

Background: RhoGEFs play important roles in regulation of the actin cytoskeleton.
Results: The FLJ00018/PLEKHG2 was phosphorylated, and activated by EGF signaling might be interpreted as a direct effect on its GEF activity (which is not established).
Conclusion: The phosphorylation of FLJ00018 was involved in the regulation of cell morphology.
Significance: This study suggests that FLJ00018 receives and integrates different stimuli as a relay point in cell signaling.
Keywords: Epidermal Growth Factor Receptor (EGFR), Guanine Nucleotide Exchange Factor (GEF), Neurite Outgrowth, Protein phosphorylation, Rho GTPases
Abstract
FLJ00018/PLEKHG2 is a guanine nucleotide exchange factor for the small GTPases Rac and Cdc42 and has been shown to mediate the signaling pathways leading to actin cytoskeleton reorganization. The function of FLJ00018 is regulated by the interaction of heterotrimeric GTP-binding protein Gβγ subunits or cytosolic actin. However, the details underlying the molecular mechanisms of FLJ00018 activation have yet to be elucidated. In the present study we show that FLJ00018 is phosphorylated and activated by β1-adrenergic receptor stimulation-induced EGF receptor (EGFR) transactivation in addition to Gβγ signaling. FLJ00018 is also phosphorylated and activated by direct EGFR stimulation. The phosphorylation of FLJ00018 by EGFR stimulation is mediated by the Ras/mitogen-activated protein kinase (MAPK) pathway. Through deletion and site-directed mutagenesis studies, we have identified Thr-680 as the major site of phosphorylation by EGFR stimulation. FLJ00018 T680A, in which the phosphorylation site is replaced by alanine, showed a limited response of the Neuro-2a cell morphology to EGF stimulation. Our results provide evidence that stimulation of the Ras/MAPK pathway by EGFR results in FLJ00018 phosphorylation at Thr-680, which in turn controls changes in cell shape.
Introduction
Heterotrimeric G-protein-coupled receptor (GPCR)2 can initiate a variety of intracellular signaling pathways. Binding of a ligand to its cognate GPCR causes a conformational change in the receptor, leading to the intracellular release of activated Gα and Gβγ subunits, which in turn signal the downstream effectors (1). GPCR signaling is involved in many cellular responses, such as proliferation, differentiation, and cytoskeletal regulation. Cytoskeletal regulation in particular is governed largely by the precise temporal and spatial modulation of the small G-proteins of the Rho family (Rho GTPase), RhoA, Rac, and Cdc42 (2). The Rho GTPases function as molecular switches. They are converted from the GDP-bound inactive form to a GTP-bound active state by a reaction catalyzed by Rho GTPase-specific guanine nucleotide exchange factors (RhoGEFs). RhoGEFs are large multidomain proteins that are tightly regulated to control their function. RhoGEFs can be subdivided into two main subfamilies. First, there are those that possess a Dbl homology (DH) domain that is found in tandem with a pleckstrin homology (PH) domain. This subfamily is currently represented by 70 members in mammalian genomes (3). Second, there are Dock180-related proteins containing the Dock homology region-2 domain (also known as the Docker-ZH2 domain), which form a subfamily of 11 mammalian members (4). The DH domain is responsible for catalytic activity, and the PH domain directs subcellular localization and can modulate the DH domain function.
A number of the DH domain-containing RhoGEFs, including PSD-95/Dlg/ZO-1 (PDZ)-RhoGEF, leukemia-associated RhoGEF (LARG), and p115-RhoGEF, have a regulator of G-protein signaling (RGS) domain in addition to a DH domain and PH domain. PDZ-RhoGEF and LARG also have a PDZ domain. These RhoGEFs are regulated by activated Gα12/13 subunits through their interaction with the RGS domain to activate GDP/GTP exchange activity for RhoA (5, 6). On the other hand, P-Rex1 and P-Rex2 are regulated by Gβγ subunits and polyphosphoinositide through direct interaction to activate the GDP/GTP exchange activity for Rac. We previously reported that one novel RhoGEF, FLJ00018/PLEKHG2, was activated by direct interaction with Gβγ subunits and regulated cell spreading through the activation of Rac1 and Cdc42 (7). We also reported that FLJ00018 interacts with β-actin and γ-actin. Both β- and γ-actin act as negative regulators of FLJ00018 (8). However, the details underlying the molecular mechanisms of FLJ00018 activation have yet to be elucidated.
A report by Chikumi et al. (9) suggested that PDZ-RhoGEF and LARG are also regulated by tyrosine phosphorylation by focal adhesion kinases. Furthermore, it is known that at least 19 RhoGEFs are activated by receptor-tyrosine kinase signaling (10). Phosphorylation of RhoGEFs is important in receptor-tyrosine kinase signaling-mediated RhoGEF activation. It has been reported that several RhoGEFs, including Vav, Tiam1, Asef, and β-Pix/Cool-1, were phosphorylated by extracellular stimulation (11–17). The phosphorylation site of Asef is Tyr-94, and this phosphorylation induces Rac1 activation (11). β-Pix is phosphorylated by fibroblast growth factor receptor signaling at Ser-525 and Thr-526 residues. The phosphorylation of β-Pix induces neurite outgrowth in PC12 cells (13). Vav2 interacts with EGFR and is phosphorylated by EGF stimulation (14). Recently, it has been shown that P-Rex1 is also regulated by ErbB signaling in breast cancer cells (18).
In contrast to these RhoGEFs, the regulation of FLJ00018 by phosphorylation has not been studied. In this report, we investigated the possibility that phosphorylation of FLJ00018 by β1-adrenagic receptor (β1-AR) stimulation might regulate the function of FLJ00018 in cells. We found that phosphorylation of FLJ00018 by EGFR-activated Ras/MAPK signaling caused the activation of FLJ00018 and controlled the cell morphological change in Neuro-2a cells independently of interaction with Gβγ subunits.
EXPERIMENTAL PROCEDURES
Materials
pcDNA3.1-β1-AR, pcDNA3.1-Gβ1, pcDNA3.1-Gγ2, pcDNA3.1-H-Ras G12V, pcDNA3.1-H-Ras T17N, pcDNA3.1-RhoA T19N, pcDNA3.1-Rac1 T17N, and pCDNA3.1-Cdc42 T17N were purchased from Missouri S&T cDNA Resource Center. These plasmids were subcloned into a pF4A-CMV or pF5A-CMV-neo Flexi vector (Promega) using polymerase chain reaction (PCR) amplification and a Flexi system (Promega). A complementary DNA (cDNA) clone for FLJ00018 genes was isolated during the Kazusa human cDNA project, which aimed to accumulate information on the coding sequence of long cDNAs for unidentified human genes (19). To construct the Myc- and Halo-tagged forms, native-form protein expression clones, the open reading frame of FLJ00018 was subcloned into a pFN21A-Myc vector or the pFN21A-Halo vector using PCR amplification and the Flexi system. The cDNA-encoding deletion mutants of FLJ00018 indicated in the corresponding figures were generated by restriction enzyme digestion or PCR amplification using pFN21A-Myc-FLJ00018. Monomeric Azami Green (mAG) is a fluorescent protein (20); an expression vector coded with this protein, phmAG1-MCLinker, was purchased from MBL. The pFN21K-mAG vector was made by PCR amplification using phmAG1-MCLinker and restriction enzyme digestion and ligation. pFN21K-mAG-FLJ00018 was made by restriction enzyme digestion of pFN21A-Myc-FLJ00018. pF1K-EGFR and pF1K-Vav3 were subcloned into pFC21A-Halo and a pFN21K-Halo vector using the Flexi system. The pSRE.L-luciferase reporter plasmid was purchased from Stratagene, and pRL-SV40 was purchased from Nippon Gene. Isoproterenol, KN-93, and SB202190 were purchased from Calbiochem, EGF was from Peprotech, and AG1478, U-0126, and Phos-tag AAL107 were from Wako. SP600125 was purchased from Sigma. Phosphatase inhibitor mixture tablet (PhosSTOP) and protease inhibitor mixture tablet (Complete mini) were purchased from Roche Applied Science. λ-Phosphatase and alkaline phosphatase were purchased from New England Biolabs.
Cell Culture and Transfection
NIH3T3, HEK293, and Neuro-2a cells were grown in DMEM supplemented with 10% calf serum (NIH3T3) or 10% FBS (HEK293 and Neuro-2a) at 37 °C. Transient transfection was performed using Lipofectamine Plus reagent according to the manufacturer's instructions (Invitrogen). The amount of plasmid DNA was 400 ng for the serum response element (SRE)-dependent gene transcription assay and 1000 ng for immunoblot analysis. Cells were transfected with DNA for 3 h then treated with 1× insulin-transferrin-selenium-X (Invitrogen) for 6–8 h. The cells were washed twice with serum-free DMEM and incubated for 16–18 h in DMEM.
Assay of SRE-dependent Gene Transcription
Cells seeded in 24-well plates were co-transfected with the indicated expression plasmids together with the pSRE.L-luciferase reporter plasmid and the pRL-SV40 control reporter plasmids. After transfection, the cells were treated with inhibitors before stimulation at the indicated times and concentrations. Cells were washed once with ice-cold PBS and lysed with passive lysis buffer. Luciferase activities were determined with a dual-luciferase reporter assay system (Promega). The activity of the experimental reporter was normalized against the activity of the control vector.
Dephosphorylation of FLJ00018 by Phosphatase
Transfected cells were stimulated at the indicated times and concentrations. Then cells were washed once with ice-cold PBS and lysed with lysis buffer (50 mm Tris-HCl, pH 7.5, 100 mm NaCl, 0.5% Nonidet P-40, and protease inhibitor solution). Equal amounts of cell lysate were incubated with alkaline phosphatase solution (10 units/dish) or λ-phosphatase solution (400 units/dish) for 2 h at 30 °C. The reaction was terminated by adding 2×SDS sample buffer. Then, the phosphorylation status of the reaction mixture was tested by immunoblot analysis as described below.
Immunoblot Analysis
Transfected cells were treated with inhibitors before stimulation at the indicated times and concentrations. Cells were washed once with ice-cold PBS and lysed with 0.5% (w/v) SDS in distilled water. The protein amount of each lysate was quantified using a bicinchoninic acid protein assay kit (Thermo Scientific) with BSA as the standard. The same amounts of protein were subjected to 7.5 or 10% SDS-PAGE and immunoblotted with various antibodies, employing a chemiluminescence reagent (PerkinElmer Life Sciences). Densitometry was performed using an LAS-4000 image analyzer and LAS-4000-mini image analyzer (GE Healthcare).
Mn2+ Phos-tag SDS-PAGE
To visualize the levels of FLJ00018 phosphorylation, a newly developed immunoblotting technique was performed (21–23). In Phos-tag gel, phosphoprotein-binding molecules called AAL-107 Phos-tag ligands retain phosphorylated proteins, thereby separating them from their unphosphorylated counterparts during electrophoresis. This results in two or more distinct protein bands representing the phosphorylated and unphosphorylated protein. Cell lysates were resolved by 6% SDS-polyacrylamide gel containing 15 μm Phos-tag and 30 μm MnCl2 or 3% SDS-polyacrylamide gel containing 13 μm Phos-tag, 26 μm MnCl2, and 1.5% agarose. After electrophoresis, the gels were washed in transfer buffer containing 1 mm EDTA for 10 min and then transferred to PVDF membranes.
Immunofluorescence Analysis
After stimulation with EGF (20 ng/ml, 24 h), transfected cells cultured on coverslips were washed once with ice-cold PBS and fixed with 4% paraformaldehyde for 30 min. The cover slips were then mounted with Perma Fluor. Fluorescence images were acquired using a fluorescence microscope (BZ-9000; KEYENCE). Ten fluorescence images were taken from random fields, and the total neurite length of 50 cells was measured in each sample. The total neurite lengths were then analyzed using Image J software.
Statistical Analysis
Data are expressed as the means ± S.D. of values from at least three independent experiments. The significance of group differences was analyzed by Student's t test. A p value of <0.05 was considered significant.
RESULTS
FLJ00018 Is Activated by β1-Adrenergic Receptor Stimulation through a Gβγ-dependent and -independent Pathway
We previously reported that FLJ00018 was activated by Gβγ and m2 muscarinic acetylcholine receptor stimulation (7). To examine whether FLJ00018 is activated by stimulation of other GPCRs, we used a β-adrenergic receptor, β1-AR, that is known to couple with multiple signaling pathways via Gs protein. NIH3T3 cells were transiently transfected with β1-AR and FLJ00018, and then luciferase assays were performed to measure FLJ00018-induced SRE-dependent gene transcription. Several reports have suggested that β1-AR signaling participates in multiple pathways including the 1) Gs/adenylate cyclase (AC)/cAMP/CNrasGEF/Ras/MAPK pathway, 2) Gs/AC/cAMP/PKA/Gi/Gβγ/Ras/MAPK pathway, and 3) GRK5 or GRK6/β-arrestin/src/EGFR pathway (24–26). As shown in Fig. 1A, stimulation with isoproterenol (20 μm) enhanced FLJ00018-induced SRE-dependent gene transcription in NIH3T3 cells expressing FLJ00018 and β1-AR. However, the protein expression levels or protein modification state of FLJ00018 was not changed by the β1-AR stimulation in FLJ00018 and β1-AR co-expressed cells (Fig. 1B). To investigate the involvement of Gβγ subunits in the enhancement of SRE-dependent gene transcription, we used the C-terminal region of β-adrenergic receptor kinase (β-ARKct), which is thought to interact with Gβγ subunits and attenuate Gβγ-dependent signaling. Co-expression of β-ARKct attenuated the enhancement of FLJ00018-induced SRE-dependent gene transcription. However, co-expression of β-ARKct did not completely inhibit the transcription (Fig. 1A). This result suggests that β1-AR signaling may be involved in both Gβγ-dependent and -independent FLJ00018 activation pathways, but only the former is attenuated by β-ARKct.
FIGURE 1.
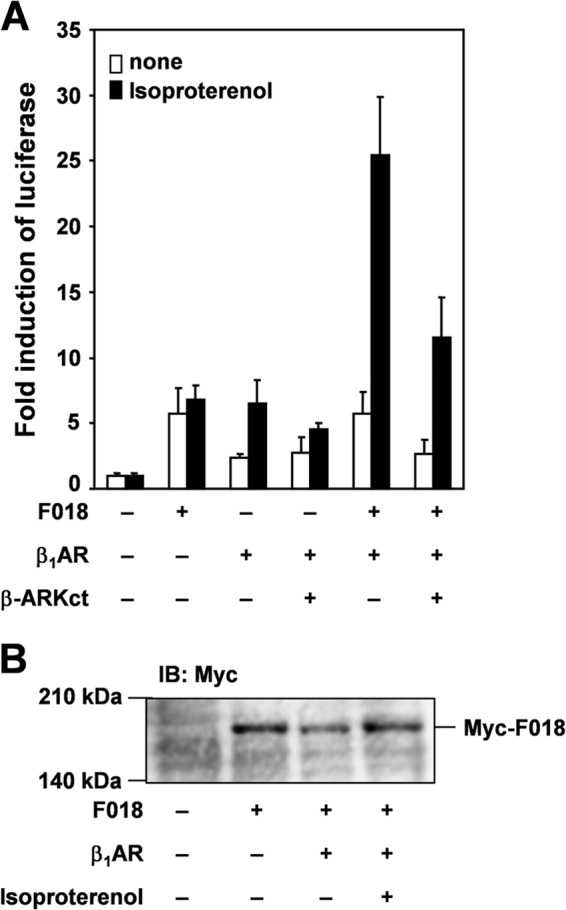
Effect of isoproterenol on FLJ00018-induced SRE-dependent gene transcription in β1-AR-expressing NIH3T3 cells. A, NIH3T3 cells were co-transfected with pSRE.L-luciferase, pRL-SV40 plasmid DNAs, and expression vectors for FLJ00018 (F018), β1-AR, and β-ARKct as indicated. Transfected cells were stimulated with 20 μm isoproterenol for 5.5 h. Luciferase activity was determined by a dual-luciferase reporter assay. Luciferase activity obtained with mock cells was taken as 1.0, and relative activities are shown. Values represent the means ± S.D. from at least three experiments. B, NIH3T3 cells were co-transfected with expression vectors for β1-AR and FLJ00018 as indicated. Transfected cells were stimulated with isoproterenol (20 μm) for 15 min. Equal amounts of protein were resolved by 7.5% SDS-PAGE. Immunoblotting (IB) was performed with antibodies against Myc.
FLJ00018 Is Activated by β1-AR-mediated Transactivation of EGF Receptor
It was previously shown that EGFR is activated through GPCR signaling, including those from β1-AR signaling (27, 28). Some papers have shown that β1-AR and β2-AR also induce the transactivation of EGFR (26, 29–31). This transactivation induces the activation of EGFR tyrosine kinase. To investigate the involvement of the EGFR transactivation in β1-AR-stimulated FLJ00018-induced SRE-dependent gene transcription, we tested the autophosphorylation of EGFR by β1-AR stimulation in cells co-expressing EGFR, β1-AR, and FLJ00018. As shown in Fig. 2A, after stimulation with isoproterenol (20 μm), tyrosine phosphorylation of EGFR was increased in a time-dependent manner, and the levels of phosphorylation peaked at 15 min. In general, it is known that constitutive (agonist-independent) activity is observed with the overexpression of many GPCRs (32). In Fig. 2A, it was thought that EGFR tyrosine phosphorylation in the absence of β1-AR stimulation may have been caused by the agonist-independent activation by overexpression of β1-AR, although the reason is not clear. To examine the involvement of transactivation in β1-AR-stimulated FLJ00018-induced SRE-dependent gene transcription, we used AG1478, a specific inhibitor of EGFR tyrosine kinase activity. Stimulation of isoproterenol (20 μm) increased FLJ00018-induced SRE-dependent gene transcription in cells expressing FLJ00018, β1-AR, and EGFR. However, treatment with AG1478 (10 μm) before isoproterenol stimulation decreased the enhancement of FLJ00018-induced SRE-dependent gene transcription (Fig. 2B). To examine the levels of expression of FLJ00018 in these cells, we performed immunoblot analysis. In isoproterenol-stimulated cells, the mobility of the FLJ00018 band was slower than that of the FLJ00018 band in unstimulated cells (Fig. 2C). However, the FLJ00018 band in cells co-expressing Gβγ and FLJ00018 did not exhibit this mobility shift. These results suggested the possibility that FLJ00018 is modified in response to β1-AR stimulation-induced EGFR transactivation. Recently, it was reported that the functions of several RhoGEFs, including Vav, Tiam-1, β-Pix, and Asef, are regulated through phosphorylation by extracellular stimulation (10–18). To investigate whether FLJ00018 is phosphorylated by β1-AR stimulation-induced EGFR transactivation, we used alkaline phosphatase and λ-phosphatase. As shown in Fig. 2D, both of the phosphatases attenuated the mobility shift of FLJ00018, whereas the mobility shift was visible in the cells not treated with phosphatase. A mobility shift of FLJ00018 by β1-AR stimulation-induced EGFR transactivation was also observed in HEK293 cells (Fig. 2E). In the same set of experiments, we also performed a Phos-tag gel analysis using the HEK293 cells. The Phos-tag gels contain AAL-107 Phos-tag ligands, which are phosphoprotein-binding molecules (21–23). AAL-107 Phos-tag ligands retain phosphorylated proteins, which thereby separate from their unphosphorylated counterparts during electrophoresis. In theory this will result in two or more distinct protein bands representing the phosphorylated and unphosphorylated status of the protein. Using this method, the mobility shift of FLJ00018 was found to be enhanced by isoproterenol stimulation in cells expressing β1-AR, EGFR, and FLJ00018 compared with cells expressing FLJ00018 alone (Fig. 2F). Even though a band shift was not observed in SDS-PAGE (Fig. 1, B and E), when we used Phos-tag gel, β1-AR stimulation caused a slight shift in the FLJ00018 band (Fig. 2F). In addition, the observed FLJ00018 mobility shift was attenuated in AG1478-treated NIH3T3 cells (Fig. 2G). These results suggest the possibility that FLJ00018 is phosphorylated and activated by β1-AR stimulation-induced EGFR transactivation and β1-AR stimulation.
FIGURE 2.

FLJ00018 is activated by β1-AR-mediated transactivation of EGF receptors. A, NIH3T3 cells were co-transfected with expression vectors for EGFR, β1-AR, and FLJ00018 (F018) as indicated. Transfected cells were stimulated with isoproterenol (20 μm) for 0–30 min. Equal amounts of protein were resolved by 7.5% SDS-PAGE. Immunoblotting (IB) was performed with antibodies against phosphotyrosine (P-Tyr) or EGFR. B, NIH3T3 cells were co-transfected with pSRE.L-luciferase, pRL-SV40 plasmid DNAs, and expression vectors for FLJ00018, β1-AR, and EGFR as indicated. Transfected cells were stimulated with 20 μm isoproterenol for 5.5 h before treatment with 10 μm AG1478 for 1 h. Luciferase activity was determined by a dual-luciferase reporter assay. Luciferase activity obtained with mock cells was taken as 1.0, and relative activities are shown. Values are the means ± S.D. from at least three experiments. #, p < 0.05. C, NIH3T3 cells were co-transfected with expression vectors for EGFR, β1-AR, Myc-tagged FLJ00018, and Gβ1γ2 as indicated. Transfected cells were stimulated with isoproterenol (20 μm) for 15 min. Equal amounts of protein were resolved by 7.5% SDS-PAGE. To detect Myc-tagged FLJ00018, immunoblotting was performed with antibodies against Myc (Myc). D, NIH3T3 cells were co-transfected with expression vectors for EGFR, β1-AR, and Myc-tagged FLJ00018 as indicated. Transfected cells were stimulated with isoproterenol (20 μm) for 15 min. After lysis, the cell lysates were incubated with alkaline phosphatase (AP; 10 units) and λ-phosphatase (λ; 400 units) for 2 h at 30 °C. The reaction was terminated by the addition of SDS sample buffer. Equal amounts of the reaction mixture were resolved by 7.5% SDS-polyacrylamide gel electrophoresis. To detect Myc-tagged FLJ00018, immunoblotting was performed with antibodies against Myc (Myc). E and G, HEK293 cells (E) or NIH3T3 cells (G) were co-transfected with expression vectors for EGFR, β1-AR, and Myc-tagged FLJ00018. Transfected cells were treated with 10 μm AG1478 for 30 min (G) before stimulation with 20 μm isoproterenol for 15 min. Equal amounts of protein were resolved by 7.5% SDS-PAGE (E and G) or 3% SDS-polyacrylamide gel containing 13 μm Phos-tag, 26 μm MnCl2 and 1.5% agarose (F). Immunoblotting was performed with anti-Myc.
FLJ00018 Is Activated and Phosphorylated by Direct EGF Stimulation
As shown in Fig. 2, FLJ00018 was phosphorylated by EGFR transactivation via GPCR stimulation. To investigate whether FLJ00018 is phosphorylated and activated by direct EGFR stimulation, we measured EGF-induced SRE-dependent gene transcription in cells co-expressing EGFR and FLJ00018. After stimulation with EGF (20 ng/ml) for 6 h, FLJ00018-induced SRE-dependent gene transcription was greatly enhanced (Fig. 3A). Previously, we showed that FLJ00018 acts as a RhoGEF for Rac1 and Cdc42 (7). To investigate which types of Rho are involved in EGF-stimulated FLJ00018-induced SRE-dependent gene transcription, we used constitutively inactive forms of RhoA, Rac1, and Cdc42. When the cells were stimulated with EGF (20 ng/ml, 6 h), FLJ00018-induced SRE-dependent gene transcription was blocked by the inactive forms of Rac1 and Cdc42 (Fig. 3B). From these results it has been suggested that EGF-stimulated FLJ00018 activated Rac1 and Cdc42. When the cells were stimulated with EGF (20 ng/ml) for 15 min, the mobility of the FLJ00018 band was slower than that of the FLJ00018 band from unstimulated cells in the immunoblot analysis (Fig. 3C). However, the FLJ00018 band in cells co-expressing Gβγ and FLJ00018 did not exhibit this mobility shift. In addition, the mobility shift of FLJ00018 was inhibited by alkaline phosphatase (10 units for 2 h) and λ-phosphatase (400 units for 2 h) treatment (Fig. 3D). Furthermore, the mobility shift of FLJ00018 also took place in HEK293 cells (Fig. 3E), and this mobility shift was enhanced in Phos-tag gel (Fig. 3F). These data suggest that FLJ00018 is phosphorylated by EGFR signaling. In addition, we performed immunoblot analysis and confirmed a mobility-shifted band of FLJ00018 at various time points. As we have shown in Fig. 3G, the shifted band of FLJ00018 was increased in a time-dependent manner and peaked at 5 min. Compared with EGFR transactivation (Fig. 2A), EGFR stimulation by EGF is a rapid response and may cause differences in cell function. Because EGFR is a receptor tyrosine kinase, we examined whether FLJ00018 is tyrosine-phosphorylated by EGFR stimulation. As shown in Fig. 3H, a FLJ00018 tyrosine phosphorylation band was not detected in cells expressing EGFR and FLJ00018 under our experimental conditions (Fig. 3H, dotted triangle), whereas Vav3 tyrosine phosphorylation in response to EGF stimulation was observed in cells expressing EGFR and Vav3 (Fig. 3H, open triangles). These results suggest that EGFR stimulation mainly induced the serine/threonine phosphorylation of FLJ00018.
FIGURE 3.

FLJ00018 is activated by EGF receptor signaling. A and B, NIH3T3 cells were co-transfected with pSRE.L-luciferase, pRL-SV40 plasmid DNAs, and expression vectors for FLJ00018 (F018), EGFR, RhoA T19N, Rac T17N, and Cdc42 T17N as indicated. Transfected cells were stimulated with 20 ng/ml EGF for 6 h. Luciferase activity was determined by a dual-luciferase reporter assay. Luciferase activity obtained with mock cells was taken as 1.0, and relative activities are shown. Values are the means ± S.D. from at least three experiments. C–G, NIH3T3 cells (C, D, and G) or HEK293 cells (E and F) were co-transfected with expression vectors for Myc-tagged FLJ00018, EGFR, and/or Gβ1γ2 as indicated. Transfected cells were stimulated with 20 ng/ml EGF for 15 min (C and E) or 0–30 min as indicated (G). After lysis, the cell lysates were incubated with alkaline phosphatase (AP; 10 units) and λ-phosphatase (λ; 400 units) for 2 h at 30 °C, and the reaction was terminated by the addition of SDS sample buffer (D). IB, immunoblot. Equal amounts of protein were resolved by 7.5% SDS-polyacrylamide gel electrophoresis (C–E and G) or by 3% SDS-polyacrylamide gel containing 13 μm Phos-tag, 26 μm MnCl2, and 1.5% agarose (F). To detect Myc-tagged FLJ00018, immunoblotting was performed with antibodies against Myc. H, NIH3T3 cells were co-transfected with expression vectors for Halo-tagged FLJ00018 or Halo-tagged Vav3 and EGFR as indicated. Transfected cells were stimulated with 20 ng/ml EGF for 15 min. Equal amounts of protein were resolved by 7.5% SDS-PAGE. To detect Halo-tagged FLJ00018 or Vav3, immunoblotting was performed with antibodies against Halo. Tyrosine phosphorylation of proteins was detected by antibodies against phosphotyrosine (P-Tyr). Dotted triangle, FLJ00018; open triangles, Vav3; closed triangles, EGFR tyrosine phosphorylation.
FLJ00018 Is Phosphorylated through H-Ras-dependent Signaling Pathways
Next, we investigated which signaling pathway of EGFR is involved in FLJ00018 phosphorylation. For this purpose, cells were co-transfected with FLJ00018 and a constitutively active form of H-Ras (H-RasCA) or a dominant-negative form of H-Ras (H-RasDN). Figs. 4, A and B, show the band corresponding to FLJ00018 from immunoblot analysis. H-RasCA induced a mobility shift of the FLJ00018 band. H-RasDN inhibited the mobility shift of the FLJ00018 band in response to EGF stimulation (20 ng/ml). These results suggest that H-Ras is involved in FLJ00018 phosphorylation through EGF stimulation.
FIGURE 4.
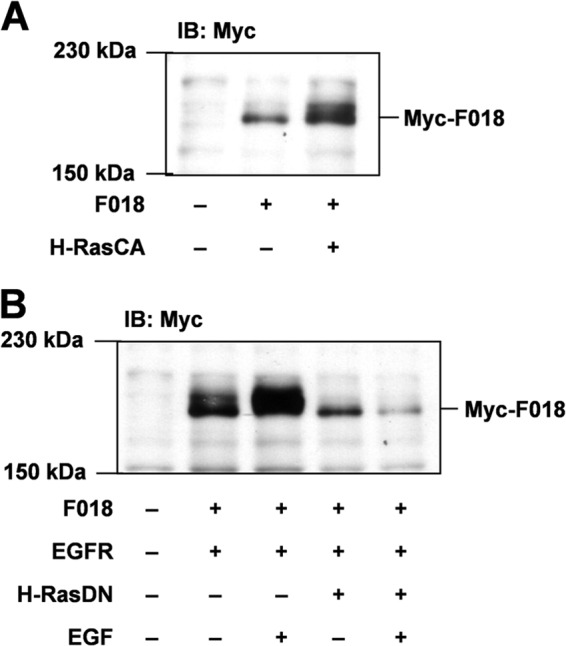
FLJ00018 is activated through H-Ras. A and B, NIH3T3 cells were co-transfected with expression vectors for FLJ00018 (F018), EGFR, and a constitutively active form of H-Ras (H-RasCA) (A) or a dominant-negative form of H-Ras (H-RasDN) (B) as indicated. Transfected cells were stimulated with 20 ng/ml EGF for 15 min. Equal amounts of the proteins were resolved by 7.5% SDS-PAGE. To detect Myc-tagged FLJ00018, immunoblotting was performed with antibodies against Myc.
FLJ00018 Is Activated through the Ras/MAPK Pathway
To determine which kinase phosphorylates FLJ00018, we examined the effect of the Ras/MAPK pathway inhibitor on FLJ00018 phosphorylation. In this experiment cells were pretreated with U-0126 (10 μm), a MEK inhibitor, for 30 min before EGF stimulation (15 min, 20 ng/ml). No mobility shift of FLJ00018 bands was detected in U-0126-treated cells, whereas an FLJ00018 mobility shift was observed in cells treated with KN-93 (10 μm), a calmodulin-dependent protein kinase II (CaMKII) inhibitor (Fig. 5A). These results suggest that FLJ00018 is phosphorylated by MEK or downstream effectors of MEK. We also measured FLJ00018-induced SRE-dependent gene transcription in cells treated with U-0126 (10 μm, 1 h) or KN-93 (10 μm, 1 h). Both inhibitors blocked FLJ00018-induced SRE-dependent gene transcription (Fig. 5, B and C). These results appeared to be in conflict with the immunoblot analysis showing that U-0126 inhibited the FLJ00018 mobility shift, whereas KN-93 did not. As a possible explanation for these disparate results, MEK or downstream effectors may regulate FLJ00018-induced SRE-dependent gene transcription through the phosphorylation of FLJ00018, whereas CaMKII may regulate FLJ00018-induced SRE-dependent gene transcription in an FLJ00018 phosphorylation-independent manner. Because the CaMKII inhibitor attenuated SRE-dependent gene transcription, we tested the possibility that the other MAPKs are also involved in FLJ00018 activation. However, neither the p38-MAPK inhibitor, SB202190 (10 μm, 30 min), nor the JNK inhibitor, SP600125 (10 μm, 30 min), affected the EGF-induced FLJ00018 mobility shift (Fig. 5D). These inhibitors failed to attenuate SRE-dependent gene transcription in FLJ00018-transfected cells (Fig. 5E). From these results, it is suggested that FLJ00018 is phosphorylated by the MAPK signaling pathway after EGF stimulation, although FLJ00018-induced SRE-dependent gene transcription may be regulated by multiple signaling pathways including the MAPK and CaMKII signaling pathways.
FIGURE 5.

FLJ00018 is activated through Ras/MAPK pathway. A and D, NIH3T3 cells were co-transfected with expression vectors for FLJ00018 (F018) and EGFR as indicated. Transfected cells were treated with 10 μm KN-93 (A), 10 μm U-0126 (A), 10 μm SB202190 (SB) (D), or 10 μm SP600125 (SP) (D) for 30 min before stimulation with 20 ng/ml EGF for 15 min. Equal amounts of proteins were resolved by 7.5% SDS-PAGE. To detect Myc-tagged FLJ00018, immunoblotting (IB) was performed with antibodies against Myc. B, C, and E, NIH3T3 cells were co-transfected with pSRE.L-luciferase, pRL-SV40 plasmid DNAs, and expression vectors for FLJ00018 and EGFR as indicated. Transfected cells were treated with KN-93 (10 μm) (B), U-0126 (10 μm) (C), SB202190 (SB) (10 μm) (E), or SP600125 (SP) (10 μm) (E) for 1 h before stimulation with 20 ng/ml EGF for 6 h. Luciferase activity was determined by dual-luciferase reporter assay. Luciferase activity obtained with mock cells was taken as 1.0, and relative activities are shown. Values are the means ± S.D. from at least three experiments.
Identification of Phosphorylation at Threonine 680 in FLJ00018 via EGFR Stimulation
To identify phosphorylation sites of FLJ00018, we co-expressed EGFR with several FLJ00018 mutants. The structures of these mutants are indicated in Fig. 6A. Fig. 6B shows that mobility shifts were observed for the FLJ00018 WT and the FLJ00018 P1 mutant but not for the P1.5, P2, P3, and ΔN mutants. These results suggest that FLJ00018 is phosphorylated between amino acid residues 615 and 970. To confirm that there is no phosphorylation site in the FLJ00018 P1.5, P2, P3, and ΔN mutants, we used Phos-tag gel and performed immunoblot analysis. The result showed a weakly shifted band in FLJ00018 P1.5 and ΔN mutants in response to EGF stimulation (15 min, 20 ng/ml), whereas FLJ00018 P2 and P3 had no shifted bands (Fig. 6C). These results suggest that the main FLJ00018 phosphorylation site exists between amino acid residues 615 and 970. However, it is likely that there is more than one phosphorylation site in FLJ00018. One of the amino acid residues in the region from 615 to 970 of FLJ00018, threonine 680, consists of the consensus sequence of extracellular signal-regulated kinase (ERK) phosphorylation (PXTP) (33). We prepared and used a FLJ00018 T680A mutant that mimics the non-phosphorylated state of FLJ00018. The mobility of FLJ00018 WT shifted in response to EGF stimulation, whereas only a slightly shifted band was observed in the FLJ00018 T680A mutant (Fig. 7A). To confirm that threonine 680 of FLJ00018 is phosphorylated, we used a Phos-tag experiment. As in the NIH3T3 cells (Fig. 7A), the mobility shift of the FLJ00018 T680A mutant was modest compared with that of the FLJ00018 WT in HEK293 cells (Fig. 7, B and C). Next, we examined whether the phosphorylation of threonine 680 affects the activity of FLJ00018. The FLJ00018 T680A mutant did not change EGF-stimulated SRE-dependent gene transcription (Fig. 7D). In addition, FLJ00018 T680A-induced SRE-dependent gene transcription was inhibited by Rac1 T17N and Cdc42 T17N (Fig. 7E). The levels of this transcription and the patterns of its inhibition by Rac1 T17N and Cdc42 T17N were not significantly different from those of the FLJ00018 WT (Fig. 3B). These results indicate that possibility that one of the phosphorylation sites of FLJ00018 is threonine 680, although threonine 680 phosphorylation did not affect any of the guanine nucleotide exchange activities of FLJ00018. Additional phosphorylation sites might be involved in EGFR stimulation-induced FLJ00018 activation. In this study, however, we did not examine the direct effects of GEF on Rho GTPase. Also, we did not detect another phosphorylation site(s) using a phospho-specific antibody. Further analysis is needed to detect the other exact phosphorylation site(s).
FIGURE 6.
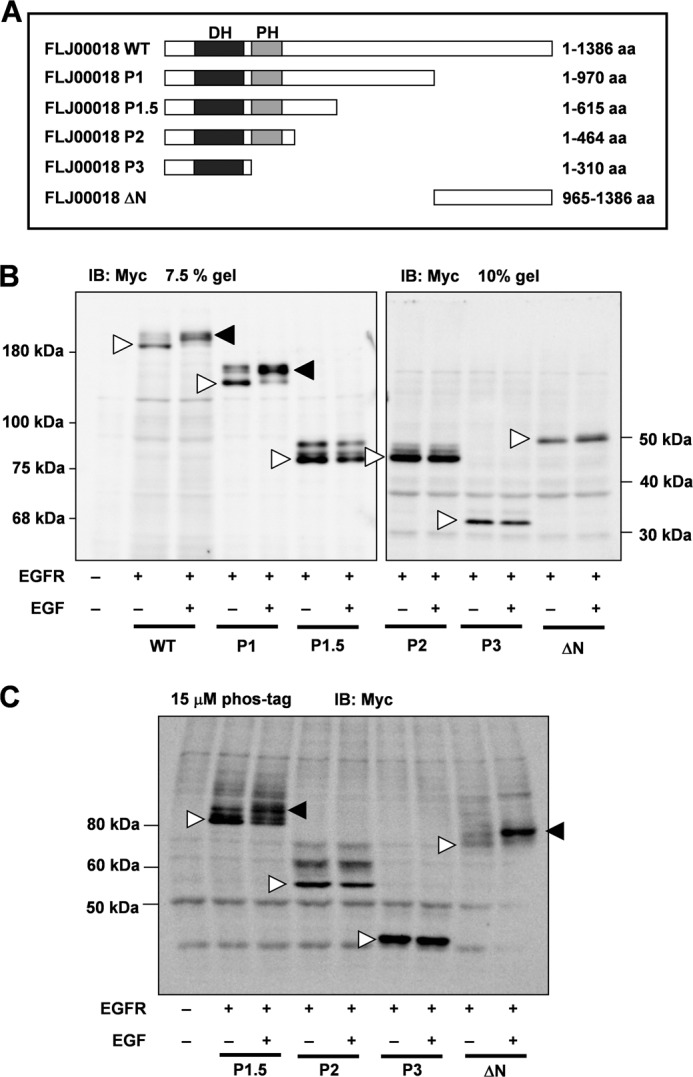
Identification of the structure involved in FLJ00018 phosphorylation by EGFR signaling. A, structure of FLJ00018 mutants. The WT, P1, P1.5, P2, P3, and ΔN constructs code for amino acid (aa) residues 1–1386, 1–970, 1–615, 1–464, 1–310, and 965–1386 of FLJ00018, respectively. B and C, NIH3T3 cells were co-transfected with expression vectors for EGFR and Myc-tagged FLJ00018 mutants as indicated. Transfected cells were stimulated with 20 ng/ml EGF for 15 min. Equal amounts of proteins were resolved by 7.5% or 10% SDS-PAGE (B) or 6% SDS-polyacrylamide gel containing 15 μm Phos-tag and 30 μm MnCl2 (C). Immunoblotting (IB) was performed with antibodies against Myc. Closed triangles, shifted bands; Open triangles, basal levels of proteins.
FIGURE 7.

FLJ00018 is phosphorylated by EGFR at amino acid residue threonine 680. A and B, NIH3T3 cells (A) or HEK293 cells (B and C) were co-transfected with expression vectors for EGFR and the Myc-tagged FLJ00018 or FLJ00018 T680A mutant as indicated. Transfected cells were stimulated with 20 ng/ml EGF for 15 min. Equal amounts of proteins were resolved by 7.5% (A and B) or 3% SDS-polyacrylamide gel containing 13 μm Phos-tag, 26 μm MnCl2, and 1.5% agarose (C). Immunoblotting (IB) was performed with antibodies against Myc). D and E, NIH3T3 cells were co-transfected with pSRE.L-luciferase, pRL-SV40 plasmid DNAs, expression vectors for FLJ00018 (F018), and EGFR, RhoA T19N, Rac T17N, and Cdc42 T17N as indicated. Transfected cells were stimulated with 20 ng/ml EGF for 6 h. Luciferase activity was determined by a dual-luciferase reporter assay. Luciferase activity obtained with mock cells was taken as 1.0, and relative activities are shown. Values are the means ± S.D. from at least three experiments. WT, FLJ00018 WT; TA, FLJ00018 T680A mutant.
Threonine 680 Phosphorylation of FLJ00018 Regulates Neurite Outgrowth via EGFR Stimulation in Neuro-2a Cells
Because some RhoGEFs have been implicated in cell morphological changes, including cell spreading and neurite outgrowth (13, 17, 34), we tested whether FLJ00018 was involved in neurite outgrowth. Neuro-2a cells were co-transfected with FLJ00018 or FLJ00018 mutants and EGFR. After stimulating the co-transfected cells with EGF, we performed immunoblot analysis and confirmed the mobility shift of FLJ00018 in the Neuro-2a cell line (data not shown). To visualize the morphology of the Neuro-2a cells, we used a fusion protein of FLJ00018 and mAG, a fluorescent protein. When the cells were stimulated with EGF, we observed neurite outgrowth and cell spreading in the cells transfected with mAG-tagged FLJ00018 and in the EGFR cells (Fig. 8A, g and h). However, no neurite outgrowth or cell spreading was observed in the FLJ00018 T680A-expressing cells (Fig. 8A, k and l). Quantitative analysis of the neurite length showed that the EGF-stimulated FLJ00018 T680A-expressing cells displayed a significant decrease in total neurite length compared with that in the EGF-stimulated FLJ00018WT-expressing cells (Fig. 8B). These results suggest that threonine phosphorylation at threonine 680 of FLJ00018 by EGFR stimulation plays a crucial role in neural outgrowth and cell spreading.
FIGURE 8.

FLJ00018 activation causes neurite outgrowth via EGFR activation. A, Neuro-2a cells were transfected with expression vectors for EGFR and the mAG-tagged FLJ00018 (F018) or FLJ00018 T680A mutant as indicated. Transfected cells were stimulated with 20 ng/ml EGF for 24 h. After stimulation, cells were fixed by 4% paraformaldehyde. Fluorescence images were acquired using a fluorescence microscope. Scale bar, 20 μm. B, 10 fluorescence images were taken from random fields and analyzed using Image J software. The total neurite length of 50 cells was measured in each sample. Total neurite lengths were analyzed using Image J software. Values are the means ± S.D. from at least three experiments. #, p < 0.05.
DISCUSSION
In our previous study FLJ00018 was activated by direct interaction with a Gβγ subunit (7), and the activation was attenuated through interaction with cytosolic actin (8). In this study we also discovered another FLJ00018 activation mechanism by threonine phosphorylation at 680 through the EGFR/Ras/MAPK pathway.
It has been reported that various GPCR agonists, including endothelin, thrombin, carbachol, and lysophosphatidic acid, stimulate EGFR through GPCR activation. Because the signaling pathway from GPCR to EGFR differs by cell type and the condition of GPCR stimulation, various different mechanisms have been proposed. The most frequently described mechanisms of EGFR transactivation involve a disintegrin and metalloproteinase (ADAM)/heparin-binding EGF pathway. It has been shown that that when the lysophosphatidic acid receptor is expressed at endogenous levels, it can activate the phospholipase Cγ1 and PI3K/AKT pathway through matrix metalloprotease- and heparin-binding EGF release-dependent EGFR activation. On the other hand, β2-AR can activate the ERK pathway in a c-Src- and EGFR-dependent manner, but it is unable to activate the phospholipase Cγ1 and PI3K/AKT pathways (35, 36). In addition to this mechanism, some other signaling pathways have been proposed to play a role in β-AR stimulation-mediated EGFR transactivation. Daaka et al. (25) have shown the role of the Gs/AC/cAMP/PKA/Gi/Gβγ/Ras/MAPK pathway in EGFR transactivation. Briefly, β2-AR can switch the signal from Gs to Gi, and the Gi signal of β2-AR can activate MAPK via Gβγ, c-Src, and Ras. However, other researchers have proposed the existence of a pathway independent of Gs/Gi switching; this pathway requires Src activation to stimulate EGFR and downstream MAPK activation (37). In addition to these pathways, a number of other pathways have been proposed, including the GRK5 and GRK6/β-arrestin/Src/EGFR pathway (26). These reports support the existence of an EGFR transactivation pathway, which activates Ras/MAPK from β-AR stimulation. However, further analysis is needed to elucidate which mechanism activates FLJ00018.
Protease-activated receptors 1 activated by thrombin have been shown to induce EGFR transactivation in vascular smooth muscle cells. After EGFR is activated by thrombin, LARG is activated, which in turn activates RhoA (38). This result indicates that RhoGEF can be activated in an EGFR transactivation-dependent manner. On the other hand, another researcher hypothesized that PI3K can directly activate the MAPK pathway via β2-AR stimulation (31). Also, it was shown that the β2-AR/Gi pathway activates platelet-derived growth factor receptor/PI3K signaling through Src activation (39). Moreover, another study has described the dependence of the interaction between β1-AR and EGFR on β-arrestin, which causes differences between the signal from direct EGF stimulation and EGFR transactivation signal (30). Indeed, when we co-transfected FLJ00018 and β1-AR with β-ARKct, β-ARKct could not completely inhibit the β1-AR stimulation-induced SRE-dependent gene transcription (Fig. 1). Also, the band shift of FLJ00018 T680A was not completely abolished (Fig. 7). These facts might suggest that the function of FLJ00018 is regulated by multiple signaling pathways after β-AR stimulation.
There are several RhoGEFs that are mainly activated through tyrosine phosphorylation by receptor-tyrosine kinase signaling (10). On the other hand, β-Pix is phosphorylated by FGF stimulation via Ras/MAPK (13). P-Rex1 is phosphorylated at serine 1169 by insulin-like growth factor receptor signaling via the kinase related to AKT phosphorylation (40). These reports indicate that RhoGEF is phosphorylated by many signaling pathways downstream of receptor-tyrosine kinase. Considering that RhoGEF phosphorylation by receptor-tyrosine kinase signaling occurs via not only the Ras/MAPK pathway but also many other pathways, including the PI3K/AKT pathway, our results that FLJ00018 T680A mutants showed reduced but still existing band shifts compared with FLJ00018 WT (Fig. 7) might indicate phosphorylation by another pathway of Ras/MAPK.
Some of the RhoGEFs, including Tiam1, p115-RhoGEF, and β-PIX, are also phosphorylated by Ca2+ signaling or PKC (15, 16, 41–43). Tiam1 is phosphorylated on threonine by PKC in lysophosphatidic acid-treated cells (15). Tiam1 is also phosphorylated by platelet-derived growth factor receptor signaling via CaMKII activation (41). It has also been reported that β-Pix was phosphorylated on Ser-516 by CaMKI in rat hippocampal neurons. The phosphorylation of β-Pix enhanced its RhoGEF activity for Rac1 and then promoted spinogenesis and synaptogenesis (43). In the present study we observed that a CaMKII inhibitor, KN-93, inhibited FLJ00018-induced SRE-dependent gene transcription but did not affect the electrophoretic mobility shift. These results suggest that CaMKII was not involved in the phosphorylation of FLJ00018 and that CaMKII participated in the FLJ00018-induced SRE-dependent gene transcription mechanism. The details of these phenomena are unknown at this time.
It has been reported that neurite outgrowth is mediated by some RhoGEFs, such as Tiam1 and β-Pix. Tiam1 is activated by TrkA and TrkB signaling. When TrkB is activated by NGF, Tiam1 interacts with Ras and itself becomes activated. This in turn results in neurite outgrowth through Rac1 activation (17). Although Tiam1 receives intracellular signals from TrkA/B, β-Pix is phosphorylated through the Ras/ERK/PAK2 pathway upon FGF stimulation. This phosphorylation also induces neurite outgrowth in PC12 cell lines (13). Furthermore, Burridge and co-workers (44) have shown that RhoG is activated independently of Rac1 by EGF stimulation. Vav family RhoGEFs and PLEKHG6, a RhoG-specific RhoGEF, have been shown to be involved in RhoG activation. On the other hand, another paper has shown that a RhoGEF, Trio, activates neurite outgrowth in a RhoG-dependent manner (45). In line with these findings, our results suggest that FLJ00018 phosphorylated by Ras/MAPK signaling participated in morphological changes in Neuro-2a cells. From these results, FLJ00018 might also be physiologically involved in neurite outgrowth in neural cells. However, FLJ00018-induced SRE-dependent gene transcription was not related to threonine 680 phosphorylation of FLJ00018 in our study (Fig. 7D). A previous report suggested that G-protein signaling modulator-3 was spatiotemporally regulated by interaction with the 14-3-3 protein through its serine phosphorylation site (46). It is thus thought that the phosphorylation of FLJ00018 may be related to the spatiotemporal regulation of FLJ00018. These points will need to be studied in detail.
In conclusion, our experiments revealed an additional activation mechanism of FLJ00018, distinct from the Gβγ binding activation mechanism, by the phosphorylation at threonine 680 of through EGFR stimulation-dependent Ras/MAPK pathways. This threonine phosphorylation induces cell morphological change in Neuro-2a cells. These insights suggest new mechanisms of neurite outgrowth (Fig. 9). In the future it will be necessary to examine whether the signaling pathway operates under more than one physiological condition. Further analysis may allow understanding of the development of neurites into nerve systems and may uncover promising pharmacological targets to reduce aberrant neural development.
FIGURE 9.
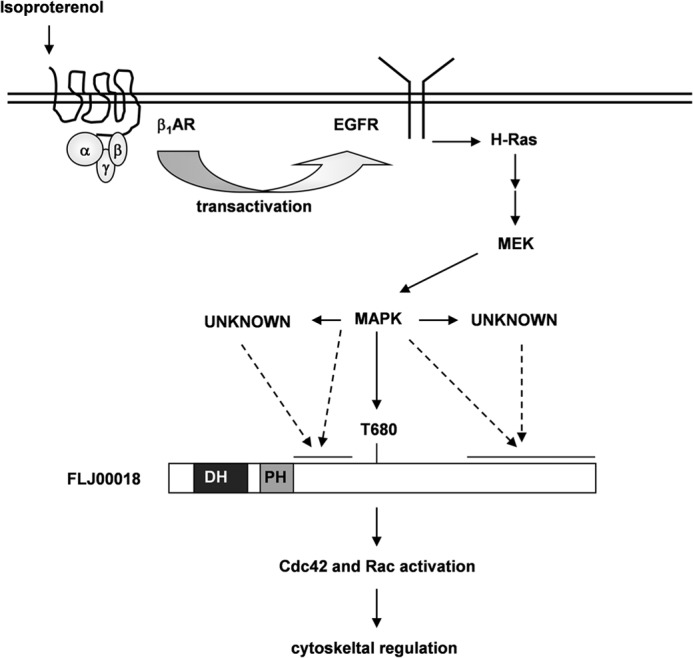
Proposed mechanisms of FLJ00018 activation through EGFR transactivation. Stimulated β1-AR induces EGFR transactivation. Through the MAPK cascade, activated EGFR regulates and phosphorylates FLJ00018. FLJ00018 may be phosphorylated by MEK or its downstream effectors, and the phosphorylated FLJ00018 may in turn activate Rho family members Cdc42 and Rac. In Neuro-2a cells, phosphorylated FLJ00018 regulates neurite outgrowth.
Acknowledgments
We thank Professor Rudolph L. Juliano and Dr. John Bauman (University of North Carolina at Chapel Hill) for critical reading of the manuscript. We also thank Hisashi Kato for technical assistance. The DNA sequence analysis was supported by the Division of Genomic Research in the Life Science Research Center at Gifu University.
This work was supported by a grant-in-aid for Scientific Research (C) from Japan Society for the Promotion of Science.
- GPCR
- heterotrimeric G-protein-coupled receptor
- AC
- adenylate cyclase
- β-ARKct
- C-terminal of β-adrenergic receptor kinase
- β1-AR
- β1-adrenergic receptor
- CaMK
- calmodulin-dependent protein kinase
- DH
- Dbl homology
- EGFR
- EGF receptor
- LARG
- leukemia-associated RhoGEF
- mAG
- monomeric Azami Green
- PDZ
- PSD-95/Dlg/ZO-1
- PH
- Pleckstrin homology
- RGS
- regulator of G protein signaling
- RhoGEF
- Rho guanine nucleotide exchange factor
- SRE
- serum response element.
REFERENCES
- 1. Neves S. R., Ram P. T., Iyengar R. (2002) G protein pathways. Science 296, 1636–1639 [DOI] [PubMed] [Google Scholar]
- 2. Hall A. (1998) Rho GTPases and the actin cytoskeleton. Science 279, 509–514 [DOI] [PubMed] [Google Scholar]
- 3. Rossman K. L., Der C. J., Sondek J. (2005) GEF means go. Turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 6, 167–180 [DOI] [PubMed] [Google Scholar]
- 4. Côté J. F., Vuori K. (2007) GEF what? Dock180 and related proteins help Rac to polarize cells in new ways. Trends Cell Biol. 17, 383–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fukuhara S., Chikumi H., Gutkind J. S. (2000) Leukemia-associated Rho guanine nucleotide exchange factor (LARG) links heterotrimeric G proteins of the G12 family to Rho. FEBS Lett. 485, 183–188 [DOI] [PubMed] [Google Scholar]
- 6. Fukuhara S., Murga C., Zohar M., Igishi T., Gutkind J. S. (1999) A novel PDZ domain containing guanine nucleotide exchange factor links heterotrimeric G proteins to Rho. J. Biol. Chem. 274, 5868–5879 [DOI] [PubMed] [Google Scholar]
- 7. Ueda H., Nagae R., Kozawa M., Morishita R., Kimura S., Nagase T., Ohara O., Yoshida S., Asano T. (2008) Heterotrimeric G protein βγ subunits stimulate FLJ00018, a guanine nucleotide exchange factor for Rac1 and Cdc42. J. Biol. Chem. 283, 1946–1953 [DOI] [PubMed] [Google Scholar]
- 8. Sato K., Handa H., Kimura M., Okano Y., Nagaoka H., Nagase T., Sugiyama T., Kitade Y., Ueda H. (2013) Identification of a Rho family specific guanine nucleotide exchange factor, FLJ00018, as a novel actin-binding protein. Cell. Signal. 25, 41–49 [DOI] [PubMed] [Google Scholar]
- 9. Chikumi H., Fukuhara S., Gutkind J. S. (2002) Regulation of G protein-linked guanine nucleotide exchange factors for Rho, PDZ-RhoGEF, and LARG by tyrosine phosphorylation. Evidence of a role for focal adhesion kinase. J. Biol. Chem. 277, 12463–12473 [DOI] [PubMed] [Google Scholar]
- 10. Schiller M. R. (2006) Coupling receptor tyrosine kinases to Rho GTPases-GEFs, what's the link. Cell. Signal. 18, 1834–1843 [DOI] [PubMed] [Google Scholar]
- 11. Itoh R. E., Kiyokawa E., Aoki K., Nishioka T., Akiyama T., Matsuda M. (2008) Phosphorylation and activation of the Rac1 and Cdc42 GEF Asef in A431 cells stimulated by EGF. J. Cell Sci. 121, 2635–2642 [DOI] [PubMed] [Google Scholar]
- 12. Feng Q., Baird D., Peng X., Wang J., Ly T., Guan J. L., Cerione R. A. (2006) Cool-1 functions as an essential regulatory node for EGF receptor- and Src-mediated cell growth. Nat. Cell Biol. 8, 945–956 [DOI] [PubMed] [Google Scholar]
- 13. Shin E. Y., Shin K. S., Lee C. S., Woo K. N., Quan S. H., Soung N. K., Kim Y. G., Cha C. I., Kim S. R., Park D., Bokoch G. M., Kim E. G. (2002) Phosphorylation of p85 β PIX, a Rac/Cdc42-specific guanine nucleotide exchange factor, via the Ras/ERK/PAK2 pathway is required for basic fibroblast growth factor-induced neurite outgrowth. J. Biol. Chem. 277, 44417–44430 [DOI] [PubMed] [Google Scholar]
- 14. Pandey A., Podtelejnikov A. V., Blagoev B., Bustelo X. R., Mann M., Lodish H. F. (2000) Analysis of receptor signaling pathways by mass spectrometry. Identification of vav-2 as a substrate of the epidermal and platelet-derived growth factor receptors. Proc. Natl. Acad. Sci. U.S.A. 97, 179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fleming I. N., Elliott C. M., Collard J. G., Exton J. H. (1997) Lysophosphatidic acid induces threonine phosphorylation of Tiam1 in Swiss 3T3 fibroblasts via activation of protein kinase C. J. Biol. Chem. 272, 33105–33110 [DOI] [PubMed] [Google Scholar]
- 16. Buchanan F. G., Elliot C. M., Gibbs M., Exton J. H. (2000) Translocation of the Rac1 guanine nucleotide exchange factor Tiam1 induced by platelet-derived growth factor and lysophosphatidic acid. J. Biol. Chem. 275, 9742–9748 [DOI] [PubMed] [Google Scholar]
- 17. Shirazi Fard S., Kele J., Vilar M., Paratcha G., Ledda F. (2010) Tiam1 as a signaling mediator of nerve growth factor-dependent neurite outgrowth. PLoS ONE 5, e9647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sosa M. S., Lopez-Haber C., Yang C., Wang H., Lemmon M. A., Busillo J. M., Luo J., Benovic J. L., Klein-Szanto A., Yagi H., Gutkind J. S., Parsons R. E., Kazanietz M. G. (2010) Identification of the Rac-GEF P-Rex1 as an essential mediator of ErbB signaling in breast cancer. Mol. Cell 40, 877–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nakajima D., Okazaki N., Yamakawa H., Kikuno R., Ohara O., Nagase T. (2002) Construction of expression-ready cDNA clones for KIAA genes. Manual curation of 330 KIAA cDNA clones. DNA Res 9, 99–106 [DOI] [PubMed] [Google Scholar]
- 20. Karasawa S., Araki T., Yamamoto-Hino M., Miyawaki A. (2003) A green-emitting fluorescent protein from Galaxeidae coral and its monomeric version for use in fluorescent labeling. J. Biol. Chem. 278, 34167–34171 [DOI] [PubMed] [Google Scholar]
- 21. Kinoshita E., Kinoshita-Kikuta E., Takiyama K., Koike T. (2006) Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteomics 5, 749–757 [DOI] [PubMed] [Google Scholar]
- 22. Kinoshita E., Kinoshita-Kikuta E., Koike T. (2009) Separation and detection of large phosphoproteins using Phos-tag SDS-PAGE. Nat. Protoc. 4, 1513–1521 [DOI] [PubMed] [Google Scholar]
- 23. Kinoshita E., Kinoshita-Kikuta E., Ujihara H., Koike T. (2009) Mobility shift detection of phosphorylation on large proteins using a Phos-tag SDS-PAGE gel strengthened with agarose. Proteomics 9, 4098–4101 [DOI] [PubMed] [Google Scholar]
- 24. Pak Y., Pham N., Rotin D. (2002) Direct binding of the β-1 adrenergic receptor to the cyclic AMP-dependent guanine nucleotide exchange factor CNrasGEF leads to Ras activation. Mol. Cell. Biol. 22, 7942–7952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Daaka Y., Luttrell L. M., Lefkowitz R. J. (1997) Switching of the coupling of the β2-adrenergic receptor to different G proteins by protein kinase A. Nature 390, 88–91 [DOI] [PubMed] [Google Scholar]
- 26. Noma T., Lemaire A., Naga Prasad S. V., Barki-Harrington L., Tilley D. G., Chen J., Le Corvoisier P., Violin J. D., Wei H., Lefkowitz R. J., Rockman H. A. (2007) β-Arrestin-mediated β1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J. Clin. Invest. 117, 2445–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Daub H., Weiss F. U., Wallasch C., Ullrich A. (1996) Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 379, 557–560 [DOI] [PubMed] [Google Scholar]
- 28. Daub H., Wallasch C., Lankenau A., Herrlich A., Ullrich A. (1997) Signal characteristics of G protein-transactivated EGF receptor. EMBO J. 16, 7032–7044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maudsley S., Pierce K. L., Zamah A. M., Miller W. E., Ahn S., Daaka Y., Lefkowitz R. J., Luttrell L. M. (2000) The β2-adrenergic receptor mediates extracellular signal-regulated kinase activation via assembly of a multi-receptor complex with the epidermal growth factor receptor. J. Biol. Chem. 275, 9572–9580 [DOI] [PubMed] [Google Scholar]
- 30. Tilley D. G., Kim I. M., Patel P. A., Violin J. D., Rockman H. A. (2009) β-Arrestin mediates β1-adrenergic receptor-epidermal growth factor receptor interaction and downstream signaling. J. Biol. Chem. 284, 20375–20386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lefkowitz R. J., Cotecchia S., Samama P., Costa T. (1993) Constitutive activity of receptors coupled to guanine nucleotide regulatory proteins. Trends Pharmacol. Sci. 14, 303–307 [DOI] [PubMed] [Google Scholar]
- 32. Kim J., Eckhart A. D., Eguchi S., Koch W. J. (2002) β-adrenergic receptor-mediated DNA synthesis in cardiac fibroblasts is dependent on transactivation of the epidermal growth factor receptor and subsequent activation of extracellular signal-regulated kinases. J. Biol. Chem. 277, 32116–32123 [DOI] [PubMed] [Google Scholar]
- 33. Sheridan D. L., Kong Y., Parker S. A., Dalby K. N., Turk B. E. (2008) Substrate discrimination among mitogen-activated protein kinases through distinct docking sequence motifs. J. Biol. Chem. 283, 19511–19520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang Y., Sawada T., Jing X., Yokote H., Yan X., Sakaguchi K. (2007) Regulation of ephexin1, a guanine nucleotide exchange factor of Rho family GTPases, by fibroblast growth factor receptor-mediated tyrosine phosphorylation. J. Biol. Chem. 282, 31103–31112 [DOI] [PubMed] [Google Scholar]
- 35. Drube S., Stirnweiss J., Valkova C., Liebmann C. (2006) Ligand-independent and EGF receptor-supported transactivation. Lessons from β2-adrenergic receptor signalling. Cell Signal 18, 1633–1646 [DOI] [PubMed] [Google Scholar]
- 36. Liebmann C. (2011) EGF receptor activation by GPCRs. An universal pathway reveals different versions. Mol. Cell. Endocrinol. 331, 222–231 [DOI] [PubMed] [Google Scholar]
- 37. Friedman J., Babu B., Clark R. B. (2002) β2-adrenergic receptor lacking the cyclic AMP-dependent protein kinase consensus sites fully activates extracellular signal-regulated kinase 1/2 in human embryonic kidney 293 cells. Lack of evidence for Gs/Gi switching. Mol. Pharmacol. 62, 1094–1102 [DOI] [PubMed] [Google Scholar]
- 38. Wang D., Paria B. C., Zhang Q., Karpurapu M., Li Q., Gerthoffer W. T., Nakaoka Y., Rao G. N. (2009) A role for Gab1/SHP2 in thrombin activation of PAK1. Gene transfer of kinase-dead PAK1 inhibits injury-induced restenosis. Circ. Res. 104, 1066–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yano N., Ianus V., Zhao T. C., Tseng A., Padbury J. F., Tseng Y. T. (2007) A novel signaling pathway for β-adrenergic receptor-mediated activation of phosphoinositide 3-kinase in H9c2 cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 293, H385–H393 [DOI] [PubMed] [Google Scholar]
- 40. Montero J. C., Seoane S., Pandiella A. (2013) Phosphorylation of P-Rex1 at serine 1169 participates in IGF-1R signaling in breast cancer cells. Cell. Signal. 25, 2281–2289 [DOI] [PubMed] [Google Scholar]
- 41. Fleming I. N., Elliott C. M., Exton J. H. (1998) Phospholipase Cγ, protein kinase C and Ca2+/calmodulin-dependent protein kinase II are involved in platelet-derived growth factor-induced phosphorylation of Tiam1. FEBS Lett. 429, 229–233 [DOI] [PubMed] [Google Scholar]
- 42. Holinstat M., Mehta D., Kozasa T., Minshall R. D., Malik A. B. (2003) Protein kinase Cα-induced p115RhoGEF phosphorylation signals endothelial cytoskeletal rearrangement. J. Biol. Chem. 278, 28793–28798 [DOI] [PubMed] [Google Scholar]
- 43. Saneyoshi T., Wayman G., Fortin D., Davare M., Hoshi N., Nozaki N., Natsume T., Soderling T. R. (2008) Activity-dependent synaptogenesis. Regulation by a CaM-kinase kinase/CaM-kinase I/βPIX signaling complex. Neuron 57, 94–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Samson T., Welch C., Monaghan-Benson E., Hahn K. M., Burridge K. (2010) Endogenous RhoG is rapidly activated after epidermal growth factor stimulation through multiple guanine-nucleotide exchange factors. Mol. Biol. Cell 21, 1629–1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Estrach S., Schmidt S., Diriong S., Penna A., Blangy A., Fort P., Debant A. (2002) The human Rho-GEF trio and its target GTPase RhoG are involved in the NGF pathway, leading to neurite outgrowth. Curr. Biol. 12, 307–312 [DOI] [PubMed] [Google Scholar]
- 46. Giguère P. M., Laroche G., Oestreich E. A., Duncan J. A., Siderovski D. P. (2012) Regulation of the subcellular localization of the G-protein subunit regulator GPSM3 through direct association with 14-3-3 protein. J. Biol. Chem. 287, 31270–31279 [DOI] [PMC free article] [PubMed] [Google Scholar]