

Summary
RNase L is an ankyrin repeat domain-containing dual endoribonuclease-pseudokinase that is activated by unusual 2,′5′-oligoadenylate (2-5A) second messengers and which impedes viral infections in higher vertebrates. Despite its importance in interferon-regulated antiviral innate immunity, relatively little is known about its precise mechanism of action. Here we present a functional characterization of 2.5 Å and 3.25 Å X-ray crystal and small-angle X-ray scattering structures of RNase L bound to a natural 2-5A activator with and without ADP or the nonhydrolysable ATP mimetic AMP-PNP. These studies reveal how recognition of 2-5A through interactions with the ankyrin repeat domain and the pseudokinase domain, together with nucleotide binding, imposes a rigid intertwined dimer configuration that is essential for RNase catalytic and antiviral functions. The involvement of the pseudokinase domain of RNase L in 2-5A sensing, nucleotide binding, dimerization, and ribonuclease functions highlights the evolutionary adaptability of the eukaryotic protein kinase fold.
Graphical Abstract
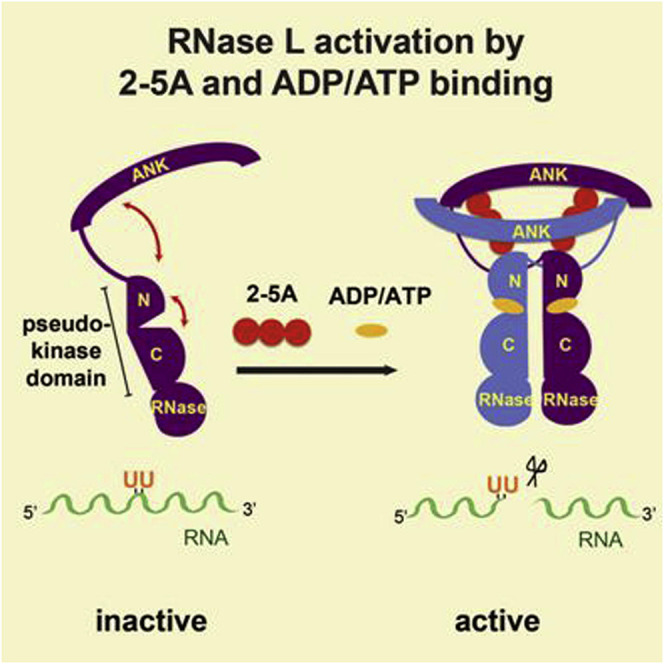
Highlights
-
•
Structural basis for RNase L regulation by 2-5A and nucleotide (ADP or ATP) binding
-
•
Recognition of 2-5A is mediated by both ankyrin repeat and protein kinase domains
-
•
Nucleotide enforces a closed conformation of the kinase domain
-
•
Nucleotide binding to the pseudokinase domain is essential for RNA cleavage function
RNase L is an ankyrin repeat domain containing dual endoribonuclease-pseudokinase that is activated by 2′,5′-oligoadenylate (2-5A) second messengers and which impedes viral infections in higher vertebrates. Huang et al. revealed the structure of RNase L bound to 2-5A activator with and without the ATP mimetic AMP-PNP.
Introduction
In higher vertebrates, RNase L is a principal mediator of the interferon (IFN)-inducible antiviral state that can determine survival of animals infected with highly pathogenic viruses (Zhou et al., 1993, Zhou et al., 1997). Virus-infected cells produce and secrete type I IFNs that bind to the cell-surface receptor, IFNAR, initiating JAK-STAT signaling to many IFN-stimulated genes (ISG), including 2′,5′-oligoadenylate (2-5A) synthetase (OAS) genes (reviewed in Kristiansen et al., 2011, Stark et al., 1998). OAS1-3 proteins are pathogen recognition receptors for the viral pathogen-associated molecular pattern, double-stranded (ds) RNA (reviewed in Silverman [2007]). Once activated by viral dsRNA, OAS uses ATP to synthesize a series of 2-5A molecules consisting of p3A(2′p5′A)n, n ≥ 2 (Kerr and Brown, 1978). The only well-established function of 2-5A is to activate RNase L. 2-5A binds to monomeric, inactive RNase L, causing it to dimerize into its active state and resulting in cleavage of single-stranded viral and cellular RNA, principally after UpU and UpA dinucleotides (Floyd-Smith et al., 1981, Wreschner et al., 1981). The triadenylate form of 2-5A is the minimal active species; however, longer 2′,5′-oligoadenylates, such as the tetraadenylate, retain the ability to activate RNase L (Dong et al., 1994). Naturally occurring species of 2-5A contain three phosphoryl groups on the 5′-OH (Kerr and Brown, 1978), although 5′-monophosphorylated 2-5A is capable of activating human RNase L in vitro (Dong et al., 1994). RNase L contains, from its N to its C termini, an ankyrin repeat (ANK) domain, a pseudo-protein kinase (PK) domain lacking catalytic function (Dong and Silverman, 1999), and a ribonuclease (RNase) domain, the latter two of which form a fused module homologous to the dual kinase-ribonuclease enzyme IRE1, a key effector of the unfolded protein response pathway (Walter and Ron, 2011).
Crystallographic analysis of a monomeric ANK domain fragment of human RNase L (Tanaka et al., 2004) provided a first view of a subset of molecular interactions required for binding to 2-5A. Subsequent analysis of the complete ANK domain revealed how concerted binding of 2-5A to two ANK domains can mediate dimerization (Han et al., 2012). Whether the PK and RNase domains also participate in 2-5A recognition and how 2-5A induces dimerization of the ANK domain imposing a specific dimer configuration on other regions of RNase L remained open questions. The function of the pseudo-PK domain has remained largely uncharacterized, but early studies indicated a possible role for ADP or ATP binding in enzyme stabilization or activation (Dong et al., 1994, Wreschner et al., 1982). In addition, a K392R mutation in the kinase ATP binding site of human RNase L (corresponding to residue 390 in porcine RNase L) resulted in greatly impaired RNase activity (Dong and Silverman, 1999). Lastly, the RNase domain of RNase L shows sequence similarity to only the RNase domain of IRE1 (Korennykh et al., 2009, Lee et al., 2008), and by inference, RNase L was predicted to share a common catalytic mechanism regulated by the imposition of a precise dimer configuration (Lee et al., 2008).
To uncover the molecular basis for the regulation of RNase L antiviral function, we solved the 2.5 Å and 3.25 Å X-ray crystal and small-angle X-ray scattering (SAXS) solution structures of near full-length porcine RNase L bound to a natural 2-5A activator with and without the ATP mimetic AMP-PNP. Together with mutational and functional studies performed in vitro and in vivo, these studies shed light on how RNase L recognizes 2-5A through concerted interactions involving the ANK domain and through an unexpected appendage to the pseudo-PK domain, and furthermore on how 2-5A recognition, together with nucleotide binding to the pseudo-PK domain, facilitates the imposition of a specific dimer configuration required for RNase catalytic function.
Results
X-Ray Crystal Structure of RNase L in Complex with 2-5A
We identified diffracting crystals for porcine (Sus scrofa) RNase L (residues 21–732) lacking 20 N-terminal and 11 C-terminal protease-labile residues dispensable for enzymatic function (Dong and Silverman, 1997). Crystals with the same space group, unit cell dimensions, and crystal packing arrangement were grown with natural 5′-triphosphorylated, 2′,5′- triadenylate activator (2′-5′)p3A3 in the presence and absence of the ATP analog AMP-PNP. Structures determined to 2.5 Å and 3.25 Å resolution, respectively, revealed a near-complete model of RNase L lacking only one N-terminal residue, three C-terminal residues, 11 residues comprising the ANK domain-PK domain linker, a four residue loop in the PK domain, and two and 19 residue loops within the RNase domain (see Table 1 for data collection and refinement statistics). Crystal packing arrangements and representative unbiased electron density centered on the 2-5A and AMP-PNP ligands are shown in Figure S1, available online. A structure-based sequence alignment of RNase L and for comparison the dual PK domain-RNase domain catalytic module of IRE1, is shown in Figure S2.
Table 1.
Summary of Data Collection, Structure Determination, and Refinement Statistics
| Ligands | 2-5A + AMP-PNP | 2-5A |
|---|---|---|
| Space group | P21 | P21 |
| Unit cell parameters (Å; °) | ||
| a | 59.20 | 59.78 |
| b | 267.4 | 268.48 |
| c | 110.21 | 111.89 |
| β | 90.04 | 90.04 |
| Molecules/asu | 4 | 4 |
| Resolution | 59.2–2.50 (2.66–2.5) | 49.7–3.25 (3.37–3.25) |
| Unique reflections | 106,347 (15,940) | 51,209 (5,016) |
| Redundancy | 3.0 (3.2) | 2.4 (2.3) |
| Completeness (%) | 95.7 (88.4) | 94.1 (92.9) |
| Wilson B (Å2) | 52.7 | 88.3 |
| I/σI | 6.8 (1.6) | 8.2 (1.2) |
| CC1/2 (%) | 69.1 | 43.9 |
| Rmeas | 0.104 (0.526) | 0.131 (0.980) |
| Rwork/Rfree | 0.196/0.231 | 0.230/0.289 |
| Rmsd from ideal geometry | ||
| Bonds (Å) | 0.007 | 0.010 |
| Angles (°) | 1.231 | 1.394 |
| Average B factors (Å2) | ||
| Protein | 52.9 | 129.5 |
| 2-5A | 41.1 | 115.0 |
| AMP-PNP:Mg2+ | 37.7 | - |
| Ramachandran plot | ||
| Favored regions (%) | 97.54 | 96.26 |
| Disallowed regions (%) | 0 | 0 |
Structure Overview
Since the structures of RNase L in the presence and absence of AMP-PNP were near identical (rmsd = 0.5 Å), we focused our analysis and discussion on the higher-resolution AMP-PNP bound structure. Four protomers of RNase L in the crystal asymmetric unit associate to form two identical (rmsd = 0.3 Å) noncrystallographic dimers with each dimer engaging 2 mole equivalents of 2-5A and of AMP-PNP ligands (Figure 1 ). Easily recognized within each RNase L protomer are the ANK, PK, and RNase domains. As observed in the isolated ANK domain crystal structures of human RNase L (Han et al., 2012, Tanaka et al., 2004), the ANK domain of porcine RNase L adopts a crescent-shaped structure consisting of a stack of nine ankyrin repeats (Figure 1B), each possessing a characteristic loop-helix1-loop-helix2-loop architecture. Situated between ANK repeats 5 and 6 lies a noncharacteristic α-helical element (corresponding to residues 156–163) denoted α-AR8′ (Figure 1A and Figure S2). Largely disordered in the isolated ANK domain structures, helix α-AR8′ features prominently in interdomain interactions.
Figure 1.

Crystal Structure of RNase L
(A) Ribbons representation of the RNase L dimer. 2-5A and AMP-PNP ligands are shown as sticks.
(B) Top view of the RNase L dimer orientation shown in (A).
(C) Bottom view of the RNase L dimer orientation shown in (A). Left, ribbons and surface representation. Inset, zoom-in view of RNase active site residues. Catalytic residues of RNase L are depicted as pink sticks, and IRE1 residues found at equivalent positions are colored beige. Disordered regions are shown as dashed lines, and the RNase L-specific helix α1′ is shown in red.
The Pseudokinase Domain
The pseudo-PK domain possesses a bilobal fold with a smaller N-terminal lobe (N-lobe) and a larger C-terminal lobe (C-lobe) (Figure 2 A). The N-lobe consists of a canonical five-strand antiparallel β sheet (denoted β1–β5) and flanking helix αC (Figure S2). Grafted onto this common core, the first 36 ordered residues of the PK domain form a noncanonical appendage, with loop (L1)-helix (αA)-loop (L2) topology, which features prominently in 2-5A activator recognition and in interdomain interactions. The PK C-lobe, which also deviates greatly from canonicality, is comprised of a three strand antiparallel β sheet (β5′, β7–β8), a two-strand β sheet (β6, β9), and seven α helices (αD−αI) (Figure S2). Positioned between β strand nine and helix αF (as shown for PKA in Figure 2B) are the expected positions of the activation segment, helix αEF and the helix αEF-αF connecting loop, elements which serve phospho-regulatory and phospho-acceptor recognition functions in prototypical protein kinases (Nolen et al., 2004). In the RNase L structure, this entire region is absent and substituted by a four-residue shunt, whose structure enables a nonstandard position of a split helix αG, an element also normally implicated in PK substrate recognition (Brennan et al., 2011, Dar et al., 2005). Other distinguishing features of the PK C-lobe include uncommon substitutions at PK conserved positions Gln484, Gln481, and Asp502 (Lys, Arg, and Gly residues in IRE1, respectively) (Figure 2C). Together, these unconventional features of the kinase C-lobe likely account for the incompetence of RNase L as a PK enzyme (Dong and Silverman, 1999).
Figure 2.

Structure of the Pseudokinase Domain of RNase L
(A) Ribbons view of the pseudo-PK domain with noncanonical elements highlighted in red, pink, and magenta.
(B) Comparison of the structural topology of RNase L to the prototypical protein kinase PKA. PKA activation segment, helix αEF, and αEF-αF loop are colored green.
(C) Ribbons view of the kinase active site region of RNase L colored as in (A). Corresponding IRE1 residues are shown as green sticks.
(D) Residues that compose the catalytic (C) and regulatory (R) spines of RNase L are shown as sticks with transparent surface. Binding of nucleotide to the kinase active site correlates with the intact regulatory and catalytic spines.
See also Figure S2.
However, suggesting a critical role for nucleotide binding in the overall biological function of RNase L, a fully ordered AMP-PNP molecule is found coordinated by a full complement of PK invariant residues, namely, Asp482 (catalytic base), Asn487 (Mg2+ coordination), Asp500 (Mg2+ coordination), Lys390 (phosphate coordination), Glu402 (salt contact with Lys390), and Ala388 (contact with adenine base), within the kinase interlobe cleft (Figure 2C). The uncommon substitution of Gly for Asp at position 502 is notable for contributing additional interactions to a magnesium ion, which tethers the γ-phosphate of AMP-PNP (Figure 2C). The binding of AMP-PNP gives rise to a rigid closed conformation of the PK domain, as evidenced by intact catalytic and regulatory spines (Figure 2D), features reflective of the active state of protein kinases (Kornev et al., 2008). The absence of nucleotide in the AMP-PNP-free structure notably disrupts the integrity of the catalytic spine and leaves the kinase C-lobe relatively untethered from the N-lobe.
The RNase Domain
Extending as a rigid protrusion from PK C-lobe is the RNase domain consisting of nine α helices (denoted α1, α1′, α2-α8, Figures 1A and 1C) with a 19-residue disordered sequence (residues 644–662, harboring a 12-residue porcine-specific insertion) between helices α3 and α4. Centered on helix α4 of each protomer at the base of a deep cleft formed by the dimerization of two RNase domains lies the ribonuclease active site with two copies composed in cis of an invariant tetrad consisting of Tyr663, Arg 675, Asn676, and His680 side chains and the conserved basic side chain of Lys672. Consistent with a shared catalytic mechanism for phosphodiester bond cleavage inferred originally from comparisons to tRNA ribonuclease (Lee et al., 2008, Xue et al., 2006), the catalytic residues of RNase L adopt conformations and placements highly similar to those adopted by the corresponding residues of IRE1 (Figure 1C). In contrast, residues composing the surrounding solvent-exposed surface are completely divergent across paralogues with RNase L also possessing a unique helical insert (helix α1′) lining the outermost edge of the catalytic cleft. This divergence of structure may account for the distinct RNA substrate repertoires of RNase L and IRE1.
Dimer Configuration of RNase L
The dimer configuration of full-length RNase L arises from the intertwining of two promoters through a 155° twisting action across the ANK domain-PK domain junction, which gives rise to the appearance of an exchange of ANK and PK domains between protomers. Crossprotomer contacts within the dimer are vastly greater than typical homodimers (Jones and Thornton, 1996) (total buried surface area = 7,894/7,102 Å2, including/excluding 2-5A contacts, respectively) (Figures S2 and S3) and consist of (1) the antiparallel interaction of two ANK domains, (2) the parallel back-to-back interaction of the dual pseudo-PK domain-RNase domain modules, (3) crossprotomer interaction between the ANK and pseudo-PK domains, and (4) coordinated binding of two 2-5A activators, each engaging a composite binding pocket involving two ANK domains and the N-lobe of one PK domain.
Dimerization of the ANK domain is facilitated by a head-to-tail arrangement with concave surfaces facing inward (SA contact without 2-5A = 795 Å2) (Figures 1, S2, and S3). In brief, ANK domain dimerization is mediated by the reciprocal interaction of ANK repeats 1–4 of one protomer with ANK repeat 9 of a second protomer at either end of the crescent shaped structure, similar to the interaction mode uncovered for the isolated human ANK domain bound to a synthetic 2-5A activator (rmsd between dimers = 2.1 Å, PDB ID 4G8L).
Back-to-Back Interaction of the Dual Pseudo-PK Domain–RNase Domain Module
Dimerization of the dual pseudo-PK domain-RNase domain module involves discontinuous contacts between the two kinase N-lobes (surface area = 1,478 Å2) and between the two RNase domains (surface area = 795 Å2) (Figures 3 A, S2, and S3). Engagement of these two surfaces is coordinated by the rigidly fused junction between the PK C-lobe and the RNase domain arising from a contiguous hydrophobic core (Figure S4A), and by the binding of AMP-PNP to the pseudo-PK active site, which is predicted to constrain the inherent interlobe flexibility of the PK domain to a single closed conformation (Masterson et al., 2011). Although only 3 (Tyr419, Arg675, and His680) of 27 back-to-back dimer contact residues on each protomer are conserved across paralogues (Figure S2), the back-to-back dimer configuration of RNase L is highly similar (difference of 25°) to that adopted by yeast IRE1 (Figure S4A). Back-to-back dimer contacts are primarily polar in nature and include a reciprocal salt bridge between Glu383 and Arg410 in the kinase N-lobes and between Lys603 and Glu683 and between Arg675 and Glu679 in the RNase domains (Figure 3A, insets). Beyond this back-to-back dimer arrangement, no other packing arrangement of RNase L in the crystal environment bears resemblance to the helical polymer arrangement uncovered for IRE1 (Figures S1B–S1D).
Figure 3.

Crossprotomer Interactions of the RNase L Dimer
(A) Back-to-back interaction of the dual pseudo-PK–RNase domain module. Right panels show zoom-in view of boxed regions.
(B) In trans interaction of the ANK domain with the pseudo-PK domain. Bottom panels show zoom-in view of the boxed regions.
See also Figure S3.
Kinase Domain-ANK Domain Contacts
Excluding the covalent link mediated by the eight-residue disordered linker sequence, only a tenuous pi-stacking interaction (buried surface = 78.2 Å2) between Arg308 and Trp352 links the ANK and PK domains within each protomer (Figures 3 and S4B). In contrast, interprotomer interactions between PK and ANK domains within the dimer are extensive (buried surface = 1,588 Å2) (Figures 3B, S2, and S3). One set of interactions is mediated by the loop regions following ANK repeats 1, 2, 4, 5, 6, and 7 and by helix α-AR8′. These elements contact the top surface of the PK N-lobe, with helix α-AR8′ engaging the G-rich loop of the PK domain through a hydrophobic interaction between ANKIle162 and PKCys427 and a salt interaction between ANKAsp158 and PKLys368. A second constellation of interactions is mediated by the noncanonical appendage of the PK N-lobe, which engages the loop regions linking ANK repeats 1–2 and 2–3. Included are three centered salt bridges between PKAsp364, ANKArg88, and PKGlu366 and between PKArg350, ANKGlu55, and PKHis347, and a hydrophobic interaction between ANKTrp58, PKIle351, and PKPhe362. A final set of interactions is mediated by the loop regions linking ANK repeats 5–6, 6–7, and 7–8, which form a constellation of charge-complementary interactions with the forward projecting end of PK helix αC and the β4-β5 linker, notably between ANKArg 269 and PKGlu394.
The 2-5A Binding Site
The RNase L dimer engages two 2-5A activator molecules through symmetric pincer-like mechanisms with each 2-5A molecule binding to a composite channel composed by the two antiparallel aligned ANK domains and one PK domain (Figure 4 A). The channel mouth oriented inward to a large internal cavity between RNase L protomers engages the triphosphate moieties of Adenine 1 (Figure 4B), while the channel mouth oriented outward to solvent accommodates the untethered end of activators of length greater than three adenine residues (Figure 4A). Remarkably, within the tight confine of the channel center, virtually every functional group of 2-5A is engaged in molecular interactions with RNase L.
Figure 4.

Composite Binding Site of the 2-5A Activator
(A) Surface view of the RNase L dimer highlighting the outward facing channel that accommodates 2-5A activators with length greater than three adenosine residues.
(B) Ribbons view of the composite binding site for the 2-5A activator (shown as sticks). Interacting residues are shown in the zoom in view as sticks with transparent surface.
See also Figures S4 and S5.
Contacts with the two ANK domains bury 711 Å2 of the 1,167 Å2 total surface area of 2-5A and are similar overall to the contacts uncovered for a synthetic, monophosphate 2-5A analog in isolated ANK domain structures (Han et al., 2012, Tanaka et al., 2004). Subtle differences in binding mode include a flip of the Adenine 3 base (Figure S5) and the repositioning of the ANKArg308 side chain to capture a pi-stacking interaction with PKTrp352 to form a clamp around the Adenine 3 ribose ring (Figure 4A). ANK repeats 1–6 of one protomer make extensive (SA = 529 Å2) contact with the back side of all three adenine residues while ANK repeat nine of the second protomer makes focused contacts (SA = 182 Å2) with the phosphate, sugar, and base of Adenine 3. Novel PK domain contacts covering 156 Å2 of 2-5A are mediated primarily by L2 of the PK N-lobe appendage, with the β and γ phosphates of Adenosine 1 coordinated by a bifurcated salt interaction with the Lys164 side chain, which in turn is coordinated by a salt interaction with Asp424 in the β4-β5 linker of the PK domain (the latter is visible in Figure 3B). Lastly, the phosphate moiety linking Adenine 1 and Adenine 2 is coordinated by a salt interaction with Arg353 in the PK N-lobe appendage and by van der Waals contacts with adjacent Pro354, Ile363, and Phe362 side chains.
Structure-Guided Mutational and In Vitro Functional Analyses of RNase L
To probe the functional interplay between ANK, PK, and RNase domains and the importance of specific contacts, we pursued functional analyses of wild-type (WT) RNase L and a comprehensive panel of structure guided mutants (Figures 5 , S6, and S7). We first investigated the effect of ligand binding on the oligomerization status of WT enzyme in solution using velocity analytical ultracentrifugation (Figure S6A). RNase L alone and in the presence of AMP-PNP was a monomer over the 0.5–10 μM protein concentration range tested. In the presence of 2-5A activator alone or with both 2-5A and AMP-PNP, RNase L was a dimer. These results with porcine RNase L are consistent with the behavior of human RNase L (Cole et al., 1997). Using fluorescein-labeled RNase L, which enabled AUC analyses at nanomolar protein concentrations, we observed that AMP-PNP had no significant effect on the ability of RNase L to dimerize when already bound to 2-5A (Figure S6B). Using surface plasmon resonance (SPR) with biotin-labeled 2-5A immobilized onto streptavidin sensor chips, we assessed the relative binding affinity of full-length versus an ANK domain-only fragment of RNase L. Consistent with the PK domain contributing direct contacts to 2-5A, full-length RNase L bound to 2-5A with ∼7-fold higher affinity than the isolated ANK domain (57 ± 16 nM versus 376 ± 10 nM, respectively) (Figure 5A). In the case of full-length RNase L, the presence of ATP had no significant effect on the ability to bind 2-5A (Figures 5A and S7A).
Figure 5.

In Vitro Functional Characterization of RNase L
(A) Binding of 2-5A to full-length RNase L and isolated RNase L ANK domain assessed by surface plasmon resonance. Response levels for specific binding of the indicated RNase L forms to immobilized biotinylated 2-5A were plotted against increasing protein concentration using, where appropriate, a variable slope model to determine the Hill coefficient and Kd. Reported Kd is the average of two independent experiments ± SD. Displayed results are representative of two independent experiments.
(B) RNase L activity profiles monitored in the indicated presence/absence of 2-5A activator and 50 μM of the indicated nucleotide using a FRET pair-labeled RNA substrate. Displayed results are representative of two independent experiments measured in duplicate (± SEM).
(C) Binding of fluorescently labeled ATP analog (BODIPY FL ATP-γ-S) to RNase L assessed by monitoring the fluorescence polarization signal in the presence of increasing concentrations of the indicated forms of RNase L. Displayed results are representative of two independent experiments measured in duplicate (± SEM).
(D–H) RNase activity profiles of WT and the indicated mutant forms of RNase L monitored as described in (B). Concentration of ADP in (D) was 2 μM and in (E)–(H) was 50 μM. Displayed results are representative of two independent experiments measured in duplicate.
See also Figures S6 and S7.
We next assessed the effect of ligand binding on the RNase activity of WT enzyme at 1.6 nM concentration. In the absence of both or either of 2-5A and nucleotide ligand, no RNase activity was detected, whereas in the presence of both ligands robust RNase activity was observed (Figure 5B). We attribute the unanticipated strict dependency of RNase L RNase function on nucleotide (Dong et al., 1994, Wreschner et al., 1982) to the purity of our protein preparations (see Experimental Procedures). We also investigated the ability of RNase L to discriminate between nucleotides, since the RNase function of yeast IRE1 responds more potently to ADP than to ATP (Sidrauski and Walter, 1997). We found that ATP, ADP, and, to a slight lesser degree, the nonhydrolysable analog AMP-PNP all potentiated RNase function (Figure 5B). Consistent with these observations, mutation of ATP-coordinating residues in RNase L that markedly reduced nucleotide binding (Figure 5C) also caused a major impairment to RNase enzyme function (Figure 5D). This impairment could be partially offset for some mutants by higher nucleotide concentration (Figure S7B). Consistent with the corollary finding, we observed that 2-5A did not potentiate RNase L binding to nucleotide (Figure S7C).
Highlighting the functional importance of the parallel back-to-back dimer configuration of the dual PK domain-RNase domain module, single or double mutations in the kinase domain (Arg410Glu) or in the RNase domain (Arg675Glu, Glu679Arg, Arg675Glu/Glu679Arg) greatly impaired RNase activity (Figure 5E). Notably, these adverse effects were comparable to the loss of function displayed by the RNase catalytic tetrad mutant His680Ala (Figure 5F). Similarly, mutation of the sole cis interaction site between ANK and PK domains (Arg308Ala or Trp352Ala) caused great to intermediate loss of RNase function, respectively (Figure 5G). The difference in severity for the two mutations may in part reflect the different roles that each position also plays in directly contacting 2-5A (Figure 4). Disruption of the ANK domain-PK domain interaction in trans by the single charge reversal substitutions Asp158Lys or Lys368Asp, by the double substitution Asp158Lys/Lys368Asp, or by deletion of helix αAR8′ (Δ156-166) also potently diminished RNase activity (Figure 5G). Lastly, Tyr310Ala, Lys164Glu, and Arg353Glu mutations affecting 2-5A contacts with each of the two ANK domains, and the PK domain, respectively, also perturbed RNase function (Figure 5H). Since all RNase L mutants tested appeared properly folded as assessed by elution volume during purification by size exclusion chromatography (data not shown), these in vitro functional studies support the notion that the dimer configuration visualized in a crystal environment reflects a functionally relevant form of RNase L in solution.
Impact of RNase L Mutagenesis on Ribonuclease Activity in Intact Cells
To determine the effects of different mutations on enzyme activity in intact cells, HeLa-M cells deficient in endogeneous RNase L were used (Xiang et al., 2003). WT or mutant forms of porcine RNase L were transfected into HeLa-M cells for 24 hr prior to transfecting with the synthetic double-stranded RNA, poly(rI):poly(rC) (pIC), for an additional 3 hr. When introduced into cells, pIC activates OAS, causing production and accumulation of 2-5A, and activation of RNase L (Zhao et al., 2012). To monitor RNase L activity in intact cells, total cellular RNA was isolated and separated on RNA chips (Agilent) (Xiang et al., 2003) (Figures 6A and 6B, upper panels). The rRNA remained intact in control HeLa-M cells transfected with empty vector following by pIC (Figures 6A and 6B, lane 1). In contrast, WT porcine RNase L expressed in HeLa-M cells was strongly activated in response to pIC as determined by monitoring highly specific and characteristic discrete cleavage products of 28S and 18S rRNA from ribosomes (Silverman et al., 1983) (Figures 6A and 6B, compare lanes 2 and 3 without and with pIC, respectively). Mutant RNase L lacking residues 156–166, an ANK segment interacting with both 2-5A and the PK domain, lacked ribonuclease activity (Figure 6A, lane 4). The Lys164Glu mutation of the binding site for natural 2-5A (through its β and γ phosphoryl groups), showed reduced ribonuclease activity (Figure 6A, lane 5). Mutant Tyr310Ala in the ANK 2-5A binding site was inactive, consistent with a prior report (Han et al., 2012) (Figure 6A, lane 6). Arg410Glu in the PK:PK interface was also defective for ribonuclease activity (Figure 6A, lane 7). Lys603Glu in the RNase:RNase interface had reduced activity (Figure 6A, lane 8). RNase domain mutants Lys672Ala, Arg675Glu, and Glu679Arg were all defective (Figure 6A, lanes 9–11). ANK mutation Asp158Lys, a residue that forms a salt bridge to Lys368 in the PK domain, had reduced activity, whereas ribonuclease activity in the double compensatory mutant Asp158Lys-Lys368Glu, that regenerates the salt bridge, was partially restored (Figure 6A, lanes 12 and 13). The latter result supports the functionality of the ANK:PK domain interface, although a partial recovery of function was not observed for a similar Asp158Lys-Lys368Asp mutant in our in vitro analysis. Five mutations in the ATP binding site of the PK domain were also evaluated for ribonuclease activity in intact cells (Figure 6B). Mutants Ala388Phe, Asp482Ala, Asn487Ala, Asp500Ala and to a lesser degree Glu402Ala all showed reduced activity (Figure 6B, lanes 4–8). Expression of the WT and the various mutant forms of porcine RNase L in HeLa-M cells occurred at similar levels as determined in western blots (Figures 6A and 6B, lower panels).
Figure 6.

Functional Analysis of RNase L in HeLa-M Cells
(A and B) Effect of RNase L mutations on RNase activity. Cells were transfected with empty vector or with plasmids expressing WT or the indicated mutant porcine RNase L. After 24 hr cells were transfected with poly(rI):poly(rC) (pIC) for 3 hr. RNA was extracted from a fraction of cells and separated on RNA chips. Western blots were performed on the cell extracts with anti-FLAG antibody for detection of recombinant porcine RNase L and with anti-β-actin antibody.
(C and D) Effect of RNase L mutations on viral infections. HeLa-M cells transfected with plasmids encoding WT or mutant forms of porcine RNase L were infected with EMCV for 14 hr (see Experimental Procedures). Viral yields were determined by plaque assays on L929 indicator cells. Data are shown as the mean ± SD from three biological replicates. ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05; ns, not significant (when each group was compared to empty vector control [Vector] with two-tailed Student’s t test).
Effect of RNase L Mutagenesis on Viral Infections
To determine if select mutations affect viral infections, WT or mutant forms of RNase L were expressed in HeLa-M cells followed by infection at an moi of 0.01 with the picornavirus, encephalomyocarditis virus (EMCV) (Hoskins and Sanders, 1957). At 14 hr postinfection, cellular supernatants containing progeny virus were collected and titered by plaque assays on monolayers of L929 indicator cells to determine viral yields. Expression levels of the WT and different mutant RNase L proteins in the HeLa-M cells were similar as determined by western blotting (Figures 6A and 6B; data not shown). WT RNase L reduced viral growth by about 12-fold (Figure 6C). In contrast, Δ156-166 (a segment in ANK that contacts both 2-5A and the PK domain), K164E (the binding site for natural 2-5A through its β and γ phosphoryl groups), Arg410Glu (PK:PK interface), and Lys603Glu (RNase:RNase interface) were either not inhibitory to viral growth or minimally inhibitory, indicating these amino acid residues are essential for the antiviral activity of RNase L. These findings validate key features of the dimeric RNase L structure in the context of an ongoing virus infection. In addition, no significant inhibition of viral replication was obtained with ATP binding site mutants Ala388Phe and Asp500Ala. However ATP-binding site mutants Glu402Ala, Asp482Ala, and Asn487Ala retained a reduced ability to suppress viral replication when compared to WT RNase L (Figure 6D).
Small-Angle X-Ray Scattering Analysis of RNase L
To investigate the effect of nucleotide binding on the structure of RNase L in solution free of confounding crystal packing interactions, we employed SAXS analyses. The Rg and Dmax derived for the scattering data for RNase L bound to 2-5A was consistent with those expected from the crystal structure (see Table S1 for data collection parameters). The SAXS envelope resulting from ten independently generated ab initio models resulted in an elongated envelope with two distinct volumes. Superimposition of the crystal structure of RNase L onto this envelope using the supcomb program revealed that the ankyrin repeat domains and kinase N-lobes readily fit in the larger volume of the envelope and that the kinase C-lobes and RNase domains fit in the smaller volume (Figure 7 A) (Kozin and Svergun, 2001). In good agreement, the Porod-calculated molecular weight (195 kDa) is consistent with RNase L existing as a dimer in solution. We then collected SAXS data for RNase L bound to 2-5A in the presence of ADP and found the sample had smaller Rg and Dmax values (Table S1). The averaged SAXS envelope for this sample was also elongated but smaller in overall volume. Superimposition of the X-ray crystal structure of RNase L onto this envelope revealed a tightening of volume along the dimerization axis most pronounced in the region corresponding to the kinase C-lobe and RNase domains. These results are consistent with ADP causing a restriction of motion between the two protein kinase lobes.
Figure 7.

Regulation of RNase L by 2-5A and Nucleotide Binding
(A) Small-angle X-ray scattering analysis of the 2-5A-induced RNase L dimer in the presence and absence of ADP (see Table S1 for data collection statistics). Top panels display the ab initio-calculated molecular envelopes. Bottom panels show envelopes with superimposed RNase L crystal coordinates.
(B) Schematic of RNase L regulation by 2-5A and nucleotide binding. 2-5A binding to RNase L through direct contact with the ANK domain and kinase N-lobe mediates molecular dimerization, while nucleotide binding to the kinase domain restrains the kinase domain to a single closed conformation required for the composition of the RNase active site.
See also Table S1.
Discussion
The dimeric structure of RNase L in a complex with 2-5A and AMP-PNP provides mechanistic insight into a fundamental molecular event in antiviral innate immunity, activation of RNase L. The presented results are consistent with the following model for RNase L regulation and function (Figure 7B). In the absence of stimuli, RNase L is a catalytically incompetent monomer. In response to the binding of 2-5A ligands to composite binding channels involving two ANK domains and one PK domain and to the binding of nucleotide to the PK active site, full-length RNase L dimerizes into a single rigid body that juxtaposes the two dual PK domain-RNase domain modules in a parallel back-to-back configuration. Dimerization of the RNase domain composes the RNase active site, enabling the cleavage of RNA substrates to mediate an antiviral response.
The RNase L regulatory model displays interesting parallels to that of IRE1, the sole protein in eukaryotes to share a homologous dual PK–RNase domain module. In place of an ANK domain, IRE1 possesses an endoplasmic reticulum (ER) resident luminal domain, which allows it to sense the accumulation of toxic misfolded proteins. The detection of misfolded protein transitions the ER luminal domain from low (n = 1 or 2) to high (n ≥ 2) stoichiometric oligomers (Credle et al., 2005, Kimata et al., 2007). Unlike RNase L, the dual PK–RNase domain module of IRE1 plays no role in the direct sensing of upstream-activating stimuli, and indeed the sensor and catalytic halves of IRE1 reside in separate organelle compartments, a feature which necessitates a less direct and precise mechanism for propagating dimerization/oligomerization signals. Lastly, the ability of the PK domain of RNase L to participate in stimuli sensing is supported by the evolution of a unique N-terminal lobe appendage not present in IRE1.
As a bona fide protein kinase, IRE1 also exploits its phosphotransfer function for autoregulation (Shamu and Walter, 1996). Oligomerization of the ER luminal domain induces the close approach of cytoplasmic domains allowing trans autophosphorylation on sites within the PK activation segment, a modification integral to the ability of the dual PK–RNase domain module to sample a productive back-to-back dimer orientation (Lee et al., 2008). RNase L, in contrast, lacks both the infrastructure to support phosphotransfer function and also the infrastructure (namely an activation segment harboring phosphoacceptor sites) to respond to autophosphorylation. In this respect, RNase L appears to have de-evolved to a structure akin to that of primitive atypical protein kinases such as Rio1 and Bud32 (Laronde-Leblanc et al., 2005, Mao et al., 2008), which are similarly lacking in canonical C-lobe features. Despite a loss of phosphotransfer function, the ATP binding function of RNase L, like that of IRE1, is preserved, enabling the enforcement of a closed conformation of the PK domain required to assemble a dimeric and functional RNase module (Lee et al., 2008). It remains to be determined, however, whether the absolute requirement of porcine RNase L for ADP or ATP extends to other species of RNase L.
The ability of IRE1 dimers to assemble into higher-order oligomers provides an additional level of control to promote RNase activity beyond that of minimal dimers (Korennykh et al., 2009). While a highly cooperative (hill coefficient > 4) dependence of RNase activity on enzyme concentration was interpreted to reflect RNase L also acting as a higher-order oligomer (Han et al., 2012), we see no evidence of oligomer formation even at the high protein concentrations employed in our analytical ultracentrifugation and SAXS analyses. Instead, we reason that this behavior may be a manifestation of the dependence of RNase function on dimerization and the coordinated binding of four (two 2-5A and two nucleotide) ligands.
IRE1 may act as a sensor of energy levels by virtue that its RNase activity is stimulated more greatly by ADP than ATP (Sidrauski and Walter, 1997). This characteristic is not shared by RNase L. Since total cellular ATP/ADP levels are generally high (3–5 mM) relative to the high affinity of RNase L for ATP (0.6–1.2 μM), and are tightly regulated to remain constant, they would be constitutively available for binding to the PK domain of RNase L. These properties likely account for our observation that some ATP binding site mutants of RNase L that display near-complete impairment of RNase function in vitro at low ATP concentration display significant residual function in cells. It is thus likely that the RNase function of RNase L is regulated primarily by 2-5A levels. However, it is conceivable that as-yet-undefined events or factors such as posttranslational modifications, cellular or viral protein products, or small molecules influence RNase function by modulating the ability of RNase L to interact with nucleotide.
Prior studies have shown that RNase L is a potent inhibitor of viral infections that can be essential for controlling viral pathogenesis, for instance as shown for viral hepatitis caused by a murine coronavirus, MHV (Zhao et al., 2012). While a number of different RNA and DNA viruses are susceptible to RNase L (reviewed in Silverman [2007]), some viruses have acquired or evolved genes that antagonize the OAS-RNase L pathway at different levels, including preventing OAS activation by sequestering dsRNA (Xiang et al., 2002), degrading 2-5A (Zhang et al., 2013, Zhao et al., 2012), or directly inhibiting RNase L (Sorgeloos et al., 2013, Townsend et al., 2008). Viral antagonism of the OAS-RNase L pathway emphasizes the threat to the virus imposed by RNase L. Activation of RNase L also has profound effects on the cell by regulating innate immune signaling (Malathi et al., 2007), autophagy (Chakrabarti et al., 2012, Siddiqui and Malathi, 2012), and apoptosis (Castelli et al., 1997, Castelli et al., 1998, Zhou et al., 1997). In addition, some human genetic studies have suggested that RNase L gene variants influence risk for different types of cancer, including hereditary prostate cancer (Carpten et al., 2002), hereditary nonpolyposis colorectal cancer (Krüger et al., 2005), and breast cancer (Madsen et al., 2008). A better understanding of RNase L activation at a molecular level may provide insight into these different facets of RNase L biology. Indeed, in these studies we show that the PK-PK interface, RNase-RNase interface, and ANK and pseudo-PK domains that interact with 2-5A are all essential for the antiviral activity of RNase L.
Experimental Procedures
Protein Expression, Crystallization, and Structure Determination
WT and mutant Sus scrofa RNase L proteins (residues 21–732 for crystallization studies and residues 1–743 for SAXS and biochemical studies) were expressed in Escherichia coli as TEV-cleavable GST fusions and purified by glutathione affinity, TEV cleavage, anion exchange, and finally size exclusion chromatography. All crystals were grown at 20°C in hanging drops by mixing one volume of protein solution with one volume of precipitant solution. See Supplemental Information for details on expression plasmids, protein expression and purification, 2-5A synthesis, protein concentrations, precipitant solutions, cryoprotectants, and data collection and structure determination methodologies.
Analytical Ultracentrifugation
Sedimentation velocity analytical ultracentrifugation was performed using unlabeled or fluorescein-labeled RNase L proteins with a Beckman ProteomeLab XL-I at 42,000 rpm with either refractive index or fluorescence (Aviv Biomedical Inc.) detector system. See Supplemental Information for details of protein preparation, experimental conditions, and data analysis methods.
In Vitro RNA Cleavage Assays
Methods are as described previously (Thakur et al., 2005) with minor deviations described in Supplemental Information.
2-5A Binding Analysis Using Surface Plasmon Resonance
SPR studies were performed using a Biacore3000 instrument, streptavidin chips (Biacore SA BR-1003-98), and biotin-labeled 2-5A. See Supplemental Information for 2-5A derivatization with biotin and experimental details. Sensograms were analyzed with the BIAevaluation version 3.0.
In Vitro ATP Binding Assay
ATP binding was determined by fluorescence polarization using BODIPY FL ATP-γ-S (25 nM, Molecular Probes Inc.) and titrating in WT RNase L or mutants in a buffer containing 25 mM HEPES, 50 mM NaCl, 1 mM DTT, 0.01% Brij 35, ± 3 μM 2-5A, in a total reaction volume of 25 μL. Polarization was measured on an HT analyst (ex:em, 485 nM:535 nM, Molecular Devices) and results analyzed in Graphpad Prism.
Cell-Based RNA Cleavage Assay
The cell-based assay for RNase L activity was performed as described previously (Carpten et al., 2002), with minor modifications described in Supplemental Information.
In Vivo EMCV Assay
HeLa-M cells (1 × 105 cells per well in 12-well plates) were transiently transfected using Lipofectamine 2000 (LifeTechnologies) with 0.6 μg/ml of plasmid pCS2 containing WT or mutant porcine RNase L cDNA for 20 hr. Media was replaced 6 hr before infection with EMCV (Hoskins and Sanders, 1957) at an moi of 0.01 in serum-free media for 1 hr. Cells were washed and placed in complete media plus fetal bovine sera. At 14 hr postinfection, media containing virus was harvested and viral titers determined by plaque assay on L929 cells.
SAXS Analyses
The scattering data for RNase L with ADP and 2-5A and RNaseL/2-5A samples were collected on a BioSAXS-1000 detector with MicroMax-007HF X-ray generator. Data collection and processing details are provided in Supplemental Information.
Acknowledgments
Diffraction experiments performed at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines were supported by a grant from the National Institute of General Medical Sciences (P41 GM103403) from the National Institutes of Health and by the U.S. DOE under contract number DE-AC02-06CH11357. We thank Dr. Gabor Bunkoczi, a lecturer at the CCP4/APS school on Macromolecular Crystallography, for assistance with the RNase L structure determination. This work was supported by a grant to F.S. from the Canadian Institutes of Health Research (MOP-84370), by a Canada Research Chair in Structural Biology to F.S., by a grant to R.H.S. from the National Institutes of Health (NIH) (grant number CA044059), by a grant to M.A.B. and A.A.P. from the Centers for Disease Control and Prevention, and by NIH grant number AI045135 to M.A.B.
Published: January 23, 2014
Footnotes
Supplemental Information includes seven figures, one table, and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2013.12.025.
Contributor Information
Robert H. Silverman, Email: silverr@ccf.org.
Frank Sicheri, Email: sicheri@lunenfeld.ca.
Accession Numbers
The coordinates of RNase L have been deposited in the Protein Data Bank under accession codes 4O1O and 4O1P.
Supplemental Information
References
- Brennan D.F., Dar A.C., Hertz N.T., Chao W.C., Burlingame A.L., Shokat K.M., Barford D. A Raf-induced allosteric transition of KSR stimulates phosphorylation of MEK. Nature. 2011;472:366–369. doi: 10.1038/nature09860. [DOI] [PubMed] [Google Scholar]
- Carpten J., Nupponen N., Isaacs S., Sood R., Robbins C., Xu J., Faruque M., Moses T., Ewing C., Gillanders E. Germline mutations in the ribonuclease L gene in families showing linkage with HPC1. Nat. Genet. 2002;30:181–184. doi: 10.1038/ng823. [DOI] [PubMed] [Google Scholar]
- Castelli J.C., Hassel B.A., Wood K.A., Li X.L., Amemiya K., Dalakas M.C., Torrence P.F., Youle R.J. A study of the interferon antiviral mechanism: apoptosis activation by the 2-5A system. J. Exp. Med. 1997;186:967–972. doi: 10.1084/jem.186.6.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelli J.C., Hassel B.A., Maran A., Paranjape J., Hewitt J.A., Li X.L., Hsu Y.T., Silverman R.H., Youle R.J. The role of 2′-5′ oligoadenylate-activated ribonuclease L in apoptosis. Cell Death Differ. 1998;5:313–320. doi: 10.1038/sj.cdd.4400352. [DOI] [PubMed] [Google Scholar]
- Chakrabarti A., Ghosh P.K., Banerjee S., Gaughan C., Silverman R.H. RNase L triggers autophagy in response to viral infections. J. Virol. 2012;86:11311–11321. doi: 10.1128/JVI.00270-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole J.L., Carroll S.S., Blue E.S., Viscount T., Kuo L.C. Activation of RNase L by 2′,5′-oligoadenylates. Biophysical characterization. J. Biol. Chem. 1997;272:19187–19192. doi: 10.1074/jbc.272.31.19187. [DOI] [PubMed] [Google Scholar]
- Credle J.J., Finer-Moore J.S., Papa F.R., Stroud R.M., Walter P. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA. 2005;102:18773–18784. doi: 10.1073/pnas.0509487102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar A.C., Dever T.E., Sicheri F. Higher-order substrate recognition of eIF2alpha by the RNA-dependent protein kinase PKR. Cell. 2005;122:887–900. doi: 10.1016/j.cell.2005.06.044. [DOI] [PubMed] [Google Scholar]
- Dong B., Silverman R.H. A bipartite model of 2-5A-dependent RNase L. J. Biol. Chem. 1997;272:22236–22242. doi: 10.1074/jbc.272.35.22236. [DOI] [PubMed] [Google Scholar]
- Dong B., Silverman R.H. Alternative function of a protein kinase homology domain in 2′, 5′-oligoadenylate dependent RNase L. Nucleic Acids Res. 1999;27:439–445. doi: 10.1093/nar/27.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong B., Xu L., Zhou A., Hassel B.A., Lee X., Torrence P.F., Silverman R.H. Intrinsic molecular activities of the interferon-induced 2-5A-dependent RNase. J. Biol. Chem. 1994;269:14153–14158. [PubMed] [Google Scholar]
- Floyd-Smith G., Slattery E., Lengyel P. Interferon action: RNA cleavage pattern of a (2′-5′)oligoadenylate-dependent endonuclease. Science. 1981;212:1030–1032. doi: 10.1126/science.6165080. [DOI] [PubMed] [Google Scholar]
- Han Y., Whitney G., Donovan J., Korennykh A. Innate immune messenger 2-5A tethers human RNase L into active high-order complexes. Cell Rep. 2012;2:902–913. doi: 10.1016/j.celrep.2012.09.004. [DOI] [PubMed] [Google Scholar]
- Hoskins J.M., Sanders F.K. Propagation of mouse encephalomyocarditis virus in ascites tumour cells maintained in vitro. Br. J. Exp. Pathol. 1957;38:268–272. [PMC free article] [PubMed] [Google Scholar]
- Jones S., Thornton J.M. Principles of protein-protein interactions. Proc. Natl. Acad. Sci. USA. 1996;93:13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr I.M., Brown R.E. pppA2’p5’A2’p5’A: an inhibitor of protein synthesis synthesized with an enzyme fraction from interferon-treated cells. Proc. Natl. Acad. Sci. USA. 1978;75:256–260. doi: 10.1073/pnas.75.1.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimata Y., Ishiwata-Kimata Y., Ito T., Hirata A., Suzuki T., Oikawa D., Takeuchi M., Kohno K. Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. J. Cell Biol. 2007;179:75–86. doi: 10.1083/jcb.200704166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korennykh A.V., Egea P.F., Korostelev A.A., Finer-Moore J., Zhang C., Shokat K.M., Stroud R.M., Walter P. The unfolded protein response signals through high-order assembly of Ire1. Nature. 2009;457:687–693. doi: 10.1038/nature07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornev A.P., Taylor S.S., Ten Eyck L.F. A helix scaffold for the assembly of active protein kinases. Proc. Natl. Acad. Sci. USA. 2008;105:14377–14382. doi: 10.1073/pnas.0807988105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozin M.B., Svergun D.I. Automated matching of high- and low-resolution structural models. J. Appl. Cryst. 2001;34:33–41. [Google Scholar]
- Kristiansen H., Gad H.H., Eskildsen-Larsen S., Despres P., Hartmann R. The oligoadenylate synthetase family: an ancient protein family with multiple antiviral activities. J. Interferon Cytokine Res. 2011;31:41–47. doi: 10.1089/jir.2010.0107. [DOI] [PubMed] [Google Scholar]
- Krüger S., Silber A.S., Engel C., Görgens H., Mangold E., Pagenstecher C., Holinski-Feder E., von Knebel Doeberitz M., Moeslein G., Dietmaier W., German Hereditary Non-Polyposis Colorectal Cancer Consortium Arg462Gln sequence variation in the prostate-cancer-susceptibility gene RNASEL and age of onset of hereditary non-polyposis colorectal cancer: a case-control study. Lancet Oncol. 2005;6:566–572. doi: 10.1016/S1470-2045(05)70253-9. [DOI] [PubMed] [Google Scholar]
- Laronde-Leblanc N., Guszczynski T., Copeland T., Wlodawer A. Structure and activity of the atypical serine kinase Rio1. FEBS J. 2005;272:3698–3713. doi: 10.1111/j.1742-4658.2005.04796.x. [DOI] [PubMed] [Google Scholar]
- Lee K.P., Dey M., Neculai D., Cao C., Dever T.E., Sicheri F. Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell. 2008;132:89–100. doi: 10.1016/j.cell.2007.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen B.E., Ramos E.M., Boulard M., Duda K., Overgaard J., Nordsmark M., Wiuf C., Hansen L.L. Germline mutation in RNASEL predicts increased risk of head and neck, uterine cervix and breast cancer. PLoS ONE. 2008;3:e2492. doi: 10.1371/journal.pone.0002492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malathi K., Dong B., Gale M., Jr., Silverman R.H. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature. 2007;448:816–819. doi: 10.1038/nature06042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao D.Y., Neculai D., Downey M., Orlicky S., Haffani Y.Z., Ceccarelli D.F., Ho J.S., Szilard R.K., Zhang W., Ho C.S. Atomic structure of the KEOPS complex: an ancient protein kinase-containing molecular machine. Mol. Cell. 2008;32:259–275. doi: 10.1016/j.molcel.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masterson L.R., Shi L., Metcalfe E., Gao J., Taylor S.S., Veglia G. Dynamically committed, uncommitted, and quenched states encoded in protein kinase A revealed by NMR spectroscopy. Proc. Natl. Acad. Sci. USA. 2011;108:6969–6974. doi: 10.1073/pnas.1102701108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolen B., Taylor S., Ghosh G. Regulation of protein kinases; controlling activity through activation segment conformation. Mol. Cell. 2004;15:661–675. doi: 10.1016/j.molcel.2004.08.024. [DOI] [PubMed] [Google Scholar]
- Shamu C.E., Walter P. Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 1996;15:3028–3039. [PMC free article] [PubMed] [Google Scholar]
- Siddiqui M.A., Malathi K. RNase L induces autophagy via c-Jun N-terminal kinase and double-stranded RNA-dependent protein kinase signaling pathways. J. Biol. Chem. 2012;287:43651–43664. doi: 10.1074/jbc.M112.399964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C., Walter P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell. 1997;90:1031–1039. doi: 10.1016/s0092-8674(00)80369-4. [DOI] [PubMed] [Google Scholar]
- Silverman R.H. Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol. 2007;81:12720–12729. doi: 10.1128/JVI.01471-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman R.H., Skehel J.J., James T.C., Wreschner D.H., Kerr I.M. rRNA cleavage as an index of ppp(A2’p)nA activity in interferon-treated encephalomyocarditis virus-infected cells. J. Virol. 1983;46:1051–1055. doi: 10.1128/jvi.46.3.1051-1055.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorgeloos F., Jha B.K., Silverman R.H., Michiels T. Evasion of antiviral innate immunity by Theiler’s virus L∗ protein through direct inhibition of RNase L. PLoS Pathog. 2013;9:e1003474. doi: 10.1371/journal.ppat.1003474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark G.R., Kerr I.M., Williams B.R., Silverman R.H., Schreiber R.D. How cells respond to interferons. Annu. Rev. Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- Tanaka N., Nakanishi M., Kusakabe Y., Goto Y., Kitade Y., Nakamura K.T. Structural basis for recognition of 2′,5′-linked oligoadenylates by human ribonuclease L. EMBO J. 2004;23:3929–3938. doi: 10.1038/sj.emboj.7600420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur C.S., Xu Z., Wang Z., Novince Z., Silverman R.H. A convenient and sensitive fluorescence resonance energy transfer assay for RNase L and 2′,5′ oligoadenylates. Methods Mol. Med. 2005;116:103–113. doi: 10.1385/1-59259-939-7:103. [DOI] [PubMed] [Google Scholar]
- Townsend H.L., Jha B.K., Han J.Q., Maluf N.K., Silverman R.H., Barton D.J. A viral RNA competitively inhibits the antiviral endoribonuclease domain of RNase L. RNA. 2008;14:1026–1036. doi: 10.1261/rna.958908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P., Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wreschner D.H., McCauley J.W., Skehel J.J., Kerr I.M. Interferon action—sequence specificity of the ppp(A2’p)nA-dependent ribonuclease. Nature. 1981;289:414–417. doi: 10.1038/289414a0. [DOI] [PubMed] [Google Scholar]
- Wreschner D.H., Silverman R.H., James T.C., Gilbert C.S., Kerr I.M. Affinity labelling and characterization of the ppp(A2’p)nA-dependent endoribonuclease from different mammalian sources. Eur. J. Biochem. 1982;124:261–268. doi: 10.1111/j.1432-1033.1982.tb06586.x. [DOI] [PubMed] [Google Scholar]
- Xiang Y., Condit R.C., Vijaysri S., Jacobs B., Williams B.R., Silverman R.H. Blockade of interferon induction and action by the E3L double-stranded RNA binding proteins of vaccinia virus. J. Virol. 2002;76:5251–5259. doi: 10.1128/JVI.76.10.5251-5259.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y., Wang Z., Murakami J., Plummer S., Klein E.A., Carpten J.D., Trent J.M., Isaacs W.B., Casey G., Silverman R.H. Effects of RNase L mutations associated with prostate cancer on apoptosis induced by 2′,5′-oligoadenylates. Cancer Res. 2003;63:6795–6801. [PubMed] [Google Scholar]
- Xue S., Calvin K., Li H. RNA recognition and cleavage by a splicing endonuclease. Science. 2006;312:906–910. doi: 10.1126/science.1126629. [DOI] [PubMed] [Google Scholar]
- Zhang R., Jha B.K., Ogden K.M., Dong B., Zhao L., Elliott R., Patton J.T., Silverman R.H., Weiss S.R. Homologous 2′,5′-phosphodiesterases from disparate RNA viruses antagonize antiviral innate immunity. Proc. Natl. Acad. Sci. USA. 2013;110:13114–13119. doi: 10.1073/pnas.1306917110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L., Jha B.K., Wu A., Elliott R., Ziebuhr J., Gorbalenya A.E., Silverman R.H., Weiss S.R. Antagonism of the interferon-induced OAS-RNase L pathway by murine coronavirus ns2 protein is required for virus replication and liver pathology. Cell Host Microbe. 2012;11:607–616. doi: 10.1016/j.chom.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou A., Hassel B.A., Silverman R.H. Expression cloning of 2-5A-dependent RNAase: a uniquely regulated mediator of interferon action. Cell. 1993;72:753–765. doi: 10.1016/0092-8674(93)90403-d. [DOI] [PubMed] [Google Scholar]
- Zhou A., Paranjape J., Brown T.L., Nie H., Naik S., Dong B., Chang A., Trapp B., Fairchild R., Colmenares C., Silverman R.H. Interferon action and apoptosis are defective in mice devoid of 2′,5′-oligoadenylate-dependent RNase L. EMBO J. 1997;16:6355–6363. doi: 10.1093/emboj/16.21.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.