

Abstract
Jab1/CSN5 is a multifunctional protein that plays an important role in integrin signaling, cell proliferation, apoptosis, and the regulation of genomic instability and DNA repair. Dysregulation of Jab1/CSN5 activity has been shown to contribute to oncogenesis by functionally inactivating several key negative regulatory proteins and tumor suppressors. In this review, we discuss our current understanding of the relationship between Jab1/CSN5 and DNA damage and summarize recent findings regarding opportunities for and challenges to therapeutic intervention.
Keywords: Jab1/CSN5, DNA damage, therapeutic approach, genomic instability, tumorigenesis
Introduction
The human genome is continuously exposed to different kinds of DNA-damaging agents. The damage can be categorized into two groups: spontaneous DNA damage generated during normal metabolism and induced DNA damage deriving from multiple physical and chemical factors, such as ionizing radiation, ultraviolet (UV) radiation, environmental toxins, and chemotherapeutic agents.1 Genomic instability induced by DNA damage can lead to cell cycle arrest, apoptosis, or tumorigenesis. Altogether, each cell could experience up to 10 000 spontaneous DNA lesions a day.2
Cells have evolved the DNA damage response (DDR) to maintain genomic integrity. This response can include mismatch repair (MMR), nucleotide excision repair, base excision repair, single-strand break repair, and double-strand break (DSB) repair. Non-homologous end joining and homologous recombination (HR) are two major DSB repair pathways.1 Increasing evidence has demonstrated that mutation of genes that respond to DNA damage greatly promotes tumorigenesis. In addition, increased DDR has been reported to be one of the mechanisms of chemotherapy or radiotherapy resistance.3,4 Therefore, studies of DNA damage and repair can elucidate the fundamental mechanisms triggering tumorigenesis and provide novel strategies for tumor therapy.
Emerging evidence supports the idea that Jab1/constitutive photomorphogenic 9 signalosome subunit 5 (CSN5) is critically involved in DDR and is linked to the maintenance of genome integrity.4-6 Furthermore, Jab1/CSN5 is essential for tumor survival and is involved in chemotherapy and radiotherapy resistance.4,7 In this review, we assess the role of Jab1/CSN5 in DDR and summarize recent findings that highlight the novel roles of Jab1/CSN5 in this process. We also summarize the effect of Jab1/CSN5 inhibition on cancer progression and suggest that inhibition of Jab1/CSN5 is a promising targeted approach to treat human cancer.
DDR and Cancer
Loss of genomic integrity owing to inactivation of DDR genes may enhance the risk of a cell to accumulate mutations in genes that promote cancer development. Somatic mutations in DDR genes have been observed in several cancer types. For a growing number of DDR genes, hereditary mutations, such as MUTYH,8 ERCC1,9 Polδ,10 Polη,11 Rad51,12 Rpa1,13 and others,14 are known to be associated with increased cancer risk. Women with heterozygous germline mutations in HR genes such as BRCA1 or BRCA2, which are involved in DSB repair via HR,15 are predisposed to breast and ovarian cancers,16,17 and the incidence of breast, pancreatic, and prostate cancers is higher in men with BRCA2 mutations.18 Mutations in the MMR genes MSH2 and MLH1 (which cause Lynch syndrome) predispose patients to a wide range of tumor types.19,20 For example, hereditary nonpolyposis colorectal cancer is associated with microsatellite instability, as well as carcinomas of the endometrium, ovary, stomach, and kidney.1 The genomic instability in hereditary nonpolyposis colorectal cancer is caused by heterozygous mutations of MMR genes, such as MLH1, MSH2, MSH6, and PMS2.21
Mutations in three additional HR genes, BACH1, PALB2, and RAD51C, have been identified in approximately 3% of familial breast cancer patients and have been associated with a 2-fold increased risk of breast cancer.22,23 Mutations of CHK2, ATM, NBS1, and RAD50 have also been associated with a doubled risk of breast cancer, indicating the importance of the ATM pathway, together with HR, in preventing breast cancer formation.23 Bi-allelic mutations of RAD51C are also associated with increased risk of myelodysplasia and acute myeloid leukemia.22,24
Slyskova et al. demonstrated that the expression of four nucleotide excision repair genes (CSB, CCNH, XPA, and XPD) and four base excision repair genes (NEIL1, APEX1, OGG1, and PARP1) was 1.08- to 1.28-fold higher in colorectal carcinomas (P < 0.05).25 In addition, it is known that deficient nucleotide excision repair greatly increases the risk of melanoma and other skin cancers because of defective repair of UV radiation-induced lesions in skin cells after sun exposure.2
Cancer is an evolutionary disease fueled by genomic instability that is often caused by chromosomal translocations, which can lead to aberrant expression of oncogenes, such as c-Myc or Jab1/CSN5, or to the generation of deregulated chimeric proteins with enhanced activity.6,26 Experiments have shown that in the absence of p53, non-homologous end joining-defective mice develop B-cell lymphomas harboring translocations between the immunoglobulin locus and c-Myc that depend on unrepaired breaks created by RAG1/RAG2.26 Importantly, Jab1/CSN5, along with Myc, was reported to act as a master regulator of a wound gene expression signature in breast cancer cells and this finding suggests that Jab1/CSN5 plays an important role in activating the transcription of stress response genes that are involved in the proliferation and matrix invasiveness of cells.27 Similarly, ATM deficiency was shown to lead to persistent RAG-induced breaks, which could join other DSBs to generate translocations and promote tumorigenesis.26
Faulty DDR genes not only predispose carriers to cancer formation by facilitating senescence, apoptosis bypass, and cellular proliferation despite the accumulation of DNA damage28,29 but also alter the sensitivity of tumors to chemotherapy. Although response to therapy may be affected by nucleotide excision repair30 and base excision repair,31 chemotherapy sensitivity has been most robustly linked to HR and MMR deficiencies. In particular, inhibiting DDR components—thereby increasing DNA damage—and administering chemotherapeutic drugs could lead to cancer cell death. DDR defects such as HR deficiency may be targeted by synthetic lethal approaches or therapeutic strategies that disrupt synthetic viability. This approach is illustrated by the sensitivity of BRCA-deficient tumor cells to poly(ADP-ribose) polymerase (PARP) inhibition.32-34 This effect has been proposed to be caused by the accumulation of unrepaired DNA breaks in the absence of both PARP-dependent single-strand break repair and HR. In addition, recent studies have demonstrated that ATM, DNA-PK, and CHK1 inhibitors have preferential toxicity toward cancer cells following treatment with genotoxic agents.35,36 Methotrexate, an inhibitor of DNA base synthesis, has been shown to selectively kill MMR-deficient cancer cells by contributing to the accumulation of oxidative DNA lesions.37
DNA repair processes are not only important for the intrinsic response of tumors to chemotherapy but are also mechanisms for cancer therapy resistance. In this respect, DDR defects in tumors may be a mixed blessing. For example, HR deficiency confers sensitivity of tumor cells to DSB-inducing agents but at the same time causes additional genomic instability and genetic alterations that may induce therapy resistance.14
Jab1/CSN5 Overexpression in Cancer
The CSN complex was first identified in plants and demonstrated to be an essential regulator of light-mediated development.38-40 The CSN complex has a mass of about 450 kDa and comprises eight core subunits—CSN1–CSN8—in order of descending size. These subunits are highly conserved across diverse species and play essential roles in multiple cancers.41,42 In a screening for genes that when downregulated are synthetic lethal with activating Ras mutations, Steve Elledge’s group identified several subunits of the CSN complex.43 They demonstrated that DLD-1 cells bearing the Ras mutation and with stable depletion of COPS4 exhibit impaired growth. Furthermore, those with lower COPS3 and CDC16 expression levels together with higher EVI5 expression levels were associated with a striking enhancement of survival in patients bearing tumors that exhibited a positive Ras signature.43
Our group originally identified Jab1/CSN5 as a c-Jun coactivator, and other groups subsequently discovered it to be the fifth member and an integral component of the CSN.44,45 In addition to being associated with the large CSN complex, Jab1/CSN5 can be found in a monomeric form or associated with a smaller non-CSN complex in various species.46,47 Of the eight CSN subunits, CSN5 is unique in that it not only harbors the catalytic center of CSN isopeptidase activity but it also stably exists independently of the CSN complex in vivo.45 Jab1/CSN5 actively participates in important biologic functions, both as part of the CSN holocomplex and independently of the CSN complex. For example, quantitative assessment of CUL1 complexes after knockdown revealed that loss of COPS5 did not result in a significant loss of association with the larger CSN complex despite a reduction in the amount of the COPS5 subunit associated with CUL1.48,49 A large proportion of Jab1/CSN5 is found in the free form.50 Whereas CSN-associated Jab1/CSN5 is mostly nuclear, the free forms appear to be both cytoplasmic and nuclear.47,50,51 The dependence of CSN5 nuclear accumulation on other subunits has been clearly demonstrated in the CSN-like complex of budding yeast.52 Although our findings shown that ectopic Jab1/CSN5 played a critical role in cancers and that Jab1/CSN5 appears to be both cytoplasmic and nuclear, whether Jab1/CSN5 acts as a complex or independently of a complex in cancers remains to be determined.6
The domains responsible for the interaction between Jab1/CSN5 and p27 and for its metalloproteinase activity have been identified. Jab1/CSN5 interacts with p27 and mediates the shuttling of p27 between the nucleus and the cytoplasm in a CRM1-dependent manner through a nuclear export signal-like sequence between amino acids 233 and 242 at its C-terminal end.50 Jab1/CSN5 has an Mpr1-Pad1-N (MPN) terminal domain that encompasses a Jab1/CSN5 MPN domain metalloenzyme (JAMM) motif (also known as the MPN+ motif). Some have suggested that the Jab1/CSN5 MPN domain functions as a protein–protein platform, whereas the JAMM motif acts as a cofactor for enzymatic activity.53 The JAMM motif within Jab1/CSN5 is essential to CSN deneddylation activity, but deneddylation still relies on the entire CSN complex, as the loss of any one of the CSN subunits results in its inactivation.53 The JAMM motif is essential for the co-activation of the oncogenic MYC transcription factor by Jab1/CSN5 and for the transformative effects Jab1/CSN5 in a breast epithelial model; these effects were also found to be dependent on the assembly of the entire CSN.54 This domain does not, however, appear to be critical to the functions of Jab1/CSN5 outside of the full CSN, such as the stabilization of HIF-1a and its role as a co-factor for E2F1-induced apoptosis.55,56 In fact, the domains responsible for many of the mechanisms of Jab1/CSN5 and the mechanism by which it performs its many functions, as well as the involvement of the CSN or smaller complex, remain to be determined.57
Jab1/CSN5 is involved in in tumorigenesis by degrading several essential targets, including p27, p53, cyclin E, Smad 4/7, and LHR.57 These substrates of Jab1/CSN5 are involved in many cellular processes, such as cell-cycle regulation, proliferation, apoptosis, angiogenesis, and survival. Indeed, accumulating evidence has shown that Jab1/CSN5 overexpression is inversely correlated with p27 expression and with poor survival in various human malignancies.57
Actually, Jab1/CSN5 is overexpressed in a variety of human cancers, such as ovarian cancer,58 hepatocellular carcinoma,59 lung cancer,60 pancreatic cancer,61 breast carcinoma,62 nasopharyngeal carcinoma (NPC),7 and many others.57 Moreover, investigators have found that Jab1/CSN5 is a prognostic marker for multiple cancers. For example, overexpression of Jab1/CSN5 is associated with lymph node metastasis of hepatocellular carcinoma59 and oral squamous cell carcinoma,63 and it leads to poor survival in patients with these cancers. Similarly, Jab1/CSN5 expression is correlated with tumor size in thyroid carcinoma cases.64 Therefore, Jab1/CSN5 overexpression could be considered a biomarker of poor prognosis for many types of cancers.
Role of Jab1/CSN5 in DDR
The mechanisms of Jab1/CSN5 activity have been reported to affect either the activity or the stability of proteins involved in DDR.4,5 CSN regulates the Skp1/cullin-1/F-box (SCF) ubiquitin ligases by removing the ubiquitin-like protein Nedd8 from the cullin subunit of SCF. SCF-dependent ubiquitination mediates the degradation of many cell-cycle regulators, including p27, CDK inhibitor p21, E2F1, β-catenin, IκBα, and cyclins E, A, and B.65 Nayak et al. found that, in Caenorhabiditis elegans, the SCF complex regulates meiosis and the Skp1-related (skr) gene family, which are required for the restrain of cell proliferation and for the formation of bivalent chromosomes at diakinesis.66 A recent study showed that, as a complex, CSN mediates the inhibition of the DDB1-DDB2-CUL4A/B-RBX1 complex.67 Knockdown of Jab1/CSN5 reduced the repair activity of DDB2 by 50%.68 A distinguishing feature of Jab1/CSN5 is that it is able to mediate the nuclear export and degradation of several nuclear proteins, including DNA damage and repair proteins.
The 9-1-1 complex, which is constructed from Rad9, Hus1, and Rad1 orthologs (using Schizosaccharomyces pombe nomenclature), is critical in the initiation of cellular responses to DNA damage. The complex is loaded around DNA by the Rad17-containing clamp loader in response to DNA damage. The DNA-bound complex then facilitates ATR-mediated phosphorylation and activation of Chk1, a protein kinase that regulates S-phase progression, G2/M arrest, and replication fork stabilization.69 Therefore, the 9-1-1 complex acts as a DNA-damage sensor for transducing the DNA damage. Recently, Huang et al. found that Jab1/CSN5 directly interacts with the Rad1 ortholog, translocates the complex from the nucleus to the cytoplasm, and rapidly degrades the complex via the 26S proteasome, indicating the importance of Jab1/CSN5 in DNA damage and repair pathways.70 Furthermore, Jab1/CSN5 overexpression significantly suppresses Chk1 phosphorylation at Ser345, which could be induced by UV radiation, γ-radiation, and hydroxyurea.70 Jab1/CSN5 significantly suppresses checkpoint signaling activation, DNA synthesis recovery from blockage, and cell viability after replication stresses. These results suggest that Jab1/CSN5 is an important regulator of the stability of the 9-1-1 complex in cells and provide novel information on the involvement of Jab1/CSN5 in checkpoint and DNA repair signaling in response to DNA damage.
Other findings have demonstrated that Mei-41—a member of the Drosophila ATM/ATR family of kinases that are required for DNA damage and recombination checkpoints in yeast, worms, and humans as well as in flies71—regulates Rad51 and Rad54, which are required for DSB repair.72 Kinases in the ATM/ATR subfamily that includes Mei-41 play a central role in checkpoint-mediated responses to DNA damage.71 These kinases are thought to act as sensors of DNA damage, as they are activated upon binding to damaged DNA. Phosphorylation of several downstream effectors, including the Chk1 and Chk2 kinases and p53, then restrains cell-cycle progression until the DNA damage is repaired, when the checkpoint kinases dissociate from the DNA. Studies in which Jab1/CSN5 phenotypes were suppressed by mei-41 mutations have demonstrated that Jab1/CSN5 acts upstream of the DNA damage checkpoint and suggest DSBs arising during meiotic recombination cannot be efficiently repaired in Jab1/CSN5-mutant cells.73 Moreover, Jab1/CSN5 was found to directly interact with mei-W68,73 which encodes the Drosophila homolog of yeast gene SPO11, a gene required for the induction of DSBs72 and the initiation of meiotic recombination.74
In agreement with these studies, our group found that Jab1/CSN5 is associated with Rad51,4,5 which is a key protein in the HR repair pathway.75 Tian and colleagues found that loss of Jab1/CSN5 results in spontaneous DNA breaks that are associated with increased expression of the histone H2AX,5 which recognizes DNA DSBs and initiates the recruitment of DDR proteins.75 This recruitment is coupled with a deficiency in HR repair due to decreased Rad51 expression and function. Exogenous Jab1/CSN5 overexpression was associated with an increase in Rad51 levels, whereas reduced endogenous Jab1/CSN5 protein expression was associated with a decrease in Rad51 levels in mouse embryonic fibroblasts5 and NPC cell lines.4 Moreover, a correlation plot of the Jab1/CSN5 and Rad51 blot intensities showed a linear correlation between the levels of the two proteins (r = 0.6824; P = 0.0072), confirming that the levels of Jab1/CSN5 are proportional to the Rad51 levels in NPC cells.4 We found that Rad51 was decreased in U2OS osteosarcoma cells treated with Jab1/CSN5 small interfering RNA (siRNA).5 In comparison, the expression levels of Ku70, an important protein in the non-homologous end joining DNA repair pathway, and of phosphorylated Chk2, a key molecule in the transduction of DNA damage signaling induced by DSBs,76 were increased after ionizing radiation exposure regardless of whether the cells had been treated with Jab1/CSN5 or control siRNA.5 Consistent with these findings, our studies showed that Jab1/CSN5 siRNA-treated HONE1 NPC cells exhibited increased levels of γ-H2AX,4 which is an early indicator of the presence of DSBs.77 Similarly, the levels of phosphorylated Chk2 increased in NPC cells after DNA damage exposure regardless of whether the cells had been treated with Jab1/CSN5 siRNA (although the phosphorylated Chk2 levels were higher in Jab1/CSN5-deficient cells).4 In contrast, Rad51 levels decreased in Jab1/CSN5 siRNA-treated cells 48 h after treatment with cisplatin or UV radiation. In addition, γ-H2AX levels were higher but Rad51 levels were lower in Jab1/CSN5 knockdown cells than in control cells upon exposure to ionizing radiation.4 Jab1/CSN5 knockdown also affected Rad51 activity: ectopic expression of Rad51 rescued the defective repair function in cells with knockdown of Jab1/CSN5 expression. These results suggest that depletion of Jab1/CSN5 reduces the expression of Rad51, thereby reducing the ability of NPC cells to repair DNA lesions.
Genotoxic stress triggers a series of posttranslational modifications in p53 that contribute to its stabilization, nuclear accumulation, and biochemical activation. Thus, p53 is considered a DNA damage sensor. Our data showed that p53 was increased after DNA damage and that the upregulation was enhanced in Jab1/CSN5-knockdown cells.4,5 Consistent with our studies, Lee and colleagues showed that Jab1/CSN5 can suppress p53 activity under stressed conditions,78 indicating an important role of Jab1/CSN5 in DDR through the p53 pathway. Moreover, p53-binding protein 1 (53BP1) is well characterized as a “mediator” of DNA damage checkpoint responses.79 Observations that Jab1/CSN5 was associated with 53BP1 in growing cells suggest that Jab1/CSN5 is involved in DDR. These reports allow us to speculate that Jab1/CSN5 participates in the signaling pathway in response to DNA damage through a functional interaction with 53BP1. Our own recent studies indicated that Jab1/CSN5 regulates Rad51 through the p53 pathway.4
Jab1/CSN5 Inhibition as a Novel Therapeutic Strategy against Cancer
A growing body of evidence strongly suggests that Jab1/CSN5 plays a role in the pathogenesis of several tumor types and in many cases has specifically correlated it with reduced levels of p27 and poor prognosis.57 Patients with Jab1/CSN5 overexpression and reduced p27 levels had poorer overall survival, and patients with lymph node metastasis and Jab1/CSN5 overexpression had poorer disease-free and overall survival rates than did patients with epithelial ovarian tumors58 or esophageal80 or laryngeal81 squamous cell carcinoma.
Recent studies have shown that depletion of Jab1/CSN5 inhibits the growth of cancer cells. For example, knockdown of Jab1/CSN5 inhibited proliferation and induced apoptosis in breast cancer cells82 and, in our research, NPC cells.7 Furthermore, Jab1/CSN5-deficient mice were found to have an embryonically lethal phenotype, suggesting that Jab1/CSN5 is critical for fetal development and survival.5 Our previous studies showed that Jab1/CSN5-null embryos were smaller than wild-type embryos and displayed growth retardation.5 Jab1/CSN5-null embryos are viable up to the blastocyst stage but begin to exhibit disrupted development at embryonic day 6 and are no longer viable at embryonic day 8.5, which is before gastrulation occurs.5,83 Other investigators have found that several targets of Jab1/CSN5, including p27, p53, c-myc, and cyclin E, are highly expressed in Jab1/CSN5−/− embryos, resulting in impaired proliferation and accelerated apoptosis.5,83 Similarly, in pro-B cells, Jab1/CSN5 deletion leads to aberrant expression of the apoptosis-triggering protein Fas ligand.84 A more recent study showed that by interacting with Jab1/CSN5, Fank1 could suppress apoptosis by activating the AP-1-induced anti-apoptotic pathway.85
Consistent with these data, we found that Jab1/CSN5 deficiency resulted in early embryonic lethality owing to accelerated apoptosis. Loss of Jab1/CSN5 expression sensitized both primary embryonic fibroblasts and osteosarcoma cells to γ-radiation-induced apoptosis.5 It is interesting to note that our NPC studies showed that inhibition of Jab1/CSN5 expression with 5 nM of siRNA for 48 h was insufficient to induce apoptosis,7 whereas higher doses of Jab1/CSN5 siRNA (10 or 20 nM) induced apoptosis (unpublished data), and knocking down Jab1/CSN5 expression in NPC cells increased UV radiation-, ionizing radiation-, and cisplatin-induced apoptosis.4 In contrast, overexpression of Jab1/CSN5 in CNE1 NPC cells blocked UV radiation- and cisplatin-induced apoptosis.4 Collectively, these results indicate that Jab1/CSN5 may promote cell proliferation and inhibit apoptosis in tumors. More thorough studies are required to fully understand the underlying molecular and signaling events by which Jab1/CSN5 influences apoptosis in cancers.
Given the prominent antineoplastic potential of Jab1, the development of specific, effective, and safe Jab1/CSN5 inhibitors is likely to have a significant impact on cancer treatment. Although specific inhibitors targeting Jab1/CSN5 are largely undetermined, Li et al. recently found that PEGylated curcumin inhibited cell proliferation in pancreatic cancer cells partially through the inactivation of Jab1/CSN5.86 In addition, PEGylated curcumin sensitized pancreatic cancer cells to gemcitabine-induced apoptosis and cell growth inhibition.86 Our recent studies also demonstrated that curcumin analog T83 exhibits potent antitumor activity and induces radiosensitivity through the inactivation of Jab1/CSN5 in NPC.87 Inhibiting the Jab1/CSN5 signaling pathway may be an effective strategy in the treatment of cancer.
Many cancer therapies use DNA-damaging agents to target tumor cells, often triggering DNA repair and rendering the cells resistant to therapy. Because Jab1/CSN5 plays essential roles in multiple DNA repair pathways, blocking Jab1/CSN5 will likely sensitize tumors to chemotherapy or radiotherapy, thereby improving the therapeutic index of such approaches. Indeed, our previous studies with mice indicated that loss of Jab1/CSN5 sensitizes cells to γ-radiation-induced apoptosis and increases spontaneous DNA damage and HR defects.5 We also recently analyzed the effects of Jab1/CSN5 on the response of NPC cell lines to treatment with cisplatin, ionizing radiation, and UV radiation. We demonstrated that knocking down Jab1/CSN5 expression in these cells sensitized them to all three forms of treatment; conversely, overexpression of Jab1/CSN5 contributed to cisplatin and radiation resistance.4 These observations suggest Jab1/CSN5 is a major contributor to the resistance of NPC to both chemotherapy and radiotherapy. Therefore, Jab1/CSN5 is a novel therapeutic target in cancer.
Conclusions and Unresolved Questions
We have provided succinct information on the state of knowledge of the role of the oncoprotein Jab1/CSN5 in DDR and cancer therapy (Fig. 1). Jab1/CSN5 is a novel target for the prevention and treatment of human cancers. Therefore, the development of agents specifically targeting Jab1/CSN5 inhibition is likely to have a significant impact on cancer treatment.
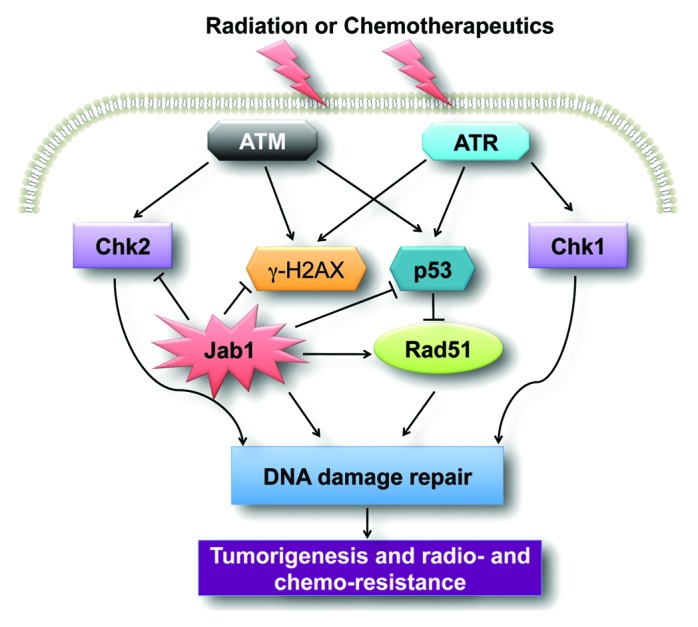
Figure 1. A schematic of the Jab1/CSN5 signaling pathway involved in cancer. Jab1/CSN5 regulates DNA damage and repairs proteins such as p53 and Rad51, leading to DNA damage repair. Overexpression of Jab1/CSN5 in cancer contributes to tumorigenesis and radio- and chemo-resistance.
The role of Jab1/CSN5 in DNA repair in normal cells remains to be determined. In our work, we observed that mouse embryonic fibroblasts lacking Jab1/CSN5 were more susceptible to radiation-induced DNA damage than their wild-type counterparts were. It will be interesting to determine whether the Jab1/CSN5 protein directly interacts with DNA damage and repair proteins and is recruited to sites of DNA damage.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Research in the laboratory is supported by funds from the National Natural Science Foundation of China (81372816) and the fellowship from the China Scholarship Council (2010638087) to YP; the National Natural Science Foundation of China (81071837) to H.Y.; the National Institutes of Health (1R01CA90853), the Cancer Prevention Research Institute of Texas (RP120451-01) and the University Cancer Foundation via the Sister Institution Network Fund at the University of Texas MD Anderson Cancer Center to F.X.C. We thank Elizabeth L Hess and Markeda L Wade in the Department of Scientific Publications at MD Anderson for editing the manuscript. This research is supported in part by the MD Anderson’s Cancer Center Support Grant CA016672.
Glossary
Abbreviations:
- CDK
cyclin-dependent kinase
- DDB2
DNA damage-binding protein 2
- DDR
DNA damage response
- DSB
double-strand break
- HR
homologous recombination
- IR
ionizing radiation
- Jab1
c-Jun activation domain-binding protein-1
- CSN5
COP9 signalosome subunit 5
- MMR
mismatch repair
- NPC
nasopharyngeal carcinoma
- PARP
poly(ADP-ribose) polymerase
- SCF
Skp1/cullin-1/F-box
- siRNA
small interfering RNA
- UV
ultraviolet radiation
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/27823
References
- 1.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361:1475–85. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- 3.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–79. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 4.Pan Y, Zhang Q, Atsaves V, Yang H, Claret FX. Suppression of Jab1/CSN5 induces radio- and chemo-sensitivity in nasopharyngeal carcinoma through changes to the DNA damage and repair pathways. Oncogene. 2013;32:2756–66. doi: 10.1038/onc.2012.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tian L, Peng G, Parant JM, Leventaki V, Drakos E, Zhang Q, Parker-Thornburg J, Shackleford TJ, Dai H, Lin SY, et al. Essential roles of Jab1 in cell survival, spontaneous DNA damage and DNA repair. Oncogene. 2010;29:6125–37. doi: 10.1038/onc.2010.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pan Y, Claret FX. Targeting Jab1/CSN5 in nasopharyngeal carcinoma. Cancer Lett. 2012;326:155–60. doi: 10.1016/j.canlet.2012.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pan Y, Zhang Q, Tian L, Wang X, Fan X, Zhang H, Claret FX, Yang H. Jab1/CSN5 negatively regulates p27 and plays a role in the pathogenesis of nasopharyngeal carcinoma. Cancer Res. 2012;72:1890–900. doi: 10.1158/0008-5472.CAN-11-3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sakamoto K, Tominaga Y, Yamauchi K, Nakatsu Y, Sakumi K, Yoshiyama K, Egashira A, Kura S, Yao T, Tsuneyoshi M, et al. MUTYH-null mice are susceptible to spontaneous and oxidative stress induced intestinal tumorigenesis. Cancer Res. 2007;67:6599–604. doi: 10.1158/0008-5472.CAN-06-4802. [DOI] [PubMed] [Google Scholar]
- 9.Matoka DJ, Yao V, Harya DS, Gregg JL, Robinson AR, Niedernhofer LJ, Parwani AV, Maier C, Bacich DJ. Deficiency of DNA repair nuclease ERCC1-XPF promotes prostate cancer progression in a tissue recombination model. Prostate. 2012;72:1214–22. doi: 10.1002/pros.22472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldsby RE, Lawrence NA, Hays LE, Olmsted EA, Chen X, Singh M, Preston BD. Defective DNA polymerase-delta proofreading causes cancer susceptibility in mice. Nat Med. 2001;7:638–9. doi: 10.1038/88963. [DOI] [PubMed] [Google Scholar]
- 11.Lin Q, Clark AB, McCulloch SD, Yuan T, Bronson RT, Kunkel TA, Kucherlapati R. Increased susceptibility to UV-induced skin carcinogenesis in polymerase eta-deficient mice. Cancer Res. 2006;66:87–94. doi: 10.1158/0008-5472.CAN-05-1862. [DOI] [PubMed] [Google Scholar]
- 12.Kuznetsov SG, Haines DC, Martin BK, Sharan SK. Loss of Rad51c leads to embryonic lethality and modulation of Trp53-dependent tumorigenesis in mice. Cancer Res. 2009;69:863–72. doi: 10.1158/0008-5472.CAN-08-3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, Putnam CD, Kane MF, Zhang W, Edelmann L, Russell R, Carrión DV, Chin L, Kucherlapati R, Kolodner RD, et al. Mutation in Rpa1 results in defective DNA double-strand break repair, chromosomal instability and cancer in mice. Nat Genet. 2005;37:750–5. doi: 10.1038/ng1587. [DOI] [PubMed] [Google Scholar]
- 14.Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer. 2012;12:587–98. doi: 10.1038/nrc3342. [DOI] [PubMed] [Google Scholar]
- 15.Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7:263–72. doi: 10.1016/S1097-2765(01)00174-5. [DOI] [PubMed] [Google Scholar]
- 16.Fackenthal JD, Olopade OI. Breast cancer risk associated with BRCA1 and BRCA2 in diverse populations. Nat Rev Cancer. 2007;7:937–48. doi: 10.1038/nrc2054. [DOI] [PubMed] [Google Scholar]
- 17.Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 18.Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol. 2010;11:196–207. doi: 10.1038/nrm2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fishel R, Lescoe MK, Rao MR, Copeland NG, Jenkins NA, Garber J, Kane M, Kolodner R. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1993;75:1027–38. doi: 10.1016/0092-8674(93)90546-3. [DOI] [PubMed] [Google Scholar]
- 20.Papadopoulos N, Nicolaides NC, Wei YF, Ruben SM, Carter KC, Rosen CA, Haseltine WA, Fleischmann RD, Fraser CM, Adams MD, et al. Mutation of a mutL homolog in hereditary colon cancer. Science. 1994;263:1625–9. doi: 10.1126/science.8128251. [DOI] [PubMed] [Google Scholar]
- 21.Spry M, Scott T, Pierce H, D’Orazio JA. DNA repair pathways and hereditary cancer susceptibility syndromes. Front Biosci. 2007;12:4191–207. doi: 10.2741/2380. [DOI] [PubMed] [Google Scholar]
- 22.Levy-Lahad E. Fanconi anemia and breast cancer susceptibility meet again. Nat Genet. 2010;42:368–9. doi: 10.1038/ng0510-368. [DOI] [PubMed] [Google Scholar]
- 23.Walsh T, King MC. Ten genes for inherited breast cancer. Cancer Cell. 2007;11:103–5. doi: 10.1016/j.ccr.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 24.Moldovan GL, D’Andrea AD. How the fanconi anemia pathway guards the genome. Annu Rev Genet. 2009;43:223–49. doi: 10.1146/annurev-genet-102108-134222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Slyskova J, Korenkova V, Collins AR, Prochazka P, Vodickova L, Svec J, Lipska L, Levy M, Schneiderova M, Liska V, et al. Functional, genetic, and epigenetic aspects of base and nucleotide excision repair in colorectal carcinomas. Clin Cancer Res. 2012;18:5878–87. doi: 10.1158/1078-0432.CCR-12-1380. [DOI] [PubMed] [Google Scholar]
- 26.Nussenzweig A, Nussenzweig MC. Origin of chromosomal translocations in lymphoid cancer. Cell. 2010;141:27–38. doi: 10.1016/j.cell.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adler AS, Lin M, Horlings H, Nuyten DS, van de Vijver MJ, Chang HY. Genetic regulators of large-scale transcriptional signatures in cancer. Nat Genet. 2006;38:421–30. doi: 10.1038/ng1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–75. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin LP, Hamilton TC, Schilder RJ. Platinum resistance: the role of DNA repair pathways. Clin Cancer Res. 2008;14:1291–5. doi: 10.1158/1078-0432.CCR-07-2238. [DOI] [PubMed] [Google Scholar]
- 31.Neijenhuis S, Verwijs-Janssen M, van den Broek LJ, Begg AC, Vens C. Targeted radiosensitization of cells expressing truncated DNA polymerase beta. Cancer Res. 2010;70:8706–14. doi: 10.1158/0008-5472.CAN-09-3901. [DOI] [PubMed] [Google Scholar]
- 32.Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, Scott C, Weitzel JN, Oaknin A, Loman N, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376:245–51. doi: 10.1016/S0140-6736(10)60893-8. [DOI] [PubMed] [Google Scholar]
- 33.Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376:235–44. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- 34.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 35.Bolderson E, Richard DJ, Zhou BB, Khanna KK. Recent advances in cancer therapy targeting proteins involved in DNA double-strand break repair. Clin Cancer Res. 2009;15:6314–20. doi: 10.1158/1078-0432.CCR-09-0096. [DOI] [PubMed] [Google Scholar]
- 36.Jiang H, Reinhardt HC, Bartkova J, Tommiska J, Blomqvist C, Nevanlinna H, Bartek J, Yaffe MB, Hemann MT. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev. 2009;23:1895–909. doi: 10.1101/gad.1815309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin SA, Hewish M, Lord CJ, Ashworth A. Genomic instability and the selection of treatments for cancer. J Pathol. 2010;220:281–9. doi: 10.1002/path.2631. [DOI] [PubMed] [Google Scholar]
- 38.Chamovitz DA, Wei N, Osterlund MT, von Arnim AG, Staub JM, Matsui M, Deng XW. The COP9 complex, a novel multisubunit nuclear regulator involved in light control of a plant developmental switch. Cell. 1996;86:115–21. doi: 10.1016/S0092-8674(00)80082-3. [DOI] [PubMed] [Google Scholar]
- 39.Karniol B, Chamovitz DA. The COP9 signalosome: from light signaling to general developmental regulation and back. Curr Opin Plant Biol. 2000;3:387–93. doi: 10.1016/S1369-5266(00)00101-1. [DOI] [PubMed] [Google Scholar]
- 40.Chamovitz DA, Segal D. JAB1/CSN5 and the COP9 signalosome. A complex situation. EMBO Rep. 2001;2:96–101. doi: 10.1093/embo-reports/kve028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu YS, Tang ZH, Pan QC, Chen XH, Liu XN, Zang GQ. Inhibition of Csn3 expression induces growth arrest and apoptosis of hepatocellular carcinoma cells. Cancer Chemother Pharmacol. 2012;69:1173–80. doi: 10.1007/s00280-011-1810-x. [DOI] [PubMed] [Google Scholar]
- 42.Yoneda-Kato N, Tomoda K, Umehara M, Arata Y, Kato JY. Myeloid leukemia factor 1 regulates p53 by suppressing COP1 via COP9 signalosome subunit 3. EMBO J. 2005;24:1739–49. doi: 10.1038/sj.emboj.7600656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–48. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Claret FX, Hibi M, Dhut S, Toda T, Karin M. A new group of conserved coactivators that increase the specificity of AP-1 transcription factors. Nature. 1996;383:453–7. doi: 10.1038/383453a0. [DOI] [PubMed] [Google Scholar]
- 45.Wei N, Deng XW. The COP9 signalosome. Annu Rev Cell Dev Biol. 2003;19:261–86. doi: 10.1146/annurev.cellbio.19.111301.112449. [DOI] [PubMed] [Google Scholar]
- 46.Freilich S, Oron E, Kapp Y, Nevo-Caspi Y, Orgad S, Segal D, Chamovitz DA. The COP9 signalosome is essential for development of Drosophila melanogaster. Curr Biol. 1999;9:1187–90. doi: 10.1016/S0960-9822(00)80023-8. [DOI] [PubMed] [Google Scholar]
- 47.Kwok SF, Solano R, Tsuge T, Chamovitz DA, Ecker JR, Matsui M, Deng XW. Arabidopsis homologs of a c-Jun coactivator are present both in monomeric form and in the COP9 complex, and their abundance is differentially affected by the pleiotropic cop/det/fus mutations. Plant Cell. 1998;10:1779–90. doi: 10.1105/tpc.10.11.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bennett EJ, Rush J, Gygi SP, Harper JW. Dynamics of cullin-RING ubiquitin ligase network revealed by systematic quantitative proteomics. Cell. 2010;143:951–65. doi: 10.1016/j.cell.2010.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sharon M, Mao H, Boeri Erba E, Stephens E, Zheng N, Robinson CV. Symmetrical modularity of the COP9 signalosome complex suggests its multifunctionality. Structure. 2009;17:31–40. doi: 10.1016/j.str.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 50.Tomoda K, Kubota Y, Arata Y, Mori S, Maeda M, Tanaka T, Yoshida M, Yoneda-Kato N, Kato JY. The cytoplasmic shuttling and subsequent degradation of p27Kip1 mediated by Jab1/CSN5 and the COP9 signalosome complex. J Biol Chem. 2002;277:2302–10. doi: 10.1074/jbc.M104431200. [DOI] [PubMed] [Google Scholar]
- 51.Bounpheng MA, Melnikova IN, Dodds SG, Chen H, Copeland NG, Gilbert DJ, Jenkins NA, Christy BA. Characterization of the mouse JAB1 cDNA and protein. Gene. 2000;242:41–50. doi: 10.1016/S0378-1119(99)00525-9. [DOI] [PubMed] [Google Scholar]
- 52.Maytal-Kivity V, Piran R, Pick E, Hofmann K, Glickman MH. COP9 signalosome components play a role in the mating pheromone response of S. cerevisiae. EMBO Rep. 2002;3:1215–21. doi: 10.1093/embo-reports/kvf235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wei N, Serino G, Deng XW. The COP9 signalosome: more than a protease. Trends Biochem Sci. 2008;33:592–600. doi: 10.1016/j.tibs.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 54.Adler AS, Littlepage LE, Lin M, Kawahara TL, Wong DJ, Werb Z, Chang HY. CSN5 isopeptidase activity links COP9 signalosome activation to breast cancer progression. Cancer Res. 2008;68:506–15. doi: 10.1158/0008-5472.CAN-07-3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Savvides SN, Scheiwein M, Bohme CC, Arteel GE, Karplus PA, Becker K, Schirmer RH. Crystal structure of the antioxidant enzyme glutathione reductase inactivated by peroxynitrite. J Biol Chem. 2002;277:2779–84. doi: 10.1074/jbc.M108190200. [DOI] [PubMed] [Google Scholar]
- 56.Hallstrom TC, Nevins JR. Jab1 is a specificity factor for E2F1-induced apoptosis. Genes Dev. 2006;20:613–23. doi: 10.1101/gad.1345006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shackleford TJ, Claret FX. JAB1/CSN5: a new player in cell cycle control and cancer. Cell Div. 2010;5:26. doi: 10.1186/1747-1028-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sui L, Dong Y, Ohno M, Watanabe Y, Sugimoto K, Tai Y, Tokuda M. Jab1 expression is associated with inverse expression of p27(kip1) and poor prognosis in epithelial ovarian tumors. Clin Cancer Res. 2001;7:4130–5. [PubMed] [Google Scholar]
- 59.Hsu MC, Huang CC, Chang HC, Hu TH, Hung WC. Overexpression of Jab1 in hepatocellular carcinoma and its inhibition by peroxisome proliferator-activated receptorgamma ligands in vitro and in vivo. Clin Cancer Res. 2008;14:4045–52. doi: 10.1158/1078-0432.CCR-07-5040. [DOI] [PubMed] [Google Scholar]
- 60.Osoegawa A, Yoshino I, Kometani T, Yamaguchi M, Kameyama T, Yohena T, Maehara Y. Overexpression of Jun activation domain-binding protein 1 in nonsmall cell lung cancer and its significance in p27 expression and clinical features. Cancer. 2006;107:154–61. doi: 10.1002/cncr.21961. [DOI] [PubMed] [Google Scholar]
- 61.Kouvaraki MA, Korapati AL, Rassidakis GZ, Tian L, Zhang Q, Chiao P, Ho L, Evans DB, Claret FX. Potential role of Jun activation domain-binding protein 1 as a negative regulator of p27kip1 in pancreatic adenocarcinoma. Cancer Res. 2006;66:8581–9. doi: 10.1158/0008-5472.CAN-06-0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kouvaraki MA, Rassidakis GZ, Tian L, Kumar R, Kittas C, Claret FX. Jun activation domain-binding protein 1 expression in breast cancer inversely correlates with the cell cycle inhibitor p27(Kip1) Cancer Res. 2003;63:2977–81. [PubMed] [Google Scholar]
- 63.Gao L, Huang S, Ren W, Zhao L, Li J, Zhi K, Zhang Y, Qi H, Huang C. Jun activation domain-binding protein 1 expression in oral squamous cell carcinomas inversely correlates with the cell cycle inhibitor p27. Med Oncol. 2012;29:2499–504. doi: 10.1007/s12032-012-0177-0. [DOI] [PubMed] [Google Scholar]
- 64.Ahn J, Hong SA, Lee SE, Kim J, Oh YS, Park SJ, Chung YJ. Cytoplasmic localization of Jab1 and p27 Kip1 might be associated with invasiveness of papillary thyroid carcinoma. Endocr J. 2009;56:707–13. doi: 10.1507/endocrj.K08E-372. [DOI] [PubMed] [Google Scholar]
- 65.Michel JJ, Xiong Y. Human CUL-1, but not other cullin family members, selectively interacts with SKP1 to form a complex with SKP2 and cyclin A. Cell Growth Differ. 1998;9:435–49. [PubMed] [Google Scholar]
- 66.Nayak S, Santiago FE, Jin H, Lin D, Schedl T, Kipreos ET. The Caenorhabditis elegans Skp1-related gene family: diverse functions in cell proliferation, morphogenesis, and meiosis. Curr Biol. 2002;12:277–87. doi: 10.1016/S0960-9822(02)00682-6. [DOI] [PubMed] [Google Scholar]
- 67.Fischer ES, Scrima A, Böhm K, Matsumoto S, Lingaraju GM, Faty M, Yasuda T, Cavadini S, Wakasugi M, Hanaoka F, et al. The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell. 2011;147:1024–39. doi: 10.1016/j.cell.2011.10.035. [DOI] [PubMed] [Google Scholar]
- 68.Groisman R, Polanowska J, Kuraoka I, Sawada J, Saijo M, Drapkin R, Kisselev AF, Tanaka K, Nakatani Y. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell. 2003;113:357–67. doi: 10.1016/S0092-8674(03)00316-7. [DOI] [PubMed] [Google Scholar]
- 69.Parrilla-Castellar ER, Arlander SJ, Karnitz L. Dial 9-1-1 for DNA damage: the Rad9-Hus1-Rad1 (9-1-1) clamp complex. DNA Repair (Amst) 2004;3:1009–14. doi: 10.1016/j.dnarep.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 70.Huang J, Yuan H, Lu C, Liu X, Cao X, Wan M. Jab1 mediates protein degradation of the Rad9-Rad1-Hus1 checkpoint complex. J Mol Biol. 2007;371:514–27. doi: 10.1016/j.jmb.2007.05.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Melo J, Toczyski D. A unified view of the DNA-damage checkpoint. Curr Opin Cell Biol. 2002;14:237–45. doi: 10.1016/S0955-0674(02)00312-5. [DOI] [PubMed] [Google Scholar]
- 72.Ghabrial A, Ray RP, Schüpbach T. okra and spindle-B encode components of the RAD52 DNA repair pathway and affect meiosis and patterning in Drosophila oogenesis. Genes Dev. 1998;12:2711–23. doi: 10.1101/gad.12.17.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Doronkin S, Djagaeva I, Beckendorf SK. CSN5/Jab1 mutations affect axis formation in the Drosophila oocyte by activating a meiotic checkpoint. Development. 2002;129:5053–64. doi: 10.1242/dev.129.21.5053. [DOI] [PubMed] [Google Scholar]
- 74.McKim KS, Hayashi-Hagihara A. mei-W68 in Drosophila melanogaster encodes a Spo11 homolog: evidence that the mechanism for initiating meiotic recombination is conserved. Genes Dev. 1998;12:2932–42. doi: 10.1101/gad.12.18.2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shinohara A, Ogawa H, Ogawa T. Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell. 1992;69:457–70. doi: 10.1016/0092-8674(92)90447-K. [DOI] [PubMed] [Google Scholar]
- 76.Buscemi G, Perego P, Carenini N, Nakanishi M, Chessa L, Chen J, Khanna K, Delia D. Activation of ATM and Chk2 kinases in relation to the amount of DNA strand breaks. Oncogene. 2004;23:7691–700. doi: 10.1038/sj.onc.1207986. [DOI] [PubMed] [Google Scholar]
- 77.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 78.Lee EW, Lee S, Song J. Jab1 has negative effects on p53-mediated genotoxic stresses. BMB Rep. 2009;42:299–303. doi: 10.5483/BMBRep.2009.42.5.299. [DOI] [PubMed] [Google Scholar]
- 79.Wang B, Matsuoka S, Carpenter PB, Elledge SJ. 53BP1, a mediator of the DNA damage checkpoint. Science. 2002;298:1435–8. doi: 10.1126/science.1076182. [DOI] [PubMed] [Google Scholar]
- 80.Wang F, Wang Y, Yu X, Yang D, Wang Z, Lu C, Yuan Z, Xiao M, Shen A. Significance of Jab1 expression in human esophageal squamous cell carcinoma. J Clin Gastroenterol. 2009;43:520–6. doi: 10.1097/MCG.0b013e3181919245. [DOI] [PubMed] [Google Scholar]
- 81.Dong Y, Sui L, Watanabe Y, Yamaguchi F, Hatano N, Tokuda M. Prognostic significance of Jab1 expression in laryngeal squamous cell carcinomas. Clin Cancer Res. 2005;11:259–66. [PubMed] [Google Scholar]
- 82.Hsu MC, Chang HC, Hung WC. HER-2/neu transcriptionally activates Jab1 expression via the AKT/beta-catenin pathway in breast cancer cells. Endocr Relat Cancer. 2007;14:655–67. doi: 10.1677/ERC-07-0077. [DOI] [PubMed] [Google Scholar]
- 83.Tomoda K, Yoneda-Kato N, Fukumoto A, Yamanaka S, Kato JY. Multiple functions of Jab1 are required for early embryonic development and growth potential in mice. J Biol Chem. 2004;279:43013–8. doi: 10.1074/jbc.M406559200. [DOI] [PubMed] [Google Scholar]
- 84.Sitte S, Gläsner J, Jellusova J, Weisel F, Panattoni M, Pardi R, Gessner A. JAB1 is essential for B cell development and germinal center formation and inversely regulates Fas ligand and Bcl6 expression. J Immunol. 2012;188:2677–86. doi: 10.4049/jimmunol.1101455. [DOI] [PubMed] [Google Scholar]
- 85.Wang H, Song W, Hu T, Zhang N, Miao S, Zong S, Wang L. Fank1 interacts with Jab1 and regulates cell apoptosis via the AP-1 pathway. Cell Mol Life Sci. 2011;68:2129–39. doi: 10.1007/s00018-010-0559-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li J, Wang Y, Yang C, Wang P, Oelschlager DK, Zheng Y, Tian DA, Grizzle WE, Buchsbaum DJ, Wan M. Polyethylene glycosylated curcumin conjugate inhibits pancreatic cancer cell growth through inactivation of Jab1. Mol Pharmacol. 2009;76:81–90. doi: 10.1124/mol.109.054551. [DOI] [PubMed] [Google Scholar]
- 87.Pan Y, Wang M, Bu X, Zuo Y, Wang S, Wang D, Liu Q, Su B, Xu T, Wang C, et al. Curcumin analogue T83 exhibits potent antitumor activity and induces radiosensitivity through inactivation of Jab1 in nasopharyngeal carcinoma. BMC Cancer. 2013;13:323. doi: 10.1186/1471-2407-13-323. [DOI] [PMC free article] [PubMed] [Google Scholar]