S2). Thus, B3LYP and 6-31G/TZVP were utilized for the remainder of the calculations.
The good agreement between the metric parameters of the QM/MM-optimized active site models of the NO adducts of Cys- and Sec-bound Fe(II)CDO and the relevant X-ray crystal structure data, as well as between the computationally predicted and experimentally determined EPR parameters, warrants a closer examination of the computational results. For the NO/Cys-bound Fe(II)CDO species, the optimized Fe-N(His86) and Fe-N(His88) bond lengths are identical (at 2.11 Å), while the Fe-N(His140) bond trans to the cysteine substrate amino group is slightly shorter (at 2.01 Å). Very similar Fe-N(His) bond lengths result for the analogous Sec-bound species. Consistent with Fe-Se bonds being inherently longer than Fe-S, the calculated Fe-Se(Sec) bond distance is 0.13 Å longer than the Fe-S(Cys) bond distance. In contrast, the Fe-N(O) and N-O bond lengths, as well as the Fe-N-O bond angle, are almost identical between the two species, further supporting the conclusion that the substitution of Sec for Cys does not significantly affect NO binding. The geometric parameters of the Fe-NO unit are also comparable to those of synthetic, crystallographically characterized S=1/2 {FeNO}7 complexes (see Table 2).58, 59
As anticipated from the experimental EPR spectrum of NO/Cys-bound Fe(II)CDO, the calculated molecular orbital (MO) diagram (Figure 4) is consistent with a pseudo-octahedral, low-spin Fe(II) complex, with the Fe 3dxy-, 3dyz-, and 3dxz-based occupied MOs considerably lower in energy than the Fe 3dx2-y2- and 3dz2-based unoccupied MOs. The formally singly occupied MO corresponds to the second-highest occupied spin-up (α) MO (α-HOMO-1) because the corresponding spin down (β) MO (i.e., the β-LUMO) is unoccupied. The α-HOMO-1 has 72% NO and 19% Fe 3d character, making it weakly σ-bonding with respect to the Fe-NO bond. The large localization of unpaired spin density on the NO ligand is consistent with the observed I=1 hyperfine coupling in the g2 region of the EPR spectrum (Figure 3). The other, out-of-plane NO π*-based MOs (α-LUMO and β-LUMO+1) have compositions similar to the α-HOMO-1 and β-LUMO, though they are obviously derived from the other NO π* orbital and are thus rotated by 90° about the N-O bond. For the NO adduct of Sec-Fe(II)CDO, the computed MO compositions and relative energies (data not shown) are very similar to those of the Cys-bound counterpart, consistent with the close correspondence between the Abs and EPR spectra exhibited by these two species.
Figure 4.

MO energy level diagram obtained from a single-point DFT calculation on the QM/MM-optimized active site model of the nitrosyl adduct of Cys-Fe(II)CDO. Boundary surface plots of the α-HOMO-1 (bottom left) and relevant β orbitals (right) are also shown. Note that the HOMO-LUMO energy gap is not to scale.
To further validate these computed electronic structure descriptions and to assign the major features in the experimental Abs and MCD spectra, the electronic transition energies and Abs intensities were computed for the active sites of the QM/MM-optimized protein models. Initial attempts to use time-dependent density functional theory (TDDFT) to calculate the Abs spectra of the NO adducts of Cys- and Sec-bound Fe(II)CDO were unsuccessful, as the computed spectra contained a large number weak, low-energy features that have no experimental counterparts (see Figure S2). Therefore, subsequent electronic excited state computations were conducted on these active site models using the semi-empirical INDO/S-CI method; the relevant results obtained from these calculations are summarized in Figure 5. Overall, the agreement between the experimental and calculated Abs spectra is quite good, thus justifying a more detailed analysis of the INDO/S-CI results. Most of the transitions producing sizeable contributions to the computed spectrum are composed of numerous one-electron excitations involving different donor and acceptor orbitals. Hence, the principal donor and acceptor orbitals for the transitions of interest were identified on the basis of electron density difference maps (see Figure 5, right). Visual inspection of these plots led to the Abs and MCD band assignments provided in Table 3.
Figure 5.
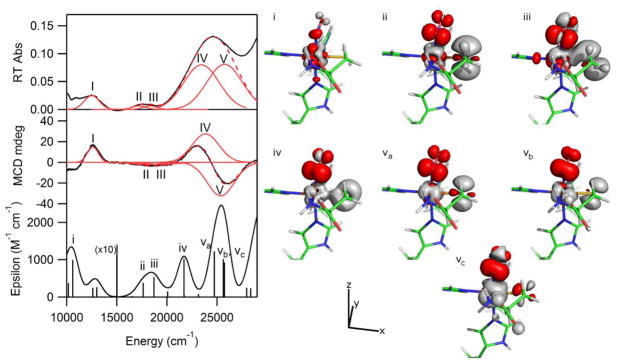
Gaussian deconvolutions and band labels for the low energy regions of the RT Abs and 4.5 K MCD spectra with the results of the INDO/S-CI calculation for comparison. Also shown are the EDDMs plotted for the transitions of interest (red and gray represent gain and loss of electron density, respectively). The model is oriented in a similar fashion as in Figure 4.
Table 3.
Band assignments for RT Abs and LT MCD spectra along with the observed and calculated band shifts upon substitution of Sec for Cys
| Obs. Band | Calc. Band | Assignment | Δobs (cm−1) | Δcalc (cm−1) |
|---|---|---|---|---|
| I | I | Fe xz → Fe z2 | −600 | −230 |
| II | ii | S 3p/Fe xz → NO π* | −550 | −170 |
| III | iii | S 3p/Fe xz → NO π* | −200 | −100 |
| IV | iv | Fe xz/S 3p → NO π* | −2,400 | −500 |
| V | va,b,c | Fe yz/S 3p → NO π* | −2,325 | −350 (ave) |
A simultaneous Gaussian deconvolution of the Abs and MCD spectra of NO/Cys-Fe(II)CDO revealed that the major feature in the RT Abs spectrum at 24,600 cm−1 is composed of two separate transitions (IV and V). In this same region, the INDO/S-CI-predicted spectrum shows two bands, whose individual transitions are labeled by iv, va, vb, and vc. Transition iv is predominantly an Fe 3dxz→NO π* electronic excitation, while transitions va, vb, and vc all have significant contributions from excitations originating from the Fe 3dyz and S 3p orbitals and terminating in the NO π* orbital that is rotated by 90° about the N-O bond vector relative to the acceptor orbital for transition iv. The roughly orthogonal polarizations predicted for transitions iv and v are consistent with the appearance of a pseudo-A term associated with transition IV and V at ~ 24,500 cm−1 in the experimental MCD spectrum. The Abs feature centered at 17,750 cm−1 similarly contains contributions from two separate transitions (II and III). The energies and relative intensities of these transitions correlate well with the INDO/S-CI predicted transitions labeled ii and iii. The corresponding EDDMs show that these transitions possess predominantly S(Cys)/Fe 3dxz→NO π* CT character. Finally, the lowest energy transition observed at 12,500 cm−1 in the Abs spectrum (I) coincides with a range of transitions in the INDO-predicted spectrum. All of the predicted transitions in this region are Fe 3d→3d excitations. As this feature in the experimental Abs and MCD spectra could be fit with a single Gaussian band, transition I is assigned as the Fe 3dxz→3dz2 transition, which is predicted to carry the highest intensity in this region.
Because the computational package used for INDO/S-CI calculations lacks suitable parameters for selenium, we were unable to calculate an Abs spectrum for NO-bound Sec-Fe(II)CDO. Thus, to corroborate our band assignments on the basis of the shifts of the relevant electronic transitions caused by substituting Sec for Cys, the six transitions that dominate the Abs and MCD spectra of the NO/Cys-Fe(II)CDO species mentioned above were further analyzed. Since numerous one-electron excitations mix to result in a single predicted transition, visual inspection of the EDDMs was used us to identify the major one-electron excitation responsible for each transition of interest for Cys/NO-bound Fe(II)CDO. As the MO compositions of the Cys/NO and Sec/NO adducts were very similar, the analogous one electron orbitals could readily be identified in the Sec/NO MO diagram. The energy differences were taken between the principal donor and acceptor MOs to compute pre-electronic relaxation transition energies for each of the two adducts, and the differences between these transitions energies are reported in Table 3. Although the absolute red-shifts are consistently underestimated, the transitions predicted to be the most affected by the substitution of Sec for Cys are indeed those displaying the largest red-shifts in the experimental Abs and MCD spectra. These red-shifts can be rationalized in terms of two mechanisms; namely, the decreased ligand field strength and the increased energies of the frontier MOs of Sec compared to Cys. Thus, the Fe 3dxz→3dz2 transition (I) is primarily affected by the stabilization of the Fe dz2-based acceptor MO and the modest destabilization of the Fe 3dxz-based donor MO upon substitution of Sec for Cys (note that the Fe 3dxz-based orbital is weakly π-bonding with respect to the Fe-S/Se bond). Alternatively, the other transitions identified all contain substantial S/Se character in the donor MOs, so the higher frontier MO energies of Sec vs. Cys will also result in red-shifted transitions.
Discussion
Since the first crystal structure of CDO was published in 2006,10 considerable interest in the catalytic mechanism of this enzyme, as well as in its overall contribution to oxidative cysteine metabolism, has been generated. The crucial developmental role this enzyme fills in mouse models60 and the epigenetic consequences of the cdo1 gene promoter methylation61 indicate that CDO plays a key part in the health of higher-order organisms. Work by Pierce and coworkers has demonstrated that an Fe(III)-superoxo species may be involved in the overall mechanism,62 but beyond this no proposed catalytic intermediates have been experimentally observed. Through the detailed investigation of the electronic structure of a species closely mimicking a proposed enzyme intermediate (i.e., oxygen-bound Cys-Fe(II)CDO), we sought to shed light on the subsequent steps of the catalytic mechanism.
As stipulated by the EPR spectrum, our computational results indicate that the NO adducts of Cys- and Sec-Fe(II)CDO are best described as low-spin Fe(II) complexes possessing a single unpaired electron in an NO π*-based MO. This electron configuration is very unusual for a {FeNO}7 non-heme iron enzyme species and is adopted by only a few synthetic {FeNO}7 complexes.58, 59, 63 Several factors may contribute to the preference for the low-spin configuration by the NO adducts of CDO, including the unusual Fe(II) binding motif and the presence of a strongly electron-donating thiolate (or selenoate) ligand. However, the NO adduct of substrate-bound isopenicillin N-synthase, which features an iron-thiolate bonding interaction, adopts the more common S=3/2 ground state,57 so the presence of the thiolate alone cannot account for the unusual spin state observed for CDO. Hence, it appears that both the 3His facial triad and a single, strongly donating anionic ligand are needed to stabilize the low-spin configuration. This hypothesis is supported by the fact that of the three known synthetic {FeNO}7 complexes that adopt an S=1/2 ground state, two have a mixed nitrogen- and oxygen-based, mono-anionic ligand set arranged in an octahedral geometry.58, 59 While the third synthetic complex features two mono-anionic dithiolene ligands in addition to the nitrosyl ligand, its square-pyramidal ligand environment bears little resemblance to the CDO active site.63
Precisely why S=1/2 rather than S=3/2 {FeNO}7 adducts are formed upon the addition of NO to both Cys-bound and Sec-bound Fe(II)CDO remains unclear; however, some clues are provided by the computed MO diagrams for these species (Figure 4). Because S=3/2 {FeNO}7 species can be described as containing a high-spin Fe(III) ion coupled antiferromagnetically to NO−,30 a hypothetical S=3/2 NO adduct of Cys-bound Fe(II)CDO would have all of its Fe 3d-based α spin MOs, as well as the in-plane and out-of-plane NO π*-derived MOs in the β manifold, filled. In such a system, the population of the Fe 3dx2-y2-based α MO (which has a substantial Fe-S(Cys) anti-bonding interaction) would result in a substantially weaker Fe-S bond than in the S=1/2 species. As such, these DFT results provide further support for our proposal that the strong Fe-S(Cys) bonding interaction is a major reason why NO/Cys-bound Fe(II)CDO adopts an S=1/2 ground state.
Extension to the Oxygen Adduct
Previous spectroscopic work has demonstrated the ability of Sec to bind to Fe(II)CDO in a manner similar to that of the native substrate Cys.29 Here we have demonstrated that NO, a commonly utilized substrate analogue for O2, can bind to both the Sec- and Cys-bound Fe(II)CDO active sites, yielding unusual S=1/2 {FeNO}7 species with virtually identical geometric and electronic structures. Despite these striking similarities, CDO is unable to oxidize Sec.28 Hence, any proposed catalytic reaction mechanism for CDO must not only provide a reasonable route for the synthesis of cysteine sulfinic acid from Cys, but also account for the inability of CDO to oxidize Sec to its seleninic acid form.
In addition to providing electronic structure descriptions for the NO adducts of Cys- and Sec-bound Fe(II)CDO species that agree well with our spectroscopic data, our computations also properly predict similar driving forces for the formation of these species. Specifically, the energies of the nitrosyl adducts are within a few kcal/mol of the precursor energies, being +4.7 and −2.2 kcal/mol for the Cys- and Sec-bound species, respectively. These results are at least qualitatively consistent with our observation that not all available Fe(II)CDO active sites bind NO, as evidenced by the presence of the prominent features at >28,000 cm−1 in the MCD spectra obtained at 4.5 K (where entropic effects not accounted for in our calculations are negligible). Hence, the combined spectroscopic and computational investigation of these nitrosyl adducts has allowed us to validate a computational methodology that is suitable for predicting the geometric and electronic structures of catalytically relevant species. To this end, we replaced NO by O2 in our computational models and investigated several viable reaction intermediates, including an end-on bound Fe-O2 species (B), one featuring a four-membered Fe-SCys-O-O ring (C), and a post O-O bond scission species where the distal oxygen atom (Od) has been transferred to the cysteinyl sulfur and the proximal oxygen atom (Op) remains coordinated to the iron center (D). In each case, several spin states were investigated (see Figure 6 and Table 4). Because the main goal of these calculations was to obtain clues as to why Fe(II)CDO is unable to convert Sec to its seleninic acid form, entropic effects that should similarly affect the free energies of the Cys- and Sec-bound intermediates were not accounted for.
Figure 6.

Calculated reaction coordinate diagrams and active site models of QM/MM-optimized reaction intermediates for the oxidation of Cys (A→D) and Sec (A′→D′) by Fe(II)CDO. All energies are given relative to those of high-spin Cys- or Sec-Fe(II)CDO plus free 3O2. In both the energy diagram and the active site overlays, the S=0, 1, and 2 species are shown in blue, red, and green, respectively, except 5A and 5A′, whose models are shown in dark blue and dark green, respectively. For clarity, only the first coordination sphere has been shown, and all hydrogen atoms have been omitted.
Table 4.
Computed relative energies (in kcal/mol) and key metric parameters (in Å) of the lowest energy intermediates in the reaction of Cys- and Sec-bound Fe(II)CDO with O2
| Species | Erel | Fe-S/Se | Fe-Op | S/Se-Od | Op-Od |
|---|---|---|---|---|---|
| 5A + 3O2 | 0.00 | 2.368 | n.a. | n.a. | 1.208 |
| 5B | −9.24 | 2.230 | 2.189 | 4.296 | 1.290 |
| 5C | −0.09 | 2.772 | 1.980 | 1.684 | 1.476 |
| 3D | −14.38 | 2.457 | 1.635 | 1.526 | 3.157 |
| 5A′+ 3O2 | 0.00 | 2.529 | n.a. | n.a. | 1.208 |
| 3B′ | 18.10 | 2.385 | 1.899 | 2.999 | 1.281 |
| 5C′ | 1.02 | 2.885 | 1.974 | 1.835 | 1.463 |
| 3D′ | −8.24 | 2.565 | 1.633 | 1.666 | 3.201 |
A summary of the proposed electronic structures for the lowest energy intermediates along the Cys reaction pathway are shown in Scheme 1, while relevant MO diagrams and Loewdin spin densities are provided in Figures S3–S6 in the Supporting Information. An examination of the initial O2- and Cys-bound Fe(II)CDO intermediate (B) reveals that the quintet spin state (in which an intermediate spin S=3/2 Fe(III) is formally coupled ferromagnetically to a superoxide radical) is stabilized by ≈ 9 kcal/mol with respect to the Cys-bound Fe(II)CDO species and free O2 (A). The 3B and 1B species are predicted to be less stable than the 5B species by 24 kcal/mol and 36 kcal/mol, respectively. In the S=2 species, the Fe center adopts a distorted octahedral geometry, with a His88-Fe-Op bond angle of 163.8°. The unpaired electron on the O2·− moiety is located in an MO that is oriented perpendicular to the Fe-O-O plane. It is worth noting that the {FeNO}7 adduct discussed earlier was assigned to be a low-spin Fe(II) with an NO·, whereas the O2-adduct is proposed to have an intermediate spin Fe(III). Nitric oxide is a relatively weak oxidant [E° (NO/3NO−) = −0.8 V vs. NHE64], whereas O2 has a much greater oxidizing power [E° (O2/O2−) = −0.16 V vs. NHE65]. With this in mind, it is perhaps not surprising why our computations predict that dioxygen can oxidize the Fe center of CDO, while NO lacks this ability.
Scheme 1.
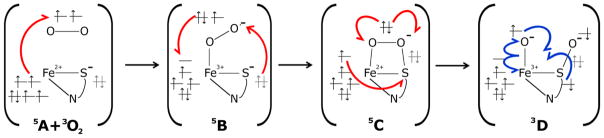
Formal electronic configurations of the proposed lowest energy reaction intermediates along the Cys reaction pathway. Electrons originating from the S(Cys) lone pair are shown in gray, and only relevant atoms are shown for clarity.
The next intermediate is formally obtained through the attack of the cysteinyl sulfur’s lone pair on Od and subsequent electron transfer from superoxide back to the Fe(III) center. For this intermediate (C), the quintet spin state is again lowest in energy. This species is best described as possessing a high-spin S=2 Fe(II) ion bound to a persulfenate moiety. A spin-crossover event to an S=1 state is predicted to occur upon moving from this intermediate via homolytic cleavage of the Op-Od bond to the singly oxygenated sulfur intermediate (3D). Based on the MO energies and compositions, this complex can be described as a resonance hybrid between an intermediate-spin Fe(III) bound to O·− (major contributor) and an Fe(IV)-oxo-like species. Unpaired β electrons are localized on both the sulfur and the distal oxygen, and the coupling of these to the α electrons on Fe(III) and Op results in the S=1 state. Although the next step of the reaction was not investigated computationally in this work, it is likely that the α electron on Op and the β electron on the sulfur can recombine to form the final S-O bond. The homolytic cleavage of the Fe-Op bond would then result in a one-electron-reduced iron center, yielding an Fe(II)-product complex.
The Cys-related catalytic intermediates A–D proposed in this work seem plausible, with all intermediates investigated having at least one low-energy spin state that should be accessible. Additionally, the geometries and energies of the analogous Sec-bound intermediates provide clues as to why Sec is not oxidized by CDO. Whereas the S=2 oxygen adduct of Cys-bound Fe(II)CDO (5B) is stabilized with respect to its precursor and free 3O2, the analogous S=2 oxygen adduct of Sec-bound Fe(II)CDO (species 5B′) is much higher in energy and is no longer the ground state O2-bound species (that being 3B′). This energetic difference is likely a direct result of the fact that the geometries of 5B and 5B′ are considerably different (see Figure 7). Rather than binding in an end-on fashion to the Fe(II) center as in 5B, the dioxygen moiety in 5B′ has shifted to insert above the Fe-Se bond instead, in a manner nearly identical to what is observed in the crystal structure of persulfenate-bound CDO (PDB file 3ELN).43 The 3ELN crystal structure was proposed to be that of a reaction intermediate of the oxidation of Cys, though the crystals from which the structure was obtained were soaked with Cys in an aerobic atmosphere for a period of 1–4 hours before diffraction data were collected, thus having ample time for an on-pathway intermediate to completely turn over.43 Having obtained a similar structure through the in silico reaction of Sec-Fe(II)CDO with O2, it seems plausible that the persulfenate-CDO complex is not a relevant reaction intermediate, but rather represents an off-reaction-pathway species. In the case of Sec-bound Fe(II)CDO, the high energy of the 5B′ species suggests that the formation of a dioxygen adduct is thermodynamically unfavorable. This hypothesis is consistent with our observation that CDO samples prepared anaerobically in the presence of Sec are spectroscopically indistinguishable from those prepared aerobically.
Figure 7.
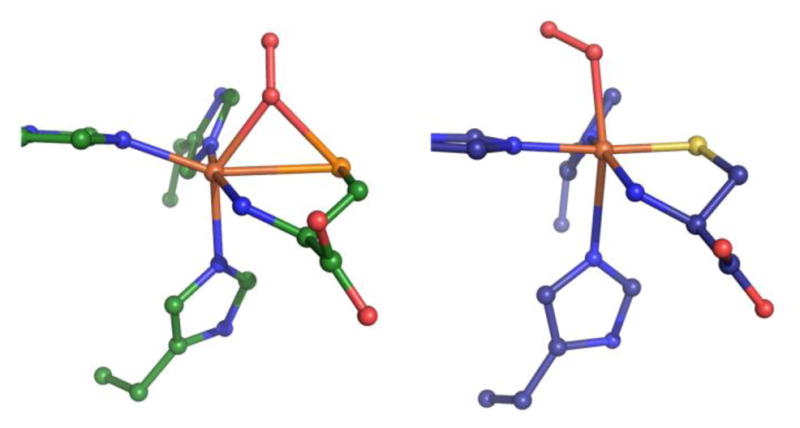
Comparison of the QM/MM-optimized structures for 5B′ (left) and 5B (right).
Two purely computational investigations into the mechanism of CDO have recently been reported in the literature, including a QM/MM study performed by Kumar et al. that employed a slightly different computational methodology from that used in our work.26 For the four-membered ring and post-O2 bond scission intermediates (C and D, respectively), the energetic ordering of the spin states, as well as the relative energies between them, are very similar to ours. However, large differences exist regarding the identity of the initial O2-bound intermediate. While we favor an S=2, pseudo-octahedral complex (5B in Figure 6), the corresponding species in the Kumar study has a nearly perfect octahedral geometry about the Fe center and is much higher in energy than both the S=0 (most stable) and S=1 species. The main difference between our 5B model and the S=0 species reported by Kumar et al concerns the relative energy of the Fe 3dz2 orbital. In the former species this orbital is sufficiently low in energy to become singly occupied, while in the latter it remains unoccupied. However, it is worth noting that the choice of functional and computational methodology utilized in calculations such as those presented here may have a profound effect on the predicted energetic ordering of spin states. 66 Keeping this in mind, we are cautiously optimistic that our computational methodology, which was validated on the basis of the experimental Abs and MCD spectra of the NO adducts of Cys- and Sec-bound Fe(II)CDO, is capable of providing molecular orbitals, energies, and compositions that are consistent with those found in this enzyme system.
A second, more recent computational investigation by Che, et al. also evaluated a possible mechanism for CDO.67 In this case, only the enzyme’s first coordination sphere was considered. Thus, all outer-sphere contributions to the energy, geometry, and electronic structure of the active site were ignored. Che’s study investigated the substitution of Cys for Sec, and it was proposed that the enzyme is incapable of oxidizing Sec because a ferric-superoxo complex is never formed. This was attributed to selenium’s superior electron donation ability, which resulted in no change in oxidation state at the iron center upon O2 binding. However, Che proposes that O2 is still capable of binding to Sec-bound Fe(II)CDO to yield a high-spin Fe(II) ion coupled ferromagnetically to a superoxo radical and coupled antiferromagnetically to a selenium-based radical. This conclusion is inconsistent with our observation that the active site is unreactive to O2 if Sec is already bound to the iron center.
Conclusions
The uncommon S=1/2 {FeNO}7 species generated upon the reaction of NO with a preformed Cys-Fe(II)CDO or Sec-Fe(II)CDO complex represents yet another unusual characteristic of the CDO enzyme. This adduct presented us with an almost unique opportunity to explore the spectral signatures and electronic properties of unusual S=1/2 {FeNO}7 species. Through the validation of our computational methodology via the successful prediction of the geometric and electronic structures of these nitrosyl adducts, we were able to investigate the geometries and energetics associated with several proposed reaction intermediates. In doing so, it was discovered that the Cys-bound active site has a low-lying S=2 intermediate after O2 binding that is inaccessible to the Sec-bound active site, providing a possible explanation for the lack of reactivity of Sec-Fe(II)CDO toward O2.
Supplementary Material
Figure 1.

Crystal structure of human CDO with substrate Cys bound to the iron center.11 The putative O2 (or NO) binding site is located trans to His86.
Acknowledgments
Funding Source: This work was supported by the National Institutes of Health Grant GM 64631 (T.C.B.), the National Science Foundation Grant MCB-0843239 (B.G.F.), and the National Science Foundation Grant CHE-0840494 (computational resources).
Abbreviations
- CDO
cysteine dioxygenase
- Cys
cysteine
- Sec
selenocysteine
- EPR
electron paramagnetic resonance
- MCD
magnetic circular dichroism
- Abs
electronic absorption
- DFT
density functional theory
- RT
room temperature
- LT
low temperature
- QM/MM
quantum mechanics/molecular mechanics
- MO
molecular orbital
- EDDM
electron density difference map
- SCF
self-consistent field
- INDO/S-CI
intermediate neglect of differential overlap/singles-configuration interaction
Footnotes
Supporting Information. Tables comparing experimental vs. calculated Abs and EPR data for synthetic {FeNO}7 complexes; SDS-PAGE gel; comparison of computational results derived from BP86- and B3LYP-optimized QM/MM models; MO diagrams and boundary surface plots for species 5A, 5B, 5C, 3D; cartesian coordinates for QM/MM-optimized protein models. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Yamaguchi K, Hosokawa Y. CYSTEINE DIOXYGENASE. Methods in Enzymology. 1987;143:395–403. doi: 10.1016/0076-6879(87)43069-3. [DOI] [PubMed] [Google Scholar]
- 2.Lombardi Jb, Singer TP, Boyer PD. CYSTEINE OXYGENASE. 2 STUDIES ON MECHANISM OF REACTION WITH 18OXYGEN. Journal of Biological Chemistry. 1969;244:1172-&. [PubMed] [Google Scholar]
- 3.Cooper AJL. BIOCHEMISTRY OF SULFUR-CONTAINING AMINO-ACIDS. Annual Review of Biochemistry. 1983;52:187–222. doi: 10.1146/annurev.bi.52.070183.001155. [DOI] [PubMed] [Google Scholar]
- 4.Stipanuk MH. Sulfur amino acid metabolism: Pathways for production and removal of homocysteine and cysteine. Annual Review of Nutrition. 2004;24:539–577. doi: 10.1146/annurev.nutr.24.012003.132418. [DOI] [PubMed] [Google Scholar]
- 5.Slivka A, Cohen G. BRAIN ISCHEMIA MARKEDLY ELEVATES LEVELS OF THE NEUROTOXIC AMINO-ACID, CYSTEINE. Brain Research. 1993;608:33–37. doi: 10.1016/0006-8993(93)90770-n. [DOI] [PubMed] [Google Scholar]
- 6.Pean AR, Parsons RB, Waring RH, Williams AC, Ramsden DB. TOXICITY OF SULFUR-CONTAINING-COMPOUNDS TO NEURONAL CELL-LINES. Journal of the Neurological Sciences. 1995;129:107–108. doi: 10.1016/0022-510x(95)00078-g. [DOI] [PubMed] [Google Scholar]
- 7.Heafield MT, Fearn S, Steventon GB, Waring RH, Williams AC, Sturman SG. PLASMA CYSTEINE AND SULFATE LEVELS IN PATIENTS WITH MOTOR-NEURON, PARKINSONS AND ALZHEIMERS-DISEASE. Neuroscience Letters. 1990;110:216–220. doi: 10.1016/0304-3940(90)90814-p. [DOI] [PubMed] [Google Scholar]
- 8.Gordon C, Bradley H, Waring RH, Emery P. ABNORMAL SULFUR OXIDATION IN SYSTEMIC LUPUS-ERYTHEMATOSUS. Lancet. 1992;339:25–26. doi: 10.1016/0140-6736(92)90144-r. [DOI] [PubMed] [Google Scholar]
- 9.Emery P, Bradley H, Gough A, Arthur V, Jubb R, Waring R. Increased prevalence of poor sulphoxidation in patients with rheumatoid arthritis: effect of changes in the acute phase response and second line drug treatment. Annals of the Rheumatic Diseases. 1992;51:318–320. doi: 10.1136/ard.51.3.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCoy JG, Bailey LJ, Bitto E, Bingman CA, Aceti DJ, Fox BG, Phillips GN. Structure and mechanism of mouse cysteine dioxygenase. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:3084–3089. doi: 10.1073/pnas.0509262103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ye S, Wu X, Wei L, Tang DM, Sun P, Bartlam M, Rao ZH. An insight into the mechanism of human cysteine dioxygenase - Key roles of the thioether-bonded tyrosine-cysteine cofactor. Journal of Biological Chemistry. 2007;282:3391–3402. doi: 10.1074/jbc.M609337200. [DOI] [PubMed] [Google Scholar]
- 12.Straganz GD, Nidetzky B. Variations of the 2-His-1-carboxylate theme in mononudear non-heme Fe-III oxygenases. Chembiochem. 2006;7:1536–1548. doi: 10.1002/cbic.200600152. [DOI] [PubMed] [Google Scholar]
- 13.Straganz GD, Glieder A, Brecker L, Ribbons DW, Steiner W. Acetylacetone-cleaving enzyme Dke1: a novel C-C-bond-cleaving enzyme from Acinetobacter johnsonii. Biochemical Journal. 2003;369:573–581. doi: 10.1042/BJ20021047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Straganz GD, Diebold AR, Egger S, Nidetzky B, Solomon EI. Kinetic and CD/MCD Spectroscopic Studies of the Atypical, Three-His-Ligated, Non-Heme Fe2+ Center in Diketone Dioxygenase: The Role of Hydrophilic Outer Shell Residues in Catalysis. Biochemistry. 2010;49:996–1004. doi: 10.1021/bi901339n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leitgeb S, Nidetzky B. Structural and functional comparison of 2-His-1-carboxylate and 3-His metallocentres in non-haem iron(II)-dependent enzymes. Biochemical Society Transactions. 2008;36:1180–1186. doi: 10.1042/BST0361180. [DOI] [PubMed] [Google Scholar]
- 16.Hegg EL, Que L. The 2-His-1-carboxylate facial triad - An emerging structural motif in mononuclear non-heme iron(II) enzymes. European Journal of Biochemistry. 1997;250:625–629. doi: 10.1111/j.1432-1033.1997.t01-1-00625.x. [DOI] [PubMed] [Google Scholar]
- 17.Koehntop KD, Emerson JP, Que L. The 2-His-1-carboxylate facial triad: a versatile platform for dioxygen activation by mononuclear non-heme iron(II) enzymes. Journal of Biological Inorganic Chemistry. 2005;10:87–93. doi: 10.1007/s00775-005-0624-x. [DOI] [PubMed] [Google Scholar]
- 18.Wachter RM, Branchaud BP. Construction and analysis of a semi-quantitative energy profile for the reaction catalyzed by the radical enzyme galactose oxidase. Biochimica Et Biophysica Acta-Protein Structure and Molecular Enzymology. 1998;1384:43–54. doi: 10.1016/s0167-4838(97)00209-4. [DOI] [PubMed] [Google Scholar]
- 19.Whittaker MM, Ekberg CA, Peterson J, Sendova MS, Day EP, Whittaker JW. Spectroscopic and magnetochemical studies on the active site copper complex in galactose oxidase. Journal of Molecular Catalysis B-Enzymatic. 2000;8:3–15. [Google Scholar]
- 20.Schnell R, Sandalova T, Hellman U, Lindqvist Y, Schneider G. Siroheme- and Fe-4-S-4 -dependent NirA from Mycobacterium tuberculosis is a sulfite reductase with a covalent Cys-Tyr bond in the active site. Journal of Biological Chemistry. 2005;280:27319–27328. doi: 10.1074/jbc.M502560200. [DOI] [PubMed] [Google Scholar]
- 21.Ito N, Phillips SEV, Stevens C, Ogel ZB, McPherson MJ, Keen JN, Yadav KDS, Knowles PF. NOVEL THIOETHER BOND REVEALED BY A 1.7-A CRYSTAL-STRUCTURE OF GALACTOSE-OXIDASE. Nature. 1991;350:87–90. doi: 10.1038/350087a0. [DOI] [PubMed] [Google Scholar]
- 22.Whittaker MM, Whittaker JW. Cu(I)-dependent biogenesis of the galactose oxidase redox cofactor. Journal of Biological Chemistry. 2003;278:22090–22101. doi: 10.1074/jbc.M300112200. [DOI] [PubMed] [Google Scholar]
- 23.Siakkou E, Rutledge MT, Wilbanks SM, Jameson GNL. Correlating crosslink formation with enzymatic activity in cysteine dioxygenase. Biochimica Et Biophysica Acta-Proteins and Proteomics. 2011;1814:2003–2009. doi: 10.1016/j.bbapap.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 24.Dominy JE, Simmons CR, Karplus PA, Gehring AM, Stipanuk MH. Identification and characterization of bacterial cysteine dioxygenases: a new route of cysteine degradation for eubacteria. Journal of Bacteriology. 2006;188:5561–5569. doi: 10.1128/JB.00291-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aluri S, de Visser SP. The mechanism of cysteine oxygenation by cysteine dioxygenase enzymes. Journal of the American Chemical Society. 2007;129:14846-+. doi: 10.1021/ja0758178. [DOI] [PubMed] [Google Scholar]
- 26.Kumar D, Thiel W, de Visser SP. Theoretical Study on the Mechanism of the Oxygen Activation Process in Cysteine Dioxygenase Enzymes. Journal of the American Chemical Society. 2011;133:3869–3882. doi: 10.1021/ja107514f. [DOI] [PubMed] [Google Scholar]
- 27.Pierce BS, Gardner JD, Bailey LJ, Brunold TC, Fox BG. Characterization of the nitrosyl adduct of substrate-bound mouse cysteine dioxygenase by electron paramagnetic resonance: Electronic structure of the active site and mechanistic implications. Biochemistry. 2007;46:8569–8578. doi: 10.1021/bi700662d. [DOI] [PubMed] [Google Scholar]
- 28.Tchesnokov EP, Wilbanks SM, Jameson GNL. A Strongly Bound High-Spin Iron(II) Coordinates Cysteine and Homocysteine in Cysteine Dioxygenase. Biochemistry. 2012;51:257–264. doi: 10.1021/bi201597w. [DOI] [PubMed] [Google Scholar]
- 29.Gardner JD, Pierce BS, Fox BG, Brunold TC. Spectroscopic and Computational Characterization of Substrate-Bound Mouse Cysteine Dioxygenase: Nature of the Ferrous and Ferric Cysteine Adducts and Mechanistic Implications. Biochemistry. 2010;49:6033–6041. doi: 10.1021/bi100189h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown CA, Pavlosky MA, Westre TE, Zhang Y, Hedman B, Hodgson KO, Solomon EI. SPECTROSCOPIC AND THEORETICAL DESCRIPTION OF THE ELECTRONIC-STRUCTURE OF S=3/2 IRON-NITROSYL COMPLEXES AND THEIR RELATION TO O-2 ACTIVATION BY NONHEME TRON ENZYME ACTIVE-SITES. Journal of the American Chemical Society. 1995;117:715–732. [Google Scholar]
- 31.Nauser T, Dockheer S, Kissner R, Koppenol WH. Catalysis of electron transfer by selenocysteine. Biochemistry. 2006;45:6038–6043. doi: 10.1021/bi0602260. [DOI] [PubMed] [Google Scholar]
- 32.Kleffmann T, Jongkees SAK, Fairweather G, Wilbanks SM, Jameson GNL. Mass-spectrometric characterization of two posttranslational modifications of cysteine dioxygenase. Journal of Biological Inorganic Chemistry. 2009;14:913–921. doi: 10.1007/s00775-009-0504-x. [DOI] [PubMed] [Google Scholar]
- 33.Hondal RJ, Nilsson BL, Raines RT. Selenocysteine in native chemical ligation and expressed protein ligation. Journal of the American Chemical Society. 2001;123:5140–5141. doi: 10.1021/ja005885t. [DOI] [PubMed] [Google Scholar]
- 34.Smith AT, Majtan T, Freeman KM, Su Y, Kraus JP, Burstyn JN. Cobalt Cystathionine beta-Synthase: A Cobalt-Substituted Heme Protein with a Unique Thiolate Ligation Motif. Inorganic Chemistry. 2011;50:4417–4427. doi: 10.1021/ic102586b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.WaveMetrics I. Igore 6.22a. 6.22. Lake Oswego; Oregon: 2011. [Google Scholar]
- 36.Neese F. QCPE Bull. 1995;15:5. [Google Scholar]
- 37.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery J, JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision C.01. Gaussian, Inc; Wallingford, CT: 2010. [Google Scholar]
- 38.Lee CT, Yang WT, Parr RG. DEVELOPMENT OF THE COLLE-SALVETTI CORRELATION-ENERGY FORMULA INTO A FUNCTIONAL OF THE ELECTRON-DENSITY. Physical Review B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 39.Becke AD. DENSITY-FUNCTIONAL THERMOCHEMISTRY. 3 THE ROLE OF EXACT EXCHANGE. Journal of Chemical Physics. 1993;98:5648–5652. [Google Scholar]
- 40.Hehre WJ, Ditchfie R, Pople JA. SELF-CONSISTENT MOLECULAR-ORBITAL METHODS. 12 FURTHER EXTENSIONS OF GAUSSIAN-TYPE BASIS SETS FOR USE IN MOLECULAR-ORBITAL STUDIES OF ORGANIC-MOLECULES. Journal of Chemical Physics. 1972;56:2257-&. [Google Scholar]
- 41.Schafer A, Horn H, Ahlrichs R. FULLY OPTIMIZED CONTRACTED GAUSSIAN-BASIS SETS FOR ATOMS LI TO KR. Journal of Chemical Physics. 1992;97:2571–2577. [Google Scholar]
- 42.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A 2ND GENERATION FORCE-FIELD FOR THE SIMULATION OF PROTEINS, NUCLEIC-ACIDS, AND ORGANIC-MOLECULES. Journal of the American Chemical Society. 1995;117:5179–5197. [Google Scholar]
- 43.Simmons CR, Krishnamoorthy K, Granett SL, Schuller DJ, Dominy JE, Begley TP, Stipanuk MH, Karplus PA. A Putative Fe2+-Bound Persulfenate Intermediate in Cysteine Dioxygenase. Biochemistry. 2008;47:11390–11392. doi: 10.1021/bi801546n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Word JM, Lovell SC, Richardson JS, Richardson DC. Asparagine and glutamine: Using hydrogen atom contacts in the choice of side-chain amide orientation. Journal of Molecular Biology. 1999;285:1735–1747. doi: 10.1006/jmbi.1998.2401. [DOI] [PubMed] [Google Scholar]
- 45.Becke AD. DENSITY-FUNCTIONAL EXCHANGE-ENERGY APPROXIMATION WITH CORRECT ASYMPTOTIC-BEHAVIOR. Physical Review A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 46.Perdew JP, Yue W. ACCURATE AND SIMPLE DENSITY FUNCTIONAL FOR THE ELECTRONIC EXCHANGE ENERGY - GENERALIZED GRADIENT APPROXIMATION. Physical Review B. 1986;33:8800–8802. doi: 10.1103/physrevb.33.8800. [DOI] [PubMed] [Google Scholar]
- 47.Neese F. An Ab Initio, DFT and Semiempirical Electronic Structure Package. 2012. Orca 2.9.1. [Google Scholar]
- 48.Schrodinger. The PyMOL Molecular Graphics System. 1.5.0.4 [Google Scholar]
- 49.Neese F. Prediction of Electron Paramagnetic Resonance g Values Using Coupled Perturbed Hartree-Fock and Kohn-Sham Theory. J Chem Phys. 2001;115:11080–11096. [Google Scholar]
- 50.Neese F. Metal and Ligand Hyperfine Couplings in Transition Metal Complexes: The Effect of Spin-orbit Coupling as Studied by Coupled Perturbed Kohn-Sham Theory. J Chem Phys. 2003;118:3939–3948. [Google Scholar]
- 51.Neese F. Prediction and interpretation of the Fe-57 isomer shift in Mossbauer spectra by density functional theory. Inorganica Chimica Acta. 2002;337:181–192. [Google Scholar]
- 52.Wachters AJ. GAUSSIAN BASIS SET FOR MOLECULAR WAVEFUNCTIONS CONTAINING THIRD-ROW ATOMS. Journal of Chemical Physics. 1970;52:1033-&. [Google Scholar]
- 53.Kutzelnigg W, Fleischer U, Schindler M, editors. The IGLO method: Ab initio calculation and interpretation of NMR chemical shifts and magnetic susceptibilities. Vol. 23. Springer-Verlag; Heidelberg. Germany: 1990. [Google Scholar]
- 54.Enemark JH, Feltham RD. PRINCIPLES OF STRUCTURE, BONDING, AND REACTIVITY FOR METAL NITROSYL COMPLEXES. Coordination Chemistry Reviews. 1974;13:339–406. [Google Scholar]
- 55.Sellmann D, Blum N, Heinemann FW, Hess BA. Synthesis, reactivity, and structure of strictly homologous 18 and 19 valence electron iron nitrosyl complexes. Chemistry-a European Journal. 2001;7:1874–1880. doi: 10.1002/1521-3765(20010504)7:9<1874::aid-chem1874>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 56.Li M, Bonnet D, Bill E, Neese F, Weyhermuller T, Blum N, Sellman D, Wieghardt K. Tuning the electronic structure of octahedral iron complexes FeL(X) (L=1-alkyl-4,7-bis(4-tert-butyl-2-mercaptobenzyl)-1,4,7-triazacyclononan e, X = Cl, CH3O, CN, NO). The S=1/2 reversible arrow S=3/2 spin equilibrium of FeLPr(NO) Inorganic Chemistry. 2002;41:3444–3456. doi: 10.1021/ic011243a. [DOI] [PubMed] [Google Scholar]
- 57.Brown CD, Neidig ML, Neibergall MB, Lipscomb JD, Solomon EI. VTVH-MCD and DFT studies of thiolate bonding to {FeNO}(7)/{FeO2}(8) complexes of isopenicillin N synthase: Substrate determination of oxidase versus oxygenase activity in nonheme Fe enzymes. Journal of the American Chemical Society. 2007;129:7427–7438. doi: 10.1021/ja071364v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Serres RG, Grapperhaus CA, Bothe E, Bill E, Weyhermuller T, Neese F, Wieghardt K. Structural, spectroscopic, and computational study of an octahedral, non-heme {Fe-NO}(6–8) series: Fe(NO)(cyclam-ac) (2+/+/O) Journal of the American Chemical Society. 2004;126:5138–5153. doi: 10.1021/ja030645+. [DOI] [PubMed] [Google Scholar]
- 59.Patra AK, Rowland JM, Marlin DS, Bill E, Olmstead MM, Mascharak PK. Iron nitrosyls of a pentadentate ligand containing a single carboxamide group: Syntheses, structures, electronic properties, and photolability of NO. Inorganic Chemistry. 2003;42:6812–6823. doi: 10.1021/ic0301627. [DOI] [PubMed] [Google Scholar]
- 60.Stipanuk Martha H, Fieselmann K, Hirschberger LL, Ueki Iori Lam J, Peters R, Roman HB, Valli A. Sulfur metabolism in the cysteine dioxygenase knockout mouse: impairment in taurine synthesis and increased formation of acid labile sulfur. Amino Acids. 2011;41:S78–S78. [Google Scholar]
- 61.Brait M, Ling S, Nagpal JK, Chang X, Park HL, Lee J, Okamura J, Yamashita K, Sidransky D, Kim MS. Cysteine dioxygenase 1 is a tumor suppressor gene silenced by promoter methylation in multiple human cancers, In. PLoS One. 2012:e44951. doi: 10.1371/journal.pone.0044951. PLoS One. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Crawford JA, Li W, Pierce BS. Single Turnover of Substrate-Bound Ferric Cysteine Dioxygenase with Superoxide Anion: Enzymatic Reactivation, Product Formation, and a Transient Intermediate. Biochemistry. 2011;50:10241–10253. doi: 10.1021/bi2011724. [DOI] [PubMed] [Google Scholar]
- 63.Ghosh P, Stobie K, Bill E, Bothe E, Weyhermuller T, Ward MD, McCleverty JA, Wieghardt K. Electronic structure of nitric oxide adducts of bis(diaryl-1,2-dithiolene)iron compounds: Four-membered electron-transfer series Fe(NO)(L)(2) (z) (z = 1+, 0, 1-, 2-) Inorganic Chemistry. 2007;46:522–532. doi: 10.1021/ic061874a. [DOI] [PubMed] [Google Scholar]
- 64.Bartberger MD, Liu W, Ford E, Miranda KM, Switzer C, Fukuto JM, Farmer PJ, Wink DA, Houk KN. The reduction potential of nitric oxide (NO) and its importance to NO biochemistry. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:10958–10963. doi: 10.1073/pnas.162095599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wood PM. THE 2 REDOX POTENTIALS FOR OXYGEN REDUCTION TO SUPEROXIDE. Trends in Biochemical Sciences. 1987;12:250–251. [Google Scholar]
- 66.Ghosh A. Transition metal spin state energetics and noninnocent systems: challenges for DFT in the bioinorganic arena. Journal of Biological Inorganic Chemistry. 2006;11:712–724. doi: 10.1007/s00775-006-0135-4. [DOI] [PubMed] [Google Scholar]
- 67.Che X, Gao J, Liu Y, Liu C. Metal vs. chalcogen competition in the catalytic mechanism of cysteine dioxygenase. Journal of Inorganic Biochemistry. 2013;122:1–7. doi: 10.1016/j.jinorgbio.2013.01.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.