

Background: During inflammation, increased levels of secretory phospholipase A2 (sPLA2) impact high density lipoprotein (HDL) cholesterol plasma concentration.
Results: sPLA2 cleaves apolipoprotein A-I (apoA-I) in the central and C-terminal regions.
Conclusion: sPLA2 selectively targets lipid-free apoA-I, whose levels are increased by HDL remodeling during inflammation.
Significance: In acute and chronic (atherosclerosis) inflammation, the proteolytic action of sPLA2 may accelerate clearance of apoA-I, thereby reducing HDL biogenesis.
Keywords: Apolipoproteins, Atherosclerosis, High Density Lipoprotein (HDL), Inflammation, Phospholipase, Apolipoprotein A-I, Proteolysis, Secretory Phospholipase A2, Inflammation, sPLA2
Abstract
In the acute phase of the inflammatory response, secretory phospholipase A2 (sPLA2) reaches its maximum levels in plasma, where it is mostly associated with high density lipoproteins (HDL). Overexpression of human sPLA2 in transgenic mice reduces both HDL cholesterol and apolipoprotein A-I (apoA-I) plasma levels through increased HDL catabolism by an unknown mechanism. To identify unknown PLA2-mediated activities on the molecular components of HDL, we characterized the protein and lipid products of the PLA2 reaction with HDL. Consistent with previous studies, hydrolysis of HDL phospholipids by PLA2 reduced the particle size without changing its protein composition. However, when HDL was destabilized in the presence of PLA2 by the action of cholesteryl ester transfer protein or by guanidine hydrochloride treatment, a fraction of apoA-I, but no other proteins, dissociated from the particle and was rapidly cleaved. Incubation of PLA2 with lipid-free apoA-I produced similar protein fragments in the range of 6–15 kDa, suggesting specific and direct reaction of PLA2 with apoA-I. Mass spectrometry analysis of isolated proteolytic fragments indicated at least two major cleavage sites at the C-terminal and the central domain of apoA-I. ApoA-I proteolysis by PLA2 was Ca2+-independent, implicating a different mechanism from the Ca2+-dependent PLA2-mediated phospholipid hydrolysis. Inhibition of proteolysis by benzamidine suggests that the proteolytic and lipolytic activities of PLA2 proceed through different mechanisms. Our study identifies a previously unknown proteolytic activity of PLA2 that is specific to apoA-I and may contribute to the enhanced catabolism of apoA-I in inflammation and atherosclerosis.
Introduction
Secretory phospholipase A2 (sPLA2)3 is an acute phase protein whose concentration in plasma and inflammatory fluids can increase several hundred-fold during the acute phase response (1). The main reactivity of sPLA2 is directed toward hydrolysis of the sn-2 ester bond of the glyceroacyl phospholipids present in lipoproteins and cell membranes. This reactivity induces structural and functional changes in lipoproteins and membranes and generates lysophospholipids and non-esterified fatty acids, which are proinflammatory signal molecules (2, 3).
Mammalian sPLA2 isoforms are classified as group IB, IIA, IIC, IID, IIE, IIF, III, V, X, and XII (4). Although the sequence homology between these sPLA2 enzymes varies, they share a Ca2+-dependent lipolytic mechanism and a well conserved three-dimensional structure with several disulfide bridges (4). Secretory PLA2 enzymes are implicated in various physiological and pathophysiological functions, including lipid absorption and cholesterol homeostasis, cell proliferation, myocardial injury, tumor formation, and inflammation (2–5). Notably, six sPLA2 isoforms (IIA, IID, IIE, III, V, and X) are present in human atherosclerotic lesions (4–6).
Atherosclerosis is increasingly recognized as a localized inflammatory condition within the vessel wall (7, 8). The observation that patients with chronic inflammatory diseases are at higher risk of cardiovascular events confirms the inflammatory nature of atherosclerosis (9, 10). Therefore, the proinflammatory properties of sPLA2 enzymes and their localization in atherosclerotic lesions suggest a direct mechanistic link between the activity of sPLA2 in the arterial subendothelial space and atherosclerosis development. Although recent human genetic association studies failed to detect a correlation between high sPLA2 levels and increase in cardiovascular events (11), this link is supported by data from human sPLA2 transgenic mice, in which high density lipoprotein cholesterol (HDL-C) and apolipoprotein A-I (apoA-I) plasma levels are significantly decreased compared with wild-type mice, and atherosclerotic lesions develop even on a chow diet (12–15).
High HDL-C levels correlate with low incidence of atherosclerosis and are believed to be a marker of an efficient reverse cholesterol transport (16). In particular, the ability of HDL to mediate reverse cholesterol transport from lipid-laden macrophages in the vascular wall and transport cholesterol back to the liver for excretion is considered to be one of the mechanisms underlying HDL antiatherogenic function (17). Furthermore, HDL reduces atherosclerosis through its general antioxidant and anti-inflammatory action (18). In plasma, HDL undergoes a continuous remodeling process mediated by a variety of plasma factors. This remodeling determines the plasma concentration and subpopulation distribution of HDL (19). The effect of acute and chronic inflammation on HDL remodeling is of great importance because it may impact both the polydisperse particles and the equilibrium between HDL-associated and lipid-free apolipoproteins (20). Insights into the regulation of HDL remodeling have been obtained by several in vitro studies via chemical and thermal perturbation techniques (21–25). These approaches have established that dissociation of lipid-free apoA-I always occurs during HDL remodeling events. The ability of cholesteryl ester transfer protein (CETP) and phospholipid transfer protein to remodel HDL with dissociation of lipid-free apoA-I corroborates this notion (26), which bears important physiological implications (27–29). Although lipid-free apoA-I accounts for less than 10% of total plasma apoA-I (30, 31), this form of the protein is central to HDL biogenesis, which requires interaction of lipid-free apoA-I with the ABCA1 (ATP-binding cassette A1) cell membrane transporter (27, 32). Progressive lipidation of apoA-I via ABCA1 generates nascent HDL and efficiently reduces the plasma concentration of lipid-free apoA-I, which is otherwise easily catabolized through glomerular filtration. Therefore, plasma levels of apoA-I are ultimately determined by the rate at which lipid-free apoA-I is produced (taking into account its generation through dissociation from HDL) and catabolized (33). Interestingly, HDL-C and apoA-I levels are significantly decreased in both acute and chronic inflammatory states (15) at least in part due to accelerated apoA-I catabolism (13).
Although inflammation includes a wide variety of metabolic changes, two inflammation-dependent events that may affect HDL and apoA-I catabolism are the increase in plasma levels of serum amyloid A (SAA) protein and secretory (non-pancreatic) PLA2 (12, 13, 34). Due to its high lipid binding affinity, SAA can displace up to 80% of apoA-I from HDL (35). In the acute phase response, when plasma levels of SAA rise severalfold, the protein composition of HDL is altered as lipid-free apoA-I is displaced form HDL by SAA (34). Increased catabolism of lipid-free apoA-I can than occur through normal glomerular filtration.
Studies in humans and animals have suggested that factors that affect HDL lipid composition modulate apoA-I catabolism (36). Secretory PLA2 can remodel HDL in vitro by hydrolyzing HDL phospholipids, thereby reducing HDL particle size (19). However, unlike binding of SAA, sPLA2-mediated HDL remodeling does not generate lipid-free apoA-I (19, 37). Hence, the mechanism that may link increased levels of sPLA2 and decreased plasma levels of HDL-C and apoA-I in inflammation is unknown. The results reported here reveal a previously undetected selective proteolytic activity of sPLA2 on lipid-free apoA-I. Our study suggests that in inflammation states, increased catabolism of apoA-I and, ultimately, reduction of HDL-C plasma levels may be mediated by direct interaction of sPLA2 with lipid-free apoA-I. This novel function of sPLA2 has potentially important implications for atherogenesis. In atherosclerotic lesions, high levels of sPLA2, in concert with increased levels of lipid-free apoA-I (30), could contribute to atherosclerosis progression by limiting the availability of functional antiatherogenic lipid-free apoA-I in the subendothelial space.
EXPERIMENTAL PROCEDURES
Materials
Porcine pancreatic PLA2 (PP-PLA2), bovine pancreatic PLA2 (BP-PLA2), bee venom PLA2 (BV-PLA2), and essentially fatty acid-free BSA were obtained from Sigma. Recombinant human secretory PLA2-IIA (>95% purity) was a generous gift from BioVendor Laboratories (Czech Republic). Unlike PP-PLA2, both BP-PLA2 and BV-PLA2 (i) are not subjected to exogenous trypsin digestion during extraction from the natural source (Sigma Technical Information Service) and (ii) are lyophilized, rather than prepared as a 3.2 m ammonium sulfate suspension. For PP-PLA2, the enzyme solution was extensively dialyzed against 10 mm Tris, pH 7.5 (buffer A), at 4 °C and used within a week. Lyophilized BP-PLA2 and BV-PLA2 were solubilized in buffer A and stored at 4 °C for no more than 1 week before use. Recombinant PLA2-IIA was reconstituted according to the manufacturer's instructions. Briefly, the lyophilized powder was dissolved in 0.1 m acetate buffer, pH 4.0, and diluted in buffer A to raise the pH to 7.5. The integrity and purity of PP-PLA2 was checked by SDS-PAGE and matrix-assisted laser-desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) (data not shown), and no contamination was detectable. As an additional purity test, commercial PP-PLA2 was purified by preparative size exclusion chromatography (SEC) on a Superdex 200 column. The lipolytic and proteolytic activities of the SEC-purified sample were not different from those of the unpurified sample. Therefore, it can be excluded that the observed activities of the commercial PP-PLA2 are due to residual trace amounts of contaminants. For the experiments reported in this paper, commercial PP-PLA2 was used without further purification.
Isolation of Human Very Low Density Lipoprotein (VLDL), HDL, and ApoA-I
Human lipoproteins were isolated from plasma of healthy volunteers. The blood was donated at the blood bank (Research Blood Components, LLC) according to the rules of the institutional review board and with written consent from the donors. Lipoproteins were isolated from fresh EDTA-treated plasma from a single donor by density gradient ultracentrifugation in the density range 0.94–1.006 g/ml for VLDL and 1.063–1.21 g/ml for HDL, as described (38). Different lipoprotein fractions migrated as a single band on agarose gel electrophoresis. The density fractions containing VLDL and HDL were dialyzed against 10 mm sodium phosphate, pH 7.5 (buffer B), degassed, and stored in the dark at 4 °C. The lipoprotein stock solutions were used (for experiments or for extraction of apoA-I from HDL) within 2–3 weeks, during which no protein degradation or changes in the lipoprotein electrophoretic mobility were detected by SDS-PAGE and agarose electrophoresis. ApoA-I was isolated and purified from HDL stock solutions as described (39). Purified apoA-I was refolded from 6 m GdnHCl after extensive dialysis against buffer B, stored at 4 °C, and used within 1 month (25).
Expression and Purification of ApoA-I Variants
The apoA-I variants in which residues 134–145 were removed (apoA-I:Δ134–145) or all four native tryptophans were mutated to phenylalanines (ΔW-apoA-I) were produced using primer-directed PCR mutagenesis as described (40). The mutations were verified by dideoxy automated fluorescent sequencing. The protein variants were expressed in BL21 Escherichia coli (Agilent Technologies) and purified by nickel affinity chromatography (40). Protein purity (>95%) was confirmed by SDS-PAGE and mass spectrometry analyses.
PLA2 Reaction with HDL
HDL was dialyzed against buffer A containing CaCl2 (final concentration, 2 mm). HDL (5 mg, in total proteins) and PP-PLA2 (25 μg, 1.8 μm) were incubated for 1 h at 37 °C (41). The reaction was stopped by cooling on ice after adding an excess of EDTA (final concentration, 15 mm) to chelate Ca2+, an essential co-factor for PLA2 lipolytic activity (42). These samples are termed PP-PLA2-HDL throughout this work. In control experiments, free fatty acids (FFAs) generated in PP-PLA2-HDL by the lipolytic action of PLA2 on HDL phospholipids were removed by performing the incubation in the presence of essentially fatty acid-free bovine serum albumin (BSA) (final concentration, 2% (w/v)). The incubation conditions were the same as described before (41) (i.e. 1 h at 37 °C and then 15 mm EDTA on ice). After incubation, FFAs were removed from PP-PLA2-HDL by separating the FFA-loaded BSA fraction by SEC on a Superose-6 10/300 GL column (elution: 10 mm PBS, 15 mm EDTA, pH 7.5, at 0.5 ml/min) to yield (PP-PLA2-HDL)-FFA. Complete removal of BSA from (PP-PLA2-HDL)-FFA was confirmed by SDS-PAGE analysis (data not shown). (PP-PLA2-HDL)-FFA was dialyzed against buffer B for further studies.
Remodeling of HDL by Guanidine Treatment
Intact HDL and PP-PLA2-HDL were incubated with 3 m GdnHCl at 37 °C for 3 h and then dialyzed extensively against buffer B and stored at 4 °C.
Remodeling of HDL by CETP
Plasma purified CETP was a generous gift from Prof. Kerry-Ann Rye (Sydney, Australia). Intact HDL and PP-PLA2-HDL (final 1 mg/ml, in total proteins) were incubated with VLDL (final 5 mm, in triglyceride) and CETP (final 5 units/ml) in buffer A at 37 °C for 24 h. The reaction was terminated by cooling samples on ice to yield the samples termed CETP-HDL and CETP-PP-PLA2-HDL, respectively. The different components in the two samples were separated by SEC with a Superose-6 10/300 GL column (elution: 10 mm PBS, pH 7.5, at a flow rate of 0.5 ml/min). The isolated fractions were concentrated and used for further biochemical characterization.
PLA2 Reaction with Lipid-free Proteins
Lipid-free proteins (0.5 mg) were incubated with PP-PLA2 (2.5 μg and 0.18 μm) (final enzyme/substrate ratio 1:200 (w/w)) at 37 °C for 1 h in the presence or absence of 15 mm EDTA. This concentration of PLA2 is comparable with the plasma concentration of the enzyme under chronic inflammatory conditions (43). The reaction was terminated by freezing the sample until further use. For immediate electrophoresis analysis, reactions were stopped by adding SDS loading buffer and boiling for 10 min before loading the samples on gel. Because Ca2+ is an essential co-factor for the lipolytic activity of PLA2 (42), to inhibit Ca2+-dependent lipolysis in some experiments, apoA-I was incubated with PLA2 in the presence of an excess (relative to Ca2+) of EDTA (15 mm). These samples are termed “inactive PLA2.”
Effect of the Protease Inhibitor Benzamidine on PLA2 Activity
PP-PLA2 was preincubated in the presence of benzamidine (final concentration, 2 mm) at room temperature for 15 min. Benzamidine-treated PP-PLA2 was then incubated with lipid-free apoA-I as described above.
Non-denaturing Gradient Gel Electrophoresis (NDGGE)
NovexTM 4–20% Tris-glycine gels (Invitrogen) were loaded with 3 μg of protein/lane and run to termination at 1,500 V-h under non-denaturing conditions in Tris-glycine buffer. The gels were stained with Denville Blue protein stain (Denville Scientific).
SDS-PAGE
NovexTM 4–20% Tris-glycine gels (Invitrogen) were loaded with 5 μg of protein/lane and run at 200 V for 1 h under denaturing conditions in SDS-Tris-glycine buffer. The gels were stained with Denville Blue protein stain (Denville Scientific).
Size Exclusion Chromatography
SEC was performed with a Superdex 200 prep grade XK 16/100 column and/or a Superose 6 10/300 GL column (GE Healthcare) controlled by an AKTA UPC 10 FPLC system (GE Healthcare). Elution by PBS (10 mm sodium phosphate, 150 mm NaCl, pH 7.5) was carried out at flow rates of 1.0 ml/min and 0.5 ml/min for Superdex 200 and Superose 6 columns, respectively.
MALDI-TOF MS
MALDI-TOF MS spectra were recorded on a Reflex-IV spectrometer (Bruker Daltonics, Billerica, MA) equipped with a 337-nm nitrogen laser. The instrument was operated in the positive ion reflection mode at 20-kV accelerating voltage with time lag focusing enabled. Calibration was performed using a standard calibration mixture containing oxidized B-chain of bovine insulin, equine cyctochrome c, equine apomyoglobin, and bovine serum albumin in linear mode. The matrix, cyano-4-hydroxycinnamic acid (α-cyano, Mr 189), was prepared as a saturated solution in 70% acetonitrile and 0.1% trifluoroacetic acid in water.
Liquid Chromatography Mass Spectrometry (LC-MS)
Intact apoA-I and PP-PLA2-apoA-I were prepared as described above and subjected to SDS-PAGE (Fig. 4B). Gel fragments corresponding to the protein bands stained with Denville Blue protein stain were excised from the gel slab and digested overnight with trypsin (0.02 μg/μl; Promega) as described (44). Tryptic digests were analyzed by nanoflow HPLC microelectrospray ionization on a Thermo Scientific LTQ Orbitrap XL. Samples were first trapped on a trap column (100 μm × 5 cm; Polymicro Technologies, Phoenix, AZ) packed with Poros 10R2 (Applied Biosystems, Foster City, CA) and subsequently eluted onto the analytical column (75-μm inner diameter × 10-cm fused silica analytical column, self-packed with Magic C18AQ, 5 μm; Michrom BioResources, Auburn, CA). Elution was performed at 200 nl/min with the following gradient: 99% to 30% Buffer A in 25 min and then 30% to 10% Buffer A in 30 min and re-equilibration to 99% Buffer A in 40 min (Buffer A: 0.1% formic acid in water; Buffer B: 0.1% formic acid in acetonitrile). The column outflow was ionized and sprayed into the LTQ Orbitrap by way of a 8-μm SilicaTip (New Objective) and Picoview source (New Objective). Spectra were acquired in a data-dependent mode throughout the gradient, and a full MS scan in the Orbi-trap analyzer was obtained, followed by seven subsequent MS/MS scans in the ion trap based on the seven most intense peaks identified in the full scan. Collision-induced dissociation fragmentation was achieved with a collision energy of 35%, a capillary temperature of 150 °C, and an electrospray voltage of 1.9 kV. Once an MS/MS spectrum was obtained two times, it was put on the exclusion list for 3 min to allow for lower intensity peptides to be analyzed. Data were analyzed using the Mascot algorithm by searching against the updated non-redundant database from NCBI.
FIGURE 4.

Characterization of lipid-free apoA-I and apoA-I:Δ134–145 after reaction with PP-PLA2. Purified plasma apoA-I was incubated with PP-PLA2 at an enzyme/substrate ratio of 1:200 at 25 °C for 1 h. Control samples represent apoA-I samples incubated under the same conditions in the absence of PP-PLA2. A, SEC chromatograms of control apoA-I, PP-PLA2-apoA-I, and PP-PLA2 only. For SEC analysis, samples were loaded at 0.5 mg/ml (in total protein concentration) on a preparative grade Superdex 200 XK 16/100 column and eluted with 10 mm PBS, pH 7.5, at a flow rate of 0.5 ml/min. B, SDS-PAGE of control apoA-I, PP-PLA2-apoA-I, and inactive PP-PLA2-apoA-I, in which PP-PLA2 was preincubated with EDTA before reaction with apoA-I. 5 μg of protein was loaded per lane. Bands 1–6 represent proteolyzed fragments of apoA-I that were excised from the gel and analyzed by MALDI-TOF MS. C, SDS-PAGE of apoA-I:Δ134–145 and PP-PLA2-apoA-I:Δ134–145. Molecular weight standards are Precision Plus Protein Dual Xtra standards (Bio-Rad). a.u., absorbance units.
Gel electrophoresis and SEC data reported in this paper are representative results of at least three independent experiments. Mass spectrometry data are the average of 3–5 independent experiments.
RESULTS
PLA2-mediated Hydrolysis of HDL under HDL-destabilizing Conditions
During the acute and chronic phase of inflammation, the plasma concentration of acute phase sPLA2 rises sharply (1). To investigate the effect of this increased load of sPLA2 on HDL and its resident proteins, we incubated HDL with PP-PLA2 (PP-PLA2-HDL), as described under “Experimental Procedures.” Furthermore, to mimic HDL remodeling that occurs in vivo, PP-PLA2-HDL was incubated with CETP or was exposed to chemical denaturation. Both treatments lead to HDL remodeling with concomitant release of lipid-free proteins (21, 45).
First, HDL was incubated with PP-PLA2 for 1 h at 37 °C. Under these conditions, nearly 10% of phospholipids were converted into lysophosphatidylcholine and FFA, as reported previously (41). By NDGGE and SEC analyses of PP-PLA2-HDL, surface hydrolysis of HDL phospholipids by PP-PLA2 produced a reduction in HDL particle size without dissociation of apoA-I from HDL (Fig. 1, A and B). Furthermore, no protein degradation was observed by SDS-PAGE (Fig. 1C). MALDI-TOF MS analysis of PP-PLA2-HDL confirmed the presence of only full-length apoA-I, apoA-II, and apoC proteins (Table 1).
FIGURE 1.

PP-PLA2-mediated hydrolysis of HDL. A, NDGGE of intact HDL (lane 1) and PP-PLA2-HDL (lane 2). B, SEC chromatograms of intact HDL and PP-PLA2-HDL. C, SDS-PAGE of intact HDL (lane 1) and PP-PLA2-HDL (lane 2). D, NDGGE of guanidine-treated HDL. Lane 1′, Gd-HDL; lane 2′, Gd-PP-PLA2-HDL. E, SEC chromatograms of Gd-treated HDL. F, SDS-PAGE of Gd-HDL (lane 1′) and Gd-PP-PLA2-HDL (lane 2′). For both NDGGE and SDS-PAGE, 5 μg of protein was loaded per lane. Molecular weight standards are the High Molecular Weight Calibration Kit (GE Healthcare) for NDGGE and Precision Plus Protein Dual Xtra standards (Bio-Rad) for SDS-PAGE, respectively. For SEC analysis, samples were loaded at 2 mg/ml (total protein concentration) on a preparative grade Superdex 200 XK 16/100 column and eluted with 10 mm PBS, pH 7.5, at a flow rate of 1 ml/min.
TABLE 1.
MALDI-TOF MS analysis of intact HDL, PP-PLA2-HDL, and HDL and PP-PLA2-HDL after incubation in the presence of GdnHCl (Gd-HDL and Gd-PP-PLA2-HDL)
For Gd-samples, analyses of total samples and SEC-separated fractions are reported.
| Molecular massa | ||||
|---|---|---|---|---|
| kDa | ||||
| Intact HDL | 28.1 | 17.3 | 9.4 | 6.6 |
| Gd-HDL-total | 28.1 | 17.3 | 9.4 | 6.6 |
| Gd-HDL-Fr1 | 28.1 | 17.3 | 9.4 | 6.6 |
| Gd-HDL-Fr2 | 28.1 | - | - | - |
| PP-PLA2-HDL | 28.1 | 17.3 | 9.4 | 6.6 |
| 13.9 | ||||
| Gd-PP-PLA2-HDL-total | 28.1 | 17.3 | 12.7 | 6.6 |
| 15.5 | 10.7 | 6.2 | ||
| 13.9 | 9.4 | 5.4 | ||
| Gd-PP-PLA2-HDL-Fr1 (SEC) | 28.1 | 17.7 | 12.7 | 6.6 |
| 10.7 | 5.4 | |||
| Gd-PP-PLA2-HDL-Fr2 (SEC) | 28.1 | 15.5 | 12.7 | 6.2 |
| 26.5 | 10.7 | 5.4 | ||
| Gd-PP-PLA2-HDL-Fr3 (SEC) | 28.1 | 13.9 | 12.7 | 6.2 |
| 26.5 | 10.7 | 5.4 | ||
| 22.2 | ||||
a Molecular mass of intact apoA-I, 28.1 kDa; apoA-II, 17.3 kDa; apoC-III, 9.4 kDa; apoC-I, 6.6 kDa; PP-PLA2, 13.9 kDa.
To investigate whether the presence of PLA2 on HDL affects the products of HDL remodeling by chemical denaturation, we incubated HDL and PP-PLA2-HDL in 3 m GdnHCl, as described under “Experimental Procedures.” After guanidine removal by extensive dialysis, the relative NDGGE mobilities of the dissociated proteins in guanidine-treated HDL (Gd-HDL)4 and Gd-PP-PLA2-HDL were significantly different (Fig. 1D), indicating that protein modifications must have occurred during guanidine treatment in the presence of PP-PLA2. Interestingly, a new SEC peak at higher elution volumes (Ve > 125 ml) than full-length lipid-free apoA-I (Ve = 125 ml; Fig. 4A) was present in Gd-PP-PLA2-HDL (Fig. 1E). Consistent with this observation, SDS-PAGE analysis of Gd-PP-PLA2-HDL revealed protein bands in the 15 to 5 kDa size range that were not present in Gd-HDL (Fig. 1F).
Isolation and Characterization of Protein Fragments
To characterize the PP-PLA2-mediated modifications of HDL proteins, Gd-HDL and Gd-PP-PLA2-HDL were separated by SEC in two and three fractions, respectively (Fig. 2A). By thin layer chromatography, HDL lipids were detectable only in fraction 1 (Fr1) of Gd-HDL and Gd-PP-PLA2-HDL (data not shown). NDGGE of the SEC fractions confirmed that no lipid-free proteins were present in Fr1 of both Gd-HDL and Gd-PP-PLA2-HDL, whereas no HDL-size-like particles were detected in Fr2 or Fr3 (Fig. 2B). These analyses support the conclusion that Fr1 contains HDL-associated proteins, whereas Fr2 and any fraction eluting at volumes higher than Fr2 are predominantly composed of lipid-free proteins. By SDS-PAGE analysis, bands corresponding to full-length apoA-I and apoA-II occurred in Fr1 of Gd-HDL, whereas only one band, corresponding to full-length lipid-free apoA-I, was present in Fr2 (Fig. 2C). Results for Gd-PP-PLA2-HDL were more complex (Fig. 2D). The HDL-associated proteins in Fr1 were full-length apoA-I, full-length apoA-II, and a smaller protein fragment with electrophoretic mobility corresponding to ∼10–12 kDa. Fr2 consisted of full-length apoA-I, a band with mobility consistent with the size of full-length apoA-II, and at least two protein fragments within ∼15–10 kDa. In Fr3, no full-length apoA-II and almost no full-length apoA-I but several protein fragments that ranged from ∼26 to 5 kDa were detected.
FIGURE 2.

Characterization of HDL proteins after guanidine treatment of HDL in the presence or in the absence of PP-PLA2. A, Gd-HDL and Gd-PP-PLA2-HDL were separated by SEC into two and three fractions, respectively. SEC conditions were the same as described in the legend to Fig. 1. B, NDGGE of Gd-HDL and Gd-PP-PLA2-HDL SEC fractions. Total lanes represent Gd-HDL and Gd-PP-PLA2-HDL before SEC separation. For NDGGE analysis, 3 μg of protein was loaded per lane. Molecular weight standards are the High Molecular Weight Calibration Kit (GE Healthcare). C, SDS-PAGE of Gd-HDL SEC fractions. D, SDS-PAGE of Gd-PP-PLA2-HDL SEC fractions. For SDS-PAGE analysis, 5 μg of protein was loaded per lane. Molecular weight standards are Precision Plus Protein Dual Xtra standards (Bio-Rad). a.u., absorbance units.
The molecular weight of the protein fragments in guanidine-treated samples was estimated by MALDI-TOF MS analysis. Although only apoA-I and apoA-II were detected in a stained SDS-polyacrylamide gel (Fig. 2, C and D), intact forms of all major HDL proteins (apoA-I, apoA-II, and apoCs) were identified in intact HDL, PP-PLA2-HDL, and Gd-PP-PLA2-HDL (total sample) (Table 1). Upon SEC fractionation, masses corresponding to full-length apoA-II and apoCs were detected only in Fr1 of Gd-PP-PLA2-HDL (Table 1). Importantly, by Western blot analysis of Gd-PP-PLA2-HDL, only one band compatible with the size of full-length apoA-II (∼17 kDa) was detected by anti-apoA-II antibodies (data not shown). The absence of anti-apoA-II reactive bands for sizes <17 kDa suggests that the protein bands observed by SDS-PAGE in the 15 to 5 kDa molecular mass range in Fr2 and Fr3 are lipid-free proteolytic fragments of apoA-I.
As shown in Fig. 1C, no apoA-I proteolysis is observed in PP-PLA2-HDL in the absence of guanidine treatment. FFAs produced during PLA2-mediated lipolysis can inhibit the lipolytic action of the enzyme (46). To test whether the absence of PLA2-mediated protein cleavage in PP-PLA2-HDL is due to similar inhibition of the proteolytic active site of PLA2 by FFAs, in an independent experiment, HDL was incubated with PLA2 in the presence of BSA to sequester FFAs produced during lipolysis (41). The FFA-loaded BSA was then separated from the PP-PLA2-HDL fraction by SEC. Similar to PP-PLA2-HDL, no proteolysis was detected in (PP-PLA2-HDL)-FFA (data not shown). These results suggest that HDL-associated proteins are not a viable substrate for PP-PLA2-mediated proteolysis and support the hypothesis that only the lipid-free form of apoA-I, generated upon guanidine treatment of PP-PLA2-HDL, is cleaved by PP-PLA2.
Consistent with this hypothesis, after SEC fractionation of Gd-PP-PLA2-HDL, PP-PLA2 was detected by MALDI-TOF MS analysis only in Fr3 (non-HDL-associated fraction) (Table 1). This observation suggests that a “lipid-free form” of PLA2 mediates proteolysis of lipid-free apoA-I in solution rather than on the surface of the HDL particles.
Furthermore, analysis of two major HDL subclasses isolated by density gradient ultracentrifugation, HDL2 and HDL3, showed similar protein fragments upon guanidine treatment of PP-PLA2-HDL (data not shown), suggesting that PLA2-mediated proteolysis of apoA-I does not depend on HDL particle size, HDL protein or lipid composition, or the conformation of apoA-I on HDL.
PLA2-mediated Proteolysis of ApoA-I upon HDL Destabilization by CETP
To test whether a physiologically relevant process that mediates HDL destabilization and lipid-free apolipoprotein release from HDL promotes PLA2-dependent proteolysis as observed in Gd-PP-PLA2-HDL, PP-PLA2-HDL was incubated with CETP as described under “Experimental Procedures.” Consistent with previous reports (45), CETP promoted the release of lipid-free apoA-I form HDL (Fig. 3), but no apoA-I proteolysis was observed under these conditions. In contrast, in the presence of PP-PLA2, proteolysis was observed in the lipid-free protein fraction (Fr2) of CETP-PP-PLA2-HDL (Fig. 3B). Interestingly, the proteolytic patterns observed in these reaction conditions and in Gd-PP-PLA2-HDL were similar (Figs. 2D and 3B).
FIGURE 3.

Characterization of HDL proteins after reaction with CETP in the presence or in the absence of PP-PLA2. A, CETP-HDL and CETP-PP-PLA2-HDL were separated by SEC into two fractions. SEC conditions were the same as described in the legend to Fig. 1. B, SDS-PAGE of CETP-HDL and CETP-PP-PLA2-HDL SEC fractions. 5 μg of protein was loaded per lane. Molecular weight standards are Precision Plus Protein Dual Xtra standards (Bio-Rad). a.u., absorbance units.
Upon guanidine treatment of PP-PLA2-HDL, PP-PLA2 was detected only as non-HDL-associated protein in SEC Fr3 of Gd-PP-PLA2-HDL (Table 1). To test whether a similar HDL remodeling occurs by action of CETP, the SEC fractions from CETP-PP-PLA2-HDL were analyzed by MALDI-TOF MS (Table 2). Interestingly, PP-PLA2 was detected only in the HDL-associated fraction (Fr1). Therefore, not only lipid-free but also HDL surface-bound PLA2 can cleave apoA-I, with similar substrate specificity.
TABLE 2.
MALDI-TOF MS analysis of SEC separated fractions of PP-PLA2-HDL after incubation in the presence of CETP (CETP-PP-PLA2-HDL)
| Molecular massa | ||||
|---|---|---|---|---|
| kDa | ||||
| CETP-PP-PLA2-HDL-Fr1 | 28.1 | 17.7 | 9.4 | 6.6 |
| 13.9 | ||||
| CETP-PP-PLA2-HDL-Fr2 | 28.5 | 15.3 | 12.7 | 6.2 |
| 26.2 | 10.7 | 5.4 | ||
a Molecular mass of intact apoA-I, 28.1 kDa; apoA-II, 17.3 kDa; apoC-III, 9.4 kDa; apoC-I, 6.6 kDa; PP-PLA2, 13.9 kDa.
In conclusion, destabilization of HDL by chemical or enzymatic means, in the presence of PP-PLA2, promotes proteolysis of apoA-I. This proteolytic activity can be mediated by both lipid-free and HDL-associated PLA2.
Lipid-free ApoA-I Is the Substrate of PLA2-mediated Proteolysis
The proteolytic action of PLA2 on apoA-I occurred exclusively after PP-PLA2-HDL destabilization by guanidine or CETP treatments, which promote dissociation of lipid-free apoA-I from the HDL particle. We hypothesized that, in these HDL-destabilizing conditions, lipid-free rather than HDL-associated apoA-I is the substrate of PLA2-mediated proteolysis. To test this hypothesis, purified lipid-free apoA-I was incubated with PP-PLA2 under the same conditions used for plasma HDL to generate PP-PLA2-apoA-I. By SEC analysis, lipid-free apoA-I eluted as a single peak at Ve = 125 ml, whereas no chromatographic signal was detectable in the 120–140-ml range (Fig. 4A). In the chromatogram of PP-PLA2-apoA-I (Fig. 4A), the intensity of the peak corresponding to full-length lipid-free apoA-I was reduced, compared with pure lipid-free apoA-I. Importantly, a new protein fraction eluted at Ve ∼135 ml. This elution volume is similar to the one observed in the SEC chromatogram of Gd-PP-PLA2-HDL (Fig. 1B). Furthermore, proteolytic fragments similar to those present in Gd-PP-PLA2-HDL (Fig. 2C, total and Fr2 + 3) were observed in PP-PLA2-apoA-I by SDS-PAGE analysis (Fig. 4B). When lipid-free apoA-I was incubated in the absence of PP-PLA2 for the same time period, no proteolysis was detected by SDS-PAGE analysis (Fig. 4B, apoA-I control). Therefore, lipid-free apoA-I is the substrate of the proteolytic action of PLA2. To further confirm this conclusion, the lipid-free protein fraction produced by GdnHCl treatment of HDL (Gd-HDL-Fr2; Fig. 2A) was incubated in the presence of PP-PLA2 under the same experimental conditions used for lipid-free apoA-I. Notably, PLA2-mediated proteolysis of apoA-I in PP-PLA2-Gd-HDL-Fr2 was similar to the proteolytic pattern observed for lipid-free apoA-I (data not shown).
Proteolysis and Lipolysis Mechanisms
To test whether the mechanisms of PLA2-mediated proteolysis of apoA-I and of the normal lipolytic activity of the enzyme are related, PP-PLA2 was incubated with apoA-I in the presence of EDTA (inactive PP-PLA2). EDTA inhibits the lipolytic activity of PLA2 by chelating Ca2+, a co-factor essential for PLA2-mediated lipolysis (42). The apoA-I proteolytic pattern generated by inactive-PP-PLA2 was not significantly different from that observed in the absence of EDTA (Fig. 4B), indicating that the proteolytic activity of PP-PLA2 is not Ca2+-dependent.
When lipid-free apoA-I and PP-PLA2 were incubated in the presence of the protease inhibitor benzamidine, apoA-I proteolysis was completely suppressed (Fig. 4B). Notably, benzamidine had no effect on the lipolytic activity of PLA2, as confirmed by the reduction in HDL size upon incubation of HDL with PP-PLA2 and benzamidine (data not shown). Taken together, these results indicate that the proteolytic and lipolytic activities of PLA2 proceed through different mechanisms.
Analysis of Proteolyzed ApoA-I
The protein fragments generated during incubation of lipid-free apoA-I with PP-PLA2 for 1 h (PP-PLA2-apoA-I) were separated by SDS-PAGE (Fig. 4B). The gel bands (numbered 1–6) were excised from the gel and digested with trypsin, and the tryptic digest was analyzed by LC-MS (Table 3). The mass of the main protein component in the undigested samples was determined by MALDI-TOF MS (Table 3).
TABLE 3.
LC-MS and MALDI-TOF MS analysis of control-apoA-I and PP-PLA2-apoA-I after SDS-PAGE separation
The gel bands numbered 1–6 in Fig. 4B were excised from the gel and digested with trypsin, and the tryptic digest was analyzed by LC-MS. The sequence of the tryptic peptides identified by LC-MS is highlighted in red. In a parallel SDS-PAGE experiment, the mass of the main protein component in undigested samples from the same gel bands was determined by MALDI-TOF MS (see “Experimental Procedures” and Table 1) and is reported in parenthesis. The sequence of the apoA-I peptide with calculated mass closest to the mass of the undigested protein fragment, as determined by MALDI-TOF, is shown in boldface type.
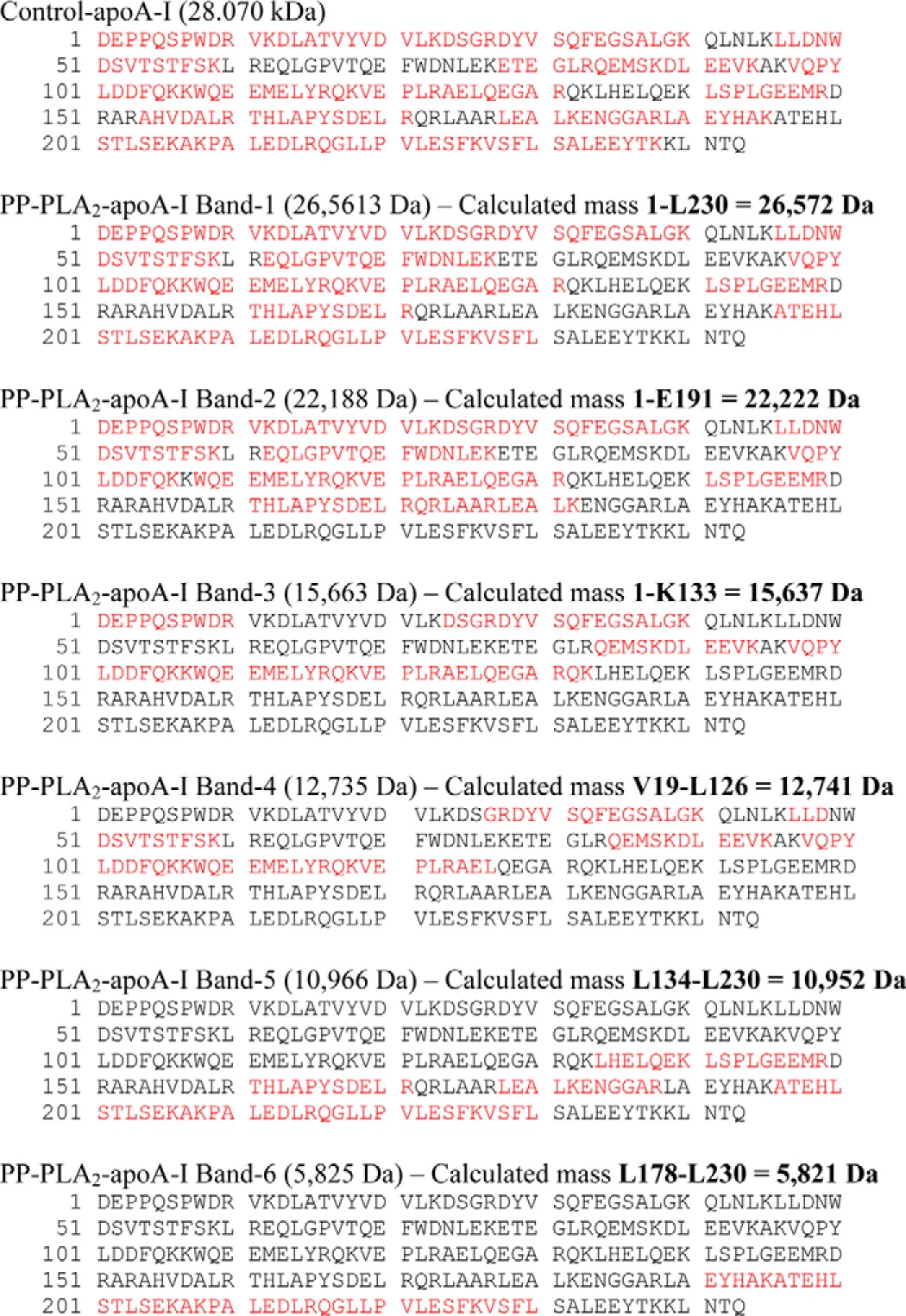
Analysis of MALDI-TOF and LC-MS data indicates that positions Lys133 and Leu230 of apoA-I are independent cleavage sites for PP-PLA2. Proteolysis at one or both of these positions produces fragments 1–230 (band 1), 1–133 (band 3), and 134–230 (band 5), respectively. A third cutting site at position Glu191 produces fragment 1–191 (band 2). From analysis of MALDI-TOF and LC-MS data, the sequence of the protein fragments in bands 4 and 6 was also predicted to be Val19–Leu126 and Leu178–Leu230, respectively.
Notably, when an apoA-I variant in which residues 134–145 were removed (apoA-I:Δ134–145) was incubated with PP-PLA2, only bands 1 and 2 were detected by SDS-PAGE analysis of PP-PLA2-apoA-I:Δ134–145 (Fig. 4C). Thus, deletion of residues 134–145 completely inhibited cleavage at Lys133, which accounts for the absence of band 3 (fragment 1–133) and band 5 (fragment 134–230). Furthermore, absence of bands 4 and 6 in PP-PLA2-apoA-I:Δ134–145 suggests that fragments 19–126 and 178–230 are produced by secondary proteolytic reactions after the protein structure is destabilized by the primary cuts at Lys133 and Leu230. Taken together, these results suggest that PP-PLA2 has specific proteolytic activity on two regions of lipid-free apoA-I: the C-terminal region (positions 191 and 230) and a segment in the protein central domain (position 133).
Structural Requirements at ApoA-I C-terminal Domain for PLA2-mediated Proteolysis
The C-terminal domain of apoA-I is essential not only for lipid binding during HDL assembly (47), but it also drives apoA-I self-association in the lipid-free state (40). To investigate the role of the structure of the C-terminal domain of apoA-I in PP-PLA2-mediated proteolysis, a tryptophan-null apoA-I variant (ΔW-apoA-I) that exhibits unique self-association behavior was incubated with PP-PLA2. The α-helical content of the higher order self-association state of ΔW-apoA-I (ΔW[high]) is significantly higher compared with WT-apoA-I (85 versus 60%, respectively) (40). Intermolecular protein-protein interactions in ΔW[high] drive the folding and thermodynamic stability of the otherwise largely unstructured C-terminal domain (48) into an α-helical structure that accounts for the overall increase in protein α-helical content. Thermal treatment of ΔW[high] dissociates the higher order self-associated species to tetramers (60 °C, ΔW[high]60C) and monomers (90 °C, ΔW[high]90C) and proportionally reverses the α-helical content of ΔW-apoA-I to that of WT-apoA-I.
Interestingly, upon incubation of ΔW[high] with PP-PLA2, none of the proteolysis bands observed with WT-apoA-I were detected by SDS-PAGE analysis (Fig. 5A). In contrast, some of the bands previously identified as the products of proteolytic cleavage at sites in the central domain of the protein were observed in PP-PLA2-ΔW[high]60C. Importantly, the proteolytic patterns of monomeric ΔW-apoA-I (PP-PLA2-ΔW[high]90C) and wild-type apoA-I (PP-PLA2-apoA-I) were indistinguishable (Figs. 5A and 4B, respectively). Taken together, the ΔW-apoA-I data and the results from Gd- and CETP-PP-PLA2-HDL indicate that when the C-terminal domain of apoA-I is engaged in either protein-lipid or protein-protein interactions, the proteolytic activity of PLA2 is inhibited.
FIGURE 5.

A, characterization of lipid-free ΔW-apoA-I after incubation in the presence or in the absence of PP-PLA2. ΔW[high] is composed of high order self-association species of the protein. Thermal treatment of ΔW[high] at 60 and 90 °C produces predominantly tetramers (ΔW[high]60C) and monomers (ΔW[high]90C) of ΔW-apoA-I, respectively (40). B, effect of PP-PLA2 on the 1–230 fragment of apoA-I (apoA-I:Δ231–243) generated by a first reaction of full-length apoA-I with PLA2 and isolated by SEC. ApoA-I:Δ231–243 was incubated in the presence of PP-PLA2 for 1 h at 37 °C (PP-PLA2-apoA-I:231–243) before SDS-PAGE analysis. C, to investigate the substrate specificity of PP-PLA2 proteolytic action, lipid-free apoA-II and bovine serum albumin (BSA, essentially fatty acid-free) were incubated with PP-PLA2 in the same conditions as described for apoA-I but for long incubation times (12 h). By SDS-PAGE analysis, no fragmentation of apo-A-II or BSA was observed. D, comparison of the proteolytic activities against apoA-I of different isoforms of PLA2 and from different animal sources, by SDS-PAGE. Porcine pancreatic (PP-PLA2, type IB) and bovine pancreatic (BP-PLA2, type IB) PLA2 were incubated with apoA-I for 1 h. Bee venom (BV-PLA2, type III) and human secreted (sPLA2, type IIA) PLA2 were incubated with apoA-I for 24 h. For all panels, 5 μg of protein was loaded per lane, and molecular weight standards were Precision Plus Protein Dual Xtra standards (Bio-Rad).
To further investigate whether modifications at the C-terminal of apoA-I can affect the proteolytic activity of PLA2 at other apoA-I sites, we used SEC to isolate the 1–230 fragment of apoA-I (apoA-I:Δ231–243) generated by a first reaction of full-length apoA-I with PLA2. The purity and mass of this fragment was confirmed by MALDI-TOF. When apoA-I:Δ231–243 was incubated in the presence of PP-PLA2 (PP-PLA2-apoA-I:231–243), no proteolysis was observed (Fig. 5B). These results indicate that the two cleavage sites at Lys133 and Leu230 are not independent. Cleavage at Lys133 occurs only on full-length apoA-I because cleavage at Leu230 inhibits further proteolysis at Lys133. Therefore, to obtain the observed proteolytic pattern, cleavage at Lys133 must precede cleavage at Leu230.
In conclusion, modifications at the extreme C terminus of apoA-I, either by truncation of the terminal amino acids or by structural modifications promoted by self-association, impair PLA2 proteolysis at the central domain of apoA-I (Lys133). Therefore, specific structural and dynamic features at the C-terminal domain of monomeric lipid-free apoA-I are required for PLA2-mediated proteolysis of apoA-I to occur.
Substrate Specificity of PLA2
Characterization of the protein fragments in Gd-PP-PLA2-HDL suggested that only apoA-I, and not apoA-II, is the substrate of PLA2-mediated proteolysis. To further investigate the substrate specificity of the proteolytic action of PP-PLA2, lipid-free apo-A-II or BSA were incubated with PP-PLA2 under the same conditions described for lipid-free apoA-I. By SDS-PAGE analysis, no fragmentation of apo-A-II or BSA was observed, even for long incubation times (12 h) (Fig. 5C), indicating that the proteolytic activity of PLA2 is specifically directed toward lipid-free apoA-I.
ApoA-I Proteolysis Is Replicated by Different PLA2 Isoforms
To investigate whether the proteolytic activity of PLA2 against apoA-I is a general property of all PLA2 enzymes or a specific activity of the pancreatic isoforms, porcine pancreatic (PP-PLA2, type IB), bee venom (BV-PLA2, type III), and human secretory PLA2 (sPLA2, type IIA) were incubated with lipid-free apoA-I, as described under “Experimental Procedures.” Although small variations in the nature and distribution of the proteolytic fragments were observed, all three enzyme isoforms cleaved apoA-I (Fig. 5D). Notably, both BV-PLA2 and sPLA2-IIA needed longer incubation times to achieve levels of protein cleavage similar to those observed with PP-PLA2 (∼12–24 h versus 1 h). In contrast, pancreatic PLA2 enzymes from different animal sources (porcine (PP-PLA2) or bovine (BP-PLA2)) had similar proteolytic efficiencies (Fig. 5D). These results suggest that the ability of PLA2 to cleave the lipid-free form of apoA-I is a general property of all PLA2 isoforms.
DISCUSSION
In inflammation states, plasma sPLA2 levels increase, whereas HDL-C levels diminish. Because sPLA2 is primarily associated with HDL in plasma (49), direct causality between these two events has been previously hypothesized, but a linking mechanism is yet to be established (13, 50). Similar to HDL-C, apoA-I levels are also reduced during inflammation, leading to the hypothesis that the decrease in HDL-C originates from a net loss of apoA-I, perhaps due to accelerated catabolism (13, 33, 51, 52). It is plausible that the combined increase in plasma levels of sPLA2 and SAA in response to inflammatory stimuli promotes HDL remodeling and generation of lipid-free apoA-I that can be rapidly catabolized, with overall reduction in plasma HDL-C levels. Notably, the fractional catabolic rate of apoA-I in human sPLA2-transgenic mice is higher than in wild-type mice, whereas the catabolic rate of apoA-II is unaffected (12). This raises the possibility that human sPLA2 specifically targets apoA-I.
Interestingly, sPLA2 is also found in atherosclerotic lesions, which are sites of chronic inflammation (53). Similar to the acute phase response, the concerted presence of SAA and sPLA2 in the extracellular matrix of atherosclerotic lesions is likely to increase apoA-I catabolism by promoting local HDL remodeling, thereby potentially reducing the antiatherogenic power of HDL (53). We previously investigated HDL remodeling strategies for the in vitro production of lipid-free/lipid-poor apoA-I (23–25). In the current study, we examined the direct effect of sPLA2 on HDL and/or its molecular components when HDL remodeling events were induced by in vitro chemical denaturation or the action of CETP, in the absence or presence of PLA2. PLA2-mediated surface hydrolysis of HDL phospholipids resulted in a reduction of HDL particle size without dissociation of apolipoproteins (Fig. 1). When HDL remodeling was stimulated through partial chemical denaturation by guanidine treatment, apoA-I, but not apoA-II, apoC-I, or apoC-III, dissociated from HDL (Fig. 2 and Table 1). Importantly, when the same remodeling experiment was performed in the presence of PLA2, apoA-I, but none of the other HDL-associated proteins, was extensively fragmented (Fig. 2 and Table 1). Similar results were observed when HDL remodeling was promoted by reaction with CETP (Fig. 3 and Table 2).
Specific PLA2-mediated proteolysis of lipid-free apoA-I was confirmed by direct incubation of the enzyme with the lipid-free protein, upon which apoA-I was cleaved in fragments similar to those observed after HDL chemical denaturation or reaction with CETP in the presence of PLA2. This activity was specific because no proteolysis was observed upon incubation of PLA2 with apoA-II or BSA. Mass spectrometric analysis of the apoA-I fragments generated by PLA2-mediated proteolysis identified two main cleavage sites in the central (Lys133) and C-terminal (Leu230) regions of apoA-I.
Amino acid 133 is proximal to a largely unstructured region of apoA-I, both in the HDL-associated (54, 55) and in the lipid-free form of the protein (48). Notably, deletion of residues 134–145 completely abolished proteolysis by PLA2 at Lys133. This result suggests that a certain degree of structural freedom in the proximity of the cleavage site is a stringent condition for proteolysis by PLA2. This notion was confirmed by the results obtained with ΔW-apoA-I, a Trp-Phe mutant in which the otherwise largely unstructured C-terminal domain of wild-type apoA-I folds into an α-helical structure due to increased protein self-association (48). Self-associated ΔW-apoA-I was completely protected from PLA2-mediated proteolysis. These results indicate that the structural and dynamic properties of lipid-free apoA-I, which is highly flexible in the central and C-terminal regions, are likely to contribute to the susceptibility of the protein to PLA2-mediated proteolysis.
Important information about the mechanism of proteolysis was derived by the observation that the proteolytic but not the lipolytic activity of PLA2 was Ca2+-independent. Similarly, the proteolytic but not the lipolytic activity of PLA2 was inhibited by benzamidine. Recently, Cavazzini et al. (56) reported that activation of the fungal sPLA2 enzyme TbSP1 is mediated by an autoproteolytic reaction. Based on the observation that autoproteolysis of TbSP1 is Ca2+-independent and inhibited by chemicals that do not affect the Ca2+-dependent lipolytic activity of the enzyme, the authors concluded that lipolysis and proteolysis are catalyzed by two independent sites. In analogy with the TbSP1 case, our data suggest that lipolysis and apoA-I proteolysis are also mediated by different catalytic sites and/or different conformational states of PLA2.
Notably, similar proteolytic activities directed toward apoA-I were observed with different classes of PLA2, including pancreatic (group IB), human plasma-secreted (group IIA), and bee venom (group III) PLA2. The susceptibility of the apoA-I C terminus to proteolysis by an array of proteases (e.g. metalloproteinases, chymotrypsin, trypsin, elastase, subtilisin, staphylococcus V8 protease, and arginine C endopeptidase) is well documented (57–60) and derives from the relatively unstructured nature of the C-terminal domain of lipid-free apoA-I (48). Because the C-terminal domain of apoA-I is critical for lipid binding (61) and deletion of the terminal 23 residues (positions 221–243) abolishes apoA-I-mediated cholesterol efflux in cultured cells (62), proteolytic degradation of the apoA-I C-terminal domain by increased levels of PLA2 may account for the production of dysfunctional apoA-I with diminished HDL biogenesis capacity, thus leading to the observed reduction in HDL-C levels during inflammation.
The current opinion is that plasma levels of enzymes of the sPLA2 family are positively associated with the pathogenesis of atherosclerosis by various mechanisms, including the generation of atherogenic lipoprotein particles (63) and the activation of several proinflammatory pathways (64). In fact, some physiological functions of sPLA2 enzymes are unrelated to their lipolytic activity and can be ascribed to the engagement of specific receptors on target cells (65). Our findings identify a direct link between PLA2 and low HDL-C levels in inflammation through generation of fragmented apoA-I that can be rapidly catabolized. This novel property of PLA2 may provide a mechanistic explanation for the contribution of sPLA2 to the development of atherosclerosis, a disease tightly linked to inflammation.
Acknowledgments
We thank Dr. Xiaohu Mei (Boston University) for useful discussions and Michael Gigliotti and Cheryl England for critical support with biochemical work. Jim Lee and Anh-Tran (Dana Farber Cancer Institute) were instrumental in mass spectrometry data collection and analysis. We especially thank Dr. Olga Gursky (Boston University) and Dr. Andrzej Witkowski (Childrens' Hospital Oakland Research Institute) for constructive discussions and critical evaluation of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant HL113059 (to G. C.). This work was also supported by a new investigator award from the Tobacco-related Disease Research Program of California (18KT-0021) (to G. C.).
Samples generated by guanidine treatment are indicated by the prefix “Gd-” throughout.
- sPLA2
- secretory phospholipase A2
- HDL-C
- high density lipoprotein cholesterol
- CETP
- cholesteryl ester transfer protein
- SAA
- serum amyloid A
- PP-PLA2
- porcine pancreatic PLA2
- BP-PLA2
- bovine pancreatic PLA2
- BV-PLA2
- bee venom PLA2
- SEC
- size exclusion chromatography
- FFA
- free fatty acid
- NDGGE
- non-denaturing gradient gel electrophoresis
- Frn
- fraction n.
REFERENCES
- 1. Vadas P., Browning J., Edelson J., Pruzanski W. (1993) Extracellular phospholipase A2 expression and inflammation: the relationship with associated disease states. J. Lipid Mediat. 8, 1–30 [PubMed] [Google Scholar]
- 2. Camejo G. (2010) Lysophospholipids: effectors mediating the contribution of dyslipidemia to calcification associated with atherosclerosis. Atherosclerosis 211, 36–37 [DOI] [PubMed] [Google Scholar]
- 3. Rodriguéz-Lee M., Bondjers G., Camejo G. (2007) Fatty acid-induced atherogenic changes in extracellular matrix proteoglycans. Curr. Opin. Lipidol. 18, 546–553 [DOI] [PubMed] [Google Scholar]
- 4. Lambeau G., Gelb M. H. (2008) Biochemistry and physiology of mammalian secreted phospholipases A2. Annu. Rev. Biochem. 77, 495–520 [DOI] [PubMed] [Google Scholar]
- 5. Zelensky A. N., Gready J. E. (2005) The C-type lectin-like domain superfamily. FEBS J. 272, 6179–6217 [DOI] [PubMed] [Google Scholar]
- 6. Rosenson R. S., Gelb M. H. (2009) Secretory phospholipase A2: a multifaceted family of proatherogenic enzymes. Curr. Cardiol. Rep. 11, 445–451 [DOI] [PubMed] [Google Scholar]
- 7. Ross R. (1999) Atherosclerosis: an inflammatory disease: reply. N. Engl. J. Med. 340, 115–126 [DOI] [PubMed] [Google Scholar]
- 8. Steinberg D. (2002) Atherogenesis in perspective: hypercholesterolemia and inflammation as partners in crime. Nat. Med. 8, 1211–1217 [DOI] [PubMed] [Google Scholar]
- 9. Libby P., Ridker P. M., Maseri A. (2002) Inflammation and atherosclerosis. Circulation 105, 1135–1143 [DOI] [PubMed] [Google Scholar]
- 10. Goodson N. (2002) Coronary artery disease and rheumatoid arthritis. Curr. Opin. Rheumatol. 14, 115–120 [DOI] [PubMed] [Google Scholar]
- 11. Holmes M. V., Simon T., Exeter H. J., Folkersen L., Asselbergs F. W., Guardiola M., Cooper J. A., Palmen J., Hubacek J. A., Carruthers K. F., Horne B. D., Brunisholz K. D., Mega J. L., van Iperen E. P., Li M., Leusink M., Trompet S., Verschuren J. J., Hovingh G. K., Dehghan A., Nelson C. P., Kotti S., Danchin N., Scholz M., Haase C. L., Rothenbacher D., Swerdlow D. I., Kuchenbaecker K. B., Staines-Urias E., Goel A., van ‘t Hooft F., Gertow K., de Faire U., Panayiotou A. G., Tremoli E., Baldassarre D., Veglia F., Holdt L. M., Beutner F., Gansevoort R. T., Navis G. J., Mateo Leach I., Breitling L. P., Brenner H., Thiery J., Dallmeier D., Franco-Cereceda A., Boer J., Stephens J. W., Hofker M. H., Tedgui A., Hofman A., Uitterlinden A. G., Adamkova V., Pitha J., Onland-Moret N. C., Cramer M. J., Nathoe H. M., Spiering W., Klungel O. H., Kumari M., Whincup P. H., Morrow D. A., Braund P. S., Hall A. S., Olsson A. G., Doevendans P. A., Trip M. D., Tobin M. D., Hamsten A., Watkins H., Koenig W., Nicolaides A. N., Teupser D., Day I. N., Carlquist J. F., Gaunt T. R., Ford I., Sattar N., Tsimikas S., Schwartz G. G., Lawlor D. A., Morris R. W., Sandhu M. S., Poledne R., Maitland-van der Zee A. H., Khaw K. T., Keating B. J., van der Harst P., Price J. F., Mehta S. R., Yusuf S., Witteman J. C., Franco O. H., Jukema J. W., de Knijff P., Tybjaerg-Hansen A., Rader D. J., Farrall M., Samani J., Kivimaki M., Fox K. A., Humphries S. E., Anderson J. L., Boekholdt S. M., Palmer T. M., Eriksson P., Pare G., Hingorani A. D., Sabatine M. S., Mallat Z., Casas J. P., Talmud P. J. (2013) Secretory phospholipase A2-IIA and cardiovascular disease: a Mendelian randomization study. J. Am. Coll. Cardiol. 62, 1966–1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tietge U. J., Maugeais C., Cain W., Grass D., Glick J. M., de Beer F. C., Rader D. J. (2000) Overexpression of secretory phospholipase A2 causes rapid catabolism and altered tissue uptake of high density lipoprotein cholesteryl ester and apolipoprotein A-I. J. Biol. Chem. 275, 10077–10084 [DOI] [PubMed] [Google Scholar]
- 13. Tietge U. J., Maugeais C., Lund-Katz S., Grass D., deBeer F. C., Rader D. J. (2002) Human secretory phospholipase A2 mediates decreased plasma levels of HDL cholesterol and ApoA-I in response to inflammation in human ApoA-I transgenic mice. Arterioscler. Thromb. Vasc. Biol. 22, 1213–1218 [DOI] [PubMed] [Google Scholar]
- 14. de Beer F. C., de Beer M. C., van der Westhuyzen D. R., Castellani L. W., Lusis A. J., Swanson M. E., Grass D. S. (1997) Secretory non-pancreatic phospholipase A2: influence on lipoprotein metabolism. J. Lipid Res. 38, 2232–2239 [PubMed] [Google Scholar]
- 15. Ivandic B., Castellani L. W., Wang X. P., Qiao J. H., Mehrabian M., Navab M., Fogelman A. M., Grass D. S., Swanson M. E., de Beer M. C., de Beer F., Lusis A. J. (1999) Role of group II secretory phospholipase A2 in atherosclerosis. 1. Increased atherogenesis and altered lipoproteins in transgenic mice expressing group IIa phospholipase A2. Arterioscler. Thromb. Vasc. Biol. 19, 1284–1290 [DOI] [PubMed] [Google Scholar]
- 16. Lewis G. F., Rader D. J. (2005) New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ. Res. 96, 1221–1232 [DOI] [PubMed] [Google Scholar]
- 17. Assmann G., Gotto A. M. (2004) HDL cholesterol and protective factors in atherosclerosis. Circulation 109, 8–14 [DOI] [PubMed] [Google Scholar]
- 18. Barter P. J., Nicholls S., Rye K. A., Anantharamaiah G. M., Navab M., Fogelman A. M. (2004) Antiinflammatory properties of HDL. Circ. Res. 95, 764–772 [DOI] [PubMed] [Google Scholar]
- 19. Baker P. W., Rye K. A., Gamble J. R., Vadas M. A., Barter P. J. (2000) Phospholipid composition of reconstituted high density lipoproteins influences their ability to inhibit endothelial cell adhesion molecule expression. J. Lipid Res. 41, 1261–1267 [PubMed] [Google Scholar]
- 20. Bashtovyy D., Jones M. K., Anantharamaiah G. M., Segrest J. P. (2011) Sequence conservation of apolipoprotein A-I affords novel insights into HDL structure-function. J. Lipid Res. 52, 435–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mehta R., Gantz D. L., Gursky O. (2003) Human plasma high-density lipoproteins are stabilized by kinetic factors. J. Mol. Biol. 328, 183–192 [DOI] [PubMed] [Google Scholar]
- 22. Pownall H. J. (2005) Remodeling of human plasma lipoproteins by detergent perturbation. Biochemistry 44, 9714–9722 [DOI] [PubMed] [Google Scholar]
- 23. Jayaraman S., Gantz D. L., Gursky O. (2006) Effects of salt on the thermal stability of human plasma high-density lipoprotein. Biochemistry 45, 4620–4628 [DOI] [PubMed] [Google Scholar]
- 24. Brooks B. R., Brooks C. L., 3rd, Mackerell A. D., Jr., Nilsson L., Petrella R. J., Roux B., Won Y., Archontis G., Bartels C., Boresch S., Caflisch A., Caves L., Cui Q., Dinner A. R., Feig M., Fischer S., Gao J., Hodoscek M., Im W., Kuczera K., Lazaridis T., Ma J., Ovchinnikov V., Paci E., Pastor R. W., Post C. B., Pu J. Z., Schaefer M., Tidor B., Venable R. M., Woodcock H. L., Wu X., Yang W., York D. M., Karplus M. (2009) CHARMM: the biomolecular simulation program. J. Comput. Chem. 30, 1545–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jayaraman S., Cavigiolio G., Gursky O. (2012) Folded functional lipid-poor apolipoprotein A-I obtained by heating of high-density lipoproteins: relevance to high-density lipoprotein biogenesis. Biochem. J. 442, 703–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rye K. A., Barter P. J. (2004) Formation and metabolism of prebeta-migrating, lipid-poor apolipoprotein A-I. Arterioscler. Thromb. Vasc. Biol. 24, 421–428 [DOI] [PubMed] [Google Scholar]
- 27. Oram J. F., Lawn R. M., Garvin M. R., Wade D. P. (2000) ABCA1 is the cAMP-inducible apolipoprotein receptor that mediates cholesterol secretion from macrophages. J. Biol. Chem. 275, 34508–34511 [DOI] [PubMed] [Google Scholar]
- 28. Acton S., Rigotti A., Landschulz K. T., Xu S., Hobbs H. H., Krieger M. (1996) Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 271, 518–520 [DOI] [PubMed] [Google Scholar]
- 29. Glass C., Pittman R. C., Weinstein D. B., Steinberg D. (1983) Dissociation of tissue uptake of cholesterol ester from that of apoprotein A-I of rat plasma high density lipoprotein: selective delivery of cholesterol ester to liver, adrenal, and gonad. Proc. Natl. Acad. Sci. U.S.A. 80, 5435–5439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. DiDonato J. A., Huang Y., Aulak K. S., Even-Or O., Gerstenecker G., Gogonea V., Wu Y., Fox P. L., Tang W. H., Plow E. F., Smith J. D., Fisher E. A., Hazen S. L. (2013) Function and distribution of apolipoprotein A1 in the artery wall are markedly distinct from those in plasma. Circulation 128, 1644–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Miyazaki O., Ogihara J., Fukamachi I., Kasumi T. (2014) Evidence for the presence of lipid-free monomolecular apolipoprotein A-1 in plasma. J. Lipid Res. 55, 214–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nagata K. O., Nakada C., Kasai R. S., Kusumi A., Ueda K. (2013) ABCA1 dimer-monomer interconversion during HDL generation revealed by single-molecule imaging. Proc. Natl. Acad. Sci. U.S.A. 110, 5034–5039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brinton E. A., Eisenberg S., Breslow J. L. (1994) Human HDL cholesterol levels are determined by apoA-I fractional catabolic rate, which correlates inversely with estimates of HDL particle size. Effects of gender, hepatic and lipoprotein lipases, triglyceride and insulin levels, and body fat distribution. Arterioscler. Thromb. 14, 707–720 [DOI] [PubMed] [Google Scholar]
- 34. Hoffman J. S., Benditt E. P. (1982) Changes in high density lipoprotein content following endotoxin administration in the mouse. Formation of serum amyloid protein-rich subfractions. J. Biol. Chem. 257, 10510–10517 [PubMed] [Google Scholar]
- 35. Coetzee G. A., Strachan A. F., van der Westhuyzen D. R., Hoppe H. C., Jeenah M. S., de Beer F. C. (1986) Serum amyloid A-containing human high density lipoprotein 3. Density, size, and apolipoprotein composition. J. Biol. Chem. 261, 9644–9651 [PubMed] [Google Scholar]
- 36. Ikewaki K., Rader D. J., Sakamoto T., Nishiwaki M., Wakimoto N., Schaefer J. R., Ishikawa T., Fairwell T., Zech L. A., Nakamura H. (1993) Delayed catabolism of high density lipoprotein apolipoproteins A-I and A-II in human cholesteryl ester transfer protein deficiency. J. Clin. Invest. 92, 1650–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jahangiri A., de Beer M. C., Noffsinger V., Tannock L. R., Ramaiah C., Webb N. R., van der Westhuyzen D. R., de Beer F. C. (2009) HDL remodeling during the acute phase response. Arterioscler. Thromb. Vasc. Biol. 29, 261–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schumaker V. N., Puppione D. L. (1986) Sequential flotation ultracentrifugation. Methods Enzymol. 128, 155–170 [DOI] [PubMed] [Google Scholar]
- 39. Wetterau J. R., Jonas A. (1982) Effect of dipalmitoylphosphatidylcholine vesicle curvature on the reaction with human apolipoprotein A-I. J. Biol. Chem. 257, 10961–10966 [PubMed] [Google Scholar]
- 40. Jayaraman S., Abe-Dohmae S., Yokoyama S., Cavigiolio G. (2011) Impact of self-association on function of apolipoprotein A-I. J. Biol. Chem. 286, 35610–35623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gao X., Yuan S., Jayaraman S., Gursky O. (2009) Differential stability of high-density lipoprotein subclasses: effects of particle size and protein composition. J. Mol. Biol. 387, 628–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yu B. Z., Berg O. G., Jain M. K. (1993) The divalent cation is obligatory for the binding of ligands to the catalytic site of secreted phospholipase A2. Biochemistry 32, 6485–6492 [DOI] [PubMed] [Google Scholar]
- 43. Pruzanski W., Stefanski E., de Beer F. C., de Beer M. C., Vadas P., Ravandi A., Kuksis A. (1998) Lipoproteins are substrates for human secretory group IIA phospholipase A2: preferential hydrolysis of acute phase HDL. J. Lipid Res. 39, 2150–2160 [PubMed] [Google Scholar]
- 44. Williams K. R., Stone K. L. (1997) Enzymatic cleavage and HPLC peptide mapping of proteins. Mol. Biotechnol. 8, 155–167 [DOI] [PubMed] [Google Scholar]
- 45. Liang H. Q., Rye K. A., Barter P. J. (1994) Dissociation of lipid-free apolipoprotein A-I from high density lipoproteins. J. Lipid Res. 35, 1187–1199 [PubMed] [Google Scholar]
- 46. Ballou L. R., Cheung W. Y. (1985) Inhibition of human platelet phospholipase A2 activity by unsaturated fatty acids. Proc. Natl. Acad. Sci. U.S.A. 82, 371–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Frank P. G., Marcel Y. L. (2000) Apolipoprotein A-I: structure-function relationships. J. Lipid Res. 41, 853–872 [PubMed] [Google Scholar]
- 48. Chetty P. S., Mayne L., Lund-Katz S., Stranz D., Englander S. W., Phillips M. C. (2009) Helical structure and stability in human apolipoprotein A-I by hydrogen exchange and mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 106, 19005–19010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gijón M. A., Pérez C., Méndez E., Sánchez Crespo M. (1995) Phospholipase A2 from plasma of patients with septic shock is associated with high-density lipoproteins and C3 anaphylatoxin: some implications for its functional role. Biochem. J. 306, 167–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jahangiri A. (2010) High-density lipoprotein and the acute phase response. Curr. Opin. Endocrinol. Diabetes Obes. 17, 156–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rader D. J., Ikewaki K. (1996) Unravelling high density lipoprotein-apolipoprotein metabolism in human mutants and animal models. Curr. Opin. Lipidol. 7, 117–123 [DOI] [PubMed] [Google Scholar]
- 52. Schaefer E. J., Zech L. A., Jenkins L. L., Bronzert T. J., Rubalcaba E. A., Lindgren F. T., Aamodt R. L., Brewer H. B. (1982) Human apolipoprotein A-I and A-II metabolism. J. Lipid Res. 23, 850–862 [PubMed] [Google Scholar]
- 53. Menschikowski M., Hagelgans A., Siegert G. (2006) Secretory phospholipase A2 of group IIA. Is it an offensive or a defensive player during atherosclerosis and other inflammatory diseases? Prostaglandins Other Lipid Mediat. 79, 1–33 [DOI] [PubMed] [Google Scholar]
- 54. Oda M. N., Budamagunta M. S., Geier E. G., Chandradas S. H., Shao B., Heinecke J. W., Voss J. C., Cavigiolio G. (2013) Conservation of apolipoprotein A-I's central domain structural elements upon lipid association on different high-density lipoprotein subclasses. Biochemistry 52, 6766–6778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sevugan Chetty P., Mayne L., Kan Z. Y., Lund-Katz S., Englander S. W., Phillips M. C. (2012) Apolipoprotein A-I helical structure and stability in discoidal high-density lipoprotein (HDL) particles by hydrogen exchange and mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 109, 11687–11692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cavazzini D., Meschi F., Corsini R., Bolchi A., Rossi G. L., Einsle O., Ottonello S. (2013) Autoproteolytic Activation of a symbiosis-regulated truffle phospholipase A2. J. Biol. Chem. 288, 1533–1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Eberini I., Calabresi L., Wait R., Tedeschi G., Pirillo A., Puglisi L., Sirtori C. R., Gianazza E. (2002) Macrophage metalloproteinases degrade high-density-lipoprotein-associated apolipoprotein A-I at both the N- and C-termini. Biochem. J. 362, 627–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kunitake S. T., Chen G. C., Kung S. F., Schilling J. W., Hardman D. A., Kane J. P. (1990) Pre-β high density lipoprotein. Unique disposition of apolipoprotein A-I increases susceptibility to proteolysis. Arteriosclerosis 10, 25–30 [DOI] [PubMed] [Google Scholar]
- 59. Dalton M. B., Swaney J. B. (1993) Structural and functional domains of apolipoprotein A-I within high density lipoproteins. J. Biol. Chem. 268, 19274–19283 [PubMed] [Google Scholar]
- 60. Ji Y., Jonas A. (1995) Properties of an N-terminal proteolytic fragment of apolipoprotein AI in solution and in reconstituted high density lipoproteins. J. Biol. Chem. 270, 11290–11297 [DOI] [PubMed] [Google Scholar]
- 61. Gorshkova I. N., Liadaki K., Gursky O., Atkinson D., Zannis V. I. (2000) Probing the lipid-free structure and stability of apolipoprotein A-I by mutation. Biochemistry 39, 15910–15919 [DOI] [PubMed] [Google Scholar]
- 62. Panagotopulos S. E., Witting S. R., Horace E. M., Hui D. Y., Maiorano J. N., Davidson W. S. (2002) The role of apolipoprotein A-I helix 10 in apolipoprotein-mediated cholesterol efflux via the ATP-binding cassette transporter ABCA1. J. Biol. Chem. 277, 39477–39484 [DOI] [PubMed] [Google Scholar]
- 63. Oörni K., Kovanen P. T. (2007) PLA2-V: a real player in atherogenesis. Arterioscler. Thromb. Vasc. Biol. 27, 445–447 [DOI] [PubMed] [Google Scholar]
- 64. Rosengren B., Jönsson-Rylander A.-C., Peilot H., Camejo G., Hurt-Camejo E. (2006) Distinctiveness of secretory phospholipase A2 group IIA and V suggesting unique roles in atherosclerosis. Biochim. Biophys. Acta 1761, 1301–1308 [DOI] [PubMed] [Google Scholar]
- 65. Ohara O., Ishizaki J., Arita H. (1995) Structure and function of phospholipase A2 receptor. Prog. Lipid Res. 34, 117–138 [DOI] [PubMed] [Google Scholar]