

Abstract
Purpose
Long-chain acyl-CoA dehydrogenase (LCAD) is a mitochondrial flavoenzyme thought to be one of the major enzymes responsible for the first step of long-chain fatty acid (LCFA) β-oxidation. Surprisingly, recent studies have shown LCAD is hardly detectable in human tissues such as liver and heart. Skeletal muscle is the largest organ in the body in terms of mass, and accounts for the majority of LCFA oxidation, especially during exercise. The purpose of this study was to investigate the expression levels of LCAD in human skeletal muscle.
Methods
Muscle biopsies were obtained from the vastus lateralis of healthy athletic men and women, and examined for mRNA abundance, protein content, and enzyme activity of LCAD. We compared LCAD content with that of very-long chain acyl-CoA dehydrogenase (VLCAD) and medium chain acyl-CoA dehydrogenase (MCAD); two mitochondrial β-oxidation enzymes that have overlapping chain-length specificity to that of LCAD. LCAD protein content and enzyme activity were also examined in enriched mitochondrial protein fractions. As controls, LCAD presence in skeletal muscle was compared to human heart, liver, and mouse skeletal muscle.
Results
The mRNA presence of LCAD in human skeletal muscle is significantly less than VLCAD and MCAD (0.08±0.01 vs 7.3±0.5 vs 2.4±0.2 respectively, P≤0.0001). LCAD protein was undetectable in human muscle homogenates, and coordinately LCAD enzyme activity was undetectable in enriched mitochondrial samples.
Conclusion
LCAD is minimally expressed in human skeletal muscle and likely does not play a significant role in LCFA oxidation.
Keywords: Long chain acyl-CoA dehydrogenase, LCAD, Fatty acid oxidation, Muscle, Men, Women
Introduction
Although all tissues are capable of oxidizing substrates, skeletal muscle is the most abundant tissue in the human body and the primary tissue responsible for the clearance of dietary lipids, proteins and carbohydrates in order to maintain metabolic homeostasis [1]. The process of lipid utilization, which occurs via mitochondrial fatty acid (FA) β-oxidation (FAO), is essential for energy production; especially during exercise, recovery from exercise, starvation, and other metabolic stresses. During metabolic demand, FA’s are sequestered from multiple stores, including circulating free FA’s, triglycerides, lipoproteins, and intramyocellular lipid (IMCL) stores, and delivered to the mitochondria in skeletal muscle. One example of metabolic demand is during exercise, where FA’s contribute on average 10–70% of substrate utilized (depending on exercise intensity) [2–4], of which ~90% of the FA’s are long-chain fatty acids (LCFA) [5]. LCFA are generally defined as FA’s with aliphatic tails of 12–22 carbons, which includes lauric acid (C12), myristic acid (C14), palmitic acid (C16), stearic acid (C18), Oleic acid (C18:2) and arachidonic acid (C20:4) [6], components of major dietary sources of lipids [7].
FAO is a catabolic pathway that generates NADH+ and FADH2 (the electron transport chain energy precursors) and removes 2-carbon units (for each cycle) in the form of acetyl-CoA for the Krebs cycle to generate additional NADH+ and FADH2. There are a number of enzymes that contribute to LCFA oxidation based on their enzymatic affinity for varying carbon lengths. For example, in the first step of β-oxidation very long chain acyl-CoA dehydrogenase (VLCAD) acts on 14–24 carbon chain length substrate with optimum activity for palmitoyl-CoA (C16-CoA) [8], and acyl-CoA dehydrogenase-9 (ACD9) acts on unsaturated long chain substrates[9]. Medium chain acyl-CoA dehydrogenase (MCAD) uses C4- to C14-CoA as substrates with optimum activity for C8-CoA [8]. Until recently long-chain acyl-CoA dehydrogenase (LCAD) was thought to be essential in the mitochondrial β-oxidation of long-chain fatty acids, and was reported to have activity in vitro extending from C6- to C20-CoA, with lauroyl-CoA (C12-CoA) being the preferred substrate [8]. The first indication that LCAD may not be an essential contributor to LCFA came when it was revealed that all patients with presumed LCAD deficiency had normal levels of LCAD, and no mutations in the LCAD cDNA [10,11]. Rather, they proved to have VLCAD deficiency. Further in vivo studies suggest that LCAD is specific for branched-chain fatty acid oxidation as it shows significant activity towards 2-methyldecanoyl-CoA and 2-methyl-palmitoyl-CoA in rat liver [12], and exclusively uses as substrate 2,6-dimethylheptanoyl-CoA, a metabolite of pristanic acid, the α-oxidation product of phytanic acid [13].
Since human LCAD deficiency is yet to be identified, and LCAD is reported to be specific for 2,6-dimethylheptanoyl-CoA, the role of LCAD in normal muscle physiology remains unclear. Rodents highly express LCAD in most tissues including skeletal muscle [14,15], and mutation models of LCAD deficiency mimic phenotypes of human VLCAD and MCAD deficiency, resulting in disruption of muscle function with myopathy and/or cardiomyopathy [14,16,17]. Both MCAD and VLCAD are highly expressed in human skeletal muscle, and exercise training increases the expression of these enzymes [18]. In contrast, LCAD has been reported to be expressed at much lower level in human tissues (fibroblasts, liver and heart) [15], calling into question its role in generating energy under physiologic stress [19]. Here we investigate mRNA expression, protein content, and enzyme activity of LCAD in human skeletal muscle. Given that LCAD localizes to the mitochondria, we also examined enriched mitochondrial samples for LCAD protein content, and enzyme activity.
Material and Methods
Subjects and samples
The study was approved by the McMaster University Hamilton Health Sciences Human Research Ethics Board and conformed to the Declaration of Helsinki guidelines. Healthy athletic men (N=12) and women (N=11) participated in this study. Subject characteristics: Age 20.2 ± 1.0 y, weight 75.2 ± 4.4 kg, body fat 24.4 ± 2.2%, fat free mass 54.7 ± 3.4 kg, VO2peak per fat free mass 57.7 ± 2.9 (mL/kgFFM/min). Subjects were asked to refrain from exercise for 48 hours prior to muscle biopsies. All subjects kept a diet record, and consumed a controlled breakfast (Ensure® Plus, and water adlib) two hours prior to biopsy. All subjects gave informed written consent prior to participation.
Human liver homogenate (ab29889) and human heart homogenate (ab29431) were purchased from Abcam Inc. (Cambridge, MA). Human liver and heart mitochondrial enriched samples were provided from MitoSciences (MitoSciences Inc., Eugene, OR).
Mouse mitochondria were isolated from the quadriceps muscles (includes vastus lateralis) of male wild-type C57BL/6J (breed in-house McMaster University Medical Centre Animal Facilities). Animal care followed strict guidelines put forth by Canadian Council of Animal Care and McMaster University Animal Research Ethics Board.
Preparation of RNA
Fifty mg of human skeletal muscle from the vastus lateralis of men and women was used to isolate total RNA using an Ambion mirVana™ isolation kit (Ambion Inc., Austin, TX #AM1561). In brief, muscle tissue was homogenized in Lysis/Binding buffer in a glass homogenizer. The RNA was extracted organically using Acid-Phenol:Chloroform and ethanol precipitation. Final isolation was done using provided filter cartridges. RNA was eluted in nuclease-free water and quantity and quality of RNA was assessed using a NanoDrop Spectrophotometer. Measurements were done in duplicate and had an average coefficient of variation (CV) of <10%. The average purity (OD260/OD280) of the samples was > 1.8.
RNA was synthesized into cDNA using Applied Biosystems High Capacity cDNA Reverse Transcription Kit (Applied Biosystems Inc., Foster City, CA Cat#4368814) and a BioRad iCycler iQ® real-time PCR detection system (Bio-Rad Laboratories, Hercules, CA) as per manufactures instructions.
TaqMan® real-time RT-PCR
Gene content was quantified using 7300 Real-time PCR System (Applied Biosystems Inc., Foster City, CA) and SYBR® Green chemistry (PerfeCTa SYBR® Green Supermix, ROX, Quanta BioSciences, Gaithersburg, MD) as previously described [20], with the exception that cDNA was not diluted in order to be able to amplify LCAD. Specific primers to each target gene were designed based on the cDNA sequence in GenBank with MIT primer 3 designer software. Specificity was checked using Blast, and RT-PCR dissociation curves. MCAD; forward 5′-TGCCAGAGAGGAAATCATCC-3′, reverse 5′-TCTCGGACCCTTGAACCAAA-3′, LCAD; forward 5′-CCCAGGATACCGCA GAACTA-3′, reverse 5′-GAAGGTGTCCTTTCCGACAA-3′, VLCAD; forward 5′-GTGGCCGCTTTCTGTCTAAC-3′, reverse 5′-CCTTCGTTCGAAACCTAGTC-3′. All samples were run in duplicate on a 96-well plate. Each target gene was run in parallel with human 2-microglobulin (β2-M) as an internal standard with RNA- and RT-negative controls.
Muscle homogenate preparation
Thirty mg of skeletal muscle was isolated from the vastus lateralis of men and women. Muscle tissue was homogenized in a phosphate lysis buffer; 50 mM K2HPO4, 1 mM EDTA, pH7.4, 0.1 mM DDT, PhosSTOP (Roach Diagnostics, Mannheim, Germany), Protease inhibitor cocktail tablets (Roach). Protein concentrations were calculated by Bradford assay (Bio-Rad). Equal concentrations of muscle homogenate from each subject were pooled to give an accurate account of total human skeletal muscle LCAD content. Human liver homogenate (ab29889) and human heart homogenate (ab29431) were purchased from Abcam Inc. (Cambridge, MA). Quality and quantity of protein was confirmed by ponseau staining of the membrane after transfer.
Muscle mitochondrial preparation
Three subjects volunteered to give extra muscle for mitochondrial enriched samples. Muscle biopsies were isolated from the vastus lateralis muscle. Human isolated liver mitochondria and human isolated heart mitochondria were acquired from MitoSciences (MitoSciences Inc., Eugene, OR). Mouse mitochondria were isolated from the quadriceps muscles of a male wild-type C57BL/6J (breed in-house McMaster University Medical Centre Animal Facilities).
Mitochondrial isolation protocol was as follows: Fresh muscle was rinsed in ice cold wash buffer (PBS plus 10 mM EDTA, pH7.4), and 100 mg of wet weight muscle was weighed out and added to 2 mls of homogenization buffer A (67 mM Sucrose, 50 mM Tris, 50 mM KCl, 10 mM EDTA, 0.2% BSA, pH7.4). Muscle was minced using a Polytron, then transferred to a Dounce homogenizer and homogenized. Homogenate was centrifuged at 700 x g for 15 min at 4°C. Supernatant was transferred to a fresh tube and centrifuged at 12,000 x g for 20 min at 4°C. The pellet was washed once in 1.5 ml buffer B (250mMSucrose, 3 mM EGTA, 10 mM Tris, pH7.4) and re-suspended in 75 μl of buffer B, flash frozen and stored at −80°C.
Immunoblotting analysis
Protein homogenates were boiled in Laemmli buffer, resolved by SDS-PAGE, and transferred to PVDF membrane. Protein presence was detected with rabbit antiserum made to recombinant human LCAD antibody [21] or human MCAD, and secondary anti-rabbit antibody conjugated to horseradish peroxidase (Amersham Bioscience, UK). Antibody binding was detected using Millipore Immobilon™ Western chemiluminescent HRP substrate (Millipore Corp., Billerica, MA). Scanned films were analyzed using ImageJ 1.40 software (Wayne Rasband National Institute of Health, USA) for quantification of protein. All membranes were stained with ponseau to control for quality and quantity of protein as well as proper transfer of the proteins from the gel onto the membrane.
Enzyme activity measurements
Acyl-CoA dehydrogenase activity was measured with the anaerobic ETF fluorescence reduction assay using an LS50B fluorescence spectrophotometer from Perkin Elmer (Norwalk, CT) with a heated cuvette block set to 32°C as previously described [22]. Thirty one micrograms of total mitochondrial extract were used in the octanoyl-, palmitoyl-CoA assays (Sigma). The final substrate concentration for octanoyl-CoA and palmitoyl-CoA was 5 μM. Double (62 μg) the amount of total mitochondrial extract was used for LCAD assay using 50 μM of 2,6-dimethylheptanoyl-CoA (a gift from Dr. Charles Hoppel, Case Western Reserve University, Cleveland, OH), as substrate. One unit of activity is defined as the amount of enzyme necessary to completely reduce 1 μmol of ETF in 1 minute.
Results
One hundred nanograms of purified RNA were used to synthesis cDNA. When cDNA was diluted 1:10 for RT-PCR amplification LCAD was undetectable. RT-PCR amplification of undiluted cDNA yielded a small but detectable amount of LCAD (0.08±0.009) in human skeletal muscle, which was less abundant than VLCAD (7.3±0.5) and less abundant than MCAD (2.4±0.2) (P≤0.001) (Fig. 1).
Fig. 1.

mRNA expression of LCAD is barely detectable in human skeletal muscle. Real-time RT-PCR expression of VLCAD, LCAD and MCAD genes in young moderately active men and women, normalized to β2-microglobulin. *LCAD is significantly different from VLCAD and MCAD (P<0.0001), & MCAD is significantly different from VLCAD and LCAD (P<0.0001). N=23, data are means ± SEM.
Comparison of LCAD protein content in human skeletal muscle using a human recombinant LCAD antibody was challenging. Initial examination of 10, 20, 40 and 80 μg of human muscle homogenate protein yielded no observable bands (data not shown). We compared 20 μg of human skeletal muscle to 20 μg of human liver and heart homogenate, as Chegary et., al., (2009) previously showed that this antibody detected LCAD in 22 μg of human heart and liver homogenate [15]. We were able to detect LCAD in 20μg of liver homogenate but unable o detect LCAD in skeletal muscle or heart homogenate (Fig. 2). Comparison of mouse muscle homogenate and isolated mitochondrial protein from mouse muscle yielded an abundant band at the predicted 43 KDa (Fig. 2), as this antibody has been shown previously to cross react with mouse LCAD [14,15]. MCAD was abundantly detected in 10 μg of the same human muscle homogenate samples (Fig. 2B). LCAD was detectable only when a large amount (30 μg) of enriched mitochondrial sample was loaded on the gel (Fig. 3).
Fig. 2.
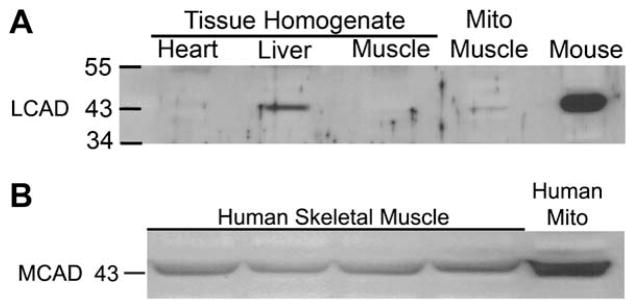
LCAD protein content in tissue homogenate. A: LCAD protein content in 20 μg of tissue homogenate from human heart (N=1), liver (N=1), and skeletal muscle (all subjects pooled) (lanes 1–3). Human and mouse skeletal muscle mitochondria are shown (lanes 4&5)(N=1) to give an appreciation of LCAD abundance in mitochondrial enriched samples. B: MCAD protein content was abundantly expressed in 10 μg human muscle homogenate (lanes 1–4; N=4 shown, representative of all 23 subjects), and an enriched mitochondrial sample (lane 5).
Fig. 3.

LCAD protein content in enriched mitochondrial samples. LCAD protein is detectable in 10 μg of human isolated mitochondria from heart and liver homogenate (lanes 1&2). Thirty μg of isolated mitochondria from human skeletal muscle was required to quantify LCAD expression (lane 3). Thirty μg of isolated mitochondria from mouse quadriceps muscle was loaded as a comparison (lane 4). Graph: The quantified bands were normalized to ponseau (total protein). LCAD expression in human mitochondrial enriched samples was most abundant in human liver. Human skeletal muscle had the least amount of LCAD protein which was 16 times less that of mouse skeletal muscle. Comparative analysis, N=1 for all samples.
Activity assays using a specific and sensitive fluorescent technique, on 62 μg of mitochondrial extracts from control human muscle samples, confirmed undetectable levels of activity with the LCAD specific substrate 2,6-dimetheylheptanoyl-CoA as compared to readily measurable activity with palmitoyl-CoA (VLCAD) and octanoyl-CoA (MCAD) (Fig. 4).
Fig. 4.

Enzyme activity of enriched mitochondrial samples from human vastus lateralis muscle. LCAD activity was undetectable in human enriched mitochondrial samples. VLCAD and MCAD activity was abundant. Thirty one micrograms of total mitochondrial extract was used to measure acyl-CoA dehydrogenase activity by ETF fluorescence reduction assay. Palmitoyl-CoA (C16:0-CoA) is specific for VLCAD activity, 2,6-dimethylheptanoyl-CoA (2mC7-CoA) is specific for LCAD activity and octanoyl-CoA is specific for MCAD (C8-CoA) activity. N=3, data are means ± SEM.
Discussion
In order to fully understand the process of long chain FAO in skeletal muscle we set out to examine LCAD expression and function in human skeletal muscle. A previous study has shown that MCAD and VLCAD are highly expressed in human skeletal muscle and exercise training increases the expression of these enzymes [18], although to our knowledge no reports on LCAD in human muscle physiology exist. Our data showed that LCAD is so lowly expressed in human skeletal muscle that it cannot be readily detected in whole muscle homogenate. Enzyme activity of LCAD in isolated skeletal muscle mitochondria also yielded no detectable activity. We were able to detect LCAD protein only when large amounts of isolated skeletal muscle mitochondria were overloaded on the gels, confirming that LCAD is expressed in skeletal muscle only at low levels. The protein studies are consistent with the observation of low mRNA expression of LCAD in skeletal muscle.
The functional importance of LCAD in human FAO remains unclear. Until 1992 it was thought that all LCFA’s were oxidized via LCAD, and deficiency of LCAD was originally reported in patients with skeletal myopathy and/or cardiomyopathy [16]. In 1992 Izai and coworkers identified VLCAD [8], which led to the recognition that VLCAD is the main, if not exclusive, enzyme involved in palmitate oxidation [12]. Only then was it discovered that the patients originally thought to be LCAD deficient had normal LCAD protein and mRNA and were instead, VLCAD deficient [10,11,23]. Subsequently, LCAD was found to oxidize branched chain acyl-CoA substrates [12,21], although this reaction is of unknown physiological relevance. Currently there are no reported cases of human LCAD deficiency [11,24].
To help elucidate the relative roles of LCAD and VLCAD in metabolism, knockout mouse models have been developed; however, studies using these animals are not strictly representative of human FAO disorders as both enzymes share a role in the oxidation of LCFA’s in mice. Specifically, LCAD−/− mice have a more sever phenotype than VLCAD−/− mice, and are more similar to human VLCAD deficiency [14,17]. Enzyme assays reveal, that VLCAD and LCAD can functionally compensate for loss of the other in mice [15,17]. This compensation leads to a less sever biochemical derangement in either mutant mouse phenotype compared with human VLCAD deficiency, where oleic acid oxidation is completely ablated causing sever disease [15]. Recently, Chegary et. al., compared EST counts in mice to those in humans and found that mice have an equal abundance of EST transcripts for LCAD and VLCAD in all tissues; whereas, VLCAD EST transcripts in humans were approximately double that of the mouse in most tissues, and LCAD EST transcripts were virtually undetectable in humans [15]. Here we compared the content of LCAD to MCAD and VLCAD. MCAD and VLCAD mRNA were easily amplified, and only small amounts of muscle homogenate protein were required to visualize MCAD and VLCAD by Western blot or demonstrate enzyme activity. In addition, LCAD protein and activity were readily detectable in human whole liver tissue homogenate as previously reported [15,25]. In contrast, LCAD mRNA, antigen, and enzymatic activity were low in human muscle. As a control, we were readily able to detect LCAD message and protein in mice as previously reported [14]. Lastly, we could not detect LCAD in human heart tissue as previously reported [25], although we were able to detect LCAD antigen in isolated mitochondria from human heart tissue when gels were overloaded. Taken together, these data indicate that LCAD does not play a significant role in LCFA oxidation in human skeletal muscle.
Although the protein coding regions of LCAD in mouse and human are well conserved [26], database searches reveal that there are significant differences in the LCAD promoter region [26,27], and the number of microRNA’s estimated to regulate LCAD expression (most likely through inhibition) is significantly higher in human (18 microRNAs) compared with mouse (2 microRNAs) (http://microrna.sanger.ac.uk). The differences in the promoter region and microRNA expression are consistent with differences in gene expression, regulation, and apparent function of LCAD in mice and humans and suggest that observed genetic alterations occurred after evolutionary divergence of rodents and primates.
Conclusions
In conclusion, LCAD is in such low abundance in skeletal and cardiac muscle that it is unlikely to play a significant role in the generation of energy through fatty acid oxidation in these tissues at rest. Rather, its physiologic function remains to be elucidated.
Acknowledgments
We thank the subjects for participating in this study. This study was primarily funded through Natural Sciences and Engineering Research Council of Canada grant held by MT. AM was supported by a Natural Sciences and Engineering Research Council of Canada Doctoral Research Award. JV was funded in part by USA NIH award number R01DK78775. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Contributor Information
Amy C. Maher, Email: maherac@mcmaster.ca.
Al-Walid Mohsen, Email: mohsaw@chp.edu.
Jerry Vockley, Email: vockleyg@upmc.edu.
Mark A. Tarnopolsky, Email: tarnopol@mcmaster.ca.
References
- 1.Janssen I, Heymsfield SB, Baumgartner RN, Ross R. Estimation of skeletal muscle mass by bioelectrical impedance analysis. J Appl Physiol. 2000;89:465–471. doi: 10.1152/jappl.2000.89.2.465. [DOI] [PubMed] [Google Scholar]
- 2.Friedlander AL, Jacobs KA, Fattor JA, Horning MA, Hagobian TA, Bauer TA, Wolfel EE, Brooks GA. Contributions of working muscle to whole body lipid metabolism are altered by exercise intensity and training. Am J Physiol Endocrinol Metab. 2007;292:E107–E116. doi: 10.1152/ajpendo.00148.2006. [DOI] [PubMed] [Google Scholar]
- 3.Jacobs KA, Krauss RM, Fattor JA, Horning MA, Friedlander AL, Bauer TA, Hagobian TA, Wolfel EE, Brooks GA. Endurance training has little effect on active muscle free fatty acid, Lipoprotein cholesterol, or triglyceride net balances. Am J Physiol Endocrinol Metab. 2006;291:E656–E665. doi: 10.1152/ajpendo.00020.2006. [DOI] [PubMed] [Google Scholar]
- 4.van Loon LJ, Thomason-Hughes M, Constantin-Teodosiu D, Koopman R, Greenhaff PL, Hardie DG, Keizer HA, Saris WH, Wagenmakers AJ. Inhibition of adipose tissue lipolysis increases intramuscular lipid and glycogen use in vivo in humans. Am J Physiol Endocrinol Metab. 2005;289:E482–E493. doi: 10.1152/ajpendo.00092.2005. [DOI] [PubMed] [Google Scholar]
- 5.Havel RJ, Naimark A, Borchgrevink CF. Turnover rate and oxidation of free fatty acids of blood plasma in man during exercise: studies during continuous infusion of palmitate-1-C14. J Clin Invest. 1963;42:1054–1063. doi: 10.1172/JCI104791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Havel RJ, Carlson LA, Ekelund LG, Holmgren A. Turnover Rate and Oxidation of Different Free Fatty Acids in Man during Exercise. J Appl Physiol. 1964;19:613–618. doi: 10.1152/jappl.1964.19.4.613. [DOI] [PubMed] [Google Scholar]
- 7.DeLany JP, Windhauser MM, Champagne CM, Bray GA. Differential oxidation of individual dietary fatty acids in humans. Am J Clin Nutr. 2000;72:905–911. doi: 10.1093/ajcn/72.4.905. [DOI] [PubMed] [Google Scholar]
- 8.Izai K, Uchida Y, Orii T, Yamamoto S, Hashimoto T. Novel fatty acid beta-oxidation enzymes in rat liver mitochondria, I. Purification and properties of very-long-chain acyl-coenzyme A dehydrogenase. J Biol Chem. 1992;267:1027–1033. [PubMed] [Google Scholar]
- 9.Ensenauer R, He M, Willard JM, Goetzman ES, Corydon TJ, Vandahl BB, Mohsen AW, Isaya G, Vockley J. Human acyl-CoA dehydrogenase-9 plays a novel role in the mitochondrial beta-oxidation of unsaturated fatty acids. J Biol Chem. 2005;280:32309–32316. doi: 10.1074/jbc.M504460200. [DOI] [PubMed] [Google Scholar]
- 10.Indo Y, Coates PM, Hale DE, Tanaka K. Immunochemical characterization of variant long-chain acyl-CoA dehydrogenase in cultured fibroblasts from nine patients with long-chain acyl-CoA dehydrogenase deficiency. Pediatr Res. 1991;30:211–215. doi: 10.1203/00006450-199109000-00001. [DOI] [PubMed] [Google Scholar]
- 11.Yamaguchi S, Indo Y, Coates PM, Hashimoto T, Tanaka K. Identification of very-long-chain acyl-CoA dehydrogenase deficiency in three patients previously diagnosed with long-chain acyl-CoA dehydrogenase deficiency. Pediatr Res. 1993;34:111–113. doi: 10.1203/00006450-199307000-00025. [DOI] [PubMed] [Google Scholar]
- 12.Mao LF, Chu C, Luo MJ, Simon A, Abbas AS, Schulz H. Mitochondrial beta-oxidation of 2-methyl fatty acids in rat liver. Arch Biochem Biophys. 1995;321:221–228. doi: 10.1006/abbi.1995.1389. [DOI] [PubMed] [Google Scholar]
- 13.Wanders RJ, Denis S, Ruiter JP, IJL, Dacremont G. 2,6-Dimethylheptanoyl-CoA is a specific substrate for long-chain acyl-CoA dehydrogenase (LCAD): evidence for a major role of LCAD in branched-chain fatty acid oxidation. Biochim Biophys Acta. 1998;1393:35–40. doi: 10.1016/s0005-2760(98)00053-8. [DOI] [PubMed] [Google Scholar]
- 14.Kurtz DM, Rinaldo P, Rhead WJ, Tian L, Millington DS, Vockley J, Hamm DA, Brix AE, Lindsey JR, Pinkert CA, O’Brien WE, Wood PA. Targeted disruption of mouse long-chain acyl-CoA dehydrogenase gene reveals crucial roles for fatty acid oxidation. Proc Natl Acad Sci U S A. 1998;95:15592–15597. doi: 10.1073/pnas.95.26.15592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chegary M, Brinke HT, Ruiter JP, Wijburg FA, Stoll MS, Minkler PE, Van Weeghel M, Schulz H, Hoppel CL, Wanders RJ, Houten SM. Mitochondrial long chain fatty acid beta-oxidation in man and mouse. Biochim Biophys Acta. 2009;1791:806–815. doi: 10.1016/j.bbalip.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wanders RJ, Vreken P, Den Boer ME, Wijburg FA, van Gennip AH, IJL Disorders of mitochondrial fatty acyl-CoA beta-oxidation. J Inherit Metab Dis. 1999;22:442–487. doi: 10.1023/a:1005504223140. [DOI] [PubMed] [Google Scholar]
- 17.Cox KB, Hamm DA, Millington DS, Matern D, Vockley J, Rinaldo P, Pinkert CA, Rhead WJ, Lindsey JR, Wood PA. Gestational, Pathologic and biochemical differences between very long-chain acyl-CoA dehydrogenase deficiency and long-chain acyl-CoA dehydrogenase deficiency in the mouse. Hum Mol Genet. 2001;10:2069–2077. doi: 10.1093/hmg/10.19.2069. [DOI] [PubMed] [Google Scholar]
- 18.Horowitz JF, Leone TC, Feng W, Kelly DP, Klein S. Effect of endurance training on lipid metabolism in women: a potential role for PPARalpha in the metabolic response to training. Am J Physiol Endocrinol Metab. 2000;279:E348–355. doi: 10.1152/ajpendo.2000.279.2.E348. [DOI] [PubMed] [Google Scholar]
- 19.He M, Rutledge SL, Kelly DR, Palmer CA, Murdoch G, Majumder N, Nicholls RD, Pei Z, Watkins PA, Vockley J. A new genetic disorder in mitochondrial fatty acid beta-oxidation: ACAD9 deficiency. Am J Hum Genet. 2007;81:87–103. doi: 10.1086/519219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mahoney DJ, Parise G, Melov S, Safdar A, Tarnopolsky MA. Analysis of global mRNA expression in human skeletal muscle during recovery from endurance exercise. Faseb J. 2005 doi: 10.1096/fj.04-3149fje. [DOI] [PubMed] [Google Scholar]
- 21.Battaile KP, McBurney M, Van Veldhoven PP, Vockley J. Human long chain, very long chain and medium chain acyl-CoA dehydrogenases are specific for the S-enantiomer of 2- methylpentadecanoyl-CoA. Biochim Biophys Acta. 1998;1390:333–338. doi: 10.1016/s0005-2760(97)00185-9. [DOI] [PubMed] [Google Scholar]
- 22.Vockley J, Mohsenal WA, Binzak B, Willard J, Fauq A. Mammalian branched-chain acyl-CoA dehydrogenases: molecular cloning and characterization of recombinant enzymes. Methods Enzymol. 2000;324:241–258. doi: 10.1016/s0076-6879(00)24236-5. [DOI] [PubMed] [Google Scholar]
- 23.Bertrand C, Largilliere C, Zabot MT, Mathieu M, Vianey-Saban C. Very long chain acyl-CoA dehydrogenase deficiency: identification of a new inborn error of mitochondrial fatty acid oxidation in fibroblasts. Biochim Biophys Acta. 1993;1180:327–329. doi: 10.1016/0925-4439(93)90058-9. [DOI] [PubMed] [Google Scholar]
- 24.Andresen BS, Olpin S, Poorthuis BJ, Scholte HR, Vianey-Saban C, Wanders R, Ijlst L, Morris A, Pourfarzam M, Bartlett K, Baumgartner ER, DeKlerk JB, Schroeder LD, Corydon TJ, Lund H, Winter V, Bross P, Bolund L, Gregersen N. Clear correlation of genotype with disease phenotype in very-long-chain acyl-CoA dehydrogenase deficiency. Am J Hum Genet. 1999;64:479–494. doi: 10.1086/302261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andresen BS, Bross P, Vianey-Saban C, Divry P, Zabot MT, Roe CR, Nada MA, Byskov A, Kruse TA, Neve S, Kristiansen K, Knudsen I, Corydon MJ, Gregersen N. Cloning and characterization of human very-long-chain acyl-CoA dehydrogenase cDNA, Chromosomal assignment of the gene and identification in four patients of nine different mutations within the VLCAD gene. Hum Mol Genet. 1996;5:461–472. doi: 10.1093/hmg/5.4.461. [DOI] [PubMed] [Google Scholar]
- 26.Kurtz DM, Tolwani RJ, Wood PA. Structural characterization of the mouse long-chain acyl-CoA dehydrogenase gene and 5′ regulatory region. Mamm Genome. 1998;9:361–365. doi: 10.1007/s003359900770. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Z, Zhou Y, Mendelsohn NJ, Bauer GS, Strauss AW. Regulation of the human long chain acyl-CoA dehydrogenase gene by nuclear hormone receptor transcription factors. Biochim Biophys Acta. 1997;1350:53–64. doi: 10.1016/s0167-4781(96)00141-8. [DOI] [PubMed] [Google Scholar]