

Abstract
Gene dosage effects have emerged as playing a central role in the pathogenesis of polycystic kidney disease. Yet, how gene dosage can ultimately have an impact on the formation of kidney cysts remains unknown. In this commentary we review the evidence for the role of gene dosage effects versus the “2-hit” mutation model in polycystic kidney disease (PKD), and also discuss how gene networks may potentially make intertwined contributions to PKD.
Introduction
PKD is characterized by the accumulation of multiple fluid-filled cysts in the kidneys and other organs. It is the most common inherited kidney disorder affecting an estimated 12.5 million people globally, and represents more than 5% of the worldwide total of end-stage renal disease [1]. PKD is transmitted as an autosomal dominant or autosomal recessive trait. Common features underlying PKD include the development of cysts and progressive impairment of renal function, but different types of PKD are distinguished from each other by different ages of onset, variable rates of renal disease progression, and a diverse array of extra-renal manifestations [1,2]. The two major classes of PKD in humans are autosomal dominant (ADPKD) and autosomal recessive (ARPKD) [1,3].
ADPKD is the most common form of PKD affecting 1 in 400–1000 live births [4], in which cyst development begins in utero and progresses slowly. The disease is often not clinically evident until after the third or fourth decade of adult life. Renal cysts also develop in utero in the less common recessive form of PKD, but in most cases recessive cystic kidney disease is more rapidly progressive, and is associated with congenital hepatic fibrosis [5].
In discussing the role of gene dosage effects in PKD, the focus of this article is on ADPKD caused by mutations in PKD1 encoding polycystin-1 (PC1; 85% of cases) or PKD2, which encodes polycystin-2 (PC2; 10-15% of cases – reviewed in [6]). The precise functional roles of PC1 and PC2 and their downstream effector pathways have yet to be determined. Current theoretical models suggest that germline mutations in PKD1 or PKD2, coupled with somatic second hit mutations, lead to absent or reduced PC1 or PC2 expression levels, which affects the cell-cell adhesion, polarity of basolateral epithelial cell membranes, promotion of primary cilia functional or length abnormalities, and/or developmental defects of tubular epithelial cells [6].
The 2-hit model of cystogenesis in ADPKD
The total number of cysts per kidney in patients who have mild to moderate ADPKD, with a mean age of 30.8 years, ranges from between 10-20 to over 200, with a mean number of 91 cysts [7]. Although every cell in the kidneys of ADPKD patients carries the same genetic germline mutation, only a very small percentage of the total number of the cells in each nephron becomes cystic. Moreover, severity of the ADPKD phenotype varies considerably from family to family, and how this clinical variability occurs in ADPKD is not very well understood [8]. Just over twenty years ago a hypothesis, known as the 2-hit model of cystogenesis, was proposed in order to try to explain why a small percentage of the total number of cells in ADPKD kidneys becomes cystic [9]. In this model, kidney cysts only begin forming upon loss of function of both alleles of PKD1 or PKD2, analogous to loss of function of both alleles of a tumour suppressor gene function in hereditary cancers, with each cyst representing a single clone of dividing cells. Therefore, in this scenario the initiating PKD1 mutation would be germline, while the second “hit” in PKD1 would be somatic, and would be a pre-requisite for focal cyst development [10].
In support of the 2-hit model, combinations of germline and somatic mutations in PKD1 have been observed in individual cysts of patients with autosomal dominant polycystic kidney disease type 1 (ADPKD1) [11], and similarly germline PKD2 mutations and somatic PKD2 mutations have been identified in individual cysts of ADPKD2 patients [12]. Furthermore, combined mutations involving germline PKD2 mutations and somatic PKD1 mutations have also been observed in a small number of human renal cysts in kidneys from PKD patients [12,13]. In addition, the direct causality between somatic inactivation of Pkd2 and cyst formation has been implicated in an adult mouse model of ADPKD [14]. In this model a modified Pkd2 allele (Pkd2Ws25) was generated in mice by insertion of a disrupted Pkd2 exon 1 in tandem with the wild type exon 1. The Pkd2Ws25 mice expressed functional PC2, but were prone to genomic rearrangements, which converted the Pkd2 allele to either a null or true wild type allele. The somatic conversion to a null allele (from Pkd2Ws25 to Pkd2-/-) correlated with cyst formation in the kidney, liver and pancreas. Furthermore, the cyst lining cells did not express PC2, suggesting that the somatic loss of Pkd2 was sufficient for cyst formation [14]. In summary, in ADPKD a germline mutation in either PKD1 or PKD2 may be combined with a somatic second hit in the remaining normal copy of the affected gene, leading to cellular recessive loss of function in both kidney tubules and bile ducts. When taken together, these observations provide strong evidence for the “2-hit” mutation model for cystogenesis in PKD.
Evidence for gene dosage effects
Despite strong supporting evidence, the 2-hit gene mutation model does not sufficiently explain a number of observations in humans or mice with ADPKD. For example, multiple studies have clearly demonstrated that PC1 expression (or PKD1 mRNA levels) was not lost and sometimes was elevated in the majority of cysts in ADPKD patients [15-18]. Furthermore, gene dosage effects were implicated in PKD mouse models characterized with either reduced or increased expression of Pkd1 or Pkd2 [19-22]. Since discrepancies between PKD mouse models and human ADPKD patients have been noted [23], it is possible that such gene dosage effects in mice could theoretically be dismissed as a discrepancy between human ADPKD and Pkd1 or Pkd2 mouse models.
Somewhat more incontrovertibly, however, in recent studies of PKD1- and/or PKD2-associated ADPKD in patients, it has clearly been shown that PKD can also be explained by gene dosage effects in humans (see Fig 1). Peter Harris's group at the Mayo Clinic, Rochester USA, has shown that heterogeneous alterations in PKD1, including incompletely penetrant mutations, could help to explain extreme variability observed in disease severity in patients with ADPKD. At the same time, these incompletely penetrant mutations raise questions about what would constitute a genetic “hit”. In the first of two related papers the authors describe consanguineous families with relatively mild phenotypes [24,25]. In one of the families in particular, two siblings were homozygous for the weakly pathogenic (so-called “hypomorphic”) mis-sense variant R3277C (RC) in the PKD1 gene and these individuals developed end-stage renal disease aged 62 and 75 years. Of course, the observation of homozygous RC allele mutations is inconsistent with a typical autosomal dominant inheritance pattern of ADPKD, suggesting that a different genetic mechanism is operating. Several other family members who were heterozygous for RC (consistent with an autosomal dominant inheritance pattern) had only a few renal cysts as adults. Moreover, in several unrelated families described by the authors, the same hypomorphic RC mis-sense mutation was again found, particularly in one family who exhibited more severe in utero onset of PKD, but this time it was combined with a more severely pathogenic mutation in PKD1 [24,25]. Thus, the RC allele in each of these individuals appeared to exhibit a low “potency” and was relatively mildly pathogenic, even when in a double dose, but it was able to induce severe in utero onset PKD when combined together with a strongly pathogenic PKD1 mutation.
Figure 1. Depiction of gene dosage changes in kidney cyst formation in ADPKD.
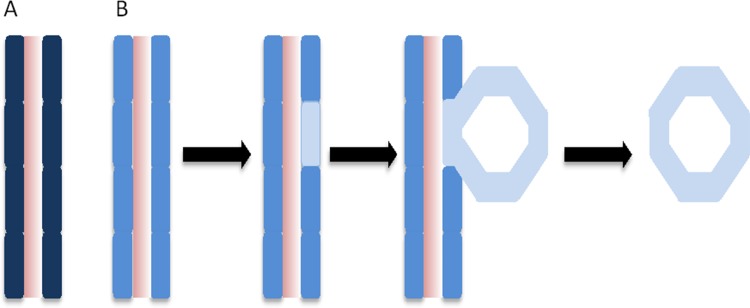
A) A segment of renal tubule from a normal individual, with a normal PKD1 gene dosage as represented by the dark blue colour of each epithelial cell of the renal tubule. B) Events leading to cyst formation in a segment of renal tubule from an individual with a PKD1 mutation, associated with reduced dosage of functional PKD1 protein as represented by the lighter blue colour of each epithelial cell of the renal tubule. Within one cell of the renal tubule segment a somatic event occurs that leads to further reduced dosage levels of PKD1 protein (represented by the very pale blue colour), which then leads to cyst formation.
In a follow-on paper the Harris group then developed a “knock-in” mouse model containing the RC variant allele and demonstrated that it was indeed a functionally hypomorphic allele capable of affecting the severity of ADPKD in mice [26]. They showed that, as expected, Pkd1+/null mice were normal, whereas in contrast Pkd1RC/null mice were viable and had rapidly progressive disease, and the Pkd1RC/RC mice developed renal cysts only gradually. These findings therefore support a gene dosage model, but ask the question how can gene dosage effects (and by inference dosage of the protein encoded by the gene) be explained?
In general, strict control of the dosage level of certain gene products is required in cells, which may be facilitated via gene networks. Several recent papers have focused on gene networks in PKD, which will be discussed in the next paragraph (see Table 1). It is also worth mentioning at this point that mice with Pkd1 haploinsufficiency develop renal anomalies over time and are not normal [27]. Moreover, progressive cystogenesis occurs in transgenic mice expressing artificial microRNAs against Pkd1 [28]. Most renal cysts in human ADPKD appear in adulthood, and interestingly, mice with renal conditional ablation of Pkd1 after postnatal day 13 or adulthood have either mild or no renal cystic phenotype [29-31]. These studies suggest that a gene dosage model may be operating within the context of a developmental window. In summary, there is now convincing evidence in both humans and mice strongly suggesting that dosage mechanisms are operating with respect to PKD1 function. However, it remains unclear how insufficient levels of PKD1 protein lead to ADPKD.
Table 1. Gene interaction networks involving Pkd1 or Pkd2 that impact dosage sensitivity and cystogenesis.
| Gene interaction network | Molecular interaction | Disease | Reference |
|---|---|---|---|
| Dicer/Pkd1 | miR200 – Pkd1 | ADPKD | [32] |
| PRKCSH/SEC63/Pkhd1/Pkd1/Pkd2 | Glucosidase IIb – polycystin 1 | Polycystic liver disease | [33] |
| PRKCSH/SEC63/Pkhd1/Pkd1/Pkd2 | SEC63p – polycystin 1 | Polycystic liver disease | [33] |
| PRKCSH/SEC63/Pkhd1/Pkd1/Pkd2 | – | ARPKD | [33] |
| Pkd1/Pkd2 | Pkd1 – Pkd2 | ADPKD | [13] |
| c-Met/NF-κB/Wnt/Pax2/Pkd1 | – | ADPKD | [36] [37] |
Abbreviations: ADPKD, autosomal dominant polycystic kidney disease; ARPKD, autosomal recessive polycystic kidney disease.
Gene networks in PKD
Interactions between two or more genes involving gene interaction networks have been shown to impact on Pkd1 gene dosage and cyst formation in several studies. Dicer 1 mutations were shown to modify the Pkd1 mutant phenotype and reduce cystogenesis by reducing the levels of certain microRNAs, including miR200, which increased the dosage levels of PC1 [32]. In other investigations, genetic interactions were studied to reveal factors that were required for adequate expression of functional protein complexes involving PC1 or PC2. For example, mutations in PRKCSH or SEC63 (which cause polycystic liver disease in humans) have been shown to modify the effect of mutations in PKD1, and were used to provide evidence that PC1 is the rate-limiting step in the progression of PKD and polycystic liver disease when Prkcsh or Sec63 were inactivated. Furthermore, the authors showed that the effects of reduced PC1 dosage differ depending on which nephron segment is involved; upon a reduced dosage of PC1 the severity of PKD is enhanced primarily by the increased growth of collecting duct cysts [33] (Table 1).
A gene network involving PKD1 and PKD2 has been suggested [12,13], and links between PC1 dosage and PC2 protein levels in the primary cilia were recently shown. Using induced pluripotent stem cells derived from PKD patient kidney cysts, Freedman et al. showed that the expression of PC2 in the primary cilia correlated with the level of expression (dosage) of PC1 [34]. The function of PC2 has been identified as an intracellular calcium release channel [2] that can interact with PC1, and so in this scenario the function of PC2 is highly dependent on the function of PC1.
Extending this theme further, not only is the Pkd1 gene known to interact with the Pkd2 gene, it appears that the dosage of PC1 can also modify the severity of cystogenesis in other forms of PKD, such as ARPKD. Fedeles et al. showed that an increase in the dosage of PC1 was able to rescue the ARPKD phenotype. According to this scenario, it is possible that ARPKD may be amenable to therapies that increase the dosage or function of PC1 [33] (Table 1).
Another example of a gene network in PKD was investigated in mice engineered to have a “del34” null mutation in Pkd1 (Pkd1del34/del34), which exhibited rapid and severe renal cyst formation in utero, and perinatal embryonic lethality [35]. Remarkably, cyst formation in the Pkd1del34/del34 null mice was much less severe when they also carried a mutation in another gene, called Pax2, which is known to play an important role in nephrogenesis [36]. The reduced dosage of functional Pax2 in these mutant mice may be genetically equivalent to inhibiting signaling pathways regulating planar cell polarity, such as hepatocyte growth factor receptor (c-Met), nuclear factor kappa B (NF-kB), or Wnt signaling; these factors were shown to form a regulatory cascade positively activating Pax2 expression in kidney tubules [37].
Summary
Gene dosage effects in PKD have emerged as playing an important role in the pathogenesis of PKD; it is likely that cyst formation within a given segment of renal tubular epithelium is a stochastic process, the probability of which increases as the functionality of PC1 and/or PC2 decreases. Following inheritance of alterations in PKD1 or PKD2 that either strongly or mildly reduce polycystin functionality, cysts may form through a stochastic process in the kidneys if additional somatic mutations are acquired in the remaining non-mutant copy of PKD1 or PKD2, or in genes that modify the function of PKD1 or PKD2 (Table 1). This raises questions about how altered functional polycystin protein levels cause the initiation of cyst formation, especially because, paradoxically, higher levels of PC1 can also lead to renal cyst formation [38]. It is very likely that other mechanisms are also associated with altered functional activity of polycystins, including epigenetic changes [39] (e.g. histone modification), or specific biochemical alterations that affect certain signaling pathways. Any of the mechanisms discussed could potentially lead to significant variation in disease severity amongst individuals with predisposing mutations. Finally, the identification of alterations in cells that occur as a result of gene dosage changes in the PKD1 network might also suggest novel routes of therapeutic intervention for PKD.
Abbreviations
- ADPKD
autosomal dominant polycystic kidney disease
- ADPKD1
autosomal dominant polycystic kidney disease type 1
- ADPKD2
autosomal dominant polycystic kidney disease type 2
- ARPKD
autosomal recessive polycystic kidney disease
- PC1
polycystin-1
- PC2
polycystin-2
- PKD
polycystic kidney disease
- RC
mis-sense variant R3277C of the PKD1 gene
Disclosures
The authors declare that they have no disclosures.
The electronic version of this article is the complete one and can be found at: http://f1000.com/prime/reports/m/6/24
References
- 1.Wilson PD. Polycystic kidney disease. N Engl J Med. 2004;350:151–64. doi: 10.1056/NEJMra022161. [DOI] [PubMed] [Google Scholar]
- 2.Igarashi P, Somlo S. Genetics and pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2002;13:2384–98. doi: 10.1097/01.ASN.0000028643.17901.42. [DOI] [PubMed] [Google Scholar]
- 3.Guay-Woodford LM. Renal cystic diseases: diverse phenotypes converge on the cilium/centrosome complex. Pediatr Nephrol. 2006;21:1369–76. doi: 10.1007/s00467-006-0164-9. [DOI] [PubMed] [Google Scholar]
- 4.Bisceglia M, Galliani CA, Senger C, Stallone C, Sessa A. Renal cystic diseases: a review. Adv Anat Pathol. 2006;13:26–56. doi: 10.1097/01.pap.0000201831.77472.d3. [DOI] [PubMed] [Google Scholar]
- 5.Zerres K, Mücher G, Becker J, Steinkamm C, Rudnik-Schöneborn S, Heikkilä P, Rapola J, Salonen R, Germino GG, Onuchic L, Somlo S, Avner ED, Harman LA, Stockwin JM, Guay-Woodford LM. Prenatal diagnosis of autosomal recessive polycystic kidney disease (ARPKD): molecular genetics, clinical experience, and fetal morphology. Am J Med Genet. 1998;76:137–44. doi: 10.1002/(SICI)1096-8628(19980305)76:2<137::AID-AJMG6>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 6.Mochizuki T, Tsuchiya K, Nitta K. Autosomal dominant polycystic kidney disease: recent advances in pathogenesis and potential therapies. Clin Exp Nephrol. 2013;17:317–26. doi: 10.1007/s10157-012-0741-0. [DOI] [PubMed] [Google Scholar]
- 7.Bae K, Park B, Sun H, Wang J, Tao C, Chapman AB, Torres VE, Grantham JJ, Mrug M, Bennett WM, Flessner MF, Landsittel DP, Bae KT. Segmentation of individual renal cysts from MR images in patients with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2013;8:1089–97. doi: 10.2215/CJN.10561012. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/717991341
- 8.Torra R, Badenas C, Darnell A, Nicolau C, Volpini V, Revert L, Estivill X. Linkage, clinical features, and prognosis of autosomal dominant polycystic kidney disease types 1 and 2. J Am Soc Nephrol. 1996;7:2142–51. doi: 10.1681/ASN.V7102142. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718303456
- 9.Reeders ST. Multilocus polycystic disease. Nat Genet. 1992;1:235–7. doi: 10.1038/ng0792-235. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718303457
- 10.Pei Y. A “two-hit” model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol Med. 2001;7:151–6. doi: 10.1016/S1471-4914(01)01953-0. [DOI] [PubMed] [Google Scholar]
- 11.Qian F, Watnick TJ, Onuchic LF, Germino GG. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell. 1996;87:979–87. doi: 10.1016/S0092-8674(00)81793-6. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718303458
- 12.Watnick T, He N, Wang K, Liang Y, Parfrey P, Hefferton D, St George-Hyslop P, Germino G, Pei Y. Mutations of PKD1 in ADPKD2 cysts suggest a pathogenic effect of trans-heterozygous mutations. Nat Genet. 2000;25:143–4. doi: 10.1038/75981. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718303460
- 13.Koptides M, Mean R, Demetriou K, Pierides A, Deltas CC. Genetic evidence for a trans-heterozygous model for cystogenesis in autosomal dominant polycystic kidney disease. Hum Mol Genet. 2000;9:447–52. doi: 10.1093/hmg/9.3.447. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718303461
- 14.Wu G, D'Agati V, Cai Y, Markowitz G, Park JH, Reynolds DM, Maeda Y, Le TC, Hou H, Kucherlapati R, Edelmann W, Somlo S. Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell. 1998;93:177–88. doi: 10.1016/S0092-8674(00)81570-6. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718303462
- 15.Lanoix J, D'Agati V, Szabolcs M, Trudel M. Dysregulation of cellular proliferation and apoptosis mediates human autosomal dominant polycystic kidney disease (ADPKD) Oncogene. 1996;13:1153–60. [PubMed] [Google Scholar]; http://f1000.com/prime/718303463
- 16.Ong AC, Harris PC, Davies DR, Pritchard L, Rossetti S, Biddolph S, Vaux DJ, Migone N, Ward CJ. Polycystin-1 expression in PKD1, early-onset PKD1, and TSC2/PKD1 cystic tissue. Kidney Int. 1999;56:1324–33. doi: 10.1046/j.1523-1755.1999.00659.x. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718303464
- 17.Ward CJ, Turley H, Ong AC, Comley M, Biddolph S, Chetty R, Ratcliffe PJ, Gattner K, Harris PC. Polycystin, the polycystic kidney disease 1 protein, is expressed by epithelial cells in fetal, adult, and polycystic kidney. Proc Natl Acad Sci USA. 1996;93:1524–8. doi: 10.1073/pnas.93.4.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718303465
- 18.Geng L, Segal Y, Peissel B, Deng N, Pei Y, Carone F, Rennke HG, Glücksmann-Kuis AM, Schneider MC, Ericsson M, Reeders ST, Zhou J. Identification and localization of polycystin, the PKD1 gene product. J Clin Invest. 1996;98:2674–82. doi: 10.1172/JCI119090. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718303466
- 19.Lantinga-van Leeuwen IS, Dauwerse JG, Baelde HJ, Leonhard WN, van de Wal A, Ward CJ, Verbeek S, Deruiter MC, Breuning MH, de Heer E, Peters DJM. Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease. Hum Mol Genet. 2004;13:3069–77. doi: 10.1093/hmg/ddh336. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718303467
- 20.Jiang S, Chiou Y, Wang E, Lin H, Lin Y, Chi Y, Wang CL, Tang M, Li H. Defining a link with autosomal-dominant polycystic kidney disease in mice with congenitally low expression of Pkd1. Am J Pathol. 2006;168:205–20. doi: 10.2353/ajpath.2006.050342. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718303468
- 21.Park EY, Sung YH, Yang MH, Noh JY, Park SY, Lee TY, Yook YJ, Yoo KH, Roh KJ, Kim I, Hwang Y, Oh GT, Seong JK, Ahn C, Lee H, Park JH. Cyst formation in kidney via B-Raf signaling in the PKD2 transgenic mice. J Biol Chem. 2009;284:7214–22. doi: 10.1074/jbc.M805890200. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718303469
- 22.Kurbegovic A, Côté O, Couillard M, Ward CJ, Harris PC, Trudel M. Pkd1 transgenic mice: adult model of polycystic kidney disease with extrarenal and renal phenotypes. Hum Mol Genet. 2010;19:1174–89. doi: 10.1093/hmg/ddp588. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718303470
- 23.Smith LA, Bukanov NO, Husson H, Russo RJ, Barry TC, Taylor AL, Beier DR, Ibraghimov-Beskrovnaya O. Development of polycystic kidney disease in juvenile cystic kidney mice: insights into pathogenesis, ciliary abnormalities, and common features with human disease. J Am Soc Nephrol. 2006;17:2821–31. doi: 10.1681/ASN.2006020136. [DOI] [PubMed] [Google Scholar]
- 24.Rossetti S, Kubly VJ, Consugar MB, Hopp K, Roy S, Horsley SW, Chauveau D, Rees L, Barratt TM, van't Hoff WG, Niaudet P, Niaudet WP, Torres VE, Harris PC. Incompletely penetrant PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease. Kidney Int. 2009;75:848–55. doi: 10.1038/ki.2008.686. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1165512
- 25.Vujic M, Heyer CM, Ars E, Hopp K, Markoff A, Orndal C, Rudenhed B, Nasr SH, Torres VE, Torra R, Bogdanova N, Harris PC. Incompletely penetrant PKD1 alleles mimic the renal manifestations of ARPKD. J Am Soc Nephrol. 2010;21:1097–102. doi: 10.1681/ASN.2009101070. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718303471
- 26.Hopp K, Ward CJ, Hommerding CJ, Nasr SH, Tuan H, Gainullin VG, Rossetti S, Torres VE, Harris PC. Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J Clin Invest. 2012;122:4257–73. doi: 10.1172/JCI64313. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718303472
- 27.Ahrabi AK, Terryn S, Valenti G, Caron N, Serradeil-Le Gal C, Raufaste D, Nielsen S, Horie S, Verbavatz J, Devuyst O. PKD1 haploinsufficiency causes a syndrome of inappropriate antidiuresis in mice. J Am Soc Nephrol. 2007;18:1740–53. doi: 10.1681/ASN.2006010052. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718077200
- 28.Wang E, Hsieh-Li H, Chiou Y, Chien Y, Ho H, Chin H, Wang CL, Liang S, Jiang S. Progressive renal distortion by multiple cysts in transgenic mice expressing artificial microRNAs against Pkd1. J Pathol. 2010;222:238–48. doi: 10.1002/path.2765. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718303473
- 29.Lantinga-van Leeuwen IS, Leonhard WN, van der Wal A, Breuning MH, de Heer E, Peters DJM. Kidney-specific inactivation of the Pkd1 gene induces rapid cyst formation in developing kidneys and a slow onset of disease in adult mice. Hum Mol Genet. 2007;16:3188–96. doi: 10.1093/hmg/ddm299. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718303474
- 30.Piontek K, Menezes LF, Garcia-Gonzalez MA, Huso DL, Germino GG. A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat Med. 2007;13:1490–5. doi: 10.1038/nm1675. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1098054
- 31.Takakura A, Contrino L, Beck AW, Zhou J. Pkd1 inactivation induced in adulthood produces focal cystic disease. J Am Soc Nephrol. 2008;19:2351–63. doi: 10.1681/ASN.2007101139. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718303475
- 32.Patel V, Hajarnis S, Williams D, Hunter R, Huynh D, Igarashi P. MicroRNAs regulate renal tubule maturation through modulation of Pkd1. J Am Soc Nephrol. 2012;23:1941–8. doi: 10.1681/ASN.2012030321. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718303476
- 33.Fedeles SV, Tian X, Gallagher A, Mitobe M, Nishio S, Lee SH, Cai Y, Geng L, Crews CM, Somlo S. A genetic interaction network of five genes for human polycystic kidney and liver diseases defines polycystin-1 as the central determinant of cyst formation. Nat Genet. 2011;43:639–47. doi: 10.1038/ng.860. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/12716956
- 34.Freedman BS, Lam AQ, Sundsbak JL, Iatrino R, Su X, Koon SJ, Wu M, Daheron L, Harris PC, Zhou J, Bonventre JV. Reduced ciliary polycystin-2 in induced pluripotent stem cells from polycystic kidney disease patients with PKD1 mutations. J Am Soc Nephrol. 2013;24:1571–86. doi: 10.1681/ASN.2012111089. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718100719
- 35.Lu W, Peissel B, Babakhanlou H, Pavlova A, Geng L, Fan X, Larson C, Brent G, Zhou J. Perinatal lethality with kidney and pancreas defects in mice with a targetted Pkd1 mutation. Nat Genet. 1997;17:179–81. doi: 10.1038/ng1097-179. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718303477
- 36.Stayner C, Iglesias DM, Goodyer PR, Ellis L, Germino G, Zhou J, Eccles MR. Pax2 gene dosage influences cystogenesis in autosomal dominant polycystic kidney disease. Hum Mol Genet. 2006;15:3520–8. doi: 10.1093/hmg/ddl428. [DOI] [PubMed] [Google Scholar]
- 37.Qin S, Taglienti M, Cai L, Zhou J, Kreidberg JA. c-Met and NF-κB-dependent overexpression of Wnt7a and -7b and Pax2 promotes cystogenesis in polycystic kidney disease. J Am Soc Nephrol. 2012;23:1309–18. doi: 10.1681/ASN.2011030277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thivierge C, Kurbegovic A, Couillard M, Guillaume R, Coté O, Trudel M. Overexpression of PKD1 causes polycystic kidney disease. Mol Cell Biol. 2006;26:1538–48. doi: 10.1128/MCB.26.4.1538-1548.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li X. Epigenetics and autosomal dominant polycystic kidney disease. Biochim Biophys Acta. 2011;1812:1213–8. doi: 10.1016/j.bbadis.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]