

Abstract
A novel temperate bacteriophage of Pseudomonas aeruginosa, phage vB_PaeP_Tr60_Ab31 (alias Ab31) is described. Its genome is composed of structural genes related to those of lytic P. putida phage AF, and regulatory genes similar to those of temperate phage PAJU2. The virion structure resembles that of phage AF and other lytic Podoviridae (S. enterica Epsilon 15 and E. coli phiv10) with similar tail spikes. Ab31 was able to infect P. aeruginosa strain PA14 and two genetically related strains called Tr60 and Tr162, out of 35 diverse strains from cystic fibrosis patients. Analysis of resistant host variants revealed different phenotypes, including induction of pigment and alginate overproduction. Whole genome sequencing of resistant variants highlighted the existence of a large deletion of 234 kbp in two strains, encompassing a cluster of genes required for the production of CupA fimbriae. Stable lysogens formed by Ab31 in strain Tr60, permitted the identification of the insertion site. During colonization of the lung in cystic fibrosis patients, P. aeruginosa adapts by modifying its genome. We suggest that bacteriophages such as Ab31 may play an important role in this adaptation by selecting for bacterial characteristics that favor persistence of bacteria in the lung.
Introduction
Cystic fibrosis (CF) is one of the most common life-threatening, autosomal recessive genetic diseases in Caucasian children. This is due to mutations that occur in a single gene encoding the CF transmembrane regulator. The life expectancy of CF patients is above all related to the development of lung disease: the persistence of abundant mucous secretion in the lungs leads to chronic coughing at a young age, followed by frequent lung infections [1]. The microorganisms that colonize the CF patients’ lungs belong to various bacterial genera. For 30% of CF patients, the predominant bacterial species during early life is Staphylococcus aureus, whereas in early adolescence, chronical infection with Pseudomonas aeruginosa is common: up to 80% of adult CF patients are colonized by this pathogen [2]. Later during colonization of the lungs, non-motile, anaerobic, mucoid variants of P. aeruginosa form a biofilm, a structure that confers resistance to several antimicrobial agents [3]. Usually, the microorganisms account for less than 10% of the dry weight of the biofilm, while 90% is composed of bacterially-produced extracellular polymeric substances (EPS) that form a matrix in which the bacterial cells are embedded [4].
The most abundant component of the EPS produced by P. aeruginosa is a polyanionic alginate, a copolymer of mannuronic and glucuronic acids [5]. Typically, P. aeruginosa mucoid strains arise in the lungs of CF patients due to mutations in the mucA gene or when MucA is degraded by regulated intramembrane proteolysis [6]. Conversion of non-mucoid P. aeruginosa strains to mucoid variants can also be the consequence of selective pressure operated by bacteriophages [7], [8]. Recent studies have shown that bacteriophages can drive the emergence of numerous variants with enhanced virulence potential [9], [10].
The majority of Pseudomonas tailed phages belong to the order Caudovirales with three main families. Strictly lytic phages are found among the Myoviridae with a long contractile tail, and the Podoviridae with a short tail, whereas members of the Siphoviridae are temperate phages, implying the possibility to undergo lytic or lysogenic interactions with their host [11]. The most striking feature emerging from phage genome comparative analyses is that they are extensively mosaic, with different segments having distinct evolutionary histories. A simple general explanation is that horizontal genetic exchanges play a dominant role in shaping these genome architectures [12], [13]. Gene modules are exchanged using host- or phage-encoded recombination machinery. Although some phages can switch host using different mechanisms, the host preferences represent a significant barrier to genetic exchange. Moreover, phages infecting a common host can also exhibit substantial diversity, creating additional barriers to genetic exchange [12], [13]. Horizontal gene transfer and the pattern of vertical, divergent evolution of phage genomes has led to the definition of different phage genera, and consequently, to a classification based on criteria related to phage genome organization and replication strategy [11]. Despite rapid phage evolution and the short generation time, viral genomes can be stably maintained over ecologically significant time and distance, and this allows their classification. Viral species can be identified and they appear to be globally widespread. Indeed, related members of specific genera with sequence identity up to 99%, can be isolated from different habitats across the globe [14], [15]. The part of the phage genome that varies greatly within each genus is confined to genes encoding the metabolic conversion proteins (early region) and the tail spikes, indicating a local adaptation necessary to infect specific hosts in specific environmental conditions.
In the present study we describe a new phage, vB_PaeP_Tr60_Ab31, whose genome is the result of recombination between two phages belonging to two different families. This phage exerts a selective pressure on P. aeruginosa, which could be deleterious to chronically infected patients.
Materials and Methods
Ethics Statement
The present project is in compliance with the Helsinki Declaration (Ethical Principles for Medical Research Involving Human Subjects). Strains were collected from sputum as part of the patients' usual care, without any additional sampling. The ethic committee “Comité Consultatif pour la Protection des Personnes dans la Recherche Biomédicale (CCPPRB) Ile-De-France”, who was consulted, specifically approved this study, and declared that patient informed consent was not needed.
Bacterial Strains
The two reference P. aeruginosa strains UCBPP-PA14 [16] and PAO1 [17] were purchased from the “Collection de l'Institut Pasteur” (CIP, Paris, France), and C50 was a gift of U. Römling (Karolinska Institute, Sweden) [18]. The other strains were isolated from sputum of French CF patients, and were previously genotyped using Variable number of tandem repeats (VNTR) analysis (MLVA) [19], [20]. Strains were considered to belong to the same clonal complex when they shared at least 10 VNTR size alleles out of 15. Serological typing was performed using 4 polyvalent and 16 monovalent antisera (Bio-Rad), as described [21]. Briefly, the slide agglutination procedure was performed on 24 h cultures of P. aeruginosa: one loop (0.01 ml) of bacterial culture (approximately 3×106 CFU) was mixed with one drop (0.01 ml) of each antiserum (firstly the four polyvalent sera, then the four monovalent sera, corresponding to the positive polyvalent serum). The slide was gently shaken with a rotary movement, and the mixture was examined with the naked eye over a dark surface. A positive reaction was defined as the appearance of agglutination in a maximum of 2 min.
Phage Amplification and Purification
Phages were amplified on fresh LB agar plates at a ratio of 1 phage for 1000 bacteria. An overnight culture of bacteria grown in LB medium was concentrated 10 times in saline magnesium (SM) phage buffer (50 mM Tris-HCl pH 7.5, 100 mM NaCl, 8.1 mM MgSO4, 0.01% gelatin). Phages were added and, after 15 min of incubation at room temperature, the mixture was poured onto a round plate together with 4 ml of soft agar. After complete bacterial lysis (≈ 8 h), 5 ml of SM phage buffer supplemented with a drop of chloroform were added to the recovered soft agar, containing phage particles. After centrifugation, the supernatant was filtered through a 0.22-μm pore size membrane, and kept at 4°C.
Small Drop and Double Agar Plate Assay
For the small drop assay, 50 μl of 10X concentrated P. aeruginosa overnight culture were added to 4 ml molten soft agar (0.7%), and poured over an LB agar plate. Then, 10 μl of serially diluted test lysate were spotted onto the bacterial lawn. For the double agar plaque assay, a mixture of 50 μl of bacterial suspension and 10 μl of phages at ten-fold serial dilutions was kept for 15 min at room temperature, and then poured onto a solid agar plate with 4 ml of soft agar. Plates were inverted and incubated overnight at 37°C.
Liquid Infection
LB medium (10 ml), supplemented with 10 mM CaCl2, was inoculated at 2.5% with an overnight culture of the indicator strain, and incubated at 37°C. When an OD600 of 0.6 was reached, phage suspensions at different multiplicities of infection (M.O.I.) were added. The OD600 was periodically measured, and when a significant reduction of the culture density was recorded, 50 μl of chloroform were added in order to facilitate bacterial lysis and release of phages. The suspension was centrifuged at 2,500×g for 10 min at 4°C to eliminate bacterial debris, and the supernatant was filtered through a 0.22 μm filter.
Electron Microscopy Examination
Phage preparations were stained with 2% potassium phosphotungstate (pH 7.0), and then visualized using an EM208S transmission electron microscope (FEI, Eindhoven, The Netherlands) operating at 80 kV.
Isolation of Resistant Bacteria
Putative resistant bacteria were isolated by simply picking bacterial colonies growing inside the lysis zone of a small drop assay, and streaking them onto new plates. Putative resistant bacteria were also recovered at the end of the liquid infections, by directly streaking 1 μl of the phage-bacterial mixture on a solid agar plate. Up to twenty colonies were picked and challenged with phages through the small drop assay. Some of them were susceptible to phage Ab31, and were thereafter called “non-resistant”. Thermolysates of both resistant and non-resistant strains were prepared by resuspending a colony in 100 μl of water, heating at 95°C for 10 min, followed by cooling on ice for 5 min. Centrifugation was performed at 2,500×g for 10 min at 4°C to pellet cell debris, and 2 μl of the supernatant were used for PCR amplification.
Phage DNA Purification
Phage DNA was purified using a rapid method adapted from [22], as described in [14]. Briefly, phages were amplified on fresh LB agar plates for 8 h at 37°C, then 5 ml of SM buffer were added to the plate, followed by overnight incubation at 4°C. The buffer was transferred to a tube, and bacterial debris were pelleted by centrifugation at 2,500×g for 10 min at 4°C. A mixture of 0.2 ml 2 M Tris-HCl pH 7.5, 0.4 ml 0.5 M EDTA, 0.2 ml 10% SDS and 10 μl diethylpyrocarbonate was added to 4 ml of supernatant. Following incubation at 65°C for 30 min, the tube was cooled on ice, and 1 ml of 5 M KOH was added. After 1 h incubation on ice, centrifugation was performed at 25,000×g for 20 min at 4°C. DNA contained in the supernatant was precipitated with 2 vol of absolute ethanol, pelleted by centrifugation, washed twice with 70% ethanol, dried and dissolved in 0.4 ml of TE buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA). Bacterial DNA was purified using the classical CTAB (cetyl-trimethylammonium bromide)-phenol extraction method as described [19]. Purified DNA was resuspended in TE buffer. The quality and concentration of DNA was measured using a ND-1000 Spectrophotometer (NanoDrop®, Labtech, Palaiseau, France).
Sequencing
Whole genome sequencing was performed by the CNRS sequencing facility in Gif sur Yvette (IMAGIF) using the Illumina platform (Illumina Genome Analyzer IIx). Assembly of short sequence reads was performed using BioNumerics tools (Applied Maths, Sint-Martens-Latem, Belgium) as described [14]. The phage genome was annotated automatically using the BaSyS annotation tools [23]. Bacterial genome contigs were annotated using BioNumerics annotation tools. Detailed methods are available on the website http://bacteriophages.igmors.u-psud.fr.
The annotated Ab31 phage sequence has been deposited at EMBL-EBI under accession number HG798806. Total reads of bacterial genome sequences have been deposited at EMBL-EBI under accession number PRJEB5001.
PCR Detection of Phage DNA
Oligonucleotides selected to test for the presence of phage DNA in resistant bacteria and to analyze the deletions in bacterial genomes, are listed in Table 1. PCR was performed using purified DNA and Taq polymerase as recommended by the supplier (VWR, Strasbourg France). PCR products were analyzed on 2% agarose gels in 0.5X TBE buffer.
Table 1. List of primers used for PCR amplification.
| Phage Ab31 | |
| Ab31-Reg1-F | GACTCAGACCACTGAGATGA |
| Ab31-Reg1-R | ACGTGTTGGCAGTTGTAGAA |
| Ab31-Term-F | TACAACGCGGATATCCGTGT |
| Ab31-Term-R | TGCTCCCTCTGATGGACAAA |
| P. aeruginosa | |
| PaTr60_Del22kb_F | TCATCCACTGTACGCCGCCG |
| PaTr60_Del22kb_R | CCGTTCCTGATGCTCGACCAGT |
| PaTr60_Del11kb_F | GACCATGACCTTGTCGCCAT |
| PaTr60_Del11kb_R | AGGAGGAAATGGGTGCGGAA |
| Porin1_PaerDel234_F | GAAATAGAGATTGCGCAGGC |
| Porin1_PaerDel234_R | CACCTTCGACGAGAGACACA |
| CupA_Paer_F | AGGATCGTCGGCGAGTAGTA |
| CupA_Paer_R | CTCTATAGCGGCTACTACAC |
| Porin2_PaerDel234_F | CTCAAGGACATCTACCGACA |
| Porin2_PaerDel234_R | AAGTCGCCGATCTGGATGAA |
| PaerDel234_Flank_F | TCCATCGCCTGCATGGCTTC |
| PaerDel234_Flank_R | CGGCATAACTTCAATCAGGC |
Results
Ab31 Virulence Spectrum
P. aeruginosa phage vB_PaeP_Tr60_Ab31, subsequently called Ab31, was isolated in Abidjan (Ivory Coast) as part of a study to determine the phage diversity in waste water of this city [Essoh et al. submitted]. Ab31 was originally enriched on P. aeruginosa strain PA14, and subsequently amplified in this strain or in Tr60 (both of serotype O10). A total of 36 P. aeruginosa strains were tested for their susceptibility to the phage, including strains from the most frequently encountered clonal complexes in CF patients, PA14 and C50 [24], and reference strain PAO1 (Table 2). Six strains were shown to belong to the PA14 clonal complex (Tr60, Tr162, C7-11, C5-17, C9-12 and C8-12), and seven strains (Tr60, Tr162, C1-3, C3-1, C3-11, C4-14 and C9-5) were of serotype O10. The latter strains were selected in case the host O antigen would serve as a receptor for the phage, as described for Vibrio cholera phage VP4 [25]. Ab31 was responsible for complete lysis of Tr60 and Tr162, two non-mucoid strains with the same genotype, isolated from two CF patients at a one year interval, in the same hospital [19]. With the other strains no significant signs of lysis were detected.
Table 2. List of the strains used and susceptibility to Ab31.
| Strain | Description | Serotypea | Source or reference | Ab31 growthb |
| PA14 | Sequenced | 10 | [16] | C+++ |
| PAO1 | Sequenced | 5 | [17] | 0 |
| C50 | Clone C | UN | [18] | 0 |
| Tr60 | PA14-clone | 10 | [19] | C+++ |
| Tr162 | PA14-clone | 10 | [19] | C+++ |
| C7-11 | PA14-clone | 15 | [14], [20] | trace |
| C5-17 | PA14-clone | 17 | [14], [20] | 0 |
| C9-12 | PA14-clone | 17 | [14], [20] | 0 |
| C8-12 | PA14-clone | 6 | [14], [20] | trace |
| C1-1 | 5 | [14], [20] | 0 | |
| C1-2 | 3 | [14], [20] | 0 | |
| C1-3 | 10 | [14], [20] | 0 | |
| C1-11 | Mucoid | 15 | [14], [20] | trace |
| C1-14 | 1 | [14], [20] | 0 | |
| C2-10 | 4 | [14], [20] | 0 | |
| C2-18 | 12 | [14], [20] | 0 | |
| C3-1 | 10 | [14], [20] | 0 | |
| C3-2 | 13 | [14], [20] | trace | |
| C3-11 | 10 | [14], [20] | 0 | |
| C3-15 | 17 | [14], [20] | 0 | |
| C3-16 | 1 | [14], [20] | 0 | |
| C3-18 | 16 | [14], [20] | 0 | |
| C3-19 | 6 | [14], [20] | 0 | |
| C4-14 | 10 | [14], [20] | 0 | |
| C5-2 | 17 | [14], [20] | 0 | |
| C5-13 | 17 | [14], [20] | 0 | |
| C7-6 | 3 | [14], [20] | 0 | |
| C8-5 | 5 | [14], [20] | 0 | |
| C8-7 | 12 | [14], [20] | 0 | |
| C8-14 | 1 | [14], [20] | 0 | |
| C8-15 | Mucoid | 1 | [14], [20] | 0 |
| C8-20 | 2 | [14], [20] | 0 | |
| C9-5 | 10 | [14], [20] | 0 | |
| C9-11 | 17 | [14], [20] | 0 | |
| C9-17 | 17 | [14], [20] | 0 | |
| C10-5 | UN | [14], [20] | 0 |
UN, unknown.
5 μL of Ab31 stock suspension (≈ 108 PFU/ml) were spotted on P. aeruginosa lawns. C+++, complete clearing; trace, a few individual plaques; 0, a turbid spot where the pipette tip touched the agar.
Phage Characteristics
The morphology of phage Ab31 was determined by transmission electron microscopy (Fig. 1). The phage possesses an icosahedral head with a diameter of approximately 60 nm, and a short non-contractile tail. Moreover, the subterminal tail spikes were similar to those of phage AF of P. putida [26].
Figure 1. Electron microscopy analysis of phage Ab31.
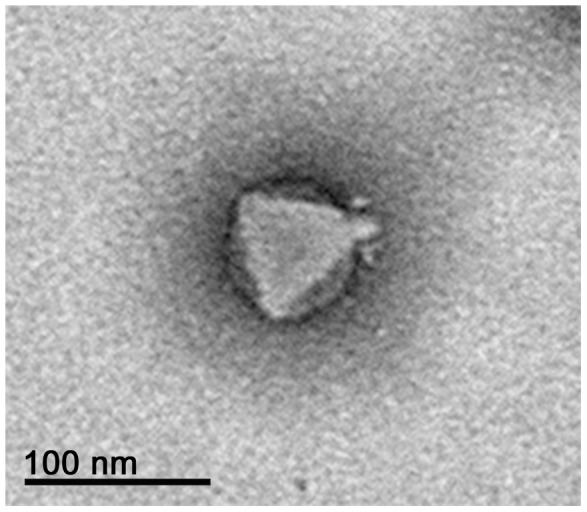
Scale bar represents 100
Infections in solid agar plates and in liquid medium were performed to analyse the Ab31 multiplication characteristics. When a phage suspension was analyzed on indicator strains using the double agar plaque assays, small clear plaques without a halo were observed. Dot assay revealed a clear zone with only a few small resistant colonies. Since it was known that many phages require CaCl2 to adsorb on the bacterial surface, the infection in liquid medium was performed using LB supplemented or not with 10 mM of CaCl2. Upon infection at an M.O.I. of 0.01, in the presence of 10 mM CaCl2, production of PFUs was stimulated 100-fold. In these conditions, the adsorption time was 4 min and the burst size was 30–50 phages per cell.
Infection in liquid LB medium at an M.O.I. of 0.1 never led to a complete clearing of the bacterial culture (Fig. 2), but we observed that not all the bacteria that survived after infection were resistant, when later challenged with Ab31. This resembles the phenomenon of persistence, in which a subset of an isogenic bacterial population occurring within a susceptible population, tolerates antibiotics [27].
Figure 2. Growth curve of uninfected PA14 (dark grey curve) and of PA14 infected by Ab31 at an M.O.I. of 0.1 (light grey curve).

Interestingly, we observed that phage infection led to a change in color of the bacterial culture from yellowish-green to green. It is likely that the presence of the phage affects the production of the pyoverdine, a virulence factor of P. aeruginosa [28].
Genome Characteristics
The Ab31 genome encompasses 45,550 bp, and the overall GC-content is ≈ 57%, which is lower than that of the P. aeruginosa PA14 genome (66.3% G+C), a characteristic shared by other P. aeruginosa phages [29]. Comparing the virtual gel obtained by in silico restriction endonuclease analysis with the experimental restriction enzyme banding pattern (Fig. 3), it was possible to establish that Ab31 DNA is apparently circular. Indeed, the number of fragments expected from in silico digestion of the circular Ab31 DNA with EcoRI, HindIII, SmaI, SspI, ClaI, SalI and SphI was 17, 12, 12, 6, 18, 23 and 19 respectively, perfectly matched, in number and in size, with the bands obtained by experimental agarose electrophoresis. Ab31 DNA digestion using NotI and PvuII, from which 3 and 2 fragments, respectively, were expected using in silico digestion analysis, produced many faint bands in addition to the expected ones. This phenomenon, which was observed repeatedly, indicates the existence of a non-specific digestion also called relaxed sequence recognition or star activity [30].
Figure 3. Restriction enzyme analysis of the Ab31 genome.

Ab31 phage DNA (2 μg) digested with EcoRI (1), HindIII (2), SmaI (3), SspI (4), NotI (5), ClaI (6), PvuII (7), SalI (8) and SphI (9) were analyzed by electrophoresis on a 0.8% agarose gel. On the left the λDNA/HindIII and on the right 1 kbp ladder are reported and they were used as molecular weight markers (Mw).
Overall the Ab31 genome did not align with any known phage sequence. However, at the nucleotide level it showed some rare regions of homology with structural genes of phage AF [26] and of a prophage of P. putida GB-1 strain [CP000926-1] (up to 70% DNA-DNA similarity), and with genes involved in replication in phage PAJU2 [31] (from 85 to 97%). At the protein level it was possible to observe additional similarities with AF (shown in red on Fig. 4) and PAJU2 (shown in blue in Fig. 4). AF and PAJU2 are two lambdoid phages whose genomes are respectively 42,689 bp and 46,872 bp long. Attempts to localize the genome ends by PCR analysis were not successful, as expected if the genome adopts a circular configuration. Therefore we were not able to determine the position of the first nucleotide, and we decided to assign it by comparison with the related phage AF.
Figure 4. Annotation of the Ab31 genome.

The Ab31 hypothetical terminal ends are at the 1 position. The morphogenesis module includes proteins similar to those encoded by phage AF (red), whereas the replication, recombination and lysis modules consist of genes similar to those of phage PAJU2 (blue). Genes encoding hypothetical proteins which have homologies with other phages are shown in purple color. Genes encoding hypothetical proteins of unknown function are shown in green color.
In the Ab31 genome, 69 putative “open reading frames” (ORFs) were identified. Thirty five ORFs were transcribed on the positive strand (Fig. 4). On the basis of sequence similarity comparisons in the GenBank database, 25 ORFs could be assigned to putative functions. The majority of the other ORFs exhibited similarity to uncharacterized bacterial or phage proteins. No tRNA genes were predicted.
Nucleotide position 1 was 60 bp upstream of the ATG codon of a putative protein containing a Helix Turn Helix domain, and sharing similarities with the small terminase subunit encoded by Escherichia coli phage phiv10 [32] and Salmonella enterica phage epsilon 15 [33]. A putative phage large terminase subunit, encoded immediately downstream of the predicted first coding sequence, shared similarities (maximal identity of 77%, E-value lower than e-200) with a prophage-encoded sequence located within the S. enterica serovar Wandsworth str. A4-580 genome. The terminase genes constitute the packaging module typically located at the beginning of the so-called late region of the phage genome. A large part of the late region usually encodes the morphogenesis proteins, whereas genes located in the early region are necessary to initiate the phage multiplication cycle. Following phage adsorption to the bacterial surface, early genes are injected first and, in some phages, they allow complete injection of the phage genome [34]. Phage Ab31 showed high similarity in the late region with the podovirus AF of P. putida and the prophage of P. putida strain GB-1: indeed, eight coding sequences reported in Fig. 4 resembled those of phage AF. These included the putative major capsid protein (maximal identity of 75%, E-value of 8e-180), internal virion protein B (maximal identity of 35%, E-value of 4e-6), the structural lysozyme (maximal identity of 33%, E-value of 8e-32) and the tail spike protein (maximal identity of 40%, E-value of 4e-37). Moreover, three hypothetical proteins located in the same region showed similarities with hypothetical proteins gp4, gp9 and gp12 of phage AF.
The second major block of genes of the Ab31 genome, downstream of the putative tail spike coding region, constitutes the so-called early/middle region. Twenty-three putative and hypothetical proteins encoded by genes located in this region shared similarities with the P. aeruginosa siphovirus PAJU2, showing maximal identity percentages that vary from 39% to 100%. This region starts with a putative acetyl-transferase sharing 46% identity with the PAJU2 acetyl-transferase (E-value lower than e-200), and a putative PAJU2-like integrase (identity 99% with PAJU2; E-value lower than e-200). Other related proteins included putative replication proteins O and P, NinB protein, phage antitermination protein Q and endolysin (Fig. 4).
Bacterial Resistance
Different conditions were used for infecting bacteria, in order to favor different resistance mechanisms. Most of the resistant and/or lysogenic bacteria were obtained from typical infection at 37°C in liquid medium of P. aeruginosa strains PA14, Tr60 and Tr162, at an M.O.I. of 1, 0.1 or 0.01. Some resistant Tr60 bacteria were also recovered from an infection in liquid medium performed at 42°C at an M.O.I. of 1. In this experiment the incubation was prolonged for 72 h, and the phage suspension was added each 24 h at the same M.O.I., for a total of three infections. Ab31-resistant variants of PA14 and Tr162 were also recovered from infections performed on solid agar plates at 37°C or 30°C, using a small drop plaque assay, and extending the incubation for 72 h in order to allow the growth of resistant bacteria inside the lysis zone. With the different approaches, about 80% (91 out of 114) of recovered bacteria were confirmed to be resistant to the phage. A majority of the resistant bacteria obtained from plates formed mucoid colonies, with entire margins and smooth surfaces, a particular characteristic often observed with P. aeruginosa strains isolated from sputa of CF patients [Sousa, 2013 #1739]. In contrast, non-resistant bacterial strains derived from the same experiment showed a non-mucoid phenotype and shared the same morphological characteristics with the wild type uninfected bacterial strain. From infected bacteria incubated on plates at 30°C, some mucoid resistant variants showing a brown pigmentation were also isolated. Ten resistant isolates were serotyped to check whether a switch had occurred, but they were all serotype O10 like the parental strains.
In order to test for the existence of lysogens, the presence of the phage DNA was searched in the Ab31-resistant bacteria after two passages on solid agar medium. Two regions of the phage genome were amplified, designated as Reg1 and Term, and predicted to produce 550 bp and 600 bp long amplicons respectively. The results for some of the resistant isolates and for Ab31 phage DNA used as a control, are shown in Fig. 5. When the DNA of the resistant bacteria was amplified using the Reg1 (Fig. 5A) or Term primers (Fig. 5B), a band of the expected size was detected with every tested isolate except for Tr60-100A. This Ab31-resistant variant was derived from infection in liquid medium at 37°C at an M.O.I. of 0.1. All the resistant variants from solid infection were positive for phage DNA, whereas only 40% of resistant from liquid infection contained phage DNA. The isolates showing a brown pigmentation were not lysogenized.
Figure 5. PCR detection of the Ab31 genome in resistant bacteria using Reg1 (A) and Term primers (B).

Tr60-E (5) and PA14-P1 (6) are derived from infection on solid agar plates at 37°C and 30°C, respectively. Tr60-10A (2), Tr60-100B (3) and Tr60-100A (4) are derived from an infection assay performed at 37°C in liquid medium at an M.O.I. of 0.1. PA14 (7), Tr60 (1) and Ab31 (8) are used as negative and positive controls, respectively. Samples were run on a 2% agarose gel for 45 min at 135 V. Mw, 100 bp ladder molecular weight markers.
Bacterial Genome Sequence Analysis
In order to study the basis of the resistance and to identify the possible integration site of the phage genome in the bacterial chromosome, the original Tr60 strain and four Ab31 resistant variants were chosen for whole genome sequencing. Tr60-10A and Tr60-100B were two putative lysogenic bacteria derived from infection in liquid medium at an M.O.I. of 1 and 0.1, respectively. Tr60-100A was a non-lysogenic isolate derived from infection in liquid medium performed at an M.O.I. of 0.1. PA14-P1 was a putative lysogenic bacteria. Tr60-10A and Tr60-100A showed a mucoid phenotype. The bacterial draft genomes were assembled and partially annotated using P. aeruginosa PA14 as a reference [16], [35].
Upon alignment of the sequenced genomes, two deletions were found in the P. aeruginosa Tr60 genome as compared to that of PA14. The first deletion encompassed approximately 22 kbp (coordinates 1919495 to 1941370). It started with 2.5 kbp of DNA of unknown function and ended inside a gene encoding a pirin-like protein (ORF PA14_22080 to ORF PA14_22280), and also contained genes for a resolvase and a recombinase. The second deletion encompassed approximately 11 kbp, and covered exactly the sequence of the Pf1 prophage of P. aeruginosa PA14 (Genbank: AY324828). Both deletions were identified in Tr60, in all Tr60 variants that were sequenced and in the genome of reference strain PAO1. The existence of the two deletions was confirmed by PCR in Tr60 and its Ab31-resistant variants using primers localised in the flanking regions (Table 1; Fig. 6). The deletions were also observed in Tr162 and in PAO1, as expected. In addition to the two deleted regions, approximately 215 “single nucleotide polymorphisms” (SNPs) were found when the genome sequences of Tr60 and PA14 were compared.
Figure 6. PCR investigation of the 11 kbp (A) and 22 kbp (B) deletions observed in Tr60.

C9-12 (1), C5-17 (2), C7-11 (3), C8-12 (4), PAO1 (5), PA14 (6), Tr162 (7), Tr60 (8), Tr60-10A (9), Tr60-100A (10). Samples were run in a 2% agarose gel for 45 min at 135 V. Mw, 100 bp ladder molecular weight markers.
We then looked at the differences between Tr60 and its Ab31-resistant variants. A deletion of about 234 kbp was found in Tr60-10A and Tr60-100A, two mucoid variants obtained from different infections performed with the same Tr60 bacterial culture. Using P. aeruginosa PA14 as a reference genome, 175 coding sequences were found to lie within this region (Table 3, genome coordinates approximately 3190870 and 3424480 in PA14). Some of these coded for enzymes involved in amino acid uptake or biosynthesis, for glucose metabolism and for transmembrane proteins. No homologous genes were found in the other regions of the bacterial genome except for the porins. A cluster of five genes involved in the CupA fimbrial organelle assembly was possibly relevant to phage resistance: the chaperone CupA1, the fimbrial subunit CupA2, the usher CupA3, CupA4, an atypical adhesin, and the chaperone CupA5 [36]. Primer pairs were selected within this region (in two porin-encoding genes and in the cupA4 gene), and in the flanking sequences, in order to confirm the existence of the deletion by PCR amplification. When amplification was performed with primers localised inside the region of deletion, an amplicon was observed for all the samples tested except for Tr60-10A and Tr60-100A (the result for cupA is shown on Fig. 7A). In contrast, amplification with the Flank234 primers, localised on both sides of the deleted region, produced a 600 bp amplicon only for Tr60-10A and Tr60-100A (Fig. 7B). In order to check whether the deletion was pre-existing in a subpopulation of the Tr60 culture before phage infection, 94 bacterial colonies were picked and PCR was performed on thermolysates with the Flank 234 and porin primers. The results showed that none of the isolates were deleted for the 234 kbp region (data not shown).
Table 3. List of coding sequences (CDS) and their position in the 234 kbp deleted region.
| CDS | Position | Product |
| 1 | 87..1418 | amino acid permease |
| 2 | 1482..2921 | gamma-aminobutyraldehyde dehydrogenase |
| 3 | 2967..4220 | diaminobutyrate–2-oxoglutarate aminotransferase |
| 4 | 4267.5229 | dehydrogenase |
| 5 | - strand (5522..7501) | acetate permease |
| 6 | - strand (7558..9828) | acyl-CoA synthetase |
| 7 | - strand (9291..10061) | dehydrogenase |
| 8 | - strand (10058..11284) | acyl-CoA dehydrogenase |
| 9 | - strand (11634..12851) | FadE36, aminoglycoside phosphotransferase |
| 10 | 12886..14838 | propionate catabolism operon regulator |
| 11 | - strand (14904..15443) | hypothetical protein |
| 12 | - strand (15466..17769) | paraquat-inducible protein B |
| 13 | - strand (17762..18619) | paraquat-inducible protein A |
| 14 | - strand (19040..20638) | aldehyde dehydrogenase |
| 15 | - strand (20752..21747) | hypothetical protein |
| 16 | - strand (21775..22950) | hypothetical protein |
| 17 | - strand (22981..24408) | MFS transporter |
| 18 | - strand (24489..25907) | porin |
| 19 | - strand (25912..26925) | 4-hydroxythreonine-4-phosphate dehydrogenase |
| 20 | - strand (26922..27881) | hypothetical protein |
| 21 | - strand (27874..29298) | MFS transporter |
| 22 | 29301..30479 | hypothetical protein |
| 23 | 30464.32554 | hypothetical protein |
| 24 | 32544..33569 | LysR family transcriptional regulator |
| 25 | 33651..34127 | hypothetical protein |
| 26 | 34342..35232 | ABC transporter substrate-binding protein |
| 27 | 35313..36029 | amino acid permease |
| 28 | 36031..36708 | amino acid ABC transporter permease |
| 29 | - strand (36724..37608) | hypothetical protein |
| 30 | - strand (37734..39329) | signal transduction protein |
| 31 | - strand (39418..40800) | dehydrogenase |
| 32 | - strand (40640..41758) | hypothetical protein |
| 33 | - strand (41808..43745) | TetR family transcriptional regulator |
| 34 | - strand (42545..44821) | hydrogen cyanide synthase HcnC |
| 35 | - strand (43801..45267) | hydrogen cyanide synthase HcnB |
| 36 | - strand (45192..45626) | hydrogen cyanide synthase HcnA |
| 37 | 45789..47114 | adenylate cyclase |
| 38 | - strand (47111..47767) | hypothetical protein |
| 39 | - strand (48272..49708) | carboxylate-amine ligase |
| 40 | 48802..51048 | hypothetical protein |
| 41 | 50993..51982 | hypothetical protein |
| 42 | - strand (51966..52247) | hypothetical protein |
| 43 | 52256..52957 | hypothetical protein |
| 44 | - strand (52961..55336) | sensor/response regulator hybrid |
| 45 | 56396..56686 | hypothetical protein |
| 46 | - strand (56717..57031) | hypothetical protein |
| 47 | - strand (56994..57581) | hypothetical protein |
| 48 | - strand (57371..58447) | hypothetical protein |
| 49 | - strand (59123..60370) | hypothetical protein |
| 50 | - strand (59701..60759) | hypothetical protein |
| 51 | - strand (60474..61766) | hypothetical protein |
| 52 | - strand (61900..62265) | hypothetical protein |
| 53 | 62570..64174 | glycogen synthase |
| 54 | 64033..65925 | glycosyl hydrolase |
| 55 | 65918..67972 | 4-alpha-glucanotransferase |
| 56 | 67752..70745 | maltooligosyl trehalose synthase |
| 57 | 71060..73210 | glycosyl hydrolase |
| 58 | 76155..77918 | cardiolipin synthase 2 |
| 59 | 76625..78910 | hypothetical protein |
| 60 | - strand (78912..81272) | glycogen branching protein |
| 61 | - strand (81107..84409) | trehalose synthase |
| 62 | - strand (84420..86534) | hypothetical protein |
| 63 | - strand (86558..87439) | KU domain-containing protein |
| 64 | - strand (87462..87704) | hypothetical protein |
| 65 | - strand (87718..88263) | hypothetical protein |
| 66 | - strand (88437..90566) | hydroperoxidase II |
| 67 | - strand (90647..90814) | hypothetical protein |
| 68 | 91301..91711 | hypothetical protein |
| 69 | - strand (91718..94156) | glycogen phosphorylase |
| 70 | - strand (94209..94496) | hypothetical protein |
| 71 | 94740..94928 | hypothetical protein |
| 72 | - strand (94948..95991) | short-chain dehydrogenase |
| 73 | - strand (95834..96352) | ompetence-damaged protein |
| 74 | - strand (96363..96608) | metallothionein |
| 75 | - strand (96913..99468) | ATP-dependent DNA ligase |
| 76 | - strand (99453..100118) | hypothetical protein |
| 77 | 99851..100615 | hypothetical protein |
| 78 | - strand (101068..102432) | transporter |
| 79 | - strand (102461..103387) | hypothetical protein |
| 80 | - strand (103126..104031) | EAL domain-containing protein |
| 81 | - strand (103980..104669) | chaperone CupA5 |
| 82 | - strand (104683..108297) | fimbrial subunit CupA4 |
| 83 | - strand (106041..108659) | usher |
| 84 | - strand (108643..109476) | chaperone CupA2 |
| 85 | 110882..112108 | fimbrial subunit CupA1 |
| 86 | 112535..113170 | hypothetical protein |
| 87 | - strand (113206..114660) | aldehyde dehydrogenase |
| 88 | - strand (114676..116313) | dehydrogenase |
| 89 | - strand (116418..117350) | LysR family transcriptional regulator |
| 90 | - strand (117387..118523) | hypothetical protein |
| 91 | 118649.119554 | LysR family transcriptional regulator |
| 92 | 119600..120130 | hypothetical protein |
| 93 | 120147..121259 | hypothetical protein |
| 94 | - strand (121794..122870) | O6-methylguanine-DNA methyltransferase |
| 95 | 123062..124045 | hypothetical protein |
| 96 | - strand (124030..124896) | hypothetical protein |
| 97 | - strand (124934..125998) | LysR family transcriptional regulator |
| 98 | 126034..127413 | major facilitator transporter |
| 99 | 127438..128667 | porin |
| 100 | 128700..129443 | LamB/YcsF family protein |
| 101 | 129529..131091 | hypothetical protein |
| 102 | 131159.131635 | hypothetical protein |
| 103 | - strand (131654..133426) | thiamine pyrophosphate protein |
| 104 | 133525..133995 | hypothetical protein |
| 105 | - strand (134394..136025) | short chain dehydrogenase |
| 106 | - strand (135137..136177) | esterase |
| 107 | - strand (136077..137552) | flavin-binding monooxygenase |
| 108 | 137699..138733 | AraC family transcriptional regulator |
| 109 | 138861..139727 | hypothetical protein |
| 110 | - strand (139729..141048) | transmembrane sensor protein |
| 111 | - strand (141294..142682) | MFS transporter |
| 112 | - strand (142477..144135) | permease |
| 113 | - strand (144849..147995) | TonB-dependent receptor |
| 114 | - strand (147592..148680) | hypothetical protein |
| 115 | - strand (148424..149845) | hypothetical protein |
| 116 | - strand (149077..150093) | hydrolase |
| 117 | - strand (150527..152425) | asparagine synthetase, glutamine-hydrolysing |
| 118 | - strand (152447..153649) | ring-hydroxylating dioxygenase, large terminal |
| 119 | - strand (153923..154405) | leucine-responsive regulatory protein |
| 120 | 154451..155173 | kynurenine formamidase, KynB |
| 121 | 155177..156427 | kynureninase |
| 122 | 155773..157989 | amino acid permease |
| 123 | 158230..160149 | hypothetical protein |
| 124 | 160165..162093 | hypothetical protein |
| 125 | - strand (162130..163506) | transcriptional regulator |
| 126 | 163546..163755 | hypothetical protein |
| 127 | 163932..165599 | hypothetical protein |
| 128 | - strand (165992..168586) | sensory box protein |
| 129 | - strand (168758..171127) | elongation factor G |
| 130 | 171142..173784 | TonB dependent receptor |
| 131 | 173892..175688 | carbamoyl transferase |
| 132 | 175732..176895 | MFS transporter |
| 133 | 176840..177565 | hydrolase |
| 134 | 176941..178200 | hypothetical protein |
| 135 | 178277..180169 | copper resistance protein A |
| 136 | 179851..181221 | copper resistance protein B |
| 137 | - strand (181241..182575) | hypothetical protein |
| 138 | - strand (182667..183848) | pyridoxal-phosphate dependent protein |
| 139 | - strand (183994..185604) | ABC transporter ATP-binding protein |
| 140 | - strand (185606..186622) | ABC transporter permease |
| 141 | - strand (186624..187697) | peptide ABC transporter permease |
| 142 | - strand (187699..189507) | ABC transporter substrate-binding protein |
| 143 | - strand (189511..192537) | TonB-dependent receptor |
| 144 | - strand (192730..193722) | LysR family transcriptional regulator |
| 145 | 193773..195158 | major facilitator transporter |
| 146 | - strand (195165..196064) | DNA-binding transcriptional regulator CynR |
| 147 | - strand (197377..198330) | Fe2+-dicitrate sensor, membrane protein |
| 148 | - strand (198327..199004) | RNA polymerase sigma factor |
| 149 | 199037..201157 | hypothetical protein |
| 150 | 203113..204468 | hypothetical protein |
| 151 | 204410..204787 | hypothetical protein |
| 152 | 204854..206836 | hypothetical protein |
| 153 | - strand (208017..209408) | serine/threonine transporter SstT |
| 154 | - strand (209734..211104) | amino acid permease |
| 155 | - strand (211261..212637) | glutamine synthetase |
| 156 | 213047..213901 | hypothetical protein |
| 157 | 213157..214200 | hypothetical protein |
| 158 | - strand (214325..215800) | hypothetical protein |
| 159 | - strand (215925..216731) | hypothetical protein |
| 160 | 217181..218839 | thiamine pyrophosphate protein |
| 161 | - strand (218847..220016) | hypothetical protein |
| 162 | - strand (219521..220477) | hypothetical protein |
| 163 | - strand (220564..222048) | transcriptional regulator |
| 164 | 222029..222676 | hypothetical protein |
| 165 | - strand (222751..223581) | hypothetical protein |
| 166 | - strand (223074..223547) | transcriptional regulator |
| 167 | 223638..224072 | hypothetical protein |
| 168 | 223754..225226 | bile acid/Na+ symporter family transporter |
| 169 | 225251..227203 | ring-cleaving dioxygenase |
| 170 | - strand (225281..226636) | glutathione reductase |
| 171 | - strand (227385..228275) | UTP-glucose-1-phosphate uridylyltransferase |
| 172 | - strand (228272..229873) | nucleotide sugar dehydrogenase |
| 173 | - strand (230015..230575) | transcriptional regulator |
| 174 | 230812..232002 | periplasmic multidrug efflux lipoprotein |
| 175 | 232018..233901 | multidrug efflux protein |
Figure 7. PCR investigation of the 234(A) and Flank234 primers (B).

Tr60 (1), Tr60-10A (2), Tr60-100A (3), Tr60-100B (4), PA14-P1 (5), PA14 (6), Negative control (7). The experimental conditions are those of Fig.5.
Compared to Tr60, Tr60-10A and Tr60-100A showed approximately the same amount of SNPs (respectively 29 and 26). The mutations occurred in the same coding sequences for both resistant bacteria. In particular, mutations were found in a transcriptional regulator, in the NADH dehydrogenase I subunit F, the pyoverdine biosynthesis protein PvcA, phenazine biosynthesis protein PhzD, C32 tRNA thiolase and in an MFS transporter. Specific mutations in a gene for an Usher protein and in a gene for a lipase chaperone, were detected in Tr60-10A and Tr60-100A, respectively. The sequence of the Tr60-100B isolate was compared with that of Tr60, and no SNPs were found.
The PA14-P1 genome showed 129 SNPs as compared to PA14. They affected genes encoding the hemolysin activator, the pyochelin synthase, an RNA methyltransferase, an acetyltransferase, the pyoverdin synthase, and several membrane proteins, including multidrug efflux pumps, type III secretion system proteins and an ABC transporter.
Search for the Phage Integration Site
To identify integrated Ab31 genomes, we first searched for phage reads among the three isolates found to possess phage DNA by PCR, but they were detected only in Tr60-100B. We then looked for phage-bacteria hybrid sequences among the total reads obtained for this isolate. The hybrid reads centered on a 64 bp region found at position 4552973–4553038 in the P. aeruginosa PA14 genome (and also in the Tr60 genome), and at position 21471–21534 on the Ab31 genome. These regions correspond in PA14 to the serine tRNA gene (PA14_51230) localized downstream of a glycosyltransferase gene, and, in the Ab31 genome, to a region that covers part of the phage integrase and part of a non-coding sequence upstream of this gene (Fig. 8).
Figure 8. Schematic representation of the Ab31 insertion region.

A 64P. aeruginosa Tr60 genome. The portion of the shared region that overlaps with the phage integrase encoding gene is underlined.
Discussion
Phage Ab31 is a temperate phage genomically related to both the virulent podovirus AF phage of P. putida and the temperate siphovirus phage PAJU2 of P. aeruginosa. The Ab31 virion structure resembles that of phage AF, S. enterica phage Epsilon 15 and E. coli phage phiv10, with similar spikes, previously shown to bind and cleave the O-antigen component of the host’s cell surface lipopolysaccharide [37]. Some Pseudomonas phages can diffuse through alginate present in Pseudomonas biofilms [38] owing to a depolymerizing enzyme that is part of the phage particles. One of the most significant examples of such an activity has been reported for phage AF. A halo surrounds the AF plaques at 30°C, due to an EPS-degrading activity within the tail spikes [26]. We did not see such a halo around Ab31 plaques, whatever the strain used or the temperature. Alignment of the tail spike protein sequences of phage Ab31 and phage AF showed limited homology only at the N-terminus (Fig. 9). Conservation of the N-terminal part is necessary for association of the spikes with the tail structure, whereas the C-terminal part of the spike protein, involved in recognition of and binding to the cell receptor, shows the highest level of variation [39], [40], [41]. This finding provides evidence that bacteria and bacteriophages have co-evolved in order to overcome the barriers that are imposed by one on the other. Recent observations suggest that bacterial resistance to phages in cystic fibrosis patients evolves with the duration of colonization [42]. The infection by Ab31 causes a slight change in the colour of the bacterial culture from yellowish-green to green, as also observed with phage PAJU2 [43]. Mutation in genes for pyoverdine biosynthesis were identified in some resistant isolates, but further analyses are necessary to determine their significance.
Figure 9. Alignment of the spike protein sequence of phages Ab31 and AF.

Among the diverse strains tested, Ab31 is specific for PA14 and for Tr60 and Tr162, two strains genetically close to PA14, and isolated from French CF patients in the same hospital. Other strains belonging to the same clonal complex but isolated at other locations were found to be resistant to Ab31, as were strains with the same O serotype. Sequencing of Tr60 revealed the presence of two regions of deletion, one of which corresponds to prophage Pf1, also designated Pf5 in the PA14 genome [44]. It was previously shown that Pf4, a Pf1-like prophage in PAO1, mediated the formation of small-colony variants, and was also a factor contributing to the bacterial virulence, but this was not confirmed for Pf5 in PA14 [45], [46]. The 22 kbp region corresponding to the second deletion encodes a resolvase and a recombinase, proteins that usually participate in DNA transfer.
Differences in the bacterial response to phage infection were observed in this study depending on the infection method used. From the infection on solid agar plate, clear lysis zones were observed, an indication that most cells were killed, whereas in liquid culture complete lysis was never obtained. This observation may be related to the formation of pseudolysogens after bacterial infection. The genomes of two sequenced Ab31-resistant variants (Tr60-10A and PA14-P1), shown to contain phage DNA when analysed by PCR, surprisingly did not contain phage reads. A possible explanation could be that the phage genome did not integrate in the bacterial chromosome but was retained in a small proportion of cells after infection. Indeed, there is evidence that some of the putative lysogenic bacteria lose the phage genome after several replatings. In contrast, Tr60-100B is a true lysogenic variant. The phage integrated within the bacterial genome through a site-specific recombination process using the shared 64 bp sequence, as shown for PAJU2 [43].
In this study, mucoid variants of P. aeruginosa Tr60 and PA14 were obtained after infection of the host in liquid medium. These variants were stably resistant to the phage, although they were not lysogenic. Miller et al. showed that temperate phages with elongated heads and flexible tails (similar to PAJU2 virions), induced from CF-associated P. aeruginosa strains, were capable of converting non-mucoid strains to the mucoid phenotype [8]. We may hypothesize that the presence of the phage induced a stress that caused mutations in genes involved in alginate production. An alternative explanation could be that the phage selected a subpopulation of mucoid bacteria, with the mucoid layer inhibiting early stages of infection. MucA is a negative regulator of alginate production through sequestration of AlgU, the primary sigma factor responsible for the expression of the alginate biosynthetic operon from the algD promoter [47]. Alternatively, conversion to mucoidy can occur when MucA is degraded by regulated intramembrane proteolysis operated by AlgW [6]. The activation of AlgW, and the consecutive proteolysis of MucA, is thought to be in response to extracellular stress, as well as the accumulation of misfolded envelope proteins. Interestingly, genome sequencing of two non-pigmented mucoid Ab31-resistant variants (Tr60-10A and Tr60-100A) revealed that no modifications of proteins involved in the alginate biosynthesis occurred. Among the Ab31 resistant variants we also obtained brown-colored, mucoid variants of PA14. The pigment which likely corresponds to pyomelanin, accumulated when the plates were kept at room temperature. It was shown that pyomelanin production is due to loss of the homogentisate gene, HmgA and this favors persistence in the lung of CF patients [48].
Notably, two mucoid variants of Tr60 carry a large deletion of 234 kbp corresponding to, among others, genes coding for proteins necessary to assemble a fimbrial organelle. This gene cluster which encodes components of the chaperone-usher pathway and a fimbrial unit, participates in biofilm formation [36], [49]. Conceivably fimbriae could be involved in phage adsorption but further investigation is required to confirm or refute this hypothesis. Other deleted genes that might act as phage receptors, are those for two porins, and one being a member of the LamB/YcsF family protein. Previous studies have shown that an outer membrane porin encoded by the ompLC gene in Edwardsiella ictaluri is required for phage sensitivity [50], while LamB is the receptor for Escherichia coli bacteriophage λ. LamB was shown to be sufficient to confer λ phage sensitivity upon transformation of the lamB gene into bacteria of different species [51], [52].
Since the two resistant isolates, in addition to the 234 kbp deletion, show a few nucleotide differences (≈ 40 SNPs) with Tr60, two hypotheses could be formulated to explain the origin of the deletion. It is possible that a variant subpopulation with the 234 kbp deletion preexisted in the Tr60 stock suspension, and that the phage infection led to its selection. We could not detect such variants by PCR analysis on 94 isolated colonies or on total DNA extracted from a Tr60 culture. Another possible explanation is that phage infection promoted rearrangement of the host genome. This hypothesis is supported by the finding that at both ends of the 234 kbp region present in the original Tr60 strain, there are sequences of 10 bp in length (ctcggcatga and ctcggcgatga) that differ by a single nucleotide insertion. Notably a similar sequence (c-cggcatga) was detected in the phage Ab31 genome at the end of the gene encoding an acetyl-transferase, upstream of the phage integrase. The 10 bp sequence “ctcggcgatga” constitutes the junction of the deleted region on the resistant bacterial genome. Moreover, the sequence upstream the 234 kbp region encodes several proteins involved in transposition, including a bacterial transposase. This suggests that the origin of the deletion in Tr60 was most probably a recombination and/or transposition event in which the phage was also involved. Large genomic deletions have been observed during early stage adaptation of P. aeruginosa in CF patients, but none were as large as 234 kbp, which represents about 3.6% of the genome [24], [53]. Rau and colleagues described a deletion of 148 kbp, encompassing the cupA cluster [53]. It is not known whether the presence of phages could play a role in the induction of such deletions. Ab31-resistant strain PA14-P1 showed no deletion corresponding to those which characterize Tr60-10A and Tr60-100A. A number of mutations in different genes were observed, but at this time it is impossible to know which one is responsible for phage resistance.
Looking at the Ab31 genome sequence it is possible to distinguish two main modules. The first, showing homologies with the AF phage genome, covers the so-called late region and contains sequences encoding the structural proteins of the phage, such as those for capsid, tail-to-head connector, tail and tail spikes. The second Ab31 genomic region encodes proteins involved in recombination and replication of the phage genome, and constitutes the so-called early/middle region. This contains several genes that show similarities with those of PAJU2 explaining why, although phage Ab31 shows a morphology typical of the virulent AF podovirus, it behaves as a temperate phage capable of lysogenizing P. aeruginosa strains. Indeed, the Ab31 insertion site in P. aeruginosa is the same as in PAJU2. Phages AF and PAJU2 infect P. putida and P. aeruginosa, respectively. These bacterial species are closely related, and phage genome exchanges probably occurred during infection of a lysogenic host by the virulent phage. As a result of their mosaic structure, some temperate phage genomes can migrate between unrelated bacteria [54]. Although genetically distant, phages AF and PAJU2 share a lambdoid genome organization which could favor genetic replacement (modular exchanges of gene blocks) [55], [56]. Similar events seem to occur between D3112-like phages morphologically identical to phage lambda, and the transposable coliphage Mu belonging to the Myoviridae family [57]. Several types of recombination events are thought to build phage genomes. There are examples of conserved sequences at gene boundaries that could serve to target homologous recombination at these positions, via transposition or site-specific recombination [13]. However a major contributor to phage genome building is illegitimate recombination, or recombination between short conserved sequences (a few bases), coupled with functional selection of genes [13], [58].
Conclusion
Our observations show that phage Ab31 is the result of a rare recombination event between genomes of two unrelated bacteriophages, normally infecting different bacterial species. It is capable of forming lysogens but its genome can also apparently persists unintegrated for a long time in the bacterial cells, with accompanying repression of virulence functions thus allowing the bacteria to escape lysis. In addition, we show that the phage exerts strong pressure on the bacteria by selecting for variants with new phenotypes, possibly improving their adaptation to chronic lung infection.
Acknowledgments
We are grateful to Barry Holland and Gilles Vergnaud for their comments on and corrections of the manuscript. This work has benefited from the facilities and expertise of the high throughput electron microscopy and sequencing platform of IMAGIF (Centre de Recherche de Gif - www.imagif.cnrs.fr).
Funding Statement
LL holds a PhD fellowship co-financed by the Direction Générale de l’Armement (DGA) and by the Paris Saclay “Initiative d’excellence” (IDEX). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Penketh AR, Wise A, Mearns MB, Hodson ME, Batten JC (1987) Cystic fibrosis in adolescents and adults. Thorax 42: 526–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brussow H (2012) Pseudomonas biofilms, cystic fibrosis, and phage: a silver lining? MBio 3. [DOI] [PMC free article] [PubMed]
- 3. Hogardt M, Heesemann J (2010) Adaptation of Pseudomonas aeruginosa during persistence in the cystic fibrosis lung. Int J Med Microbiol 300: 557–562. [DOI] [PubMed] [Google Scholar]
- 4. Flemming HC, Wingender J (2010) The biofilm matrix. Nat Rev Microbiol 8: 623–633. [DOI] [PubMed] [Google Scholar]
- 5. Schurks N, Wingender J, Flemming HC, Mayer C (2002) Monomer composition and sequence of alginates from Pseudomonas aeruginosa . Int J Biol Macromol 30: 105–111. [DOI] [PubMed] [Google Scholar]
- 6. Qiu D, Eisinger VM, Rowen DW, Yu HD (2007) Regulated proteolysis controls mucoid conversion in Pseudomonas aeruginosa . Proc Natl Acad Sci U S A 104: 8107–8112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Al-Dujaili AH, Harris DM (1975) Pseudomonas aeruginosa infection in hospital: a comparison between 'infective' and 'environmental' strains. J Hyg (Lond) 75: 195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Miller RV, Rubero VJ (1984) Mucoid conversion by phages of Pseudomonas aeruginosa strains from patients with cystic fibrosis. J Clin Microbiol 19: 717–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hosseinidoust Z, Tufenkji N, van de Ven TG (2013) Predation in homogeneous and heterogeneous phage environments affects virulence determinants of Pseudomonas aeruginosa . Appl Environ Microbiol 79: 2862–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hosseinidoust Z, van de Ven TG, Tufenkji N (2013) Evolution of Pseudomonas aeruginosa virulence as a result of phage predation. Appl Environ Microbiol 79: 6110–6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ceyssens PJ, Lavigne R (2010) Bacteriophages of Pseudomonas . Future Microbiol 5: 1041–1055. [DOI] [PubMed] [Google Scholar]
- 12. Hatfull GF (2008) Bacteriophage genomics. Curr Opin Microbiol 11: 447–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hatfull GF, Hendrix RW (2011) Bacteriophages and their genomes. Curr Opin Virol 1: 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Essoh C, Blouin Y, Loukou G, Cablanmian A, Lathro S, et al. (2013) The Susceptibility of Pseudomonas aeruginosa Strains from Cystic Fibrosis Patients to Bacteriophages. PLoS One 8: e60575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lu S, Le S, Tan Y, Zhu J, Li M, et al. (2013) Genomic and proteomic analyses of the terminally redundant genome of the Pseudomonas aeruginosa phage PaP1: establishment of genus PaP1-like phages. PLoS One 8: e62933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rahme LG, Stevens EJ, Wolfort SF, Shao J, Tompkins RG, et al. (1995) Common virulence factors for bacterial pathogenicity in plants and animals. Science 268: 1899–1902. [DOI] [PubMed] [Google Scholar]
- 17. Holloway BW (1955) Genetic recombination in Pseudomonas aeruginosa . J Gen Microbiol 13: 572–581. [DOI] [PubMed] [Google Scholar]
- 18. Romling U, Wingender J, Muller H, Tummler B (1994) A major Pseudomonas aeruginosa clone common to patients and aquatic habitats. Appl Environ Microbiol 60: 1734–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vu-Thien H, Corbineau G, Hormigos K, Fauroux B, Corvol H, et al. (2007) Multiple-locus variable-number tandem-repeat analysis for longitudinal survey of sources of Pseudomonas aeruginosa infection in cystic fibrosis patients. J Clin Microbiol 45: 3175–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Llanes C, Pourcel C, Richardot C, Plesiat P, Fichant G, et al. (2013) Diversity of beta-lactam resistance mechanisms in cystic fibrosis isolates of Pseudomonas aeruginosa: a French multicentre study. J Antimicrob Chemother 68: 1763–1771. [DOI] [PubMed] [Google Scholar]
- 21. Brokopp CD, Gomez-Lus R, Farmer JJ 3rd (1977) Serological typing of Pseudomonas aeruginosa: use of commercial antisera and live antigens. J Clin Microbiol 5: 640–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cameron JR, Philippsen P, Davis RW (1977) Analysis of chromosomal integration and deletions of yeast plasmids. Nucleic Acids Res 4: 1429–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Van Domselaar GH, Stothard P, Shrivastava S, Cruz JA, Guo A, et al. (2005) BASys: a web server for automated bacterial genome annotation. Nucleic Acids Res 33: W455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cramer N, Klockgether J, Wrasman K, Schmidt M, Davenport CF, et al. (2011) Microevolution of the major common Pseudomonas aeruginosa clones C and PA14 in cystic fibrosis lungs. Environ Microbiol 13: 1690–1704. [DOI] [PubMed] [Google Scholar]
- 25. Xu J, Zhang J, Lu X, Liang W, Zhang L, et al. (2013) O antigen is the receptor of Vibrio cholerae serogroup O1 El Tor typing phage VP4. J Bacteriol 195: 798–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cornelissen A, Ceyssens PJ, Krylov VN, Noben JP, Volckaert G, et al. (2012) Identification of EPS-degrading activity within the tail spikes of the novel Pseudomonas putida phage AF. Virology. [DOI] [PubMed]
- 27. Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S (2004) Bacterial persistence as a phenotypic switch. Science 305: 1622–1625. [DOI] [PubMed] [Google Scholar]
- 28. Crosa JH (1989) Genetics and molecular biology of siderophore-mediated iron transport in bacteria. Microbiol Rev 53: 517–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kwan T, Liu J, Dubow M, Gros P, Pelletier J (2006) Comparative genomic analysis of 18 Pseudomonas aeruginosa bacteriophages. J Bacteriol 188: 1184–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wei H, Therrien C, Blanchard A, Guan S, Zhu Z (2008) The Fidelity Index provides a systematic quantitation of star activity of DNA restriction endonucleases. Nucleic Acids Res 36: e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Uchiyama J, Rashel M, Takemura I, Kato S, Ujihara T, et al. (2012) Genetic characterization of Pseudomonas aeruginosa bacteriophage KPP10. Arch Virol 157: 733–738. [DOI] [PubMed] [Google Scholar]
- 32. Perry LL, SanMiguel P, Minocha U, Terekhov AI, Shroyer ML, et al. (2009) Sequence analysis of Escherichia coli O157:H7 bacteriophage PhiV10 and identification of a phage-encoded immunity protein that modifies the O157 antigen. FEMS Microbiol Lett 292: 182–186. [DOI] [PubMed] [Google Scholar]
- 33. Kropinski AM, Kovalyova IV, Billington SJ, Patrick AN, Butts BD, et al. (2007) The genome of epsilon15, a serotype-converting, Group E1 Salmonella enterica-specific bacteriophage. Virology 369: 234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roos WH, Ivanovska IL, Evilevitch A, Wuite GJ (2007) Viral capsids: mechanical characteristics, genome packaging and delivery mechanisms. Cell Mol Life Sci 64: 1484–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee DG, Urbach JM, Wu G, Liberati NT, Feinbaum RL, et al. (2006) Genomic analysis reveals that Pseudomonas aeruginosa virulence is combinatorial. Genome Biol 7: R90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ruer S, Stender S, Filloux A, de Bentzmann S (2007) Assembly of fimbrial structures in Pseudomonas aeruginosa: functionality and specificity of chaperone-usher machineries. J Bacteriol 189: 3547–3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jiang X, Jiang H, Li C, Wang S, Mi Z, et al. (2011) Sequence characteristics of T4-like bacteriophage IME08 benome termini revealed by high throughput sequencing. Virol J 8: 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hanlon GW, Denyer SP, Olliff CJ, Ibrahim LJ (2001) Reduction in exopolysaccharide viscosity as an aid to bacteriophage penetration through Pseudomonas aeruginosa biofilms. Appl Environ Microbiol 67: 2746–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Steven AC, Trus BL, Maizel JV, Unser M, Parry DA, et al. (1988) Molecular substructure of a viral receptor-recognition protein. The gp17 tail-fiber of bacteriophage T7. J Mol Biol 200: 351–365. [DOI] [PubMed] [Google Scholar]
- 40. Cornelissen A, Ceyssens PJ, T'Syen J, Van Praet H, Noben JP, et al. (2011) The T7-related Pseudomonas putida phage phi15 displays virion-associated biofilm degradation properties. PLoS One 6: e18597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Casjens SR, Molineux IJ (2012) Short noncontractile tail machines: adsorption and DNA delivery by podoviruses. Adv Exp Med Biol 726: 143–179. [DOI] [PubMed] [Google Scholar]
- 42. Friman VP, Ghoul M, Molin S, Johansen HK, Buckling A (2013) Pseudomonas aeruginosa Adaptation to Lungs of Cystic Fibrosis Patients Leads to Lowered Resistance to Phage and Protist Enemies. PLoS One 8: e75380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Uchiyama J, Rashel M, Matsumoto T, Sumiyama Y, Wakiguchi H, et al. (2009) Characteristics of a novel Pseudomonas aeruginosa bacteriophage, PAJU2, which is genetically related to bacteriophage D3. Virus Res 139: 131–134. [DOI] [PubMed] [Google Scholar]
- 44. Mooij MJ, Drenkard E, Llamas MA, Vandenbroucke-Grauls CM, Savelkoul PH, et al. (2007) Characterization of the integrated filamentous phage Pf5 and its involvement in small-colony formation. Microbiology 153: 1790–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Webb JS, Lau M, Kjelleberg S (2004) Bacteriophage and phenotypic variation in Pseudomonas aeruginosa biofilm development. J Bacteriol 186: 8066–8073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rice SA, Tan CH, Mikkelsen PJ, Kung V, Woo J, et al. (2009) The biofilm life cycle and virulence of Pseudomonas aeruginosa are dependent on a filamentous prophage. ISME J 3: 271–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wozniak DJ, Ohman DE (1994) Transcriptional analysis of the Pseudomonas aeruginosa genes algR, algB, and algD reveals a hierarchy of alginate gene expression which is modulated by algT. J Bacteriol 176: 6007–6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rodriguez-Rojas A, Mena A, Martin S, Borrell N, Oliver A, et al. (2009) Inactivation of the hmgA gene of Pseudomonas aeruginosa leads to pyomelanin hyperproduction, stress resistance and increased persistence in chronic lung infection. Microbiology 155: 1050–1057. [DOI] [PubMed] [Google Scholar]
- 49. Vallet-Gely I, Sharp JS, Dove SL (2007) Local and global regulators linking anaerobiosis to cupA fimbrial gene expression in Pseudomonas aeruginosa . J Bacteriol 189: 8667–8676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hossain MJ, Rahman Kh S, Terhune JS, Liles MR (2012) An outer membrane porin protein modulates phage susceptibility in Edwardsiella ictaluri . Microbiology 158: 474–487. [DOI] [PubMed] [Google Scholar]
- 51. Randall-Hazelbauer L, Schwartz M (1973) Isolation of the bacteriophage lambda receptor from Escherichia coli . J Bacteriol 116: 1436–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. de Vries GE, Raymond CK, Ludwig RA (1984) Extension of bacteriophage lambda host range: selection, cloning, and characterization of a constitutive lambda receptor gene. Proc Natl Acad Sci U S A 81: 6080–6084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rau MH, Marvig RL, Ehrlich GD, Molin S, Jelsbak L (2012) Deletion and acquisition of genomic content during early stage adaptation of Pseudomonas aeruginosa to a human host environment. Environ Microbiol 14: 2200–2211. [DOI] [PubMed] [Google Scholar]
- 54. Krylov SV, Kropinski AM, Pleteneva EA, Shaburova OV, Burkal'tseva MV, et al. (2012) Properties of the new D3-like Pseudomonas aeruginosa bacteriophage phiPMG1: genome structure and prospects for the use in phage therapy. Genetika 48: 1057–1067. [PubMed] [Google Scholar]
- 55. Brussow H, Hendrix RW (2002) Phage genomics: small is beautiful. Cell 108: 13–16. [DOI] [PubMed] [Google Scholar]
- 56. Casjens SR (2005) Comparative genomics and evolution of the tailed-bacteriophages. Curr Opin Microbiol 8: 451–458. [DOI] [PubMed] [Google Scholar]
- 57. Braid MD, Silhavy JL, Kitts CL, Cano RJ, Howe MM (2004) Complete genomic sequence of bacteriophage B3, a Mu-like phage of Pseudomonas aeruginosa . J Bacteriol 186: 6560–6574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Juhala RJ, Ford ME, Duda RL, Youlton A, Hatfull GF, et al. (2000) Genomic sequences of bacteriophages HK97 and HK022: pervasive genetic mosaicism in the lambdoid bacteriophages. J Mol Biol 299: 27–51. [DOI] [PubMed] [Google Scholar]