

Abstract
Variants in the growth factor receptor-bound protein 10 (GRB10) gene were in a GWAS meta-analysis associated with reduced glucose-stimulated insulin secretion and increased risk of type 2 diabetes (T2D) if inherited from the father, but inexplicably reduced fasting glucose when inherited from the mother. GRB10 is a negative regulator of insulin signaling and imprinted in a parent-of-origin fashion in different tissues. GRB10 knock-down in human pancreatic islets showed reduced insulin and glucagon secretion, which together with changes in insulin sensitivity may explain the paradoxical reduction of glucose despite a decrease in insulin secretion. Together, these findings suggest that tissue-specific methylation and possibly imprinting of GRB10 can influence glucose metabolism and contribute to T2D pathogenesis. The data also emphasize the need in genetic studies to consider whether risk alleles are inherited from the mother or the father.
Author Summary
In this paper, we report the first large genome-wide association study in man for glucose-stimulated insulin secretion (GSIS) indices during an oral glucose tolerance test. We identify seven genetic loci and provide effects on GSIS for all previously reported glycemic traits and obesity genetic loci in a large-scale sample. We observe paradoxical effects of genetic variants in the growth factor receptor-bound protein 10 (GRB10) gene yielding both reduced GSIS and reduced fasting plasma glucose concentrations, specifically showing a parent-of-origin effect of GRB10 on lower fasting plasma glucose and enhanced insulin sensitivity for maternal and elevated glucose and decreased insulin sensitivity for paternal transmissions of the risk allele. We also observe tissue-specific differences in DNA methylation and allelic imbalance in expression of GRB10 in human pancreatic islets. We further disrupt GRB10 by shRNA in human islets, showing reduction of both insulin and glucagon expression and secretion. In conclusion, we provide evidence for complex regulation of GRB10 in human islets. Our data suggest that tissue-specific methylation and imprinting of GRB10 can influence glucose metabolism and contribute to T2D pathogenesis. The data also emphasize the need in genetic studies to consider whether risk alleles are inherited from the mother or the father.
Introduction
Type 2 diabetes (T2D) is a multifactorial polygenic disease, in which genes interact with environmental and genetic factors to unmask the disease. To date, genome-wide association studies (GWAS) have identified more than 65 variants increasing risk of T2D [1]–[8]. Many of the identified variants seem to influence the capacity of β-cells to cope with increased insulin demands imposed by insulin resistance [9], [10]. Despite this, no GWAS to date has explored the extent to which common genetic variants influence dynamic measures of insulin secretion and action. Therefore, we have here performed the first large-scale meta-analysis for glucose-stimulated insulin secretion (GSIS) during an oral glucose tolerance test (OGTT). Association analyses included a GWAS-based discovery stage and a replication stage using a custom-designed iSelect CardioMetabochip array containing 93,896 SNPs overlapping with the discovery GWAS imputed from the HapMap2 reference panel in up to 10,831 non-diabetic individuals. We identified variants in the growth factor receptor-bound protein 10 (GRB10) gene (NCBI Gene ID: 2887) to be associated with impaired β-cell function at a genome-wide significant level (p<5×10−8). Since GRB10 has been shown to have parent-of-origin specific effects on expression in various tissues [11], [12], we investigated the transmission patterns of the risk alleles and their effects on insulin and glucose levels during an OGTT, risk for T2D, expression of GRB10 and methylation status in human pancreatic islets, as well as evaluated the effects on islet function through disruption of GRB10 in human pancreatic islets.
Results
Meta-Analysis of GWAS and CardioMetabochip Studies for Insulin Secretion and Action during an OGTT
We conducted a meta-analysis for dynamic measures of insulin response to glucose during an OGTT using GWAS, CardioMetabochip and de novo genotyping. The combined analysis in up to 26,037 non-diabetic individuals provided genome-wide significant association (p<5×10−8) with insulin secretion measured as corrected insulin response (CIR) to glucose at 30 min during an OGTT for the locus within the GRB10 gene (lead SNP rs933360 located in intron 2) (Figure 1A, B), and at 7 previously reported T2D and glycemic trait variants, including MTNR1B, HHEX/IDE/KIF11, CDKAL1, GIPR/QPCTL, C2CD4A (NLF1), GCK and ANK1 (Table 1, Table S2A, Figure S1, S2). These associations remained virtually unchanged when CIR was adjusted for insulin sensitivity (disposition index) (Table S2A). In addition, nominal (p<0.05) association with reduced insulin response to glucose (CIR) during the OGTT was seen for the risk alleles in 24 out of 65 reported T2D loci [4], as well as for 20 glucose and 7 insulin loci out of 53 reported being associated with these traits [6]. Notably, the risk alleles in 5 of them (ANKRD55, GRB14, PPP1R3B, IRS1 and ARAP1 (fasting glucose variant)) were associated with higher insulin response to glucose (p<0.05) (Table S2B).
Figure 1. GWAS plots identifying GRB10 rs933360.

Genome-wide quantile-quantile (Q-Q) (A) and Manhattan (B) plots for CIR in the meta-analysis. Regional plots of identified GRB10 genome-wide significant association for CIR in men (C) and women (D). Directly genotyped and/or imputed SNPs of GRB10 are plotted with their meta-analysis p-values (as −log10 values) as a function of genomic position (NCBI Build 36). In each panel, the strongest associated SNP is represented by a purple diamond (with meta-analysis p-value). Estimated recombination rates (taken from HapMap) are plotted to reflect the local linkage disequilibrium structure around the associated SNPs and their correlated proxies (according to a blue to red scale from r2 = 0 to 1, based on pairwise r2 values from HapMap CEU). Gene annotations were taken from the University of California, Santa Cruz, genome browser, as implemented in LocusZoom [41].
Table 1. SNPs associated with primary insulin secretion traits at genome-wide significance levels.
| SNP | Nearest gene | N | Alleles (effect/other) | Freq (effect allele) | CIR | AUCIns/AUCGluc | ||||
| Effect | SE | p | Effect | SE | p | |||||
| Identified in the present study | ||||||||||
| rs933360 | GRB10 | 26,037 | A/G | 0.62 | −0.051 | 0.0086 | 3.14×10−9 | −0.043 | 0.0087 | 8.13×10−7 |
| Previously reported loci | ||||||||||
| rs10830963 | MTNR1B | 10,651 | G/C | 0.69 | −0.17 | 0.016 | 6.71×10−28 | −0.057 | 0.017 | 0.00062 |
| rs7923866 | HHEX/IDE | 10,418 | C/T | 0.62 | −0.12 | 0.015 | 4.16×10−16 | −0.093 | 0.015 | 1.37×10−9 |
| rs7756992 | CDKAL1 | 10,829 | G/A | 0.3 | −0.11 | 0.015 | 3.07×10−13 | −0.067 | 0.016 | 4.16×10−5 |
| rs11671664 | GIPR | 9,453 | A/G | 0.11 | −0.17 | 0.025 | 2.64×10−11 | −0.17 | 0.026 | 2.63×10−11 |
| rs4502156 | C2CD4A (NLF1/VPS13C) | 10,787 | T/C | 0.52 | −0.092 | 0.014 | 1.14×10−10 | 0.0035 | 0.015 | 0.82 |
| rs3757840 | GCK | 10,322 | T/G | 0.45 | −0.090 | 0.015 | 1.34×10−9 | −0.083 | 0.016 | 1.30×10−7 |
| rs12549902 | ANK1 | 14,834 | A/G | 0.58 | −0.060 | 0.010 | 1.01×10−8 | −0.036 | 0.011 | 0.00083 |
The results are from the meta-analyses of the discovery GWAS, CardioMetabochip and de novo genotyping. Results are reported for the directly genotyped and imputed SNPs tested for association with insulin secretion measured as CIR and AUCIns/AUCGluc (trait abbreviations are listed in the Methods “Phenotype definition” section). Freq denotes the allele frequency of the insulin secretion-reducing allele. N = sample size. Since the index SNP rs933360 (A/G) from the discovery GWAS was not present on the CardioMetabochip platform, a variant (rs6943153 (C/T)) in strong LD with the former (r2 = 0.82) was used as a proxy SNP for the meta-analyses.
In line with a previous report [6], we confirmed the association of the index GRB10 SNP rs933360 with fasting glucose levels (N = 24,608, β = −0.016, p = 0.007) (Figure S3). However, we did not observe a significant effect of SNP rs2237457, previously associated with glucose concentrations and risk for T2D in an Amish population [13].
Since GRB10 is differentially expressed when transmitted from mothers and fathers, we explored whether the GRB10 variant would have sex-specific effects on insulin and glucose levels. The sex-stratified analysis showed a greater insulin-reducing effect of the GRB10 variant in women (CIR: N = 6,202; β = −0.110±0.019; p = 1.52×10−8) than in men (CIR: N = 15,192; β = −0.038±0.012; p = 0.0012; sex heterogeneity p = 0.0016) (Figure 1C, D).
Complex Pattern of Genetic Inheritance for Variants in the GRB10 Gene: Evidence for Parent-of-Origin Effects
Given the complex parent-of-origin imprinting pattern described for GRB10, we next turned to families to explore in detail the inheritance patterns of identified variants and their potential effect on risk of T2D and insulin/glucose levels. Using a cohort of 2,322 parents-offspring trios with 4,182 individuals from Finland and Sweden, we first performed a transmission disequilibrium test, taking into account the parental phenotype being either T2D or not, to increase power (parenTDT) [14]. We also used gene-dropping permutation to control for stratification and the dependence of related individuals [14], or restricting the analysis to independent trios, including only the youngest affected offspring (N = 1,055; 182 with T2D) and oldest unaffected offspring (N = 1,019; 873 unaffected). We observed an increased transmission of the A-allele of rs933360 from parents to diabetic offspring (p = 0.0063) (Table S3A-I), particularly from diabetic fathers (p = 0.049) (Table S3A-V), and the G-allele was preferentially transmitted from non-diabetic parents to non-diabetic offspring (p = 0.026) (Table S3A-II). In accordance with these findings, when simply counting transmission of the A- and G-alleles, we observed an increased transmission of the major A-allele of rs933360 from a diabetic parent to a diabetic offspring (Chi-square p = 0.017) (Table S3A-V). This effect was even stronger when we relaxed the definition of hyperglycemia to include IFG and IGT in addition to T2D (p = 0.006) (Table S3A-VI).
We observed a similar pattern of an increased transmission of the T-allele of rs6943153 (LD with rs933360; D′ = 0.99, r2 = 0.82) to a hyperglycemic offspring (IFG/IGT/T2D) (p = 0.0045) (Table S3A-III). This latter association was confirmed using the Family Based Association Test (FBAT) [15] (p = 0.035), which can accommodate any type of genetic model and family construction. Consistent with previous findings [6], we confirmed the association of the SNP rs6943153 with fasting glucose levels (1,083 nuclear families, p = 0.02) (Table S3A-VII).
To explore whether GRB10 rs933360 would show a stronger effect on insulin secretion when inherited from either parent, we examined its effect on GSIS in 3,117 non-diabetic individuals from parents-offspring trios from Finland and Sweden [16] and USA [13]. In these families, the maternally transmitted A-allele of rs933360 was associated with reduced GSIS (CIR β = −0.127, p = 0.014; Ins30adjBMI β = −0.125, p = 0.005; Ins30 β = −0.112, p = 0.014; AUCIns β = −0.095, p = 0.016; AUCIns/AUCGluc β = 0.107, p = 0.005) (Figure 2A, B, Table S3B). No significant effect was observed for the paternally transmitted A-allele on GSIS. Surprisingly, the maternally transmitted A-allele was associated with reduced rather than elevated fasting glucose levels (β = −0.139, p = 0.0009). In contrast, the paternally transmitted A-allele was associated with elevated glucose levels (β = 0.102, p = 0.002) (Figure 2C, D, Table S3B). Thereby, the A-allele of rs933360 exerted virtually opposite effects on glucose metabolism if transmitted from the father than the mother. It is very likely that the association with risk or protection from T2D would be missed or diluted in any traditional association study, which does not take the transmission pattern into account.
Figure 2. Parent-of-origin effect of GRB10 rs933360 on insulin secretion and glucose levels.

(A) No significant effect for CIR was observed from the paternally transmitted A-allele. (B) Carriers of the maternally transmitted A-allele showed lower CIR compared to the G-allele. (C) Carriers of the paternally transmitted A-allele had elevated fasting plasma glucose levels, whereas (D) the maternally transmitted A-allele was associated with lower fasting plasma glucose levels. Fin-Swe = Trios from Finland and Sweden, Amish = Amish Family Diabetes Study, Kuopio = Kuopio Offspring Study.
In support of this, we did not observe any association between the SNP rs933360 and T2D in 16,715 non-diabetic individuals, of whom 2,637 developed T2D during a mean 25-year follow-up period (Table S4) [17]. Also, in the recent DIAGRAM+ meta-analysis, none of the evaluated GRB10 SNPs were associated with T2D [4].
A potential explanation for the paradoxical reduction in glucose levels despite reduced insulin secretion could be that the variant also enhances insulin sensitivity or reduces glucagon levels. In fact, the maternally transmitted A-allele was associated with enhanced, whereas the paternally transmitted A-allele was associated with decreased insulin sensitivity as measured by ISI during the OGTT (p<0.05 for difference between parental alleles). Although we could not observe any significant effect of rs933360 on fasting or 2 hr glucagon levels in a Finnish cohort with glucagon data available (Table S5B), we identified several GRB10 SNPs from the same haplotype block which were in weak LD with rs933360 and nominally (p<0.05) associated with fasting and 2 hr glucagon levels in the DGI GWAS (Table S6). Unfortunately, there was no glucagon data available for the trios.
Effects of GRB10 rs933360 on Gene Expression
GRB10 protein was detected in human α-, β- and δ-cells by immunofluorescence (Figure 3A). We observed strong expression of GRB10 mRNA in total human islets, with no significant difference between islets from normoglycemic and hyperglycemic individuals (Figure S4A), or between carriers of different GRB10 genotypes (Figure S4B).
Figure 3. GRB10 expression in human islets.

(A) Immunostainings demonstrating that GRB10 (green, panel A, E) is abundantly expressed in β- (panel B, D), α- (panel C, D) and δ-cells (panel F, G) in human pancreatic islets. Arrows indicate co-localization with insulin (panel D) and somatostatin (panel G). Arrowheads indicate co-localization with glucagon (panel D). Scale bar = 50 µm. (B) Schematic representation of the GRB10 gene and SNPs investigated in the present study. Grey boxes = untranslated exons. Black boxes = translated exons. (C) Examples of RT-PCR on islet cDNA (top six rows) and PCR on genomic DNA (gDNA, bottom row) from two individuals heterozygous for the reporter SNP rs1800504. The first column states the forward primer location of each PCR and a forward primer in exon 3 captures all transcripts. The peaks show the Sanger sequencing trace across rs1800504, which is underlined (A: green trace, G: black trace). Percentages indicate the contribution from the paternal allele (P.A.) (G-allele in the first case, A-allele in the second case). The paternal genotype is identified assuming complete maternal imprinting of the UN2 promoter, in line with previous findings [12]. A sequence of heterozygous genomic DNA (gDNA) is shown on the bottom for comparison (50%-50%). (D) To study if DNA methylation of GRB10 is tissue-specific, the degree of methylation was analyzed at 3 CpG sites located ∼31.7 kb downstream of rs933360 in both human pancreatic islets of 98 donors and PBL from 6 trios using EpiTYPER. The exact position of each analyzed CpG site in relation to rs933360 is given in the figure. Data are presented as mean ± SEM. * p<0.05 for difference in methylation between human islets and PBL. (E) The GRB10 mRNA levels correlated negatively with the degree of methylation at the CpG site located 31,675 bp downstream of GRB10 rs933360.
While we did not observe any correlation between the amount of GRB10 and INS (insulin) mRNA, nor between GRB10 mRNA and in vitro GSIS, there was an inverse correlation between GRB10 and GCG (glucagon) mRNA in human pancreatic islets (all donors: rho = −0.267, p = 0.017; normoglycemic: rho = −0.228, p = 0.10; hyperglycemic: rho = −0.651, p = 0.00003), suggesting that higher GRB10 expression is associated with lower glucagon (Figure S4C).
Although there was no effect of rs933360 on total GRB10 mRNA expression in human islets, we cannot exclude that the variant could influence splicing or methylation, especially as 3 different transcriptional start sites (UN1, UN1a and UN2) and tissue-specific expression have been described for the GRB10 gene (Figure 3b, S5) [12].
We tested for allelic imbalance, i.e. deviation from the expected equal expression of both alleles. For this purpose, we used the SNP rs1800504 (A→G) located in exon 3 as a reporter SNP, as it is the nearest coding variant located 16 kb from the index SNP rs933360 (D′ = 1, r2 = 0.5). This reporter SNP indicated a clear allelic imbalance with A- to G-allele ratios ranging from 35% to 75% in pancreatic islets (Table S7). We therefore examined whether the observed allelic imbalance could be related to specific transcripts arising from different promoters. Transcripts containing exon UN2 were monoallelically expressed from either the A- or G-allele, indicating imprinting of the promoter giving rise to the UN2 transcripts (pUN2) from one parent. Until now, paternally expressed (i.e. maternally imprinted) UN2 transcripts have only been observed in the brain [12]. This differential imprinting was recurring in all tissues analyzed (Figure S6). Our findings extend this expression pattern to human islets. In contrast, transcripts containing exon UN1 showed great variation (from 50% to 80%), but were mainly expressed from the other allele than those containing exon UN2 (Figure 3B, C), in line with the maternally expressed/paternally imprinted transcripts observed in placental trophoblasts [12]. It can be hypothesized that these SNPs might regulate usage of alternative promoters and thereby influence the expression of GRB10. The opposite effects of maternally and paternally inherited rs933360 allele could then be attributed to different effects of rs933360 on the promoters pUN2 and pUN1, e.g. especially if promoter preferences differ strongly between α- and β-cells.
Although allelic imbalance is an attractive model to explain differences in expression of different GRB10 transcripts, as well as the observed differences in effects of the A-allele on risk of diabetes in the offspring when transmitted from father or from mother, the above data can only point at this possibility, as we did not have enough human islets for this kind of analysis. We therefore also tested for allelic imbalance using an alternative method, i.e. by comparing data from exome and RNA sequencing. We found that another coding variant, SNP rs11555134, in the GRB10 gene was associated with allelic imbalance in 8 human pancreatic islet samples (p<0.05, Fisher's exact test).
Since GRB10 is imprinted and methylated in humans and rodents in a tissue-specific fashion [12], [18], [19], we studied whether GRB10 would be methylated in human islets or in DNA from human peripheral blood lymphocytes (PBL) and whether the degree of the DNA methylation would correlate with gene expression in human islets. We found tissue-specific differences in DNA methylation of GRB10 in human islets compared to PBL and the degree of methylation in the region analyzed with the Sequenom MassARRAY EpiTYPER ranged from 56.9% to 99.8% (Figure 3D). Although we did not observe any significant effect of rs933360 on the degree of DNA methylation in human islets (Figure S7A), there was a nominal association between rs933360 and DNA methylation in PBL in the region analyzed using EpiTYPER (p = 0.07) (Figure S7B). Moreover, we observed an inverse correlation between DNA methylation at a CpG site located 31.7 kb downstream of rs933360 and GRB10 mRNA expression (N = 81, rho = −0.335, p = 0.002) (Figure 3E) in human islets, particularly in islets from diabetic donors (N = 24, rho = −0.656, p = 0.001), and at a CpG site 8,196 bp downstream from rs933360 (N = 66, rho = −0.23, p = 0.047) (Figure S7C), suggesting that decreased methylation in this region is associated with increased GRB10 mRNA.
Expression of GRB10 in Human Muscle and Adipose Tissue
Given that we observed differences in insulin sensitivity when the risk A-allele of SNP rs933360 was inherited from the mother compared to the father, and that GRB10 is an inhibitor of insulin signaling, we also explored whether the SNP would influence expression of the GRB10 gene in human skeletal muscle and adipose tissue [20], [21]. We observed that carriers of the A-allele had decreased GRB10 mRNA level in muscle (N = 38, β = −0.405, p = 0.013) and adipose tissue (N = 1,375, β = −0.038, p = 0.005).
Effects of GRB10 on Islet Function
To gain insight into the mechanisms by which GRB10 influences pancreatic β- and α-cell function, we disrupted Grb10 expression in rat insulinoma INS-1 cells by siRNA and in human islets by shRNA achieved by lentiviral transfection. There was a clear reduction in GSIS after siRNA-disruption of Grb10 in the INS-1 cell line lacking glucagon (Figure 4A). In human pancreatic islets, decreased GRB10 expression resulted in a reduction of both insulin and glucagon secretion and expression (Figure 4B, C). In addition, GRB10 knock-down was also associated with a decrease in forskolin- and K+-stimulated glucagon secretion (Figure S8A).
Figure 4. Effects of disrupted GRB10 through knock-down on islet function.
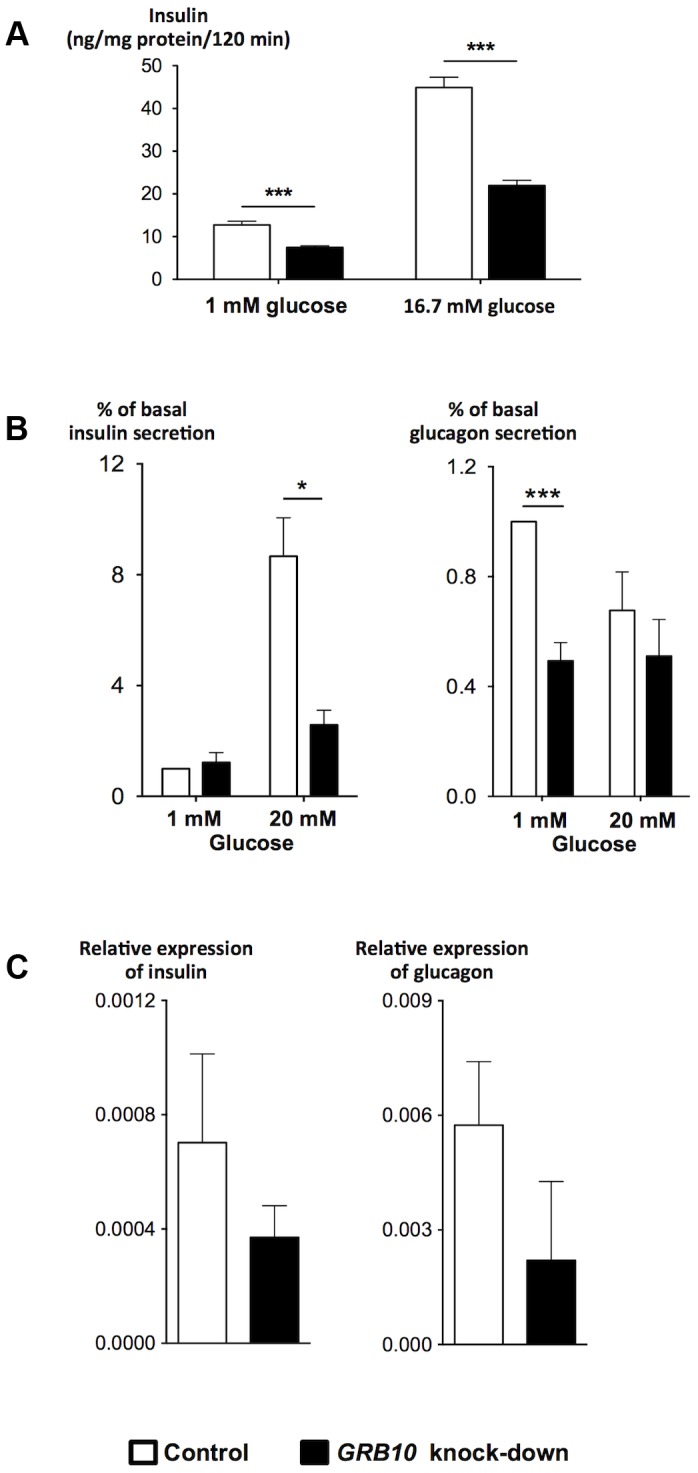
(A) Disrupted GRB10 in INS-1 rat β-cells markedly reduced glucose-stimulated insulin secretion. (B) GRB10 knock-down showed reduced glucose-stimulated insulin secretion at 20 mM glucose and glucagon secretion at 1 mM glucose in human pancreatic islets (Ninsulin = 7, Nglucagon = 6 donors of human pancreatic islets; 3–6 measurements in each experiment for each donor). (C) GRB10 knock-down resulted in a reduction of insulin and glucagon mRNA expression (N = 3 donors of human pancreatic islets; 3 measurements in each experiment for each donor). * p<0.05; ** p<0.01, *** p<0.001. Error bars denote SEM.
Grb10 has been reported to both increase [22] and decrease [23] apoptosis in islets, in addition to its effects on insulin signaling [22]. Disruption of GRB10 was associated with a significant reduction in the number of viable human pancreatic islets, as assessed by an MTS technique, supporting recent data in mice using shRNA to disrupt Grb10 in pancreatic islets [22] (Figure S8C).
Discussion
The present study provides mechanistic insights into the role of GRB10 in the regulation of islet function and glucose metabolism in man. First, the A-allele of the GRB10 rs933360 variant was associated with different effects on insulin secretion, sensitivity and glucose concentrations, if transmitted from fathers or mothers. Second, disruption of GRB10 in human islets resulted in a reduction of both insulin and glucagon secretion, the latter of which can provide an additional explanation for the reduction in glucose levels despite reduced insulin levels. Third, metabolic effects of GRB10 on glucose homeostasis involved tissue-specific methylation and parental imprinting.
GRB10 is an adaptor protein, which interacts with a number of receptor tyrosine kinases and signaling molecules and serves as a down-regulator of insulin receptor activity [24]. GRB10 participates in phosphorylation and activation of the mTORC1 protein, which is a central regulator of cellular metabolism, growth and survival [25]. Studies in mice have shown an abundant expression of Grb10 in brain, fat, muscle and heart, and with the highest expression in pancreas [26].
An interesting observation in the present study was that the common variant rs933360 in the GRB10 gene was associated with reduced GSIS, enhanced insulin sensitivity and reduced glucose levels when inherited from the mother. On the contrary, the paternally transmitted allele was associated with elevated glucose level and increased risk for T2D. These findings might partially explain the modest or lack of effects seen in several studies for variants in the GRB10 gene on T2D risk [27]. The effect on insulin sensitivity is indirectly supported by the view that carriers of the A-allele showed decreased expression of GRB10 in skeletal muscle and adipose tissue. Another potential explanation could be the effect of GRB10 on glucagon secretion. Unfortunately, due to lack of samples for glucagon measurements, we were not able to test this directly in the trios.
To further explore whether GRB10 influences glucagon secretion/expression and thereby mechanisms by which GRB10 might influence glucose metabolism, we disrupted GBR10 in human pancreatic islets. While Grb10 knock-down in the rat insulinoma cell line INS-1 lacking glucagon resulted in marked reduction of insulin, GRB10 knock-down in human pancreatic islets resulted in reduction in both insulin and glucagon secretion and expression. The effect on glucagon seen in human islets is in line with data from mice, showing reduced glucagon levels after Grb10 disruption using shRNA [22].
Since rs933360 is located in intron 2 of the GRB10 gene, one could envision an effect on gene expression. This could be a possible mechanism, as monoallelic and isoform-specific expression of GRB10 has been reported in fetal brain, skeletal muscle and in the placental trophoblasts [11], [12]. Unfortunately, we could not observe any effect of the SNP on expression of the GRB10 transcript in islets, nor could we examine parent-of-origin effects in the islets from human cadaver donors, as we lacked information on the parents. However, using two different strategies we could demonstrate allelic imbalance for SNPs in the GRB10 gene, leaving this possibility open. In line with earlier findings, we also identified three transcripts arising from alternative GBR10 promoters, with the paternally expressed UN2 isoform being imprinted from one parent in islets. However, there is sufficient amount of the maternally expressed UN1a isoform in islets to allow an effect of the maternally transmitted allele, which could contribute to the stronger effect of the maternal allele as described above.
Genetic imprinting is usually a consequence of increased DNA methylation. In line with previous reports [11], [12], we observed tissue-specific methylation of GRB10, showing highest degree of methylation in human pancreatic islets, which was associated with decreased expression of GRB10. Although this relationship was not influenced by the SNP in human pancreatic islets, there was a tendency for an effect of the SNP on degree of methylation in PBL.
Our study has some limitations. Despite large sample size for measurements of insulin secretion, we did neither have samples with measurements of glucagon in the trios, nor did we have parental information for islet donors. Also, tissue limitations prevented us from directly exploring methylation patterns and imprinting in trios. Finally, although this is the largest meta-analysis for insulin secretion to date, we discovered only one novel locus influencing GSIS. While this could be a consequence of limited power, another plausible explanation is that many homeostatic mechanisms, as well as parent-of-origin specific effects, would dilute and neutralize such effects.
In conclusion, our data demonstrate a complex genetic regulation of GRB10 function in human islets with different effects of paternally and maternally transmitted alleles. Together, these findings suggest that tissue-specific methylation and possibly imprinting of GRB10 can influence glucose metabolism and contribute to T2D pathogenesis. The data also emphasize the need in genetic studies to consider whether disease risk alleles are inherited from the mother or the father.
Methods
Ethics Statement
All studies were approved by local research ethics committees, and all participants gave informed consent. All procedures in human islets were approved by the ethics committees at the Uppsala and Lund Universities and informed consent obtained by appropriate measures from donors or their relatives.
Meta-Analyses of Genome-Wide Association and CardioMetabochip Studies
Association analyses of insulin secretion and action traits were performed within 11 cohorts participating in the Meta-Analysis of Glucose- and Insulin-related traits Consortium (MAGIC) in a total of up to 10,831 individuals. In the discovery stage 1, we performed a meta-analysis of 6 GWASs (Diabetes Genetics Initiative (DGI), Amish Family Diabetes Study, Sorbs, Helsinki Birth Cohort Study (HBCS), French Obese Adults, and Relationship between Insulin Sensitivity and Cardiovascular disease Study (RISC)) for glucose-stimulated insulin secretion (GSIS) during an oral glucose-tolerance test (OGTT) at 3 time points (fasting, 30 min, 120 min) for primary traits measured as (1) insulin response to glucose after the first 30 min estimated as corrected insulin response (CIR), and (2) overall insulin response to glucose estimated as area under the curve (AUC) for insulin over a total AUC for glucose (AUCIns/AUCGluc) in up to 5,318 non-diabetic individuals (Table S1A).
As none of the traits gave genome-wide significant association, we selected the top 50 independent signals from both primary and secondary traits (see “Phenotype Definition” below) after LD pruning (r2<0.2). Signals prioritized for replication were ranked by the number of associations observed at primary traits and/or secondary traits, association p-value and number of times the signal was observed across the traits (more than 2). We selected 14 SNPs for replication genotyping and follow-up analyses, out of which 3 loci were based on biological relevance: GRB10 [13] (rs933360, discovery p-value (CIR) = 5.09×10−6), UCN3 (rs11253130, discovery p-value (Ins30adjBMI) = 9.46×10−7) and INADL (rs2476186, discovery p-value (AUCIns/AUCGluc) = 1.88×10−6). Replication stage 2A de novo genotyping was undertaken in five population-based studies (Botnia-PPP, ULSAM, METSIM, BPS and Haguenau; only GWAS index SNP rs933360 in the latter three; max N = 15,273) (Table S1A, S2A). Replication stage 2B in silico was undertaken using an iSelect CardioMetabochip array (CM) (Illumina, San Diego, CA, USA) to genotype data in 5 independent population-based studies (Botnia-PPP, ULSAM, Ely, DR's Extra and METSIM) including up to 5,513 individuals (Table S1A).
The GWAS/CM (stage 1 and stage 2B) data including 93,896 SNPs were pooled together with the de novo genotyping results from stage 2A for non-overlapping individuals. In this meta-analysis, we defined all independent (r2<0.2) genome-wide significant (p-value<5×10−8) association signals for insulin secretion traits at 8 genomic loci (Table 1).
Phenotype Definitions
The primary insulin secretion and action indices were: (i) Corrected Insulin Response (CIR) = (100× insulin at 30 min)/(glucose at 30 min×(glucose at 30 min–3.89)), and (ii) ratio of the area under the curve (AUC) for AUC insulin/AUC glucose (AUCIns/AUCGluc, mU/mmol) calculated using the trapezium rule [28]. Insulin sensitivity index (ISI) = 10,000/√ (fasting plasma glucose (mg/dl)×fasting insulin×mean glucose during OGTT (mg/dl)×mean insulin during OGTT). Secondary insulin secretion and action indices during OGTT were: (i) disposition index (DI) = CIR×ISI; (ii) insulin at 30 min (Ins30); (iii) incremental insulin at 30 min (Increm30) = insulin 30 min – fasting insulin; (iv) insulin response to glucose during the first 30 min adjusted for BMI (Ins30adjBMI) = insulin at 30 min/(glucose at 30 min×BMI); (v) area under the curve (AUC) of insulin levels during OGTT (AUCIns, mU*min/l). Individuals with missing data on any of the three time points included in the AUC calculation were excluded.
Statistical Analysis
Linear regression models were used for association of phenotypes (z-score residuals of insulin secretion and action traits) with genotypes coded additively. Discovery (stage 1) GWAS analyses were carried out using a statistical tool that was able to account for genotype uncertainty, SNPTEST [29], or by using allele dosages in the linear regression model in MACH2QTL [30], [31], probABEL [32], corrected for residual inflation of the test statistics using the genomic control method [33]. The meta-analyses of effect sizes were performed with the fixed-effect inverse-variance method using GWAMA [34]. The GC correction was applied only once to cohort-specific results before including them into the meta-analyses. Sex-differentiated analyses were performed using GWAMA, with an assumed heterogeneity p-value of <0.05. Effect sizes for glucose levels were estimated using a fixed-effect model using the metaphor package for R version 2.14.2 (http://www.r-project.org/).
Parent-of-Origin Effect Analysis on Insulin Secretion and Glucose Levels
The Trios from Finland and Sweden, Amish Family Diabetes Study and Kuopio Offspring Study (Table S1A, S3A, B) consisted of a father, a mother and an offspring. Genotype phase was determined using Merlin and then analyzed using Solar, which uses the kinship matrix to account for family. Meta-analysis on insulin secretion and action and glucose levels during OGTT (as earlier described) was performed using a fixed-effect model. Analyses were performed on IBM SPSS Statistics 20.0 (IBM Corp., Chicago, IL, USA), R version 2.14.2 with the metaphor package (http://www.r-project.org/) and MMAP (MMAP: mixed models analysis for pedigrees and populations, http://edn.som.umaryland.edu/mmap/index.php).
The Transmission Disequilibrium Test (TDT) used to compare frequencies of transmission of the two alleles from heterozygote parents to an affected offspring was performed using PLINK (http://pngu.mgh.harvard.edu/purcell/plink/) [35] (Table S3A). The deviations from Mendelian transmissions were assessed and the power of the test was enhanced by incorporating information from phenotypically discordant parents (ParenTDT) [14]. To confirm the association, another independent test was performed, which can accommodate any type of genetic model and family construction, i.e. the Family Based Association Test (FBAT) [15]. Quantitative traits related to glucose metabolism and insulin secretion were assessed in non-diabetic individuals using qTDT and parent-of-origin effect tests. IBD estimates were calculated using Merlin. Permutations were performed using QFAM (PLINK). OGTT values were natural log-transformed and adjusted for BMI.
Association of GRB10 rs933360 with Glucagon Levels
The Botnia Prevalence, Prediction and Prevention of diabetes (Botnia-PPP) study is a population-based study from the Botnia region of Western Finland and has previously been described [36]. For this study, we selected 4,641 non-diabetic individuals above the age of 18. Linear regression analysis assuming an additive genetic risk model was performed to evaluate genotype-phenotype association. Hyperglycemic individuals were identified based on previous diagnosis, fasting plasma glucose levels of 5.5–6.9 mmol/l and 2 hr plasma glucose levels of 7.8–11.1 mmol/l (Table S5).
Association of GRB10 rs933360 with GRB10 mRNA in Human Muscle
A subgroup of 203 men with IGT at screening visit selected from the MPP study participated 20 years later in more extensive metabolic studies, including a new OGTT, a euglycemic-hyperinsulinemic clamp combined with indirect calorimetry and infusion of [3-3H]glucose [37], [38]. The men were similar in age, but had varying degrees of glucose tolerance; 69 were in the normal range, 52 had IFG and/or IGT, and 82 had T2D. T2D patients were either treated with diet alone (42%) or with oral hypoglycemic agents, which were withheld the day before the test. Microarray expression data were analyzed as previously described [20].
Association of GRB10 rs933360 with Future Risk for T2D
The Malmö Preventive Project (MPP) is a large population-based prospective study from the city of Malmö, Sweden, and has previously been described [17]. For this study, we selected 16,715 non-diabetic subjects, of whom 2,637 developed T2D during a 24.1 year mean follow-up period. The odds ratio for risk of developing T2D was calculated using logistic regression analysis assuming an additive genetic risk model. The analysis was adjusted for age, sex, BMI, participation period and an interaction term (participation period x sex) [10]. IBM SPSS Statistics 20.0 (IBM Corp.) was used for the statistical analysis.
Human Pancreatic Islets
Islets from cadaver donors were provided by the Nordic Islet Transplantation Program (www.nordicislets.org) by the courtesy of Prof. Olle Korsgren, Uppsala University, Sweden. The microarray experiments (Human Gene 1.0 ST whole transcript) were performed on islets isolated from 81 normoglycemic (mean±SEM; age 56.3±1.3 yrs, BMI 25.7±0.4 kg/m2, HbA1c 5.5±0.04%) and 46 hyperglycemic (age 60.3±1.2 yrs, BMI 27.8±0.6 kg/m2, HbA1c 6.6±0.1%) islet donors. RNA products were fragmented and hybridized to the GeneChip Human HG U 133A Array (Affymetrix, Santa Clara, CA, USA) [39]. Statistical analyses of expression data were performed using two-tailed Spearman's T-test.
Immunocytochemistry
Immunocytochemistry with antibodies for GRB10 (K20)18 (code sc-1026, Santa Cruz Biotech. Inc., CA, USA), insulin, glucagon and somatostatin was performed on human pancreatic sections as previously described [40].
RNA Sequencing of Human Islets
RNA from 48 human pancreatic islets donors (24 with HbA1c <5.5% and 24 donors with HbA1c >6.0%) was isolated and purified using miRNeasy kit (Qiagen, Hilden, Germany). As quality thresholds for RNA samples, we demanded RIN values >8, 28S/18S ratio >1.5 and the absorbance ratios 260/280 >1.8 and 260/230 >1. Sample preparation for sequencing reactions was performed using TrueSeq sample prep kit (Illumina). Fragmentation was performed using inbuilt fragmentation in the sample prep kit to obtain fragments of approximately 300 bp in length. Sequencing was performed on the Illumina HiSeq 2000 platform. Obtained sequenced reads were transformed into .qseq files using the Illumina pipeline. Alignment of the reads was performed using the TopHat short read aligner (tophat.cbcb.umd.edu). Cufflinks (cufflinks.cbcb.umd.edu) was used for splice variant calling.
Identification of GRB10 Splice Variants and Measurement of Allelic Imbalance
Analytical RT-PCRs were performed on cDNA from human islet, visceral fat, subcutaneous fat, liver and muscle in 15 µl reactions using 7.5 µl AmpliTaq Gold PCR Master Mix (Applied Biosystems, Foster City, CA, USA) supplemented with 0.75 µl DMSO and 1.5 µmol/l of forward and reverse primers. The following primers were used in various combinations: UN1fw: CAAACGCCTGCCTGACGACTG, UN1Afw: GCCCGGGACAGTCTTGAGC, UN2fw: GGCGCACACGCAGCGAC, UN3fw: ACCACCTACATCAGAGCTGACTGCC, 1bfw: CCTGGGCTACCCTCTGCTTC, 3fw: GCCTGTACTCGGCCTGCAGC, 9fw: GCCCCTACAGACCACGGGCT, 11fw: GCTGTCCCCGTTCTCGACGC, 3rv: ATGTGCACAGGCTGGGAGCG, 7rv: CTGGCTGTCATGTCTGCT, 11rv: CTGCTGAGGGATTCGGT, 16rv: GGATGCAGTGGTGCTTGA, the names referring to the target exon. PCR reactions were carried out with 53°C annealing temperature and over 50 cycles. Products were analyzed on 2% agarose. Prior to sequencing, 2.5 µl PCR product was treated with 0.5 µl ExoSAP-IT (USB, Cleveland, OH, USA) at 37°C for 30 min followed by deactivation at 80°C for 15 min. Subsequently, 1 µl was sequenced in both directions using BigDye 3.1 according to the manufacturer's protocol (Applied Biosystems). The sequence reactions were purified and analyzed by GATC Biotech AG (Konstanz, Germany).
Allelic imbalance measurements were performed by RT-PCR using the reverse primer 3rv and either of the forward primers UN1afw, UN1fw, UN2fw, UN3fw, 1bfw or 3fw in samples heterozygous for the common SNP rs1800504 (GRB10 exon3). The individual contribution from each allele was measured at the position of rs1800504 using Sanger sequence traces and the software Mutation Surveyor (Soft Genetics, PA, USA). Allelic imbalance in GRB10 was also detected by a different method: After extensive quality and coverage filtering, we did a Fisher exact test for comparing the ratio of reference/alternative alleles in the exome sequencing vs. RNA-seq for each sample. Exome sequencing was performed using the Illumina exome sequencing protocols (TruSeq DNA sample preparation Kit v2).
Methylation Studies
Sequenom's MassARRAY EpiTYPER protocol was applied to measure DNA methylation (Sequenom, San Diego, CA, USA) in human islets of 96 donors and peripheral blood lymphocytes (PBL) of 6 diabetic offspring trios (18 individuals). EpiDesigner was used for assay design at GRB10 and the primer sequences were the following; forward: aggaagagagGGGAAAGGGTGTTAAATTGTTTATG, reverse: cagtaatacgactcactatagggagaaggctTTTTAAACCCCTCAAATTCAAAAAT. 500 ng genomic DNA was bisulfite-treated with the EZ DNA Methylation kit (Zymo Research, Orange, CA, USA). The spectra were analyzed and the methylation ratios were obtained by the EpiTYPER software v.1.0.1. Global methylation analyses were performed on DNA extracted from PBL on the Illumina Infinium 450 Bead Chip and the chips were scanned on an Illumina iScan as per protocol (Illumina). 1 µg of DNA was bisulfite-treated according to protocol (EZ DNA Methylation Kit, Zymo research). For analysis, the Genome Studio Methylation Module of the Genome studio Genome Browser was used, which facilitates integration of the SNP and CpG location data (NCBI build 37). Methylation status was assessed after normalization to internal controls and background subtraction and expressed as β. The β values for the CpG sites were mapped to the gene and plotted to give an overview of methylation status for the region of interest (Figure S7D).
Disruption of GRB10 for Insulin and Glucagon Secretion Analyses
shRNA-mediated knock-down of GRB10 in human pancreatic islets
Specific silencing of endogenous hGRB10 was achieved using a lentiviral-based shRNA-silencing technique (Santa Cruz Biotech. Inc.). Isolated human islets were incubated at 2.8 mmol/l glucose + Polybree for 90 min. Thereafter, the medium was removed and the islets were washed before culture medium + lentiviral particles containing GRB10 shRNA (5 µl/ml) was added, and the islets were cultured for 36 h at 5 mmol/l glucose. For comparison, a scramble (lentiviral particles without targeting any specific region) served as control. Insulin and glucagon were measured using a radioimmunoassay kit (Electrobox, Stockhom, Euro-Diagnostica, Malmö, Sweden).
siRNA-mediated knock-down of Grb10 in INS-1 832/13 cells
For Grb10 small interfering RNA (siRNA) experiments, 20–25-nucleotide stealth-prevalidated siRNA duplex designed for rat Grb10 (Santa Cruz Biotech. Inc.) was used. INS-1 832/13 cells were seeded in 24-well plates at a density of ∼5×105 cells in culture media without antibiotics and transfected with DharmaFECT 1 (Dharmacon, Lafayette, CO, USA) according to the manufacturer's instructions.
Assessment of β-cell Viability
Human pancreatic β-cell viability assay was performed using a CellTiter 96 AQueous One Solution Cell Proliferation Assay Reagent (Promega, Stockholm, Sweden) according to the manufacturer's instructions. The actual performance is based on the spectrophotometric detection of a colored formazan product converted from a 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) compound by NADPH or NADH via metabolically active cells.
Supporting Information
Regional plots for the top 8 hits reaching genome-wide significance level. Association with insulin secretion measured as corrected insulin response (CIR) at 30 min of OGTT for the novel genetic variant GRB10 rs933360 (A) and the previously reported T2D and glycemic trait variants HHEX/IDE/KIF11 (B), MTNR1B (C), CDKAL1 (D), C2CD4A (NLF1) (E), ANK1 (F), GCK (G) and GIPR (H).
(TIFF)
Genome-wide quantile-quantile (Q-Q) and Manhattan plots for insulin secretion and action traits analyzed in the present study. Corrected insulin response (CIR) to glucose at 30 min of OGTT (A, B), insulin sensitivity index (C, D), insulin response adjusted for insulin sensitivity (CIR adj ISI) (E, F), disposition index (G, H), insulin level at 30 min of OGTT (I, J), incremental insulin at 30 min of OGTT (K, L), insulin level at 30 min of OGTT adjusted for BMI (M, N), area under the insulin curve (AUC ins) (O, P) and ratio between area under the insulin and glucose curves (AUC ins/AUC gluc) (Q, R).
(TIFF)
Meta-analysis for association of GRB10 rs933360 with fasting plasma glucose in the participating cohorts. The insulin-reducing allele was associated with lower fasting plasma glucose levels in all individuals.
(TIFF)
GRB10 mRNA levels in human islets. (A) GRB10 expression levels did not significantly differ between normoglycemic (HbA1c <5.4%) and hyperglycemic (HbA1c >6%) islet donors (T-test, p = 0.32). (B) There was no significant difference in GRB10 mRNA expression levels between carriers of different GRB10 genotypes (linear regression, p normoglycemic = 0.11; p hyperglycemic = 0.25). (C) Correlation between GRB10 and GCG (glucagon), and GRB10 and INS (insulin) mRNA expression in islets from normoglycemic (HbA1c <6%, N = 51) and hyperglycemic donors (HbA1c >6%, N = 27). Error bars denote SE.
(TIFF)
Schematic representation of the GRB10 gene and its transcripts. The 5′ exon arrangements of GRB10 splice variants were identified in human fat and islet samples by RT-PCR and Sanger sequencing. The exon order at the gene level is shown in the top row. UCSC IDs of matching transcripts are indicated to the right.
(TIFF)
Allele-specific expression analysis of the GRB10 5′-end in islet, muscle, liver and fat tissue. Transcripts containing exon UN2 are exclusively expressed from one allele (presumably paternal), whereas transcripts containing the upstream exons UN1 or UN1a derive from both alleles to a varying degree. UN1a, UN1 and UN2 are mutually exclusive exons. A sequence of heterozygous genomic DNA (gDNA) is shown on top for comparison (50%-50%). rs1800504 in exon 3 was used as a reporter SNP for the intronic index SNP rs933360.
(TIFF)
Pictorial representation of GRB10 methylation status in human islets and peripheral blood lymphocytes. The impact of rs933360 on DNA methylation of three CpG sites analyzed using EpiTYPER in human islets (N = 96) (A) and peripheral blood lymphocytes (PBL, N = 18) (B). (C) The GRB10 mRNA levels correlated negatively with the degree of methylation at CpG sites located 8,196 bp downstream of rs933360 of the GRB10 gene in human pancreatic islets (N = 66). (D) Methylation status was tested for 44 CpG sites using the Illumina Infinium 450 K global methylation assay and is represented in the graph above the gene structure. The numbers on the Y-axis on the right indicate the mean β values showing methylation status. The colored boxes represent the exons of the four isoforms annotated in the NCBI. Arrows indicate position of the SNPs tested for association. Distances between nearest CpG site tested and: (i) rs6943153 = 6.9 kb, (ii) rs933360 = 8.2 kb and (iii) rs2237457 = 13.8 kb. Hapmap does not show the linkage of the tested SNPs with any of the SNPs tested for methylation status. The CpG sites in the proximity were methylated (β>0.8). GRB10 is presented on a reverse strand.
(TIFF)
Effect of GRB10 disruption on islet function and cell survival in human pancreatic islets. (A) GRB10 knock-down showed reduced glucagon secretion at 1 mM glucose, particularly stronger for forskolin- and K+-stimulated glucagon secretion. Reduced GRB10 expression in human pancreatic islets had modest effects on insulin secretion, i.e. a slight increase in insulin levels at 1 mM glucose. Ninsulin = 4 and Nglucagon = 3 donors of human pancreatic islets; up to 6 measurements in each experiment for each donor. (B) Effect of GRB10 disruption on insulin and glucagon content (1 non-diabetic donor, 6 measurements in each experiment). (C) GRB10 knock-down resulted in a reduction of the number of viable cells in human pancreatic islets. (D) Effect of GRB10 disruption on caspase-3 mRNA expression (2 non-diabetic donors, 3 measurements in each experiment). * p<0.05, *** p<0.001.
(TIFF)
(A) Characteristics of cohorts and study details of analysis metrics and methods. (B) Heritability estimates of parameters of insulin secretion and action indices used in the study. (C) Spearman correlations between insulin secretion and action indices for the non-diabetic participants of the Botnia-PPP study.
(XLS)
(A) Association results of selected SNPs from the discovery GWAS, the CardioMetabochip and the de novo replication genotyping. (B) Meta-analysis of GWAS and CardioMetabochip studies: association results with parameters of insulin secretion and insulin action during OGTT for loci previously associated with type 2 diabetes (T2D), 2 hour plasma glucose during OGTT (2hPG), fasting plasma glucose (FGlu), fasting insulin (FIns) and obesity.
(XLS)
(A) Assessing transmissions of GRB10 rs933360 and rs6943153 alleles in the Trios from Finland and Sweden. (B) Parent-of-origin effect of GRB10 rs933360 on parameters of insulin secretion and insulin action, and glucose levels during OGTT in the non-diabetic offspring from the Trio cohorts.
(XLS)
Odds ratios for GRB10 rs933360 for risk of T2D in the MPP study.
(XLS)
(A) Descriptive statistics for the Botnia-PPP cohort. (B) Association between GRB10 rs933360 and glucagon levels in the Botnia-PPP cohort.
(XLS)
GRB10 SNPs associated with fasting and 2 hr glucose levels in 361 non-diabetic individuals from the DGI GWAS study.
(XLS)
Measurements of allelic imbalance in the 8 heterozygous carriers of GRB10 rs1800504 in human islets.
(XLS)
Acknowledgments
DGI, BPS, PPP, MPP, human islets: Human islets were provided by the Nordic network for clinical islets transplantation by the courtesy of Dr. Olle Korsgren, Uppsala, Sweden. Microarrays were performed at SCIBLU Genomics at Lund University. We thank the patients for their participation and the Botnia Study Group for clinically studying the patients. French obese adults: We thank Jacques Weill for recruitment of obesity families at Jeanne de Flandres Hospital, Marianne Deweirder and Frédéric Allegaert for DNA management, and Stefan Gaget for database management. Amish family diabetes study: We thank our Amish community and research volunteers for their long-standing partnership in research, and acknowledge the dedication of our Amish liaisons, field workers and the Amish Research Clinic staff, without which these studies would not have been possible. ULSAM: Genotyping was performed by the SNP&SEQ Technology Platform in Uppsala, Sweden (www.genotyping.se). We thank Tomas Axelsson, Ann-Christine Wiman and Caisa Pöntinen for their excellent assistance with genotyping. Sorbs: We thank all those who participated in the study. Sincere thanks are given to Knut Krohn (Microarray Core Facility of the Interdisciplinary Centre for Clinical Research, University of Leipzig) for the genotyping support. HBCS: We thank all study participants and everyone involved in the Helsinki Birth Cohort Study.
Funding Statement
Studies at LUDC in Malmö (DGI, BPS, PPP, MPP and human islets) were supported by grants from the Swedish Research Council (SFO EXODIAB: Dnr 2009-1039), Linnaeus grant (LUDC Dnr 349-2008-6589), project grants to VLy, LG, NW, and CLi, and financial support from the Swedish Research Council (Dnr 521-2010-3490, Dnr 521-2010-3490, Dnr 521-2010-3490, Dnr 521-2007-4037, Dnr 521-2008-2974, ANDIS Dnr 825-2010-5983), the Knut & Alice Wallenberg foundation (KAW 2009.0243), the Torsten och Ragnar Söderbergs Stiftelser (MT33/09), the Inga Britt och Arne Lundberg's Research Foundation (grant nr. 359), the Heart and Lung Foundation, the Novo Nordisk foundation, Diabetes Research & Wellness, Sydvästra Skånes Diabetesförening. The PPP-Botnia study was supported by grants from the Sigrid Juselius Foundation, the Finnish Diabetes Research Society, the Signe and Ane Gyllenberg Foundation, the Swedish Cultural Foundation in Finland, the Ollqvist Foundation, the Foundation for Life and Health in Finland, Jakobstad Hospital, the Medical Society of Finland, the Närpes Research Foundation and the Vasa and Närpes Health centers. The MPP study was supported by a grant from the Swedish Heart and Lung Foundation, the E. Lundström Foundation and from the Region Skåne County Council funds. The RISC Study was supported by European Union grant QLG1-CT-2001-01252 and AstraZeneca. The Amish Family Diabetes Study was supported by NIH research grants R01 DK54261, R01 DK68495, U01 HL84756, the University of Maryland General Clinical Research Center (M01 RR16500), Hopkins Bayview General Clinical Research Center (M01 RR02719), the Mid-Atlantic Nutrition Obesity Research Center (P30 DK072488), the Baltimore Diabetes Research and Training Center grant (P60 DK79637), the Baltimore Veterans Administration Geriatric Research and Education Clinical Center, and by USDA NIFA NRI Competitive Grant 2007-35205-17883. The DR's EXTRA Study was supported by grants to RR by the Ministry of Education and Culture of Finland (627;2004-2011), Academy of Finland (102318; 123885), Kuopio University Hospital, Finnish Diabetes Association, Finnish Heart Association, Päivikki and Sakari Sohlberg Foundation and by grants from European Commission FP6 Integrated Project (EXGENESIS), LSHM-CT-2004-005272, City of Kuopio and Social Insurance Institution of Finland (4/26/2010). The METSIM Study was supported by grants from the Academy of Finland (141069, 141226), Diabetes Research Foundation, and Sigrid Juselius Foundation. The ULSAM project was supported by grants from the Swedish Research Council, the Swedish Heart-Lung Foundation, the Swedish Foundation for Strategic Research, the Royal Swedish Academy of Sciences, Swedish Diabetes Foundation, Swedish Society of Medicine, and Novo Nordisk Foundation. Part of the genotyping in ULSAM for this project was supported through the ENGAGE (European Network for Genetic and Genomic Epidemiology) Consortium, which is funded through the European Community's Seventh Framework Programme (HEALTH-F4-2007- 201413). The SNP & SEQ Technology Platform in Uppsala was supported by Science for Life Laboratory and the Swedish Research Council for Infrastructures (RFI). The Sorbs work was supported by grants from the German Research Council (DFG - SFB 1052 “Obesity mechanisms”; A01, B03, C01), from the German Diabetes Association and from the DHFD (Diabetes Hilfs- und Forschungsfonds Deutschland). IFB Adiposity Diseases is supported by the Federal Ministry of Education and Research (BMBF), Germany, FKZ: 01EO1001. PK is funded by Boehringer Ingelheim Foundation. RM acknowledges financial support from the European Commission under a Marie Curie Intra-European Fellowship, EFSD New Horizons grant, and Development Fund of the University of Tartu, in the frame of the Centre of Transitional Genomics (grant SP1GVARENG) and by Estonian Government (grant #SF0180142s08). The research of IP and VLa was funded in part through the European Community's Seventh Framework Programme (FP7/2007-2013), ENGAGE project, grant agreement HEALTH-F4-2007-201413. Helsinki Birth Cohort Study (HBCS) has been supported by grants from the Academy of Finland (Grant No. 120386 and 125876 to JGE), the Finnish Diabetes Research Society, Folkhälsan Research Foundation, Novo Nordisk Foundation, Finska Läkaresällskapet, Liv och Hälsa, the Wellcome Trust (Grant No. 89061/Z/09/Z and 089062/Z/09/Z), Samfundet Folkhälsan, Finska Läkaresällskapet, the Signe and Ane Gyllenberg foundation, the Univeristy of Helsinki, Ministry of Education, Ahokas Foundation, Emil Aaltonen Foundation, European Science Foundation (EUROSTRESS), Juho Vainio Foundation, and Wellcome Trust (grant WT089062). MIM is a Wellcome Trust Senior Investigator, and an NIHR Senior Investigator, and has also received funding from EU Framework 7: ENGAGE Consortium HEALTH-F4-2007-201413, Wellcome Trust: 083270, 090367, 090532, 098381, Medical Research Council: G0601261 and Diabetes UK: RD08/0003704. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Diabetes Genetics Initiative of Broad Institute of Harvard and MIT and Novartis Institutes of BioMedical Research, Lund University (2007) Saxena R, Voight BF, Lyssenko V, Burtt NP, et al. (2007) Genome-Wide Association Analysis Identifies Loci for Type 2 Diabetes and Triglyceride Levels. Science 316: 1331–1336 doi:10.1126/science.1142358 [DOI] [PubMed] [Google Scholar]
- 2. Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, et al. (2010) New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 42: 105–116 Available: http://www.nature.com/ng/journal/v42/n2/abs/ng.520.html. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Manning AK, Hivert M-F, Scott RA, Grimsby JL, Bouatia-Naji N, et al. (2012) A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet 44: 659–669 doi:10.1038/ng.2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Morris AP, Voight BF, Teslovich TM, Ferreira T, Segrè AV, et al. (2012) Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet 44: 981–990 doi:10.1038/ng.2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, et al. (2007) A Genome-Wide Association Study of Type 2 Diabetes in Finns Detects Multiple Susceptibility Variants. Science 316: 1341–1345 doi:10.1126/science.1142382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Scott RA, Lagou V, Welch RP, Wheeler E, Montasser ME, et al. (2012) Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet 44: 991–1005 doi:10.1038/ng.2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sladek R, Rocheleau G, Rung J, Dina C, Shen L, et al. (2007) A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445: 881–885 doi:10.1038/nature05616 [DOI] [PubMed] [Google Scholar]
- 8. Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, et al. (2007) Replication of Genome-Wide Association Signals in UK Samples Reveals Risk Loci for Type 2 Diabetes. Science 316: 1336–1341 doi:10.1126/science.1142364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ingelsson E, Langenberg C, Hivert M-F, Prokopenko I, Lyssenko V, et al. (2010) Detailed physiologic characterization reveals diverse mechanisms for novel genetic Loci regulating glucose and insulin metabolism in humans. Diabetes 59: 1266–1275 doi:10.2337/db09-1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lyssenko V, Jonsson A, Almgren P, Pulizzi N, Isomaa B, et al. (2008) Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med 359: 2220–2232. [DOI] [PubMed] [Google Scholar]
- 11. Blagitko N, Mergenthaler S, Schulz U, Wollmann H, Craigen W, et al. (2000) Human GRB10 is imprinted and expressed from the paternal and maternal allele in a highly tissue- and isoform-specific fashion. Human Molecular Genetics 9: 1587–1595. [DOI] [PubMed] [Google Scholar]
- 12. Monk D, Arnaud P, Frost J, Hills FA, Stanier P, et al. (2009) Reciprocal imprinting of human GRB10 in placental trophoblast and brain: evolutionary conservation of reversed allelic expression. Human Molecular Genetics 18: 3066–3074 doi:10.1093/hmg/ddp248 [DOI] [PubMed] [Google Scholar]
- 13. Rampersaud E, Damcott CM, Fu M, Shen H, McArdle P, et al. (2007) Identification of Novel Candidate Genes for Type 2 Diabetes From a Genome-Wide Association Scan in the Old Order Amish: Evidence for Replication From Diabetes-Related Quantitative Traits and From Independent Populations. Diabetes 56: 3053–3062 doi:10.2337/db07-0457 [DOI] [PubMed] [Google Scholar]
- 14. Purcell S, Sham P, Daly MJ (2005) Parental phenotypes in family-based association analysis. Am J Hum Genet 76: 249–259 doi:10.1086/427886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Horvath S, Xu X, Laird NM (2001) The family based association test method: strategies for studying general genotype–phenotype associations. Eur J Hum Genet 9: 301–306 doi:10.1038/sj.ejhg.5200625 [DOI] [PubMed] [Google Scholar]
- 16. Altshuler D, Hirschhorn JN, Klannemark M, Lindgren CM, Vohl MC, et al. (2000) The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet 26: 76–80 doi:10.1038/79216 [DOI] [PubMed] [Google Scholar]
- 17. Nilsson PM, Nilsson JA, Berglund G (2006) Population-attributable risk of coronary heart disease risk factors during long-term follow-up: the Malmo Preventive Project. J Intern Med 260: 134–141 doi:10.1111/j.1365-2796.2006.01671.x [DOI] [PubMed] [Google Scholar]
- 18. Arnaud P (2003) Conserved methylation imprints in the human and mouse GRB10 genes with divergent allelic expression suggests differential reading of the same mark. Human Molecular Genetics 12: 1005–1019 doi:10.1093/hmg/ddg110 [DOI] [PubMed] [Google Scholar]
- 19. Nitert MD, Dayeh T, Volkov P, Elgzyri T, Hall E, et al. (2012) Impact of an exercise intervention on DNA methylation in skeletal muscle from first-degree relatives of patients with type 2 diabetes. Diabetes 61: 3322–3332 doi:10.2337/db11-1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, et al. (2003) PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34: 267–273 doi:10.1038/ng1180 [DOI] [PubMed] [Google Scholar]
- 21. Stančáková A, Civelek M, Saleem NK, Soininen P, Kangas AJ, et al. (2012) Hyperglycemia and a common variant of GCKR are associated with the levels of eight amino acids in 9,369 Finnish men. Diabetes 61: 1895–1902 doi:10.2337/db11-1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Doiron B, Hu W, Norton L, DeFronzo RA (2012) Lentivirus shRNA Grb10 targeting the pancreas induces apoptosis and improved glucose tolerance due to decreased plasma glucagon levels. Diabetologia 55: 719–728 doi:10.1007/s00125-011-2414-z [DOI] [PubMed] [Google Scholar]
- 23. Zhang J, Zhang N, Liu M, Li X, Zhou L, et al. (2012) Disruption of Growth Factor Receptor-Binding Protein 10 in the Pancreas Enhances β-Cell Proliferation and Protects Mice From Streptozotocin-Induced β-Cell Apoptosis. Diabetes 61: 3189–3198 doi:10.2337/db12-0249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Holt L, Siddle K (2005) Grb10 and Grb14: enigmatic regulators of insulin action - and more? Biochem J 388: 393–406 doi:10.1042/BJ20050216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yu Y, Yoon S-O, Poulogiannis G, Yang Q, Ma XM, et al. (2011) Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science Signaling 332: 1322–1326 Available: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=21659605&retmode=ref&cmd=prlinks. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang L, Balas B, Christ-Roberts CY, Kim RY, Ramos FJ, et al. (2007) Peripheral disruption of the grb10 gene enhances insulin signaling and sensitivity in vivo. Molecular and Cellular Biology 27: 6497–6505 Available: http://mcb.asm.org/cgi/doi/10.1128/MCB.00679-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, et al. (2010) Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet 42: 579–589 doi:10.1038/ng.609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Matthews JNS, Altman DG, Campbell MJ, Royston P (1990) Analysis of Serial Measurements in Medical-Research. Brit Med J 300: 230–235 Available: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1662068/pdf/bmj00163-0030.pdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Marchini J, Howie B, Myers S, McVean G, Donnelly P (2007) A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet 39: 906–913 doi:10.1038/ng2088 [DOI] [PubMed] [Google Scholar]
- 30. Li Y, Willer C, Sanna S, Abecasis G (2009) Genotype imputation. Annu Rev Genomics Hum Genet 10: 387–406 doi:10.1146/annurev.genom.9.081307.164242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR (2010) MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol 34: 816–834 doi:10.1002/gepi.20533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aulchenko YS, Ripke S, Isaacs A, van Duijn CM (2007) GenABEL: an R library for genome-wide association analysis. [DOI] [PubMed]
- 33. Devlin B, Roeder K (2004) Genomic Control for Association Studies. Biometrics 55: 997–1004 doi:10.1111/j.0006-341X.1999.00997.x [DOI] [PubMed] [Google Scholar]
- 34. Mägi R, Morris AP (2010) GWAMA: software for genome-wide association meta-analysis. BMC Bioinformatics 11: 288 doi:10.1186/1471-2105-11-288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, et al. (2007) PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. The American Journal of Human Genetics 81: 559–575 doi:10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Isomaa B, Forsén B, Lahti K, Holmström N, Wadén J, et al. (2010) A family history of diabetes is associated with reduced physical fitness in the Prevalence, Prediction and Prevention of Diabetes (PPP)–Botnia study. Diabetologia 53: 1709–1713 doi:10.1007/s00125-010-1776-y [DOI] [PubMed] [Google Scholar]
- 37. Eriksson KF, Lindgarde F (1990) Impaired glucose tolerance in a middle-aged male urban population: a new approach for identifying high-risk cases. Diabetologia 33: 526–531. [DOI] [PubMed] [Google Scholar]
- 38. Eriksson KF, Lindgarde F (1991) Prevention of type 2 (non-insulin-dependent) diabetes mellitus by diet and physical exercise. The 6-year Malmö feasibility study. Diabetologia 34: 891–898. [DOI] [PubMed] [Google Scholar]
- 39. Taneera J, Lang S, Sharma A, Fadista J, Zhou Y, et al. (2012) A systems genetics approach identifies genes and pathways for type 2 diabetes in human islets. Cell Metabolism 16: 122–134 doi:10.1016/j.cmet.2012.06.006 [DOI] [PubMed] [Google Scholar]
- 40. Riva M, Nitert MD, Voss U, Sathanoori R, Lindqvist A, et al. (2011) Nesfatin-1 stimulates glucagon and insulin secretion and beta cell NUCB2 is reduced in human type 2 diabetic subjects. Cell Tissue Res 346: 393–405 doi:10.1007/s00441-011-1268-5 [DOI] [PubMed] [Google Scholar]
- 41. Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, et al. (2010) LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26: 2336–2337 doi:10.1093/bioinformatics/btq419 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Regional plots for the top 8 hits reaching genome-wide significance level. Association with insulin secretion measured as corrected insulin response (CIR) at 30 min of OGTT for the novel genetic variant GRB10 rs933360 (A) and the previously reported T2D and glycemic trait variants HHEX/IDE/KIF11 (B), MTNR1B (C), CDKAL1 (D), C2CD4A (NLF1) (E), ANK1 (F), GCK (G) and GIPR (H).
(TIFF)
Genome-wide quantile-quantile (Q-Q) and Manhattan plots for insulin secretion and action traits analyzed in the present study. Corrected insulin response (CIR) to glucose at 30 min of OGTT (A, B), insulin sensitivity index (C, D), insulin response adjusted for insulin sensitivity (CIR adj ISI) (E, F), disposition index (G, H), insulin level at 30 min of OGTT (I, J), incremental insulin at 30 min of OGTT (K, L), insulin level at 30 min of OGTT adjusted for BMI (M, N), area under the insulin curve (AUC ins) (O, P) and ratio between area under the insulin and glucose curves (AUC ins/AUC gluc) (Q, R).
(TIFF)
Meta-analysis for association of GRB10 rs933360 with fasting plasma glucose in the participating cohorts. The insulin-reducing allele was associated with lower fasting plasma glucose levels in all individuals.
(TIFF)
GRB10 mRNA levels in human islets. (A) GRB10 expression levels did not significantly differ between normoglycemic (HbA1c <5.4%) and hyperglycemic (HbA1c >6%) islet donors (T-test, p = 0.32). (B) There was no significant difference in GRB10 mRNA expression levels between carriers of different GRB10 genotypes (linear regression, p normoglycemic = 0.11; p hyperglycemic = 0.25). (C) Correlation between GRB10 and GCG (glucagon), and GRB10 and INS (insulin) mRNA expression in islets from normoglycemic (HbA1c <6%, N = 51) and hyperglycemic donors (HbA1c >6%, N = 27). Error bars denote SE.
(TIFF)
Schematic representation of the GRB10 gene and its transcripts. The 5′ exon arrangements of GRB10 splice variants were identified in human fat and islet samples by RT-PCR and Sanger sequencing. The exon order at the gene level is shown in the top row. UCSC IDs of matching transcripts are indicated to the right.
(TIFF)
Allele-specific expression analysis of the GRB10 5′-end in islet, muscle, liver and fat tissue. Transcripts containing exon UN2 are exclusively expressed from one allele (presumably paternal), whereas transcripts containing the upstream exons UN1 or UN1a derive from both alleles to a varying degree. UN1a, UN1 and UN2 are mutually exclusive exons. A sequence of heterozygous genomic DNA (gDNA) is shown on top for comparison (50%-50%). rs1800504 in exon 3 was used as a reporter SNP for the intronic index SNP rs933360.
(TIFF)
Pictorial representation of GRB10 methylation status in human islets and peripheral blood lymphocytes. The impact of rs933360 on DNA methylation of three CpG sites analyzed using EpiTYPER in human islets (N = 96) (A) and peripheral blood lymphocytes (PBL, N = 18) (B). (C) The GRB10 mRNA levels correlated negatively with the degree of methylation at CpG sites located 8,196 bp downstream of rs933360 of the GRB10 gene in human pancreatic islets (N = 66). (D) Methylation status was tested for 44 CpG sites using the Illumina Infinium 450 K global methylation assay and is represented in the graph above the gene structure. The numbers on the Y-axis on the right indicate the mean β values showing methylation status. The colored boxes represent the exons of the four isoforms annotated in the NCBI. Arrows indicate position of the SNPs tested for association. Distances between nearest CpG site tested and: (i) rs6943153 = 6.9 kb, (ii) rs933360 = 8.2 kb and (iii) rs2237457 = 13.8 kb. Hapmap does not show the linkage of the tested SNPs with any of the SNPs tested for methylation status. The CpG sites in the proximity were methylated (β>0.8). GRB10 is presented on a reverse strand.
(TIFF)
Effect of GRB10 disruption on islet function and cell survival in human pancreatic islets. (A) GRB10 knock-down showed reduced glucagon secretion at 1 mM glucose, particularly stronger for forskolin- and K+-stimulated glucagon secretion. Reduced GRB10 expression in human pancreatic islets had modest effects on insulin secretion, i.e. a slight increase in insulin levels at 1 mM glucose. Ninsulin = 4 and Nglucagon = 3 donors of human pancreatic islets; up to 6 measurements in each experiment for each donor. (B) Effect of GRB10 disruption on insulin and glucagon content (1 non-diabetic donor, 6 measurements in each experiment). (C) GRB10 knock-down resulted in a reduction of the number of viable cells in human pancreatic islets. (D) Effect of GRB10 disruption on caspase-3 mRNA expression (2 non-diabetic donors, 3 measurements in each experiment). * p<0.05, *** p<0.001.
(TIFF)
(A) Characteristics of cohorts and study details of analysis metrics and methods. (B) Heritability estimates of parameters of insulin secretion and action indices used in the study. (C) Spearman correlations between insulin secretion and action indices for the non-diabetic participants of the Botnia-PPP study.
(XLS)
(A) Association results of selected SNPs from the discovery GWAS, the CardioMetabochip and the de novo replication genotyping. (B) Meta-analysis of GWAS and CardioMetabochip studies: association results with parameters of insulin secretion and insulin action during OGTT for loci previously associated with type 2 diabetes (T2D), 2 hour plasma glucose during OGTT (2hPG), fasting plasma glucose (FGlu), fasting insulin (FIns) and obesity.
(XLS)
(A) Assessing transmissions of GRB10 rs933360 and rs6943153 alleles in the Trios from Finland and Sweden. (B) Parent-of-origin effect of GRB10 rs933360 on parameters of insulin secretion and insulin action, and glucose levels during OGTT in the non-diabetic offspring from the Trio cohorts.
(XLS)
Odds ratios for GRB10 rs933360 for risk of T2D in the MPP study.
(XLS)
(A) Descriptive statistics for the Botnia-PPP cohort. (B) Association between GRB10 rs933360 and glucagon levels in the Botnia-PPP cohort.
(XLS)
GRB10 SNPs associated with fasting and 2 hr glucose levels in 361 non-diabetic individuals from the DGI GWAS study.
(XLS)
Measurements of allelic imbalance in the 8 heterozygous carriers of GRB10 rs1800504 in human islets.
(XLS)