

Abstract
Common gastrointestinal diseases such as radiation enteritis (RE), acute pancreatitis, inflammatory bowel diseases (IBD) and drug-induced hepatotoxicity share pathophysiological mechanisms at the molecular level, mostly involving the activation of many pathways of the immune response, ultimately leading to tissue injury. Increased oxidative stress, inflammatory cytokine release, inflammatory cell infiltration and activation and the up-regulation of inflammatory transcription factors participate in the pathophysiology of these complex entities. Treatment varies in each specific disease, but at least in the cases of RE and IBD immunosuppressors are effective. However, full therapeutic responses are not always achieved. The pathophysiology of ischemia-reperfusion (IR) injury shares many of these mechanisms. Brief and repetitive periods of ischemia in an organ or limb have been shown to protect against subsequent major IR injury in distant organs, a phenomenon called remote ischemic preconditioning (RIP). This procedure has been shown to protect the gut, pancreas and liver by modulating many of the same inflammatory mechanisms. Since RIP is safe and tolerable, and has shown to be effective in some recent clinical trials, I suggest that RIP could be used as a physiologically relevant adjunct treatment for non-ischemic gastrointestinal inflammatory conditions.
Keywords: Ischemia reperfusion, Ischemic preconditioning, Remote ischemic preconditioning, Acute pancreatitis, Radiation enteritis, Inflammatory bowel diseases, Hepatotoxicity, Inflammation
Core tip: Ischemia-reperfusioin injury, which commonly occurs during transplant procedures, is associated with inflammatory cytokine release, endothelial dysfunction and oxidative stress. Remote ischemic-preconditioning is a procedure that can attenuate all of these alterations. The pathophysiology of many non-ischemic gastrointestinal disorders, such as radiation-induced enteritis and inflammatory bowel disease, among others, involves many of these same mechanisms. Future research should address the question of whether remote ischemic-preconditioning could also favorably alter the course of these diseases.
INTRODUCTION
Ischemia-reperfusion (IR) injury occurs when blood flow to an organ is interrupted, such as in arterial embolism or thrombosis, during transplant procedures or shock, followed by restoration of blood flow[1,2]. The initial injury (ischemia) causes depletion of tissue energy resources, activation of proteases and increased calcium influx into ischemic cells, and reperfusion exacerbates the extent of injury through the stimulation of an intense systemic inflammatory response. This leads to tissue microcirculatory failure, necrosis and apoptosis. Marked pro-inflammatory cytokine release, inflammatory cell infiltration, production of reactive oxygen species (ROS), increased expression of nitric oxide (NO), Toll-like receptor (TLR)-4 signaling and activation of inflammatory transcription factors, among other complex pathways, are centrally involved in the events leading to IR-injury in various organs[2-4]. The gut and the liver are among the most ischemia-sensitive tissues.
Treatments aimed at attenuating IR-injury often involve the inhibition of one or many of these inflammatory pathways. Experimental success has been achieved with single cytokine blockers as well as with pleiotropic compounds with various immunomodulatory effects in both intestinal and liver IR-injury[5,6]. Recently, another strategy has proven useful in reducing IR-injury, ischemic preconditioning (IP). IP consists of a brief episode of ischemia that precedes the major ischemic event, and leads to the modulation of the innate organ defenses and inflammatory response, leading to decreased resulting injury[7]. The molecular mechanisms of IP are complex, and involve various pathways. IP leads to increased natural antioxidants such as glutathione, superoxide dismutase, heme-oxygenase-1 (HO-1), and reduced levels of lipid peroxidation[8]. Nuclear transcription factors such as nuclear factor-kappaB (NF-KappaB) are also regulated by IP, leading to reduced pro-inflammatory cytokines production and release[7]. Additionally, NO production is modulated by IP, leading to preserved sinusoidal blood flow, oxygenation and mitochondrial function[8]. These events prepare tissues to withstand IR injury, with impressive efficacy.
Remote ischemic-preconditioning (RIP) has emerged as an interesting alternative to direct IP. RIP involves the induction of protection against IR injury in an organ by brief episodes of ischemia and reperfusion in an entirely different organ or limb[9]. These protective effects begin few hours after preconditioning and may persist for up to 72 h. Circulating mediators and/or signals are hypothesized to mediate this effect, and some mechanisms involved so far are similar to those that applied in direct IP, including modulation of inflammatory cells, cytokines and oxidative stress[9,10].
Specific mechanisms involved in RIP-induced protection are also being uncovered. The induction of intestinal or hepatic RIP was shown to protect against subsequent renal IR by the attenuation of oxidative stress, lipid peroxidation, cytokine release, NO production and the upregulation of endogenous antioxidant mechanisms[11,12], and further studies have established that NO is an important mediator of RIP as well[13,14]. RIP normalizes NO production in mitochondria, and attenuates oxidative stress-mediated mitochondrial injury (24). HO-1, heat shock proteins (hsp), antioxidant and cytoprotective defense systems, are also upregulated by RIP and also attenuate tissue injury[9,10,15].
RIP also leads to reduced levels of tumor necrosis factor alpha (TNF-α) and inhibits crosstalk between TNF-α receptors and the induction of NF-KappaB[9,10,16]. RIP leads to reduced production and release of other proinflammatory cytokines and suppression of NF-KappaB-induced inflammation, and RIP has been shown to reduce long term transforming growth factor-beta (TGF-β) expression and fibrosis in kidneys damaged by IR injury[17,18]. Signal transduction and activator of transcription (STAT) pathways have also been involved in the pathophysiology of IR injury, and RIP is known to modulate STAT activity, leading to regulation of inflammation and cell survival[19,20]. Rats that are deficient in TLR-4 do not show RIP liver protection, suggesting a role for TLRs in the induction of preconditioning-associated ischemic tolerance[21,22].
In experimental studies, both IP and RIP have been shown to be beneficial in IR-injury of the gut, liver and pancreas[23-25]. The traditional view is that both IP and RIP protect tissues from a subsequent episode of full blown IR-injury. However, the variety of the inflammatory pathways regulated by these interventions suggests that they have wide-ranging immunomodulatory properties (Figure 1). Experimental studies have shown that IP and RIP are able to attenuate the inflammatory response and the severity of injury in various animal models of non-ischemic insults such as lipopolysaccharide-induced sepsis[26], traumatic brain injury[27] and cerulein-induced pancreatitis[28]. An interesting question that arises is whether these properties could be beneficial in non-ischemic but inflammatory conditions of the gastrointestinal system.
Figure 1.
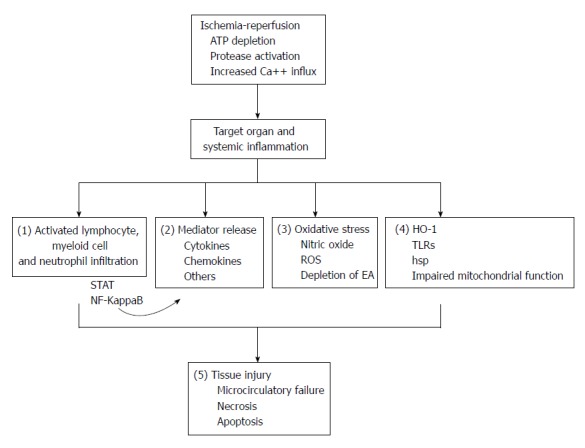
Pathophysiology of ischemia-reperfusioin injury and mechanisms modulated by remote ischemic preconditioning. There are multiple pathways involved in the pathophysiology of ischemia-reperfusion (IR) injury. This leads to end organ damage as discussed in the text. Inflammatory cell infiltration and activation during IR is driven by multiple signaling pathways and lead to tissue inflammation. (1) Remote ischemic preconditioning (RIP) can modulate inflammatory cell infiltration and activity. Inflammatory cells and resident cells produce inflammatory cytokines and chemokines, partly in response to signal transducers and activators of transcription factor (STAT) and nuclear factor kappa B (NF-Kappa B) signaling; (2) RIP can reduce cytokine and chemokine production and modulate STAT activity. Local and infiltrating inflammatory cells produce reactive oxygen species (ROS) that deplete endogenous antioxidants (EA), resulting in tissue distruction; (3) RIP reduces production of ROS and increases levels of EA. During IR, heme-oxygenase-1 (HO-1) and heat shock proteins (hsp) are upregulated, Toll-like receptors (TLRs) are activated and mitochondrial function is disrupted; (4) RIP modulates TLR signaling, protects mitochondria and regulates HO-1 production. All of these events lead to microcirculatory failure and finally cellular apoptosis; and (5) RIP preserves microcirculation and reduces pro-apoptotic signals. ATP: Adenosine triphosphate.
In this review, I propose that given the similarity in the physiopathology of IR-injury, RIP-induced protection and some gastrointestinal inflammatory conditions, RIP could show beneficial effects in their clinical course. That is, the idea of RIP as a general “immunomodulatory” strategy, outside of its application solely to limit IR-injury. I focus on drug-induced hepatotoxicity, inflammatory bowel diseases (IBD), radiation enteritis (RE) and acute pancreatitis (AP). Although it is outside the scope of this paper to review in detail the pathophysiology of these diseases, in the following sections I discuss relevant evidence in support for the current proposal.
HUMAN STUDIES OF REMOTE ISCHEMIC PRECONDITIONING
RIP is less invasive than IP and has led to great clinical interest for its application[10]. RIP is safe, tolerable, with no clear side effects, and has been used effectively in several clinical trials[29]. The induction of forearm or leg IR by the inflation of a blood-pressure cuff for brief episodes (5 min) is the preferred method in human trials, that has shown promising results in the setting of myocardial ischemia[10]. The possibility of using it on multiple occasions or intermittently remains open. A recent meta-analysis of 23 clinical trials including 1878 patients undergoing RIP, mostly for cardiac surgery, was recently published[30]. Although it found no mortality benefit, it did find reduced levels of myocardial infarction and confirmed the procedure’s safety. RIP has no known interaction with drug treatment, so it could be safely used as an adjunct to standard therapy. A recent small randomized controlled trial showed that arm RIP provided protection against contrast-medium induced nephrotoxicity[31]. This is the first clinical example of a study that used RIP with the aim of providing protection against a non-ischemic insult (contrast nephropathy).
RIP is associated with few side effects apart from slight discomfort, and published protocols can be easily replicated. One of the most common protocols involves performing 4 cycles of alternating 5-min inflation and 5-min deflation of a standard upper-arm blood pressure cuff to the individual’s systolic blood pressure plus 50 mmHg to induce transient and repetitive arm ischemia and reperfusion[31]. Number of cycles, ischemia time, and selected limb (arm or leg) can vary.
DRUG-INDUCED HEPATOTOXICITY
Drug induced acute liver failure (ALF) is one of the most severe and feared adverse reactions associated with drugs. Drug induced hepatotoxicity in general population has been estimated to be around 14/100000 inhabitants in Western countries, and contributes to 10%-50% of all causes of ALF[32]. Acetaminophen (ACP) intoxication, either accidental or intentional, is by far the most common, due to the universal access to this analgesic, and has a mortality rate of 32%-50%[32]. The specific treatment consist of n-acetylcysteine administration, an antioxidant that also affects ACP metabolism[33]. However, it requires complex dosing regimens, and the search for alternative therapeutic strategies is an active field of research.
The pathophysiology of ACP induced hepatotoxicity is well understood. ACP is metabolized by cytochrome P450 into N-acetyl-p-benzoquinone imine (NAPQI), a toxic metabolite, which is then conjugated to glutathione and detoxified under physiologic circumstances to mercapturic acid[34]. With ACP overdose, glutathione is depleted and excess NAPQI binds to intracellular proteins causing mitochondrial dysfunction and permeability, leading to increased oxidative stress and the induction of a sterile inflammatory response[35]. Hepatocytes, Kupffer cells, and other circulating non parenchymal cells resident synthesized other pro-inflammatory and injurious substances in response to these events. Among these, NO has been shown to play a major role in controlling lipid peroxidation and oxidative stress[36,37]. Induction of proteins that act as natural antioxidant defenses, such as HO-1 and heat shock protein 70 (hsp70), are increased by ACP in hepatocytes[38,39] in an attempt to reduce oxidative stress injury. Pro-inflammatory cytokines such as TNF-α are produced and other mediators such as TLR-4 are activated by ACP intoxication and are responsible for further damage and the perpetuation of the inflammatory response[40,41].
All of these pathways have been shown to be modulated by RIP in the liver. In a rodent model of hindlimb RIP, RIP led to the suppression of pro-inflammatory genes and the expression of antioxidant genes, leading to increased levels of glutathione, HO-1 and reduced TNF-α release in hepatocytes[22,24]. NO induced in the liver by RIP leads to preservation of the sinusoidal structure and maintenance of blood flow through the hepatic microcirculation, as well as improved mitochondrial oxygenation and reduced acidosis[4,42]. Finally, TLR-4 knockout mice have failed to induce hepatoprotection in RIP, suggesting a role for TLR-4 as well[22].
The overlap between the mechanisms responsible for ACP hepatotoxicity and those of RIP is striking (Table 1). Hypothetically, upon receiving a patient with suspected ACP intoxication, besides conventional treatment, forearm RIP could be initiated, which would hypothetically lead to reduced pro-inflammatory mediators production and increased antioxidant defense in the liver. This could partially mitigate the hepatotoxic effects of ACP, a common entity with high morbidity and mortality. Thus RIP could function as a physiologically reasonable adjunct treatment for ACP intoxication.
Table 1.
Pathways participating in the physiopathology of some gastrointestinal diseases and effects of remote ischemic preconditioning
| Some mechanisms of ACP-induced hepatotoxicity, RE, IBD and AP | Effects of RIP |
| Inflammatory cytokine production | Reduced inflammatory cytokines |
| Increased oxidative stress | Inhibits oxidative stress |
| Depletion of glutathione | Increased glutathione synthesis |
| NO expression and release | Modulation of NO function |
| iNOS expression | Modulation of iNOS |
| HO-1 and hsp70 production | Promotes HO-1 and hsp70 production |
| TLR signaling | Modulates TLR signaling |
| Th1 and Th2 cytokine imbalance | Reduced inflammatory cytokines |
| STAT and NF-kappaB signaling | Modulation of STAT and NF-KappaB |
| TGF-β production | Regulation of TGF-β production |
Note: Adapted from text. ACP: Acetaminophen; RE: Radiation-induced enteritis; IBD: Inflammatory bowel disease; AP: Acute pancreatitis; RIP: Remote ischemic preconditioning; HO-1: Heme oxygenase-1; hsp70: Heat shock protein-70; TLR: Toll-like receptor; NO: Nitric oxide; iNOS: Inducible nitric oxide synthase; STAT: Signal transduction and activator of transcription; NF-kappaB: Nuclear factor-kappaB; TGF-β: Transforming growth factor-beta.
INFLAMMATORY BOWEL DISEASE
IBD encompass a wide variety of conditions, of which ulcerative colitis and Crohn’s disease are the best understood. Their combined incidence and prevalence is approximately 5 cases per 100000 and 100 cases per 100000 population, respectively[43]. They constitute chronic inflammatory disorders of the gastrointestinal tract that share clinical and pathophysiological similarities, and can present with diarrhea, bloody stools, abdominal pain, weight loss and systemic signs of inflammation (depending on specific disease type). Alterations in innate immunity and in resident and infiltrating inflammatory cells such as neutrophils, CD4 lymphocytes, macrophages and mast cells lead to increased production of inflammatory cytokines of both the Th1 and Th2 response are the main effectors in these diseases[44]. Although initiator cytokines such as interleukin (IL)-17 and IL-13 dominate the initial response, the downstream mediators IL-1, IL-6 and TNF-α are the central regulators of tissue damage in IBD[45]. Besides immunosuppressive therapy, TNF-α antagonist therapy has shown tremendous effectiveness in the management of IBD. However, definitive treatment only exists in the form of surgery (in some cases), and disease can progress even under optimal pharmacologic therapy.
It is known that RIP is effective in reducing intestinal IR injury[23], and I have previously reviewed evidence that suggests RIP can modulate inflammatory cytokine production, including robust effects over TNF-α. Intestinal IP in particular has been shown to reduce the mRNA levels of many pro-inflammatory cytokines, including TNF-α and IL-1[46].
STAT proteins participate in cytokine-induced T-cell activation in IBD, and activation of STAT-4 and STAT-3 in T cells is an essential step in the physiopathology of Crohn’s disease[47]. The transcription factor NF-kappaB actively participates in the excessive inflammatory response observed in IBD, using TLR pathways, both in intestinal mucosal and inflammatory cells[48]. As I discussed, RIP is also able to modulate both of these transcription factors. Additionally, during IBD, endothelial cells are modified by intestinal inflammation, leading to altered responses to leukocyte-cell interactions and inflammatory cytokines, contributing to the initiation and propagation of IBD-related pathology[49]. RIP is known to protect endothelial cells from various insults. Intestinal IP can directly reduce NF-KappaB activation after IR-injury[46], and can also modulate leukocyte-endothelial interactions[50].
Recent evidence has shown that in response to inflammatory cytokine production, ROS are produced during disease activity in IBD, and that oxidative stress could be a triggering factor, rather than a concomitant occurrence during the pathogenesis of these diseases[51]. Not only are oxidants overproduced but antioxidants also are overexpressed as a natural defense mechanism against oxidative injury during IBD activity[52]. HO-1 is also a main endogenous defense mechanism against intestinal inflammation that has been hypothesized to participate in the pathophysiology of IBD[53]. Experimental studies have also established a protective role for Hsp70 against colitis trough suppression of inflammatory cytokines and apoptosis[54]. Constitutive and inducible NO synthetase (iNOs) participates in the initiation and regulation of inflammation during IBD[55], and has complex immune-modulating effects in normal intestinal homeostasis. Of course, RIP is associated with antioxidant effects, HO-1 and Hsp70 expression and modulation of NO responses. In rat models of intestinal IR, both IP and RIP were able to increase serum levels of HO-1 conferring cytoprotection[23,50], and RIP has been shown to modulate NO production and decrease lipid peroxidation in the colon[56].
Intestinal inflammatory diseases such as IBD also share many pathophysiological mechanisms with IR injury (Table 1), and there is direct experimental evidence suggesting that the mediators discussed above are capable of being regulated by RIP in intestinal tissues, making IBD another important candidate for RIP therapy. Of course, the ideal phase of intervention would be an acute flare of IBD, since these conditions have clear chronic courses. Some form of “pulse” therapy or periprocedural (in endoscopic procedures) use of RIP could also be promising.
RADIATION-INDUCED ENTERITIS
Radiotherapy for pelvic and abdominal tumors causes severe gastrointestinal complications. The intestines are very sensitive to radiation injury and they constitute a dose-limiting organ. As many as 16% of patients receiving abdominal or pelvic radiotherapy will develop chronic RE, and even more will suffer from the acute phase of this entity[57]. Acute symptoms include diarrhea, nausea and vomiting, and chronic manifestations include stricture formation, obstruction, inflammation, bleeding and fibrosis. Both phases of RE are thought to occur due to an imbalance of inflammatory and anti-inflammatory mediators. There is scarce evidence favoring an effective therapeutic regimen for RE, and the search for novel treatment or preventive strategies is ongoing[58]
Experimental evidence points towards an important role for oxidative stress in the physiopathology of RE. After radiation exposure, there is an important imbalance in the oxidative status of the intestine[59]. Increased myeloperoxidase, malonaldehide and decreased levels of glutathione have been associated with the acute inflammatory phase of RE[60]. RE also induces HO-1 expression, and the over-expression of HO-1 leads to tissue protection in acute RE[61]. The administration of antioxidants, like vitamin-E, diminish experimental RE by decreasing lipid peroxidation and increasing glutathione levels in the gut[62,63]. Increased induction of iNOS has also been observed in the colon after acute RE, and iNOS inhibitors diminish some of the alterations associated to RE[64,57].
The coordinated activity of several adhesion molecules participate in the recruitment of leukocytes into inflamed tissue in the setting of RE[65]. RE also induces the expression of adhesion molecules and other inflammatory mediators, such as cytokines and chemokines, both locally and systemically[66]. There are many lines of evidence that implicate cytokine imbalances directly in the pathogenesis of RE. In a rodent model of RE, the ileal expression of IL-1, IL-6, TNF-α, TGF-β was markedly increased, while the levels of the anti-inflammatory cytokine IL-10 were found to be diminished, all in association with activation of NF-KappaB[67]. NF-KappaB inhibition, in turn, was able to diminish the severity of RE, as well as the associated inflammatory response[68]. The shift towards a Th-2 like response, with release of inflammatory cytokines IL-1 and TNF-α, was shown to persist for up to 6 mo after RE, suggesting a role for this mechanisms in the chronic phase as well[64]. This same study also showed the induction of the IL-10/STAT 3 pathway by RE. At least one study has confirmed elevated levels of inflammatory cytokines in the mucosa of patients with radiation-induced proctitis[69].
The chronic phase of RE, characterized by fibrosis, is also associated to an inflammatory response. In rodent models, TGF-β immunoreactivity was found to be increased in the intestines after RE and to correlate well with both acute and chronic histopathologic lesions[70]. TGF-β and its pathways are central mediators of fibrogenesis and currently constitute an important therapeutic target for RE[71]. In experimental studies, IP could reduce the chronic elevations of TGF-β after renal IR-injury[17], but these results have not been replicated in models of liver IR[72].
RE constitutes another pathology that shares many pathophysiological mechanisms with IR-injury (Table 1). It is therefore unsurprising that many of these mechanism are affected by RIP as well, including regulation of cytokine balance, attenuation of oxidative stress and the modulation of transcription factors such as NF-KappaB. There is another important aspect to RE which makes it ideal for intervention with RIP: the fact that it is an expected complication, a side effect. This allows for the use of RIP as pre-treatment, or as a preventive measure. It is in this setting that RIP has shown greater clinical benefit in human trials.
ACUTE PANCREATITIS
AP is one of the most common emergency department admission gastrointestinal diagnoses in the world, with increasing incidences of up to 30 cases per 100000 population, with an overall mortality of around 2%[73]. Many etiologies account for AP, but gallstones and alcohol constitute for nearly 70% of cases. Irrespective of cause, a common pathogenesis seems to involve pancreatic hyperstimulation, enzyme (including trypsin) activity upregulation in acinar cells and enzyme reflux, leading to auto-digestion of the gland and local inflammation[74,75]. Although the full details of these pathogenic processes remain unknown, an intense local and systemic inflammatory response is characteristic of AP and accounts for most aspects of disease severity, systemic complications and clinical outcomes. Treatment is mostly supportive, and no specific pharmacologic treatment is currently available.
Unsurprisingly, many of the inflammatory mechanisms I have discussed in previous sections again turn out to play essential roles in the pathophysiology of AP. NF-KappaB is upregulated in AP, leading to the activation of many inflammatory genes and the upregulation of various pro-inflammatory mediators[76]. Monocytes form patients with AP show impaired NF-KappaB and STAT3 regulation[77]. The STAT signal transducers also promote the expression of TNF-α, IL-1 and IL-6 in acute pancreatitis[78]. Human and experimental studies have confirmed the central role of these pro-inflammatory cytokines in the regulation and perpetuation of a systemic inflammatory response in AP[79]. Experimental inhibition of these molecules leads to attenuation of the severity AP, and their serum levels have even been useful as predictors of severity in the clinical setting.
Interactions between pro-inflammatory cytokines and oxidative stress occurs in the development of the inflammatory response in AP, activating common signal transduction pathways that lead to amplification of the inflammatory cascade such as NF-KappaB[80]. Increased oxidative stress, lipid peroxidation and production of free radicals are important steps in the development of pancreatic tissue injury during AP[80,81]. HO-1 is upregulated in pancreatic tissue after experimental AP, contributing to intercellular defenses against increased oxidative stress[82], and neutrophils and monocytes derived from patients with AP have shown to be primed for HO-1 production, suggesting that HO-1 upregulation could be a viable therapeutic target[83]. Hsp’s and TLRs also participate in an interplay between pancreas tissues and etiological agents, modulating the local and systemic inflammatory cascade[84,85]. Although the role of NO in the pathophysiology of AP remains controversial, it can regulate many molecular targets and crucial steps of the inflammatory response[86].
Despite advances in the understanding of these processes, there is no evidence of a clinical benefit of antioxidant or anti-cytokine therapies in AP. We have already seen that both IP and RIP modulate all of these mechanisms in general, but particular evidence concerning the pancreas is available. Pancreatic IP is effective in reducing the severity of IR-induced pancreatitis, partly through the modulation of IL-1 signaling[87-89]. IP also reduces the severity of caerulein-induced pancreatitis and can also accelerate pancreatic repair, also partly through a reduction of the inflammatory process and induction of hsp70[28,90]. IP can also preserve microcirculation, reduce neutrophil infiltration and necrosis after IR-induced pancreatitis[91]. Some of these findings have been extended to RIP. RIP via hepatic IP also reduced the severity of IR-induced pancreatitis, in part through reduced IL-1 production[92], but these findings could not be replicated by renal RIP[93].
Considering that a systemic inflammatory response is the hallmark of severe acute pancreatitis and its associated systemic complications, RIP again emerges as an intriguing therapeutic possibility (Table 1). Its wide-ranging immunomodulatory effects, including those that target essential pathways known to participate in the physiopathology of AP, could turn out to be beneficial. Prevention of systemic complications and subsequent repair could also be achieved by RIP. RIP could also be specially effective in post-endoscopic retrograde cholangiopancreatography pancreatitis, as a periprocedural preventive measure.
CONCLUSION
The clinical application of RIP is an active field of research. It is a safe, tolerable and simple procedure, with wide-ranging immunomodulatory effects. Most clinical studies concern the utility of RIP in preventing IR-injury in the setting of vascular surgery or myocardial infarction, but at least one study has found a beneficial effect of RIP in reducing a non-ischemic injury, that of contrast nephropathy[31]. Recent developments also suggest that gastrointestinal protection via RIP might be clinically effective. Serum from human subjects subjected to RIP was able to protect human cultured intestinal cells from hypoxia-induced damage[94]. And in a recent randomized control trial, RIP was shown to protect the intestine against IR-injury associated to open abdominal aneurism repair, as evidenced by decreased levels of intestinal injury markers such as serum intestinal fatty acid-binding protein, endotoxin levels and diamine oxidase activity[95].
Considering that the protective effects of RIP are thought to depend on its modulation of the inflammatory response and oxidative status, its therapeutic potential could be expanded to other non-ischemic inflammatory conditions. I have shown that inflammatory gastrointestinal conditions such as IBD, RE, AP and drug-induced hepatotoxicity could be ideal candidates for RIP treatment, considering that their pathophysiological processes mirror to an extent those of IR-injury, and have themselves been shown to be modulated by RIP. RIP is most often thought of as a preventive strategy, since it is usually applied before the development of injury or disease. This makes RE and post-endoscopic retrograde colangiopancreatography pancreatitis the most logical candidates. However, the immunomodulatory effects of ischemic conditioning are known to persist even after an initial ischemic injury is established, a phenomenon known as ischemic post-conditioning[96]. Of course, clinical applicability would have to be determined following extensive pre-clinical experimentation and appropriate clinical trials. Precise timing, preconditioning technique and route would have to be determined as well. However, the current evidence suggests RIP has a great potential in treating these and other pathologic conditions.
Footnotes
P- Reviewers: Krishnan T, Rakonczay Z S- Editor: Wen LL L- Editor: A E- Editor: Wang CH
References
- 1.Berland T, Oldenburg WA. Acute mesenteric ischemia. Curr Treat Options Gastroenterol. 2008;11:3–10. doi: 10.1007/s11938-008-0001-2. [DOI] [PubMed] [Google Scholar]
- 2.de Groot H, Rauen U. Ischemia-reperfusion injury: processes in pathogenetic networks: a review. Transplant Proc. 2007;39:481–484. doi: 10.1016/j.transproceed.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 3.Montalvo-Jave EE, Escalante-Tattersfield T, Ortega-Salgado JA, Piña E, Geller DA. Factors in the pathophysiology of the liver ischemia-reperfusion injury. J Surg Res. 2008;147:153–159. doi: 10.1016/j.jss.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abu-Amara M, Yang SY, Tapuria N, Fuller B, Davidson B, Seifalian A. Liver ischemia/reperfusion injury: processes in inflammatory networks--a review. Liver Transpl. 2010;16:1016–1032. doi: 10.1002/lt.22117. [DOI] [PubMed] [Google Scholar]
- 5.Iñiguez M, Dotor J, Feijoo E, Goñi S, Prieto J, Berasain C, Avila MA. Novel pharmacologic strategies to protect the liver from ischemia-reperfusion injury. Recent Pat Cardiovasc Drug Discov. 2008;3:9–18. doi: 10.2174/157489008783331643. [DOI] [PubMed] [Google Scholar]
- 6.Mallick IH, Yang W, Winslet MC, Seifalian AM. Ischemia-reperfusion injury of the intestine and protective strategies against injury. Dig Dis Sci. 2004;49:1359–1377. doi: 10.1023/b:ddas.0000042232.98927.91. [DOI] [PubMed] [Google Scholar]
- 7.Eisen A, Fisman EZ, Rubenfire M, Freimark D, McKechnie R, Tenenbaum A, Motro M, Adler Y. Ischemic preconditioning: nearly two decades of research. A comprehensive review. Atherosclerosis. 2004;172:201–210. doi: 10.1016/S0021-9150(03)00238-7. [DOI] [PubMed] [Google Scholar]
- 8.Alchera E, Dal Ponte C, Imarisio C, Albano E, Carini R. Molecular mechanisms of liver preconditioning. World J Gastroenterol. 2010;16:6058–6067. doi: 10.3748/wjg.v16.i48.6058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tapuria N, Kumar Y, Habib MM, Abu Amara M, Seifalian AM, Davidson BR. Remote ischemic preconditioning: a novel protective method from ischemia reperfusion injury--a review. J Surg Res. 2008;150:304–330. doi: 10.1016/j.jss.2007.12.747. [DOI] [PubMed] [Google Scholar]
- 10.Kharbanda RK, Nielsen TT, Redington AN. Translation of remote ischaemic preconditioning into clinical practice. Lancet. 2009;374:1557–1565. doi: 10.1016/S0140-6736(09)61421-5. [DOI] [PubMed] [Google Scholar]
- 11.Song T, Peng YF, Guo SY, Liu YH, Liul LY. Brief small intestinal ischemia lessens renal ischemia-reperfusion injury in rats. Comp Med. 2007;57:200–205. [PubMed] [Google Scholar]
- 12.Lee JA, Choi JW, In JH, Jung HS, Kim YS, Jeon YS, Kang YJ, Kim DW, Lim YG, Park JH, et al. Hepatic ischemic preconditioning provides protection against distant renal ischemia and reperfusion injury in mice. J Korean Med Sci. 2012;27:547–552. doi: 10.3346/jkms.2012.27.5.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen H, Xing B, Liu X, Zhan B, Zhou J, Zhu H, Chen Z. Similarities between ozone oxidative preconditioning and ischemic preconditioning in renal ischemia/reperfusion injury. Arch Med Res. 2008;39:169–178. doi: 10.1016/j.arcmed.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 14.Liu X, Chen H, Zhan B, Xing B, Zhou J, Zhu H, Chen Z. Attenuation of reperfusion injury by renal ischemic postconditioning: the role of NO. Biochem Biophys Res Commun. 2007;359:628–634. doi: 10.1016/j.bbrc.2007.05.129. [DOI] [PubMed] [Google Scholar]
- 15.Plotnikov EY, Kazachenko AV, Vyssokikh MY, Vasileva AK, Tcvirkun DV, Isaev NK, Kirpatovsky VI, Zorov DB. The role of mitochondria in oxidative and nitrosative stress during ischemia/reperfusion in the rat kidney. Kidney Int. 2007;72:1493–1502. doi: 10.1038/sj.ki.5002568. [DOI] [PubMed] [Google Scholar]
- 16.Jia RP, Xie JJ, Luo FY, Zhu JG. Ischemic preconditioning improves rat kidney allograft function after ischemia/reperfusion injury: the role of tumor necrosis factor-alpha. Transplant Proc. 2008;40:3316–3320. doi: 10.1016/j.transproceed.2008.06.113. [DOI] [PubMed] [Google Scholar]
- 17.Jiang SH, Liu CF, Zhang XL, Xu XH, Zou JZ, Fang Y, Ding XQ. Renal protection by delayed ischaemic preconditioning is associated with inhibition of the inflammatory response and NF-kappaB activation. Cell Biochem Funct. 2007;25:335–343. doi: 10.1002/cbf.1395. [DOI] [PubMed] [Google Scholar]
- 18.Jiang S, Chen Y, Zou J, Xu X, Zhang X, Liu C, Fang Y, Ding X. Diverse effects of ischemic pretreatments on the long-term renal damage induced by ischemia-reperfusion. Am J Nephrol. 2009;30:440–449. doi: 10.1159/000239574. [DOI] [PubMed] [Google Scholar]
- 19.Yang N, Luo M, Li R, Huang Y, Zhang R, Wu Q, Wang F, Li Y, Yu X. Blockage of JAK/STAT signalling attenuates renal ischaemia-reperfusion injury in rat. Nephrol Dial Transplant. 2008;23:91–100. doi: 10.1093/ndt/gfm509. [DOI] [PubMed] [Google Scholar]
- 20.Hausenloy DJ, Yellon DM. Survival kinases in ischemic preconditioning and postconditioning. Cardiovasc Res. 2006;70:240–253. doi: 10.1016/j.cardiores.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 21.Karikó K, Weissman D, Welsh FA. Inhibition of toll-like receptor and cytokine signaling--a unifying theme in ischemic tolerance. J Cereb Blood Flow Metab. 2004;24:1288–1304. doi: 10.1097/01.WCB.0000145666.68576.71. [DOI] [PubMed] [Google Scholar]
- 22.Wang F, Birch SE, He R, Tawadros P, Szaszi K, Kapus A, Rotstein OD. Remote ischemic preconditioning by hindlimb occlusion prevents liver ischemic/reperfusion injury: the role of High Mobility Group-Box 1. Ann Surg. 2010;251:292–299. doi: 10.1097/SLA.0b013e3181bfda8c. [DOI] [PubMed] [Google Scholar]
- 23.Saeki I, Matsuura T, Hayashida M, Taguchi T. Ischemic preconditioning and remote ischemic preconditioning have protective effect against cold ischemia-reperfusion injury of rat small intestine. Pediatr Surg Int. 2011;27:857–862. doi: 10.1007/s00383-010-2810-3. [DOI] [PubMed] [Google Scholar]
- 24.Lai IR, Chang KJ, Chen CF, Tsai HW. Transient limb ischemia induces remote preconditioning in liver among rats: the protective role of heme oxygenase-1. Transplantation. 2006;81:1311–1317. doi: 10.1097/01.tp.0000203555.14546.63. [DOI] [PubMed] [Google Scholar]
- 25.Oehmann C, Benz S, Drognitz O, Pisarski P, Hopt UT, Obermaier R. Remote preconditioning reduces microcirculatory disorders in pancreatic ischemia/reperfusion injury. Pancreas. 2007;35:e45–e50. doi: 10.1097/mpa.0b013e318073d1b7. [DOI] [PubMed] [Google Scholar]
- 26.Tamion F, Richard V, Renet S, Thuillez C. Intestinal preconditioning prevents inflammatory response by modulating heme oxygenase-1 expression in endotoxic shock model. Am J Physiol Gastrointest Liver Physiol. 2007;293:G1308–G1314. doi: 10.1152/ajpgi.00154.2007. [DOI] [PubMed] [Google Scholar]
- 27.Pérez-Pinzón MA, Alonso O, Kraydieh S, Dietrich WD. Induction of tolerance against traumatic brain injury by ischemic preconditioning. Neuroreport. 1999;10:2951–2954. doi: 10.1097/00001756-199909290-00014. [DOI] [PubMed] [Google Scholar]
- 28.Warzecha Z, Dembinski A, Ceranowicz P, Konturek SJ, Dembinski M, Pawlik WW, Tomaszewska R, Stachura J, Kusnierz-Cabala B, Naskalski JW, et al. Ischemic preconditioning inhibits development of edematous cerulein-induced pancreatitis: involvement of cyclooxygenases and heat shock protein 70. World J Gastroenterol. 2005;11:5958–5965. doi: 10.3748/wjg.v11.i38.5958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alreja G, Bugano D, Lotfi A. Effect of remote ischemic preconditioning on myocardial and renal injury: meta-analysis of randomized controlled trials. J Invasive Cardiol. 2012;24:42–48. [PubMed] [Google Scholar]
- 30.Brevoord D, Kranke P, Kuijpers M, Weber N, Hollmann M, Preckel B. Remote ischemic conditioning to protect against ischemia-reperfusion injury: a systematic review and meta-analysis. PLoS One. 2012;7:e42179. doi: 10.1371/journal.pone.0042179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Er F, Nia AM, Dopp H, Hellmich M, Dahlem KM, Caglayan E, Kubacki T, Benzing T, Erdmann E, Burst V, et al. Ischemic preconditioning for prevention of contrast medium-induced nephropathy: randomized pilot RenPro Trial (Renal Protection Trial) Circulation. 2012;126:296–303. doi: 10.1161/CIRCULATIONAHA.112.096370. [DOI] [PubMed] [Google Scholar]
- 32.Larrey D, Pageaux GP. Drug-induced acute liver failure. Eur J Gastroenterol Hepatol. 2005;17:141–143. doi: 10.1097/00042737-200502000-00002. [DOI] [PubMed] [Google Scholar]
- 33.Abdel Salam OM, Baiuomy AR, El-Shenawy SM, Hassan NS. Effect of pentoxifylline on hepatic injury caused in the rat by the administration of carbon tetrachloride or acetaminophen. Pharmacol Rep. 2005;57:596–603. [PubMed] [Google Scholar]
- 34.Ruepp SU, Tonge RP, Shaw J, Wallis N, Pognan F. Genomics and proteomics analysis of acetaminophen toxicity in mouse liver. Toxicol Sci. 2002;65:135–150. doi: 10.1093/toxsci/65.1.135. [DOI] [PubMed] [Google Scholar]
- 35.Jaeschke H. Role of inflammation in the mechanism of acetaminophen-induced hepatotoxicity. Expert Opin Drug Metab Toxicol. 2005;1:389–397. doi: 10.1517/17425255.1.3.389. [DOI] [PubMed] [Google Scholar]
- 36.Gardner CR, Heck DE, Yang CS, Thomas PE, Zhang XJ, DeGeorge GL, Laskin JD, Laskin DL. Role of nitric oxide in acetaminophen-induced hepatotoxicity in the rat. Hepatology. 1998;27:748–754. doi: 10.1002/hep.510270316. [DOI] [PubMed] [Google Scholar]
- 37.Hinson JA, Bucci TJ, Irwin LK, Michael SL, Mayeux PR. Effect of inhibitors of nitric oxide synthase on acetaminophen-induced hepatotoxicity in mice. Nitric Oxide. 2002;6:160–167. doi: 10.1006/niox.2001.0404. [DOI] [PubMed] [Google Scholar]
- 38.Chiu H, Brittingham JA, Laskin DL. Differential induction of heme oxygenase-1 in macrophages and hepatocytes during acetaminophen-induced hepatotoxicity in the rat: effects of hemin and biliverdin. Toxicol Appl Pharmacol. 2002;181:106–115. doi: 10.1006/taap.2002.9409. [DOI] [PubMed] [Google Scholar]
- 39.Tolson JK, Dix DJ, Voellmy RW, Roberts SM. Increased hepatotoxicity of acetaminophen in Hsp70i knockout mice. Toxicol Appl Pharmacol. 2006;210:157–162. doi: 10.1016/j.taap.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 40.Blazka ME, Wilmer JL, Holladay SD, Wilson RE, Luster MI. Role of proinflammatory cytokines in acetaminophen hepatotoxicity. Toxicol Appl Pharmacol. 1995;133:43–52. doi: 10.1006/taap.1995.1125. [DOI] [PubMed] [Google Scholar]
- 41.Yohe HC, O’Hara KA, Hunt JA, Kitzmiller TJ, Wood SG, Bement JL, Bement WJ, Szakacs JG, Wrighton SA, Jacobs JM, et al. Involvement of Toll-like receptor 4 in acetaminophen hepatotoxicity. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1269–G1279. doi: 10.1152/ajpgi.00239.2005. [DOI] [PubMed] [Google Scholar]
- 42.Abu-Amara M, Yang SY, Quaglia A, Rowley P, de Mel A, Tapuria N, Seifalian A, Davidson B, Fuller B. Nitric oxide is an essential mediator of the protective effects of remote ischaemic preconditioning in a mouse model of liver ischaemia/reperfusion injury. Clin Sci (Lond) 2011;121:257–266. doi: 10.1042/CS20100598. [DOI] [PubMed] [Google Scholar]
- 43.Loftus EV. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology. 2004;126:1504–1517. doi: 10.1053/j.gastro.2004.01.063. [DOI] [PubMed] [Google Scholar]
- 44.Roberts-Thomson IC, Fon J, Uylaki W, Cummins AG, Barry S. Cells, cytokines and inflammatory bowel disease: a clinical perspective. Expert Rev Gastroenterol Hepatol. 2011;5:703–716. doi: 10.1586/egh.11.74. [DOI] [PubMed] [Google Scholar]
- 45.Strober W, Fuss IJ. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology. 2011;140:1756–1767. doi: 10.1053/j.gastro.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takeshita M, Tani T, Harada S, Hayashi H, Itoh H, Tajima H, Ohnishi I, Takamura H, Fushida S, Kayahara M. Role of transcription factors in small intestinal ischemia-reperfusion injury and tolerance induced by ischemic preconditioning. Transplant Proc. 2010;42:3406–3413. doi: 10.1016/j.transproceed.2010.06.038. [DOI] [PubMed] [Google Scholar]
- 47.Mudter J, Neurath MF. The role of signal transducers and activators of transcription in T inflammatory bowel diseases. Inflamm Bowel Dis. 2003;9:332–337. doi: 10.1097/00054725-200309000-00008. [DOI] [PubMed] [Google Scholar]
- 48.Wullaert A. Role of NF-kappaB activation in intestinal immune homeostasis. Int J Med Microbiol. 2010;300:49–56. doi: 10.1016/j.ijmm.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 49.Cromer WE, Mathis JM, Granger DN, Chaitanya GV, Alexander JS. Role of the endothelium in inflammatory bowel diseases. World J Gastroenterol. 2011;17:578–593. doi: 10.3748/wjg.v17.i5.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mallick IH, Winslet MC, Seifalian AM. Ischemic preconditioning of small bowel mitigates the late phase of reperfusion injury: heme oxygenase mediates cytoprotection. Am J Surg. 2010;199:223–231. doi: 10.1016/j.amjsurg.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 51.Iborra M, Moret I, Rausell F, Bastida G, Aguas M, Cerrillo E, Nos P, Beltrán B. Role of oxidative stress and antioxidant enzymes in Crohn’s disease. Biochem Soc Trans. 2011;39:1102–1106. doi: 10.1042/BST0391102. [DOI] [PubMed] [Google Scholar]
- 52.Zhu H, Li YR. Oxidative stress and redox signaling mechanisms of inflammatory bowel disease: updated experimental and clinical evidence. Exp Biol Med (Maywood) 2012;237:474–480. doi: 10.1258/ebm.2011.011358. [DOI] [PubMed] [Google Scholar]
- 53.Takagi T, Naito Y, Uchiyama K, Yoshikawa T. The role of heme oxygenase and carbon monoxide in inflammatory bowel disease. Redox Rep. 2010;15:193–201. doi: 10.1179/174329210X12650506623889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tanaka K, Mizushima T. Protective role of HSF1 and HSP70 against gastrointestinal diseases. Int J Hyperthermia. 2009;25:668–676. doi: 10.3109/02656730903213366. [DOI] [PubMed] [Google Scholar]
- 55.Kolios G, Valatas V, Ward SG. Nitric oxide in inflammatory bowel disease: a universal messenger in an unsolved puzzle. Immunology. 2004;113:427–437. doi: 10.1111/j.1365-2567.2004.01984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Colak T, Turkmenoglu O, Dag A, Polat A, Comelekoglu U, Bagdatoglu O, Polat G, Kanik A, Akca T, Aydin S. The effect of remote ischemic preconditioning on healing of colonic anastomoses. J Surg Res. 2007;143:200–205. doi: 10.1016/j.jss.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 57.MacNaughton WK. Review article: new insights into the pathogenesis of radiation-induced intestinal dysfunction. Aliment Pharmacol Ther. 2000;14:523–528. doi: 10.1046/j.1365-2036.2000.00745.x. [DOI] [PubMed] [Google Scholar]
- 58.Zimmerer T, Böcker U, Wenz F, Singer MV. Medical prevention and treatment of acute and chronic radiation induced enteritis--is there any proven therapy? a short review. Z Gastroenterol. 2008;46:441–448. doi: 10.1055/s-2008-1027150. [DOI] [PubMed] [Google Scholar]
- 59.Haton C, François A, Vandamme M, Wysocki J, Griffiths NM, Benderitter M. Imbalance of the antioxidant network of mouse small intestinal mucosa after radiation exposure. Radiat Res. 2007;167:445–453. doi: 10.1667/RR0581.1. [DOI] [PubMed] [Google Scholar]
- 60.Hepgül G, Tanrikulu S, Unalp HR, Akguner T, Erbil Y, Olgaç V, Ademoğlu E. Preventive effect of pentoxifylline on acute radiation damage via antioxidant and anti-inflammatory pathways. Dig Dis Sci. 2010;55:617–625. doi: 10.1007/s10620-009-0780-x. [DOI] [PubMed] [Google Scholar]
- 61.Giriş M, Erbil Y, Oztezcan S, Olgaç V, Barbaros U, Deveci U, Kirgiz B, Uysal M, Toker GA. The effect of heme oxygenase-1 induction by glutamine on radiation-induced intestinal damage: the effect of heme oxygenase-1 on radiation enteritis. Am J Surg. 2006;191:503–509. doi: 10.1016/j.amjsurg.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 62.Mutlu-Türkoğlu U, Erbil Y, Oztezcan S, Olgaç V, Toker G, Uysal M. The effect of selenium and/or vitamin E treatments on radiation-induced intestinal injury in rats. Life Sci. 2000;66:1905–1913. doi: 10.1016/s0024-3205(00)00516-6. [DOI] [PubMed] [Google Scholar]
- 63.Abbasoğlu SD, Erbil Y, Eren T, Giriş M, Barbaros U, Yücel R, Olgaç V, Uysal M, Toker G. The effect of heme oxygenase-1 induction by octreotide on radiation enteritis. Peptides. 2006;27:1570–1576. doi: 10.1016/j.peptides.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 64.Gremy O, Benderitter M, Linard C. Acute and persisting Th2-like immune response after fractionated colorectal gamma-irradiation. World J Gastroenterol. 2008;14:7075–7085. doi: 10.3748/wjg.14.7075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Panés J, Granger DN. Leukocyte-endothelial cell interactions: molecular mechanisms and implications in gastrointestinal disease. Gastroenterology. 1998;114:1066–1090. doi: 10.1016/s0016-5085(98)70328-2. [DOI] [PubMed] [Google Scholar]
- 66.Van der Meeren A, Monti P, Vandamme M, Squiban C, Wysocki J, Griffiths N. Abdominal radiation exposure elicits inflammatory responses and abscopal effects in the lungs of mice. Radiat Res. 2005;163:144–152. doi: 10.1667/rr3293. [DOI] [PubMed] [Google Scholar]
- 67.Linard C, Ropenga A, Vozenin-Brotons MC, Chapel A, Mathe D. Abdominal irradiation increases inflammatory cytokine expression and activates NF-kappaB in rat ileal muscularis layer. Am J Physiol Gastrointest Liver Physiol. 2003;285:G556–G565. doi: 10.1152/ajpgi.00094.2003. [DOI] [PubMed] [Google Scholar]
- 68.Linard C, Marquette C, Mathieu J, Pennequin A, Clarençon D, Mathé D. Acute induction of inflammatory cytokine expression after gamma-irradiation in the rat: effect of an NF-kappaB inhibitor. Int J Radiat Oncol Biol Phys. 2004;58:427–434. doi: 10.1016/j.ijrobp.2003.09.039. [DOI] [PubMed] [Google Scholar]
- 69.Indaram AV, Visvalingam V, Locke M, Bank S. Mucosal cytokine production in radiation-induced proctosigmoiditis compared with inflammatory bowel disease. Am J Gastroenterol. 2000;95:1221–1225. doi: 10.1111/j.1572-0241.2000.02013.x. [DOI] [PubMed] [Google Scholar]
- 70.Richter KK, Langberg CW, Sung CC, Hauer-Jensen M. Association of transforming growth factor beta (TGF-beta) immunoreactivity with specific histopathologic lesions in subacute and chronic experimental radiation enteropathy. Radiother Oncol. 1996;39:243–251. doi: 10.1016/0167-8140(95)01735-6. [DOI] [PubMed] [Google Scholar]
- 71.Zhu Y, Zhou J, Tao G. Molecular aspects of chronic radiation enteritis. Clin Invest Med. 2011;34:E119–E124. doi: 10.25011/cim.v34i3.15183. [DOI] [PubMed] [Google Scholar]
- 72.Knudsen AR, Kannerup AS, Grønbæk H, Andersen KJ, Funch-Jensen P, Frystyk J, Flyvbjerg A, Mortensen FV. Effects of ischemic pre- and postconditioning on HIF-1α, VEGF and TGF-β expression after warm ischemia and reperfusion in the rat liver. Comp Hepatol. 2011;10:3. doi: 10.1186/1476-5926-10-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Steinberg WM. Acute pancreatitis. In: Feldman M, Friedman LS, Brant LJ, editors. Sleisinger and Fordtran’s gastrointestinal and liver disease: pathophysiology, diagnosis, management. 8th ed. Philadelphia: WB Saunders; 2006. pp. 1241–1270. [Google Scholar]
- 74.Cruz-Santamaría DM, Taxonera C, Giner M. Update on pathogenesis and clinical management of acute pancreatitis. World J Gastrointest Pathophysiol. 2012;3:60–70. doi: 10.4291/wjgp.v3.i3.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang GJ, Gao CF, Wei D, Wang C, Ding SQ. Acute pancreatitis: etiology and common pathogenesis. World J Gastroenterol. 2009;15:1427–1430. doi: 10.3748/wjg.15.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rakonczay Z, Hegyi P, Takács T, McCarroll J, Saluja AK. The role of NF-kappaB activation in the pathogenesis of acute pancreatitis. Gut. 2008;57:259–267. doi: 10.1136/gut.2007.124115. [DOI] [PubMed] [Google Scholar]
- 77.Oiva J, Mustonen H, Kylänpää ML, Kyhälä L, Alanärä T, Aittomäki S, Siitonen S, Kemppainen E, Puolakkainen P, Repo H. Patients with acute pancreatitis complicated by organ failure show highly aberrant monocyte signaling profiles assessed by phospho-specific flow cytometry. Crit Care Med. 2010;38:1702–1708. doi: 10.1097/CCM.0b013e3181e7161c. [DOI] [PubMed] [Google Scholar]
- 78.Yu JH, Kim H. Role of janus kinase/signal transducers and activators of transcription in the pathogenesis of pancreatitis and pancreatic cancer. Gut Liver. 2012;6:417–422. doi: 10.5009/gnl.2012.6.4.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Norman J. The role of cytokines in the pathogenesis of acute pancreatitis. Am J Surg. 1998;175:76–83. doi: 10.1016/s0002-9610(97)00240-7. [DOI] [PubMed] [Google Scholar]
- 80.Pereda J, Sabater L, Aparisi L, Escobar J, Sandoval J, Viña J, López-Rodas G, Sastre J. Interaction between cytokines and oxidative stress in acute pancreatitis. Curr Med Chem. 2006;13:2775–2787. doi: 10.2174/092986706778522011. [DOI] [PubMed] [Google Scholar]
- 81.Esrefoglu M. Experimental and clinical evidence of antioxidant therapy in acute pancreatitis. World J Gastroenterol. 2012;18:5533–5541. doi: 10.3748/wjg.v18.i39.5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sato H, Siow RC, Bartlett S, Taketani S, Ishii T, Bannai S, Mann GE. Expression of stress proteins heme oxygenase-1 and -2 in acute pancreatitis and pancreatic islet betaTC3 and acinar AR42J cells. FEBS Lett. 1997;405:219–223. doi: 10.1016/s0014-5793(97)00191-9. [DOI] [PubMed] [Google Scholar]
- 83.Habtezion A, Kwan R, Yang AL, Morgan ME, Akhtar E, Wanaski SP, Collins SD, Butcher EC, Kamal A, Omary MB. Heme oxygenase-1 is induced in peripheral blood mononuclear cells of patients with acute pancreatitis: a potential therapeutic target. Am J Physiol Gastrointest Liver Physiol. 2011;300:G12–G20. doi: 10.1152/ajpgi.00231.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Feng JY, Li YY. Alteration and role of heat shock proteins in acute pancreatitis. J Dig Dis. 2010;11:277–283. doi: 10.1111/j.1751-2980.2010.00450.x. [DOI] [PubMed] [Google Scholar]
- 85.Hoque R, Malik AF, Gorelick F, Mehal WZ. Sterile inflammatory response in acute pancreatitis. Pancreas. 2012;41:353–357. doi: 10.1097/MPA.0b013e3182321500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hegyi P, Rakonczay Z. The role of nitric oxide in the physiology and pathophysiology of the exocrine pancreas. Antioxid Redox Signal. 2011;15:2723–2741. doi: 10.1089/ars.2011.4063. [DOI] [PubMed] [Google Scholar]
- 87.Warzecha Z, Dembinski A, Ceranowicz P, Dembinski M, Sendur R, Cieszkowski J, Sendur P, Tomaszewska R. Heparin inhibits protective effect of ischemic preconditioning in ischemia/reperfusion-induced acute pancreatitis. J Physiol Pharmacol. 2012;63:355–365. [PubMed] [Google Scholar]
- 88.Nikeghbalian S, Mansoorian MR, Hosseini SM, Mardani P, Geramizadeh B, Hosseini SA. Reduction of the severity of ischemia reperfusion-induced pancreatitis by ischemic pre-conditioning of the liver. Saudi J Kidney Dis Transpl. 2009;20:1010–1014. [PubMed] [Google Scholar]
- 89.Dembiński A, Warzecha Z, Ceranowicz P, Dembiński M, Cieszkowski J, Pawlik WW, Tomaszewska R, Konturek SJ, Konturek PC. Effect of ischemic preconditioning on pancreatic regeneration and pancreatic expression of vascular endothelial growth factor and platelet-derived growth factor-A in ischemia/reperfusion-induced pancreatitis. J Physiol Pharmacol. 2006;57:39–58. [PubMed] [Google Scholar]
- 90.Warzecha Z, Dembiński A, Ceranowicz P, Dembiński M, Cieszkowski J, Kuśnierz-Cabala B, Naskalski JW, Jaworek J, Konturek SJ, Pawlik WW, et al. Influence of ischemic preconditioning on blood coagulation, fibrinolytic activity and pancreatic repair in the course of caerulein-induced acute pancreatitis in rats. J Physiol Pharmacol. 2007;58:303–319. [PubMed] [Google Scholar]
- 91.Obermaier R, von Dobschuetz E, Drognitz O, Hopt UT, Benz S. Ischemic preconditioning attenuates capillary no-reflow and leukocyte adherence in postischemic pancreatitis. Langenbecks Arch Surg. 2004;389:511–516. doi: 10.1007/s00423-003-0443-x. [DOI] [PubMed] [Google Scholar]
- 92.Nikeghbalian S, Mardani P, Mansoorian MR, Salahi H, Bahador A, Geramizadeh B, Kakaei F, Johari HG, Malekhosseini SA. The effect of ischemic preconditioning of the pancreas on severity of ischemia/reperfusion-induced pancreatitis after a long period of ischemia in the rat. Transplant Proc. 2009;41:2743–2746. doi: 10.1016/j.transproceed.2009.07.026. [DOI] [PubMed] [Google Scholar]
- 93.Warzecha Z, Dembiński A, Ceranowicz P, Cieszkowski J, Konturek SJ, Dembiński M, Kuśnierz-Cabala B, Tomaszewska R, Pawlik WW. Ischemic preconditioning of the hindlimb or kidney does not attenuate the severity of acute ischemia/reperfusion-induced pancreatitis in rats. J Physiol Pharmacol. 2008;59:337–352. [PubMed] [Google Scholar]
- 94.Zitta K, Meybohm P, Bein B, Heinrich C, Renner J, Cremer J, Steinfath M, Scholz J, Albrecht M. Serum from patients undergoing remote ischemic preconditioning protects cultured human intestinal cells from hypoxia-induced damage: involvement of matrixmetalloproteinase-2 and -9. Mol Med. 2012;18:29–37. doi: 10.2119/molmed.2011.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li C, Li YS, Xu M, Wen SH, Yao X, Wu Y, Huang CY, Huang WQ, Liu KX. Limb remote ischemic preconditioning for intestinal and pulmonary protection during elective open infrarenal abdominal aortic aneurysm repair: a randomized controlled trial. Anesthesiology. 2013;118:842–852. doi: 10.1097/ALN.0b013e3182850da5. [DOI] [PubMed] [Google Scholar]
- 96.Vinten-Johansen J, Shi W. The science and clinical translation of remote postconditioning. J Cardiovasc Med (Hagerstown) 2013;14:206–213. doi: 10.2459/JCM.0b013e32835cecc6. [DOI] [PubMed] [Google Scholar]