

Background: PHF6 gene is mutated in BFLS and adult acute myeloid and T-cell acute lymphoblastic leukemias.
Results: Crystal structure of the second extended PHD domain of PHF6 was solved.
Conclusion: PHF6-ePHD2 is a novel structural module and binds dsDNA.
Significance: PHF6 may function as a transcriptional repressor using its ePHD domains binding to DNA and recruiting NuRD complex through its NoLS region to regulate gene transcription.
Keywords: Genetic Diseases, Protein-DNA Interaction, Protein Folding, Transcription Regulation, Tumor Suppressor Gene, BFLS, NuRD, PHF6, RBBP4, Extended PHD
Abstract
The plant homeodomain finger 6 (PHF6) was originally identified as the gene mutated in the X-linked mental retardation disorder Börjeson-Forssman-Lehmann syndrome. Mutations in the PHF6 gene have also been associated with T-cell acute lymphoblastic leukemia and acute myeloid leukemia. Approximately half of the disease-associated mutations are distributed in the second conserved extended plant homeodomain (ePHD2) of PHF6, indicating the functional importance of the ePHD2 domain. Here, we report the high resolution crystal structure of the ePHD2 domain of PHF6, which contains an N-terminal pre-PHD (C2HC zinc finger), a long linker, and an atypical PHD finger. PHF6-ePHD2 appears to fold as a novel integrated structural module. Structural analysis of PHF6-ePHD2 reveals pathological implication of PHF6 gene mutations in Börjeson-Forssman-Lehmann syndrome, T-cell acute lymphoblastic leukemia, and acute myeloid leukemia. The binding experiments show that PHF6-ePHD2 can bind dsDNA but not histones. We also demonstrate PHF6 protein directly interacts with the nucleosome remodeling and deacetylation complex component RBBP4. Via this interaction, PHF6 exerts its transcriptional repression activity. Taken together, these data support the hypothesis that PHF6 may function as a transcriptional repressor using its ePHD domains binding to the promoter region of its repressed gene, and this process was regulated by the nucleosome remodeling and deacetylation complex that was recruited to the genomic target site by NoLS region of PHF6.
Introduction
The plant homeodomain (PHD)3 finger 6 (PHF6) gene, located on Xq26-q27, was first discovered mutated in patients with the Börjeson-Forssman-Lehmann syndrome (BFLS, MIM 301900) (1, 2). BFLS is an X-chromosome linked mental retardation disorder characterized by moderate to severe mental retardation, epilepsy, short stature, hypogonadism, hypometabolism, marked gynecomastia, truncal obesity, tapered fingers, narrow palpebral fissure, and large ears (3–8). Various mutations affecting the coding region of PHF6 gene or the splicing of the transcript, including missense, truncation, and deletion mutations, have been linked to BFLS (1, 6, 9–13).
The PHF6 gene encodes a 365-amino acid protein, which contains four nuclear localization signals and two imperfect PHD zinc finger domains with a proposed role in gene transcription regulation (1, 14). PHF6 is highly conserved among vertebrate species, including Mus musculus and Xenopus laevis, but it has no obvious orthologs in nonvertebrates, suggesting the conservative functions of PHF6 in the cell (1). Northern blot and in situ hybridization analyses have revealed that PHF6 is highly expressed during embryonic, fetal, and postnatal stages of brain development and predominantly present in the developing embryonic central nervous system (1, 15). Therefore, it appears that PHF6 plays an important role in human brain development. In addition, RT-PCR analysis demonstrates that PHF6 gene is ubiquitously expressed in human tissues and most highly expressed in thymus, ovary, and thyroid (16). ChIP-on-chip analysis identifies PHF6 as a direct target gene of NOTCH1 that is frequently mutated in T-cell acute lymphoblastic leukemia (T-ALL) (17). Indeed, a reported BFLS patient has developed T-ALL disease (18). Meanwhile, somatic mutations and deletions of the PHF6 gene have been found in patients with T-ALL and acute myeloid leukemia (16, 19–22). Moreover, a small set of microRNAs has been found to produce cooperative suppression effects on several tumor suppressor genes implicated in T-ALL, including the PHF6 gene (23). It has been suggested that PHF6 may function as an X-linked tumor suppressor gene and play a role in the pathogenesis of hematologic tumors.
Concerning the molecular mechanism, recently PHF6 has been identified as a novel protein partner interacting with the nucleosome remodeling and deacetylation (NuRD) complex in nucleus (24). PHF6 regulates cell cycle progression by suppressing ribosomal RNA synthesis in the nucleolus (25). PHF6 physically associates with the PAF1 transcription elongation complex and plays an essential role in neuronal migration in the brain (26). Nevertheless, despite that the PHF6 is implicated in chromatin regulation, neural development, and tumor suppression, very little is known regarding how PHF6 plays its functions and how PHF6 mutations contribute to disease pathogenesis. To gain more insights into the structure-function relationship of the PHF6 and its role in the pathogenesis of BFLS and leukemia diseases, we determined the high resolution crystal structure of the second extended PHD domain (C2HC zinc finger-PHD, refer to ePHD2) from the human PHF6 protein and found that C2HC zinc finger-PHD folds as a novel integrated structural module. PHF6-ePHD2 domain can bind to dsDNA. Structural analysis of PHF6-ePHD2 reveals pathological implication of disease-associated mutations of PHF6. In addition, we demonstrated that PHF6 can directly interact with the RBBP4, a component of the NuRD complex, through its NoLS region that is important for the transcriptional suppression activity of PHF6 protein.
EXPERIMENTAL PROCEDURES
Protein Cloning, Expression, and Purification
DNA fragments encoding the full-length (365 amino acids) and the second extended PHD domain (ePHD2, residues 208–333) of human PHF6 were amplified by PCR from the human brain cDNA library and cloned into the pGEX-4T-1 (GE Healthcare) expression vector between the NdeI and XhoI sites. The resulting plasmid was transformed into Escherichia coli Rosetta (DE3) cells for protein expression and induced with 0.25 mm isopropyl 1-thio-β-d-galactopyranoside at 16 °C overnight supplemented with 0.1 mm ZnSO4 in the LB medium. 15N- or 15N/13C-labeled proteins were produced in SV40 medium supplemented with 15NH4Cl or 15NH4Cl and [13C6]glucose. The fusion protein was purified on glutathione-Sepharose beads (GE Healthcare). The proteins were eluted with buffer containing 20 mm reduced l-glutathione. The eluted fraction was concentrated to 5 ml using Amicon Ultra-15 centrifugal filter units (10-kDa cutoff, Millipore) and further purified by gel filtration chromatography using a 16/26 Superdex 200 column (GE Healthcare) pre-equilibrated in 25 mm phosphate sodium buffer, pH 6.5, containing 400 mm NaCl. After cleavage of the GST tag by thrombin (GE Healthcare) at 16 °C, an extra cation-exchange purification step was used.
Full-length RBBP4 (residues 1–425; UniProt accession number Q09028) was cloned into a pFastBac vector encoding an N-terminal His6 tag and a tobacco etch virus protease cleavage site. Recombinant baculovirus was generated in Tn5 cells using the Bac-to-Bac (Invitrogen) expression methodology according to the manufacturer's instructions.
Crystallization Conditions, Data Collection, and Structure Determination
Crystals were grown at 16 °C by vapor diffusion in sitting drops by mixing equal volumes of the well solution containing 200 mm ammonium acetate, 100 mm tri-sodium citrate dehydrate, 30% (w/v) polyethylene glycol 4000, pH 5.6, and protein solution at 10 mg/ml.
The diffraction data were collected on the beam line 17U at Shanghai Synchrotron Radiation Facility. The initial diffraction data were indexed and integrated with iMosflm (27) and scaled with SCALA (28) from CCP4 program suite (29). All of the three zinc atom positions were determined by SHELX C/D (30), and the initial phases of calculations and density modification were performed with SHELX E (30). An initial model was automatically built by the program ARP/wARP (31). The model was further built and refined using Coot (32) and Refmac (33), respectively.
NMR Spectroscopy, 15N Relaxation Experiments, and Residual Dipolar Couplings (RDCs)
All NMR experiments were collected at 298 K on a Bruker DMX 600-MHz spectrometer equipped with a cryoprobe. Sequence-specific backbone assignments were carried out by analysis of three-dimensional HNCO, HN(CA)CO, CBCANH, and CBCA(CO)NH experiments. 1H and 13C side chain assignments were obtained from three-dimensional HBHA(CBCACO)NH, C(CO)NH-TOCSY, H(CCO)NH-TOCSY, HCCH-COSY, and HCCH-TOCSY. NMR distance information was obtained form 15N- and 13C-edited NOESY-HSQC spectra. One-dimensional spectra of PHF6-ePHD2 domain mutants are collected at 298 K on a Varian 500-MHz spectrometer. All NMR data were processed using the NMRPipe (34) and analyzed by Sparky (T. D. Goddard and D. G. Kneller, University of California at San Francisco).
15N relaxation measurements were performed on a Bruker DMX-600 equipped with a cryoprobe using a 15N-labeled PHF6-ePHD2 sample with a concentration of 0.6 mm at 298 K. The 1H-15N heteronuclear NOE experiments were recorded in an interleaved fashion, alternately with and without proton presaturation in the recovery delay. A 2-s relaxation delay following a 3-s 1H presaturation delay were used in the NOE experiment, and a 5-s relaxation delay was used in the reference experiment. 15N T1 and T2 relaxation times were derived from nine spectra with different values for the relaxation delay (11, 61, 142, and 242 (run twice) and 362, 523, 753, and 1147 ms) and seven relaxation delays (17.6, 35.2, and 52.8 (run twice) and 70.4, 105.6, and 140.8 ms), respectively. T1 and T2 values were extracted and fitted using a curve-fitting subroutine included in the program Sparky.
Polyacrylamide gels were prepared and squeezed into an open-ended NMR tube (35). Apparent JNH constants of 15N-labeled PHF6(208-333) were measured at Agilent 700-MHz spectrometer using the in-phase and anti-phase scheme (36). RDC values were obtained by subtracting the reference value of the protein in isotropic solution. Two duplicate datasets were collected at a temperature of 25 °C, and PALES was used for data analysis (37). Dipolar couplings from residues having lower 1H-15N NOEs (<0.65) were excluded.
GST Pulldown Experiments
GST fusion PHF6 ePHD2 was bound to glutathione-Sepharose beads (GE Healthcare), and then the beads were incubated with calf thymus histones (Worthington) in buffer containing 25 mm BisTris, pH 6.5, 1 m NaCl, 1% Nonidet P-40 for 1 h at 4 °C. After washing six times with the buffer described above, results were analyzed on Coomassie-stained SDS-polyacrylamide gels. GST-tagged PHF6(145–207) and PHF6(152–171) were bound to beads, and the beads were subsequently mixed with purified RBBP4 in 1× PBS buffer for 1 h at 4 °C. The glutathione-Sepharose was sedimented by centrifugation and washed five times with 1× PBS buffer. The bound materials were analyzed on SDS-PAGE and Coomassie Blue staining.
Transient Transfections and Reporter Assays
HEK293T cells were transfected using Lipofectamine Plus (Invitrogen) with the following plasmids: 1) GAL-TK-Luc (0.3 μg); 2) pRK5 (GAL4DBD) or pRK5-PHF6 wide-type or deletion mutants (GAL4DBD-PHF6 fusion) (0.4 μg); 3) Renilla reporter vector pRL-Luciferase (25 ng), as normalization control for transfection efficiency. The cells were harvested 24 h after transfection, and luciferase activity was measured using the luciferase assay system (Promega). The GAL-TK-Luc, pRK5 (GAL4DBD), and Renilla reporter vector are a gift from Professor Yide Mei.
Site-directed and Deletion Mutagenesis
Site-directed point mutations in the PHF6-ePHD2 domain and deletion mutations in full-length PHF6 were performed using TaKaRa MutanBEST kit (TaKaRa) according to the manufacturer's instructions. The point mutagenic primers for the described mutations contained the following codons: C215F, TGC to TTC; H229R, CAT to CGT; K234E, AAG to GAG; R257G, AGA to GGA; G275R, GGA to AGA; C280Y, TGT to TAT; C283R, TGC to CGC; A288T, GCT to ACT; I294R, ATA to AGA; T300A, ACT to GCT; H302Y, CAT to TAT; A311P, GCT to CCT; I314V, ATT to GTT; Y325H, TAC to CAC; H329R, CAT to CGT; R209E/K211E, AGG to GAG, AAA to GAA; K234E/K235E, AAGAAG to GAGGAG; K273E/R274E/K276E/R277E, AAA to GAA, CGA to GAA, AAA to GAA, AGA to GAA; and K276E/R277E/M278A/K279E, AAAAGAATGAAA to GAAGAAGCGGAA. All constructs created by PCR were confirmed by DNA sequence analysis of both strands.
DNA Titration
The 14-bp dsDNA was dissolved in the NMR buffer (100 mm NaCl, 20 mm Na2HPO4, pH 6.5) and titrated into 0.1 mm 15N-PHF6(208-333) or 0.2 mm 15N-PHF6(208-365). The 1H-15N HSQC spectra were recorded and analyzed. The chemical shift perturbation was calculated using Equation 1,
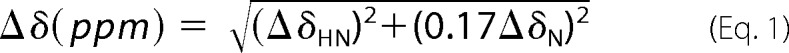 |
ΔδHN and ΔδN are the chemical shift variations in the proton and nitrogen dimensions, respectively.
Fluorescence Polarization Assays
Fluorescence polarization assays (FPA) were performed in buffer (20 mm Tris, pH 7.4, 100 mm NaCl) at 20 °C using a SpectraMax M5 Microplate Reader system (Molecular Devices). The wavelengths of fluorescence excitation and emission were 485 and 522 nm, respectively. Each 96-well contained 100 nm fluorescently labeled (5′-FAM) DNA probe and different amounts of PHF6(208-333), PHF6(208-365), or PHF6(208-333) mutants with a final volume of 200 μl. The fluorescence polarization data were analyzed essentially as described previously (38). The sequences of 5′-FAM-labeled DNA probes are described as follows: 14 bp, 5′-FAM-CCTTACAGCAAAGC-3′ and 5′-FAM-GCTTTGCTGTAAGG-3′; 18 bp, 5′-FAM-CCTTACAGCAAAGCTACT-3′ and 5′-FAM-AGTAGCTTTGCTGTAAGG-3′; 20 bp, 5′-FAM-CCTTACAGCAAAGCTACTTT-3′ and 5′-FAM-AAAGTAGCTTTGCTGTAAGG-3′; AT-rich, 5′-FAM-ATAATCGATATTTATTATGCTATTATACGTTAT-3′ and 5′-FAM-ATAACGTATAATAGCATAATAAATATCGATTAT-3′; GC-rich, 5′-FAM-ATCGCCCGCGCACGCCGCTCCGCCGCAGCGCGT-3′ and 5′-FAM-ACGCGCTGCGGCGGAGCGGCGTGCGCGGGCGAT-3′.
Electrophoretic Mobility Shift Assay
33-bp GC-rich dsDNA was incubated with increasing amounts of GST-PHF6(208-333) or GST-PHF6(208-365) for 30 min on ice in the buffer containing 20 mm Tris, pH 7.4, 100 mm NaCl. The nucleoprotein complexes were analyzed using 5% PAGE and visualized with ethidium bromide.
RESULTS
PHF6 Contains Two Highly Conserved Extended PHD Domains
PHD finger is a C4HC3 zinc finger-like motif, first noted in two closely related plant homeodomain proteins HAT3.1 and HOX1A (39). This evolutionarily conserved cysteine-rich motif was also observed for various proteins involved in epigenetics and chromatin-mediated transcriptional regulation in human, Drosophila, Caenorhabditis elegans, and yeast, etc. The PHD finger binds two zinc ions using the so-called “cross-brace” motif to form a stable unit (40). It can function as a protein-protein interaction domain and in many cases as a reader of histone marks to recognize methylated or unmethylated lysines (41, 42). It is reported that the PHF6 protein contains two imperfect PHD zinc finger domains and may play a role in chromatin transcription regulation.
Unexpectedly, in our experiments we found that the second PHD finger region of PHF6 alone was unstable in solution. So we extended the N terminus of the second PHD finger and got a stable construct (residues 208–333). It is 70 amino acids longer than the predicted PHD finger region (residues 277–329) in protein knowledge database (UniProtKB) and we refer to is as the extended PHD domain (ePHD2) (Figs. 1 and 2A).
FIGURE 1.

Sequence alignment of orthologs of PHF6 protein from diverse vertebrate species. Conserved amino acid residues are shaded with a red background. The ePHD domains are indicated in a box.
FIGURE 2.

PHF6 extended PHD2 domain is composed of a pre-PHD zinc finger and a noncanonical PHD finger. A, schematic representation of the functional domains and locations of mutations in the human PHF6 protein. Four nuclear localization signals are shown in yellow. Filled triangles represent PHF6 mutations found in BFLS patients. PHF6 mutations identified in acute myeloid leukemia patients are depicted as open circles. Mutations in T-ALL samples are shown above the box. B, secondary structural elements in the PHF6-ePHD2 domain. The N-terminal pre-PHD zinc finger is indicated by a green bar, the PHD finger by a sky blue bar, and the middle region by a magenta bar. The conserved zinc-chelating residues are marked as orange vertical bars. Residues that are involved in zinc ions coordination are connected by solid lines (the first, second, and third tetrads are connected by gray, purple, and orange lines, respectively). C, schematic representation of the PHF6-ePHD2 domain highlights the secondary structural elements, color scheme as for B, and spheres are zinc ions. D, schematic showing the zinc-binding topology and secondary structural elements of the ePHD2 domain, color scheme as for B. β-Strands are labeled β1–β6 and helices α1–α5. Cys and His residues are shown as circles and squares. E, surface view of the ePHD2 domain structure shown in C (right), color scheme as for B.
To further characterize structural domains in PHF6, we performed sequence alignment of many PHF6 proteins from different vertebrate species, which shows conserved extended PHD domains at the N and C termini, respectively, that contain a C2HC zinc finger and the previously reported imperfect PHD finger (Fig. 1). That means the ePHD2 entity may have important function.
Overall Structure of the Second Extended PHD Domain of PHF6
To understand the molecular structure of the ePHD domain, we solved the high resolution crystal structure of the ePHD2 domain of PHF6 (Table 1). In the final model, PHF6-ePHD2 domain includes two molecules in an asymmetric unit with 124 amino acid residues for molecule A (residues 208–331) and 118 amino acid residues for molecule B (residues 209–255 and 260–330). The two molecules display a root-mean-square deviation of 0.296 Å, reflecting the slight differences in the flexible loops of the protein.
TABLE 1.
Data collection and refinement statistics
| Data collection | |
| Space group | P321 |
| Cell dimensions | |
| a, b, and c | 102.78, 102.78, 51.25 Å |
| α, β, and γ | 90, 90, 120° |
| Wavelength | 0.979 Å |
| Resolution | 44.5 to 1.47 Å (1.55-1.47 Å)a |
| Rmerge | 9.2% (50.5%) |
| I/σI | 10.2 (2.8) |
| Completeness | 99.4% (99.8%) |
| Redundancy | 5.0 (4.6) |
| Refinement | |
| Resolution | 36.29 to1.47 |
| No. of reflections | 52,630 |
| Rwork/Rfree | 15.88/17.64% |
| No. of atoms | |
| Protein | 1898 |
| Ligand (zinc) | 6 |
| Water | 264 |
| B-Factors | |
| Protein | 13.05 Å2 |
| Ligand (zinc) | 8.01 Å2 |
| Water | 24.81 Å2 |
| Root mean square deviations | |
| Bond length | 0.006 Å |
| Bond angles | 1.128° |
| Ramachandran values | |
| Most favored | 98% |
| Additional allowed | 2% |
| Disallowed | 0% |
a Values in parentheses are for highest resolution shell.
The tertiary structure of PHF6-ePHD2 domain displays a globular conformation (Fig. 2, C and D). It contains two different motifs. The N-terminal part of the PHF6-ePHD2 domain (residues 208–247) includes a Cys-2–His–Cys motif constituting a putative zinc finger (designated pre-PHD), consisting of two α-helices (α1, residues 220–225; α2, residues 241–244) separated by an anti-parallel β-sheet (β1, residues 228–230; β2, residues 237–239) (Figs. 2, B and C, and 3A). Three cysteine residues and one histidine residue from the N-terminal loop, β2-strand, and α2-helix coordinate one zinc ion (designated Zn1) to stabilize the pre-PHD (Fig. 2, B and C). The C-terminal part of the ePHD2 domain (residues 279-330) as expected is a PHD finger, consisting of one short anti-parallel β-sheet (β3, residues 290-292; β4, residues 300–301) and one long anti-parallel β-sheet (β5, residues 312-316; β6, residues 321-325) that are linked by one α-helix (α4, residues 303-308) (Figs. 2, B and C, and 3A). Like other PHD fingers, the PHD finger of PHF6-ePHD2 domain consists of two interleaved zinc fingers. A pair of bound zinc ions (designated Zn2 and Zn3) specifically stabilizes the characteristic cross-braced folding topology of the PHD finger (Fig. 2, B–D). Each zinc ion is coordinated by a combination of four cysteine and histidine residues in which the Zn3 ion is coordinated by a C3H motif instead of a Cys-4 motif in PHF6-ePHD2 (Fig. 2, B and D), which is similar with BRPF2-PHD2 (38). For three zinc-chelating histidines, the zinc coordination occurs all through the Nδ1 of imidazole rings (data not shown). Moreover, in the ePHD2 structure, one long α-helix (α3, residues 265–275) and one long reversed flexural loop (L1, residues 248–264) connect the pre-PHD motif and the imperfect PHD finger to form an integrated fold unit (Fig. 2, D and E).
FIGURE 3.

Pre-PHD zinc finger and PHD2 finger of PHF6 fold as an integrated structural module. A, overall view of PHF6-ePHD2 domain, with regions of interest in boxes. B, detailed interactions between the pre-PHD zinc finger and PHD2 finger of PHF6-ePHD2 domain, color scheme as for Fig. 1. Secondary elements are labeled accordingly. C, detailed hydrophobic interactions at the interface between the pre-PHD zinc finger and helix α3 and loop L1. D and E, network of hydrogen-bonding interactions between the pre-PHD zinc finger, helix α3, and PHD2 finger of the PHF6-ePHD2 domain. Water molecules are represented by red dots. The hydrogen bonds are displayed as red dashed lines. F, structural superposition of the pre-PHD zinc finger of the ePHD2 domain of PHF6 (shown in green, the only zinc ion is shown in orange) with the PHD1 finger of BRPF2 (shown in blue, two zinc ions are shown in gray). G, structure-based sequence alignment of the pre-PHD zinc finger of PHF6-ePHD2 with AIRE-PHD and BRPF2-PHD1 fingers. The conserved zinc-chelating residues are highlighted on an orange background, the absent zinc-chelating residues in a pink and the absent acidic residues in a gray background. Secondary structural elements of pre-PHD zinc finger of PHF6-ePHD2 and BRPF2-PHD1 finger are shown above and below the sequences, respectively.
ePHD2 Domain of PHF6 Folds as an Integrated Structural Module
In ePHD2 domain, the pre-PHD and PHD finger associated with each other via extensive hydrophobic interactions and numerous hydrogen bonding interactions to form a compact globular protein module. Moreover, the long loop L1 and the helix α3 have widespread interactions with the pre-PHD and PHD finger, especially with the former (Figs. 2E and 3A). In particular, residues Leu-244 and Phe-245 of helix α2 in pre-PHD and Ile-290, Tyr-303, Tyr-313, and Tyr-322 from the β3 strand, helix α4, β5, and β6 strands in PHD finger, respectively, form a hydrophobic intermolecular interface (Fig. 3B). In addition, residues Tyr-240 and Met-243 of helix α2 and Phe-214, Phe-231, and Ala-236 from loops in pre-PHD hydrophobically interact with residues Phe-260 and Phe-263 of the long loop L1 and Ile-265, Val-268, Leu-269, and Ile-272 of the helix α3, respectively (Fig. 3C). Besides hydrophobic contacts, a network of hydrogen bonds further stabilizes the pre-PHD-PHD finger fold unit. The hydroxyl oxygen of residue Ser-246 and the main chain amino group and hydroxyl oxygen of residue Ser-247 in pre-PHD form hydrogen bonds with the hydroxyl oxygen of residue Thr-289, and the main chain amino group of residue Gly-291 in β3 strand and the main chain carbonyl group of residue Thr-289 of PHD finger, respectively (Fig. 3D). Furthermore, the side chain of residue Glu-271 in helix α3 forms two hydrogen bonds with the hydroxyl oxygen of residue Ser-246 and the main chain amino group of residue Gly-248 (Fig. 3D). A nitrogen atom on the side chain of residue Arg-274 in helix α3 forms a hydrogen bond with the main chain carbonyl group of residue Thr-289 and another nitrogen atom from Arg-274 side chain via a water molecule bridges the polar contact with the hydroxyl oxygen of residue Thr-300 in β4 strand of PHD finger (Fig. 3D). Additionally, the main chain carbonyl group of residue Phe-214 and the Nϵ2 of imidazole ring of residue His-216 in pre-PHD form hydrogen bonds with the Nϵ2 of imidazole ring of residue His-302 and the main chain carbonyl group of residue Pro-286 separately (Fig. 3E). The main chain carbonyl group of residue Gly-275 in helix α3 forms two hydrogen bonds with the main chain amino groups of residue Gly-287 and Met-278 of PHD finger (Fig. 3E). The main chain carbonyl group of residue Gly-213 via a water molecule bridges with the main chain carbonyl of residue Ile-272. These numerous interactions fix the relative orientation between the pre-PHD and PHD finger and allow PHF6-ePHD2 domain to adopt a compact structure resembling a single folded unit. The importance of the network of hydrogen bonding and hydrophobic interactions for stabilizing the structure of PHF6-ePHD2 domain is confirmed by our efforts to prepare the PHD finger region alone, and several other construct samples yielded unstable products in solution, suggesting its integrate structure is critical for the biological function of the PHF6 protein.
Structure of PHF6-ePHD2 Represents a Novel Structural Module
To identify structural similarity, we performed a structural homology search using the DALI server (43). Unexpectedly, no reported protein structures have been found to be similar with the whole PHF6-ePHD2 structure. So the structure of PHF6-ePHD2 represents a novel structural module.
The DALI results show that only the PHD finger of PHF6-ePHD2 has reasonable structural identity with the other PHD domain, such as BRPF2-PHD2 (Z-score 5.4), Pygo1-PHD (Z-score 3.8), NSD3-PHD5-C5HCH (Z-score 3.7), and Trim33-PHD (Z-score 3.5), etc. The N-terminal pre-PHD exhibits no significant structural similarity to other known structures. Interestingly, the pre-PHD motif in PHF6-ePHD2 can superimpose well with the part of BRPF2-PHD1 (Protein Data Bank code 2KU3) with a root mean square deviation value of 2.78 Å for Cα atoms over 28-amino acid residues (Fig. 3F). However, sequence alignment shows the pre-PHD motif of PHF6 lacks half of the conserved C4HC3 part that chelate two zinc ions in BRPF2-PHD1 finger (Fig. 3G). Therefore, the pre-PHD motif in PHF6-ePHD2 binds to only one zinc ion through the first half of the structure.
Structural comparison of the PHD finger of PHF6-ePHD2 with the PHD fingers of BRPF2 (2LQ6) (38), hPYGO1 (2VPB) (44), NSD3 (4GND) (45), Trim33 (3U5M) (46), and ING1 (2QIC) (47) reveal that the PHD finger of PHF6-ePHD2 closely resembles those of known PHD domain structures (Fig. 4A), but they do not superpose well, which is primarily due to the presence of the long antiparallel β-sheet (β5, β6) segment in PHF6-ePHD2 in place of the C-terminal loop in PHD fingers of ING1, Trim33, and an α-helix in the PHD finger of hPYGO1 (Fig. 4B). In the PHD2 finger of BRPF2, the secondary element of this region is one extended antiparallel β-sheet that is conserved in the BRPF protein family (38), similar to the PHD finger of PHF6-ePHD2 domain (Fig. 4B).
FIGURE 4.

Comparison of the PHD fingers of PHF6 and other reported PHD fingers. A, multiple sequence alignment of PHDs of PHF6 with other PHD fingers searched from DALI. Zinc ligands (Cys or His) are highlighted on an orange background; residues interacting with H3K4 or methylated H3K4 are shown on a red or bright blue background, and residues interacting with R2 are shown on a green background. Secondary structural elements of PHD2 finger of PHF6 are indicated above the sequences. B, structure comparison of the PHD fingers from PHF6, hPYGO1 (Protein Data Bank code 2VPB), ING1 (2QIC), and BRPF2 (2LQ6), respectively. These PHD fingers were chosen for comparison with PHD2 of PHF6, because they represent three types of PHD fingers in terms of the position and orientation of helical, loop, or β-strand elements. C and D, steric hindrance between the PHF6-ePHD2 domain and histone H3 peptide observed in Trim33-PHD-H3 complex (C) and ING1-PHD-H3 complex (D). PHF6-ePHD2 domain is colored in cyan. Trim33-PHD and H3 peptide are shown in magenta and yellow in C, respectively. ING1-PHD and H3 peptide are shown in yellow and magenta in D, respectively. The side chains of residues Arg-2, Lys-4, K4me3, and K9me3 from H3 peptide are shown in stick representation.
PHF6-ePHD2 Does Not Bind Histones in Vitro
Because PHD fingers often recognize histone marks, we investigated whether the PHF6-ePHD2 domain binds to histones. No interaction was detected between calf thymus histones and the PHF6-ePHD2 domain in high salt (1 m NaCl) buffer using an in vitro GST pulldown assay (data not shown). NMR titration experiments were also performed to explore the interaction of PHF6-ePHD2 with histone H3 and H4 N-terminal peptides; however, no obvious chemical shift perturbation was observed in 15N-HSQC spectra (data not shown), implying that PHF6-ePHD2 does not bind histones. Based on the structural information of PHF6-ePHD2, we could give the explanation for why it does not bind to histones. Despite the overall structural similarity, the PHD finger of PHF6-ePHD2 exhibits several different structural features compared with other histone-binding PHD fingers. First, all aromatic residues that are required to form the methyl-lysine recognition cage are absent in the PHD finger of PHF6-ePHD2, or acidic residues used to recognize unmodified lysine are replaced by the oppositely charged residue in the PHF6-ePHD2 PHD finger (Fig. 4A). Second, PHF6-ePHD2 contains one long helix α3 that sterically hindered any histone tail peptide binding to the histone-binding site at PHD (Fig. 4, C and D). These analyses suggest the PHD finger of PHF6 is a noncanonical PHD finger and lacks the structural features for binding to histone tails. These results indicate that although the PHD domain of PHF6-ePHD2 shows overall structural similarities with other PHD domains, the two domains of the protein are fused to produce a unique structure.
Dynamic Properties of the PHF6-ePHD2 Module
The 1H, 13C, and 15N chemical shifts of backbone resonances of PHF6-ePHD2 domain were assigned by multidimensional heteronuclear NMR experiments for the 15N,13C-labeled sample (data not shown). Using uniformly 15N-labeled PHF6-ePHD2, we measured the 15N spin relaxation data R1 and R2 and 1H-15N heteronuclear NOE values to investigate the internal mobility of the secondary structural elements and loops in PHF6-ePHD2 domain by NMR. Analysis of backbone relaxation data R1, R2, and the 1H-15N NOEs show a remarkably uniform distribution over most of the amino acid sequence (Fig. 5A). The overall average values of R1, R2, and NOE are 1.28 ± 0.02, 13.74 ± 0.17, and 0.76 ± 0.02 s−1, respectively, implying that overall molecular architecture of the PHF6-ePHD2 domain is rather rigid and might fold as an integrated structural unit. Except for the segment from 253 to 260 amino acid residues of loop L1, the increased R1 value and decreased R2 and 1H-15N NOEs values that reveal this is a flexible region compared with the core region (Fig. 5A). The dynamic property of this segment is consistent with its bad electron density map in one molecule in the asymmetric unit.
FIGURE 5.

Backbone NMR relaxation data and RDC analysis for PHF6-ePHD2 domain. A, R1, R2, and heteronuclear 1H-15N NOE of PHF6-ePHD2 domain. The secondary structural elements are shown at top. The similar relaxation characters between the pre-PHD and PHD2 fingers indicate two domains fold as an integrated unit. However, the segment from 253 to 260 amino acid residues of loop L1, enclosed by a gray box, is relatively more flexible than the core region (relatively lower R2 1H-15N NOE values and higher R1 values than those of other regions). B, characteristics of the alignment tensors for pre-PHD, PHD2, and ePHD2 domain. The Euler angles (α, β, and γ), in degrees, characterize the orientation of the principal axes' frame of the alignment tensors of pre-PHD, PHD2, and ePHD2 with respect to the Protein Data Bank coordinate frame for each domain. C, panels represent the agreement between the measured and calculated data for protein core residues. The correlation coefficient is 0.98 (pre-PHD, left, filled circle), 0.98 (PHD2, middle, open circle), and 0.98 (ePHD2, right, filled triangle).
To assess the orientation of the pre-PHD and PHD2 zinc fingers in PHF6-ePHD2 in solution further, we measured their 1H-15N RDCs in 5% stretched polyacrylamide gel medium. Alignment tensors were then determined for the pre-PHD, PHD2, and ePHD2 domain using the crystal structure, respectively. The magnitude, rhombicity, and Euler angles of the alignment tensors from the individual fits are indistinguishable (Fig. 5B), and the experimental RDC values agree well with the back-calculated values (Fig. 5C), indicating the tandem domains can be fit simultaneously to a single tensor just as well as fits to each individual domain. These results demonstrate the rigidity of the two PHD zinc fingers.
PHF6-ePHD2 Domain Basic Surface Nonspecifically Binds to DNA
We noticed that the positively charged region of the PHF6-ePHD2 domain that covers one side of the structure consisted of highly conserved arginine and lysine residues. To investigate the possibility that the PHF6-ePHD2 domain function may involve a DNA binding activity, we conducted an FPA and an electrophoretic mobility shift assay (EMSA) to test whether PHF6-ePHD2 domain directly binds to dsDNA. It has been shown that the C-terminal tail (from 334 or 343 to 365 amino acids) of PHF6 is missing in several BFLS patients, indicating an important role of the tail for the function of PHF6. We therefore performed the dsDNA binding assays using purified PHF6(208-333) and PHF6(208-365) proteins.
As shown in Fig. 6, A and B, the value of fluorescence polarization anisotropy (in millipascal units) was concurrently increased with the increasing ratio of protein/DNA, and the binding affinities of PHF6(208-333) and PHF6(208-365) to dsDNA oligonucleotides were similar. The Kd values for PHF6(208-333) binding to 14–20-bp dsDNA were about 12–17 μm, but for PHF6(208-365), the Kd values were decreased to 7–8 μm, indicating a stronger binding affinity of PHF6(208-365) to dsDNA (Table 2).
FIGURE 6.

PHF6-ePHD2 domain interacts with dsDNA. A and B, FPA of PHF6(208-333) (A) and PHF6(208-365) (B) interaction with dsDNA oligonucleotides with different lengths. Protein PHF6(208-333) and PHF6(208-365) concentrations are as follows, respectively: 0, 1.45, 2.9, 5.8, 11.6, 23.2, 46.4, 58, 74.25, 87; 0, 0.65, 1.3, 2.6, 5.2, 10.4, 20.8, 39, 52, 65, and 78 μm. C and D, FPA of PHF6(208-333) (C) and PHF6(208-365) (D) interaction with AT-rich and GC-rich dsDNA. Protein PHF6(208-333) concentrations are same as A. Protein PHF6(208-365) concentrations are as follows: 0, 0.65, 1.3, 2.6, 5.2, 10.4, 26, 39, 52, 65, and 78 μm. E and F, EMSA of GST-PHF6(208-333) (E) and GST-PHF6(208-365) (F) interaction with 33 bp of GC-rich dsDNA (2 μm) in 5% native gel. Protein GST-PHF6(208-333) and GST-PHF6(208-365) concentrations are as follows, respectively: 15.4, 30.8, 61.7, 123.4, 185, 247, 14.2, 28.3, 56.7, 113.4, 226.8, and 283.5 μm.
TABLE 2.
Kd value (μm) of PHF6-ePHD2 domain binding to different types of DNA
| DNA type | PHF6(208-333) | PHF6(208-365) | R209E/K211E | K234E/K235E | K273E/R274E/K276E/R277E | K276E/R277E/M278A/K279E |
|---|---|---|---|---|---|---|
| 14 bp | 12.84 ± 0.88 μm | 7.26 ± 0.71 μm | 62.03 ± 16.84 μm | No binding | No binding | No binding |
| 18 bp | 16.54 ± 1.56 μm | 7.92 ± 0.70 μm | ||||
| 20 bp | 16.44 ± 1.60 μm | 7.09 ± 0.58 μm | ||||
| AT-rich | 20.51 ± 1.93 μm | 4.15 ± 0.55 μm | ||||
| GC-rich | 30.54 ± 2.34 μm | 5.24 ± 0.66 μm |
We next examined whether PHF6(208-333) and PHF6(208-365) exhibit DNA-binding sequence preferences, but no obvious preference for an AT- or GC-rich sequence was observed (Fig. 6, C and D). The Kd values for PHF6(208-333) binding to AT- and GC-rich dsDNA were about 20.5 and 30.5 μm, respectively (Table 2). The Kd values for PHF6(208-365) binding to AT- or GC-rich dsDNA were about 4.2 and 5.2 μm, respectively (Table 2), suggesting the C-terminal tail of PHF6 may contribute to a high binding affinity of PHF6(208-365) to dsDNA. We also performed EMSA)to confirm the interaction of dsDNA with either PHF6(208-333) or PHF6(208-365). As shown in Fig. 6, E and F, with the increasing concentration of GST-fused PHF6(208-333) or PHF6(208-365), the amounts of shifted dsDNA were increased correspondingly.
To determine the PHF6(208-333) or PHF6(208-365) interfaces that are responsible for DNA binding, we monitored chemical shift perturbation changes upon titrating DNA into the 15N-labeled PHF6(208-333) and PHF6(208-365) protein samples. The NMR perturbation experiments showed lots of the residues with obvious chemical shift changes (Fig. 7, A–D), including the backbone amides of Arg-209, Lys-211, Phe-214, Cys-215, His-216, Val-217, Glu-219, Ile-230, Phe-231, Lys-235, Ala-236, Ala-237, Ala-238, Tyr-240, Met-243, Gly-248, Thr-249, Val-250, Thr-254, Phe-260, Phe-263, Ile-265, Gln-270, Glu-271, Lys-273, Arg-274, Arg-277, Met-278, Lys-279, Ala-288, and Thr-289 and the side chain of Asn-232 and Gln-251 (Fig. 7, E and F). The residues with the most perturbed amides were distributed across the basic face of the pre-PHD finger and helix α3 in PHF6(208-333) (Fig. 7G). The perturbed residues due to DNA binding are same for PHF6(208-333) and PHF6(208-365) (Fig. 7, E and F).
FIGURE 7.

Identification of PHF6-ePHD2 domain residues interacting with dsDNA. A and B, overlay of small regions of the 1H-15N HSQCs of the titration of DNA into PHF6(208-333). Free PHF6(208-333) is in red and in the presence of dsDNA in black. The mole ratio of DNA/protein is 1.7:1. C and D, overlay of small regions of the 1H-15N HSQCs of the titration of DNA into PHF6(208-365). Free PHF6(208-365) is in red and in the presence of dsDNA in black. The mole ratio of DNA/protein is 0.85:1. E, chemical shift perturbation values for the titration of DNA into PHF6(208-333). Secondary structural elements of PHF6-ePHD2 are shown at top. F, chemical shift perturbation values for the titration of DNA into PHF6(208-365) observed in the region of PHF6(208-333). Secondary structural elements of PHF6-ePHD2 are shown at top. G, molecular electronic potential surface of PHF6-ePHD2 domain. H, FPAs of PHF6(208-333) mutants with a 5′-FAM-labeled 14-bp dsDNA. PHF6(208-333) mutant concentrations are same as in Fig. 6A. I and J, EMSA of GST-PHF6(208-333) mutants with a 33-bp GC-rich dsDNA (2 μm) in 5% native gel. GST-PHF6(208-333) mutants concentrations are as follows, respectively. R209E/K211E: 30.8, 61.7, 123.4, and 246.7 μm; K234E/K235E, K273E/R274E/K276E/R277E, and K276E/R277E/M278A/K279E are the same concentrations as follows: 61.7, 123.4, and 246.7 μm.
To investigate whether PHF6-ePHD2 interacts with dsDNA via the positively charged region (Fig. 7G), we examined the dsDNA binding activities of four mutants of PHF6-ePHD2, R209E/K211E, K234E/K235E, K273E/R274E/K276E/R277E, and K276E/R277E/M278A/K279E. Despite that these mutations do not appear to affect overall protein structure of PHF6-ePHD2 (data not shown), the Kd value of R209E/K211E mutant was increased to 62.0 ± 16.8 μm, and the dsDNA-binding abilities of other three mutants were almost completely abolished (Fig. 7, H–J; Table 2). These results indicate that the positively residues located in pre-PHD and helix α3 are crucial for the ability of PHF6-ePHD2 to bind dsDNA.
Structural Analysis of PHF6-ePHD2 Reveals Pathological Implication of Amino Acid Point Mutations
To relate the structural information to function, we mapped the previously reported PHF6 point mutations present in BFLS, T-ALL, and acute myeloid leukemia to the three-dimensional structure of the ePHD2 domain (Fig. 8, A–D). These mutations are dispersedly located in pre-PHD finger, helix α3, loop L1, and PHD finger. Several point mutations have been found in BFLS patients. H229R, K234E, R257G, and I314V were identified within the PHF6-ePHD2 domain (1, 8, 10). Residue His-229 is located on the β1 strand; residue Lys-234 is located on the loop-linked β1 and β2 strands; residue Arg-257 is located on the loop L1, and residue Ile-314 is located on the β6 strand. To obtain the structural impact of these substitutions, we expressed, purified, and analyzed the corresponding PHF6-ePHD2 mutants by NMR spectroscopy. All H229R, K234E, R257G, and I314V mutants do not appear to have obvious impact on the structure based on the one-dimensional NMR spectra analysis (data not shown). These results indicate that residues His-229, Lys-234, Arg-257, and Ile-314 do not affect structural integrity of the PHF6-ePHD2 domain, and they may interfere with the interaction of PHF6 with its binding partners.
FIGURE 8.
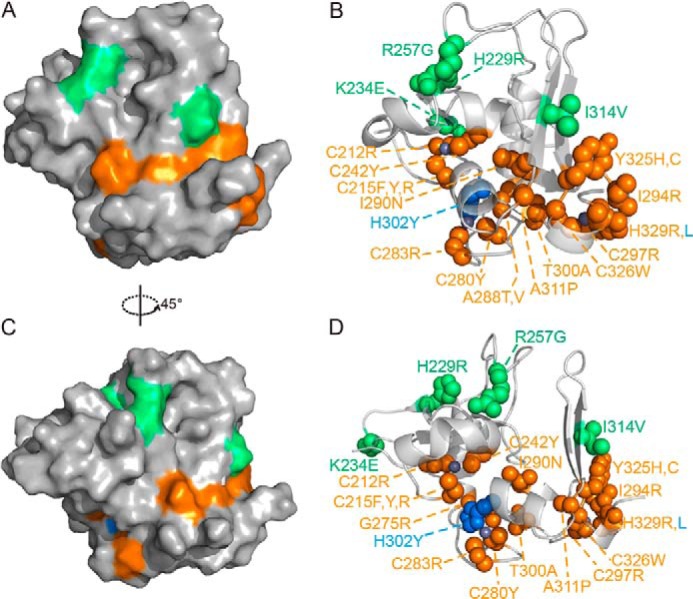
Location and structural effects of previously identified PHF6-ePHD2 domain mutations. A–D, surface (A and C) and sphere (B and D) representations of PHF6-ePHD2 domain mutations that found in BFLS (green), in acute myeloid leukemia (blue), and in T-ALL (orange). Zinc ions are in gray.
Multiple point mutations in PHF6-ePHD2 domain found in T-ALL and acute myeloid leukemia patients affect nine zinc-coordinating amino acids residues (C212R, C215F/C215Y/C215R, C242Y, C280R/C280N, C283Y, C297R, H302Y, C326W, and H329R/H329L) (1, 16, 19–22), and position Cys-215 within the pre-PHD finger appears to be a mutational hot spot. PHF6-ePHD2 mutants harboring the affecting zinc-coordinating cysteines and histidines mutations are expressed only in trace amounts, mostly in the inclusion body, indicating these mutants are misfolded. Additional point mutations change deeply buried amino acid residues (G275R, A288T/A288V, and I290N) (20, 22) in the hydrophobic core of the structure and partially solvent-exposed amino acid residues (I294R, T300A, A311P, and Y325H) (16, 20, 22). Buried amino acid mutations destabilize the structural core resulting in low soluble protein, and most are unfolded in the inclusion body. The partially solvent-exposed Y325H and I294R mutations adjacent to the third zinc-coordinating position are also mostly expressed as inclusion bodies. The T300A and A311P mutations have slight effect on the structure of the PHF6-ePHD2 domain (data not shown). The dsDNA binding abilities of these point mutants were different, but only the K234E mutant was reduced significantly (Kd = 38.4 ± 5.4 μm).
Thus, these mutations that affect the structure core or zinc coordination of the PHF6-ePHD2 domain destabilize the correct protein fold, which are likely to affect its normal biological function. Another class of mutations that locate on the surface of the ePHD2 domain also affects the protein's structural and electrical characters or stability to some extent, and they are most likely to affect interactions with specific partners directly.
PHF6 Directly Interacts with NuRD Complex Component RBBP4
Recently, it has been identified that PHF6 interacts with the NuRD (also known as Mi-2) complex. PHF6 can be co-purified with CHD4, RBBP4, and HDAC1 (24). However, it remains unknown the component of the NuRD complex to which PHF6 directly binds. We therefore determined whether RBBP4 could directly interact with PHF6. We expressed GST-fused full-length PHF6 and several PHF6 fragments, and we carried out GST pulldown experiments with recombinant RBBP4 protein. Several conclusions can be drawn from the pulldown results (Fig. 9). First, RBBP4 interacts with full-length PHF6 directly but not with GST alone (Fig. 9A). Second, the extended PHD2 domain of PHF6 is not involved in the interaction with RBBP4 (Fig. 9B). Third, RBBP4 directly binds to two PHF6 fragments, PHF6(145–207) and PHF6(152–171) (Fig. 9C), both of which contain a putative NoLS composed of nuclear localization sequences 3 and 4 (NLS3 and NLS4). This region is rich in positively charged amino acid residues (Lys and Arg), which is similar to the N-terminal region of several co-repressors and transcription factors, including SALL1, BCL11A/B, and FOG1/2 (48). A sequence alignment of PHF6 with SALL1, BCL11A/B, and FOG1/2 is shown in Fig. 9D. The high sequence similarity indicates that PHF6 likely recruit the NuRD complex in a similar manner as do SALL1, BCL11A/B, and FOG1/2.
FIGURE 9.

PHF6 directly interacts with RBBP4 through its NoLS region. A, full-length PHF6 protein interacts with the recombinant RBBP4 protein. The pulled down RBBP4 protein band is marked with an asterisk. The band migrating higher than GST-PHF6FL is a nonspecific protein band during the protein purification. GST fusion full-length PHF6 protein is unstable and easily degradable. B, PHF6-ePHD2 domain does not interact with the RBBP4 protein. C, RBBP4 directly interacts with GST fusion PHF6 fragments (residues 145–207, lane 6; residues 152–171, lane 8). Gels were stained with Coomassie Blue. D, sequence alignment of the N-terminal residues of human Bcl11A/B, Sal1, and FOG1/2 with residues 152–171 of PHF6 protein. High similarity residues are shown on a red background. E, transcriptional repression by PHF6 protein is dependent on its NoLS region. WT, wild-type PHF6; ΔePHD2, absent ePHD2 domain from full-length PHF6; R342X, absent region 342–365 amino acid residues; ΔNoLS, absent region 157–171 amino acids from full-length PHF6. Efficiency of transfection was normalized to Renilla luciferase, as described under “Experimental Procedures.” The quantifications represent means of three independent experiments ± S.D.
PHF6-mediated Transcriptional Repression Is Dependent on the RBBP4 Interaction
To investigate the function of the PHF6 protein, we used a reporter system in which PHF6 was expressed as a fusion with the GAL4 DNA binding domain (GAL4-DBD) and co-expressed with reporters controlled by GAL4-response elements (GAL-TK-luciferase). The results showed that full-length PHF6 strongly repressed the transcription of the reporter gene by more than 5-fold (Fig. 9E). However, we found the transcriptional repression function of PHF6 is not associated with the ePHD2 domain and the C-terminal region of PHF6 (Fig. 9E). When the putative NoLS region was deleted from full-length PHF6, the transcriptional repression by PHF6 was recovered (Fig. 9E), suggesting that PHF6-mediated transcriptional repression may be dependent on this RBBP4-interacting sequence.
DISCUSSION
Mutations in the PHF6 gene was first discovered in the BFLS-associated patients (1). BFLS is a recessive X-linked disorder characterized by mental retardation, hypogonadism, hypometabolism, and obesity (1, 15). Somatic mutations in the PHF6 gene were also found associated with T-ALL and acute myeloid leukemia patients, implicating PHF6 as a tumor suppressor (16, 19–22). However, the pathogenesis implication for these mutations is poorly understood. It has been reported that PHF6 can interact with both the NuRD complex and the PAF1 transcription elongation complex (24, 26). Knockdown of PHF6 profoundly impairs neuronal migration in the mouse cerebral cortex in vivo, leading to the formation of white matter heterotopias displaying neuronal hyperexcitability (26). PHF6 is implicated in chromatin regulation and neural development (1, 24), but the molecular mechanisms in these processes are still unclear. Here, we report the high resolution crystal structure of the ePHD2 domain of PHF6. Structural analysis of PHF6-ePHD2 reveals pathological implication of PHF6 gene mutations in BFLS, T-ALL, and acute myeloid leukemia. We showed that PHF6-ePHD2 could bind dsDNA. We also demonstrated that the PHF6 protein directly interacted with RBBP4 through its NoLS region that is important for the transcriptional suppression activity of PHF6 protein.
ePHD Domain of PHF6 Represents a Novel Structural Module
To better understand the function of PHF6, we have determined the structure of the ePHD2 domain from human PHF6 protein. The pre-PHD and PHD fingers in ePHD2 are associated with each other via extensive hydrophobic interactions and numerous hydrogen bonding interactions and folded as an intact structural module.
Given the sequence similarity, it is likely that the essential features of this structure are also shared by other extended PHD domain-containing proteins. We performed a Psi-blast search in NCBI, and we found several human chromatin-associated proteins indeed contain similar variational PHD domains, including MLL1/2/3/4, JMJD2A/B/C, and several PHD finger proteins (Fig. 10). Intriguingly, unlike other PHD domain-containing proteins, PHF6-ePHD2 is not able to interact with histones, suggesting that the extended PHD domains of PHF6 may have unique functions that different from other typical PHD fingers. We showed here that PHF6-ePHD2 could bind dsDNA; nevertheless, it is possible that the ePHD2 domain could interact with RNA, given the role of PHF6 in regulating rRNA synthesis (25).
FIGURE 10.

Sequence alignment of homologically extended PHD domains of proteins from a Psi-Blast search in NCBI. Zinc-chelating cysteines, histidines, and identical amino acid residues are highlighted on a red background. Similar residues that may associate with hydrophobic interactions between pre-PHD and linker region, pre-PHD and PHD, are highlighted on a yellow and cyan background, respectively. The secondary elements of the PHF6-ePHD2 domain are shown at top.
Previous studies have reported that the extended LAP/PHD finger mediates homo-oligomerization of recombinant AF10, and this structural module is conserved in several proteins, including MLL (49). However, the ePHD2 domain of PHF6 behaves as a monomer in our experiments, so despite similarity within the extended PHD domains, different proteins have their unique functional and structural features.
Implication of Pathological Mutants Relevant to BFLS, T-ALL, or Acute Myeloid Leukemia Revealed by Structure
The important role of the ePHDs of PHF6 is highlighted by its mutations found in BFLS, T-ALL, and acute myeloid leukemia. The ePHD2 domain of PHF6 harbors 50% of missense mutations found in the PHF6 gene in BFLS and more than 50% mutations in the PHF6 gene found in T-ALL. However, previously very little is known about the function of PHF6 or the pathophysiological mechanism underlying PHF6 mutations. The structural analysis presented here shows that the point mutations associated with BFLS mainly affect the electrostatic characters of the ePHD2 domain, not affecting the structural integrity but probably disrupt its ability to interact with specific binding partners. We demonstrate that the point mutations in the crucial zinc-coordinating residues or buried residues will affect the folding of the ePHD2 domain and therefore will certainly affect the function of this protein.
PHF6 through Its NoLS Region Directly Interacts with RBBP4, a Component of NuRD Complex, and Plays the Function of Transcriptional Suppression
Somatic inactivating mutations in PHF6 have been linked to T-ALL. Loss of PHF6 is also associated with leukemias driven by aberrant expression of the homeobox transcription factor oncogenes TLX1 and TLX3 (16). These findings suggest that PHF6 is an X-linked tumor suppressor (16). However, the underlying molecular mechanisms remain obscure.
Recently, PHF6 has been identified to interact with the NuRD complex and PHF6 as a chromatin-associated protein was also confirmed (24). We showed that PHF6 is able to directly interact with RBBP4, a component of the NuRD complex, through the NoLS region. We also demonstrated this NoLS region is associated with the transcriptional suppression activity of PHF6 protein. Lack of this region obviously reduces the suppression effect of PHF6 on the reporter gene activity.
Taken together, our data support the hypothesis that PHF6 may function as a transcription repressor using its ePHD domains binding to the promoter region of its repressed gene, and the gene transcription was regulated by the NuRD complex that was recruited to the target site in the genome by the NoLS region of PHF6. Mutations in PHF6 relevant to BFLS, T-ALL, or acute myeloid leukemia may disturb the expression of normal genes during brain development or T-cell development and cause disease. Further biochemical and structural studies will be aimed to identify the target genes and interacting partners of the PHF6 protein and help to further elucidate and understand the molecular functions of the PHF6 protein in human BFLS and leukemia diseases.
This work was supported by National Basic Research Program of China, 973 Program, Grants 2011CB966302, 2012CB917201, and 2011CB911104, Chinese National Natural Science Foundation Grants 31170693 and 31330018, Chinese Academy of Science Grant KJZD-EW-L05, and Strategic Priority Research Program of the Chinese Academy of Sciences Grant XDB08010100.
The atomic coordinates and structure factors (code 4NN2) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PHD
- plant homeodomain
- BFLS
- Börjeson-Forssman-Lehmann syndrome
- T-ALL
- T-cell acute lymphoblastic leukemia
- ePHD
- extended PHD
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- RDC
- residual dipolar coupling
- NuRD
- nucleosome remodeling and deacetylation
- FAM
- 6-carboxyfluorescein
- FPA
- fluorescence polarization assay
- NoLS
- nucleolar localization sequence.
REFERENCES
- 1. Lower K. M., Turner G., Kerr B. A., Mathews K. D., Shaw M. A., Gedeon A. K., Schelley S., Hoyme H. E., White S. M., Delatycki M. B., Lampe A. K., Clayton-Smith J., Stewart H., van Ravenswaay C. M., de Vries B. B., Cox B., Grompe M., Ross S., Thomas P., Mulley J. C., Gécz J. (2002) Mutations in PHF6 are associated with Borjeson-Forssman-Lehmann syndrome. Nat. Genet. 32, 661–665 [DOI] [PubMed] [Google Scholar]
- 2. Gedeon A. K., Kozman H. M., Robinson H., Pilia G., Schlessinger D., Turner G., Mulley J. C. (1996) Refinement of the background genetic map of Xq26-q27 and gene localisation for Borjeson-Forssman-Lehmann syndrome. Am. J. Med. Genet. 64, 63–68 [DOI] [PubMed] [Google Scholar]
- 3. Borjeson M., Forssman H., Lehmann O. (1962) An X-linked, recessively inherited syndrome characterized by grave mental deficiency, epilepsy, and endocrine disorder. Acta Med. Scand. 171, 13–21 [DOI] [PubMed] [Google Scholar]
- 4. Turner G., Lower K. M., White S. M., Delatycki M., Lampe A. K., Wright M., Smith J. C., Kerr B., Schelley S., Hoyme H. E., De Vries B. B., Kleefstra T., Grompe M., Cox B., Gecz J., Partington M. (2004) The clinical picture of the BoForssman-Lehmann syndrome in males and heterozygous females with PHF6 mutations. Clin. Genet. 65, 226–232 [DOI] [PubMed] [Google Scholar]
- 5. Visootsak J., Rosner B., Dykens E., Schwartz C., Hahn K., White S. M., Szeftel R., Graham J. M. (2004) Clinical and behavioral features of patients with Borjeson-Forssman-Lehmann syndrome with mutations in PHF6. J. Pediatr. 145, 819–825 [DOI] [PubMed] [Google Scholar]
- 6. Gécz J., Turner G., Nelson J., Partington M. (2006) The Borjeson-Forssman-Lehman syndrome (BFLS, MIM #301900). Eur. J. Hum. Genet. 14, 1233–1237 [DOI] [PubMed] [Google Scholar]
- 7. Carter M. T., Picketts D. J., Hunter A. G., Graham G. E. (2009) Further clinical delineation of the Borjeson-Forssman-Lehmann syndrome in patients with PHF6 mutations. Am. J. Med. Genet. 149A, 246–250 [DOI] [PubMed] [Google Scholar]
- 8. de Winter C. F., van Dijk F., Stolker J. J., Hennekam R. C. (2009) Behavioural phenotype in Borjeson-Forssman-Lehmann syndrome. J. Intellect. Disabil. Res. 53, 319–328 [DOI] [PubMed] [Google Scholar]
- 9. Lower K. M., Solders G., Bondeson M. L., Nelson J., Brun A., Crawford J., Malm G., Börjeson M., Turner G., Partington M., Gécz J. (2004) 1024C > T (R342X) is a recurrent PHF6 mutation also found in the original Borjeson-Forssman-Lehmann syndrome family. Eur. J. Hum. Genet. 12, 787–789 [DOI] [PubMed] [Google Scholar]
- 10. Vallée D., Chevrier E., Graham G. E., Lazzaro M. A., Lavigne P. A., Hunter A. G., Picketts D. J. (2004) A novel PHF6 mutation results in enhanced exon skipping and mild Borjeson-Forssman-Lehmann syndrome. J. Med. Genet. 41, 778–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mangelsdorf M., Chevrier E., Mustonen A., Picketts D. J. (2009) Borjeson-Forssman-Lehmann syndrome due to a novel plant homeodomain zinc finger mutation in the PHF6 gene. J. Child Neurol. 24, 610–614 [DOI] [PubMed] [Google Scholar]
- 12. Berland S., Alme K., Brendehaug A., Houge G., Hovland R. (2011) PHF6 deletions may cause Borjeson-Forssman-Lehmann syndrome in females. Mol. Syndromol. 1, 294–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Baumstark A., Lower K. M., Sinkus A., Andriuskeviciute I., Jurkeniene L., Gécz J., Just W. (2003) Novel PHF6 mutation p.D333del causes Borjeson-Forssman-Lehmann syndrome. J. Med. Genet. 40, e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aasland R., Gibson T. J., Stewart A. F. (1995) The PHD finger implications for chromatin-mediated transcriptional regulation. Trends Biochem. Sci. 20, 56–59 [DOI] [PubMed] [Google Scholar]
- 15. Voss A. K., Gamble R., Collin C., Shoubridge C., Corbett M., Gécz J., Thomas T. (2007) Protein and gene expression analysis of Phf6, the gene mutated in the Borjeson-Forssman-Lehmann syndrome of intellectual disability and obesity. Gene Expr. Patterns 7, 858–871 [DOI] [PubMed] [Google Scholar]
- 16. Van Vlierberghe P., Palomero T., Khiabanian H., Van der Meulen J., Castillo M., Van Roy N., De Moerloose B., Philippé J., González-García S., Toribio M. L., Taghon T., Zuurbier L., Cauwelier B., Harrison C. J., Schwab C., Pisecker M., Strehl S., Langerak A. W., Gecz J., Sonneveld E., Pieters R., Paietta E., Rowe J. M., Wiernik P. H., Benoit Y., Soulier J., Poppe B., Yao X., Cordon-Cardo C., Meijerink J., Rabadan R., Speleman F., Ferrando A. (2010) PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat. Genet. 42, 338–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Palomero T., Lim W. K., Odom D. T., Sulis M. L., Real P. J., Margolin A., Barnes K. C., O'Neil J., Neuberg D., Weng A. P., Aster J. C., Sigaux F., Soulier J., Look A. T., Young R. A., Califano A., Ferrando A. A. (2006) NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. U.S.A. 103, 18261–18266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chao M. M., Todd M. A., Kontny U., Neas K., Sullivan M. J., Hunter A. G., Picketts D. J., Kratz C. P. (2010) T-cell acute lymphoblastic leukemia in association with Borjeson-Forssman-Lehmann syndrome due to a mutation in PHF6. Pediatr. Blood Cancer 55, 722–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Van Vlierberghe P., Patel J., Abdel-Wahab O., Lobry C., Hedvat C. V., Balbin M., Nicolas C., Payer A. R., Fernandez H. F., Tallman M. S., Paietta E., Melnick A., Vandenberghe P., Speleman F., Aifantis I., Cools J., Levine R., Ferrando A. (2011) PHF6 mutations in adult acute myeloid leukemia. Leukemia 25, 130–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang Q., Qiu H., Jiang H., Wu L., Dong S., Pan J., Wang W., Ping N., Xia J., Sun A., Wu D., Xue Y., Drexler H. G., Macleod R. A., Chen S. (2011) Mutations of PHF6 are associated with mutations of NOTCH1, JAK1 and rearrangement of SET-NUP214 in T-cell acute lymphoblastic leukemia. Haematologica 96, 1808–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huh H. J., Lee S. H., Yoo K. H., Sung K. W., Koo H. H., Jang J. H., Kim K., Kim S. J., Kim W. S., Jung C. W., Lee K. O., Kim S. H., Kim H. J. (2013) Gene mutation profiles and prognostic implications in Korean patients with T-lymphoblastic leukemia. Ann. Hematol. 92, 635–644 [DOI] [PubMed] [Google Scholar]
- 22. Grossmann V., Haferlach C., Weissmann S., Roller A., Schindela S., Poetzinger F., Stadler K., Bellos F., Kern W., Haferlach T., Schnittger S., Kohlmann A. (2013) The molecular profile of adult T-cell acute lymphoblastic leukemia: mutations in RUNX1 and DNMT3A are associated with poor prognosis in T-ALL. Genes Chromosomes Cancer 52, 410–422 [DOI] [PubMed] [Google Scholar]
- 23. Mavrakis K. J., Van Der Meulen J., Wolfe A. L., Liu X., Mets E., Taghon T., Khan A. A., Setty M., Setti M., Rondou P., Vandenberghe P., Delabesse E., Benoit Y., Socci N. B., Leslie C. S., Van Vlierberghe P., Speleman F., Wendel H. G. (2011) A cooperative microRNA-tumor suppressor gene network in acute T-cell lymphoblastic leukemia (T-ALL). Nat. Genet. 43, 673–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Todd M. A., Picketts D. J. (2012) PHF6 interacts with the nucleosome remodeling and deacetylation (NuRD) complex. J. Proteome Res. 11, 4326–4337 [DOI] [PubMed] [Google Scholar]
- 25. Wang J., Leung J. W., Gong Z., Feng L., Shi X., Chen J. (2013) PHF6 regulates cell cycle progression by suppressing ribosomal RNA synthesis. J. Biol. Chem. 288, 3174–3183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang C., Mejia L. A., Huang J., Valnegri P., Bennett E. J., Anckar J., Jahani-Asl A., Gallardo G., Ikeuchi Y., Yamada T., Rudnicki M., Harper J. W., Bonni A. (2013) The X-linked intellectual disability protein PHF6 associates with the PAF1 complex and regulates neuronal migration in the mammalian brain. Neuron 78, 986–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Battye T. G., Kontogiannis L., Johnson O., Powell H. R., Leslie A. G. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 29. Collaborative Computational Project No. 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 30. Sheldrick G. M. (2008) A short history of SHELX. Acta Crystallogr. A 64, 112–122 [DOI] [PubMed] [Google Scholar]
- 31. Langer G., Cohen S. X., Lamzin V. S., Perrakis A. (2008) Automated macromolecular model building for x-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 34. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 35. Chou J. J., Gaemers S., Howder B., Louis J. M., Bax A. (2001) A simple apparatus for generating stretched polyacrylamide gels, yielding uniform alignment of proteins and detergent micelles. J. Biomol. NMR 21, 377–382 [DOI] [PubMed] [Google Scholar]
- 36. Ottiger M., Delaglio F., Bax A. (1998) Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J. Magn. Reson. 131, 373–378 [DOI] [PubMed] [Google Scholar]
- 37. Zweckstetter M. (2008) NMR: prediction of molecular alignment from structure using the PALES software. Nat. Protoc. 3, 679–690 [DOI] [PubMed] [Google Scholar]
- 38. Liu L., Qin S., Zhang J., Ji P., Shi Y., Wu J. (2012) Solution structure of an atypical PHD finger in BRPF2 and its interaction with DNA. J. Struct. Biol. 180, 165–173 [DOI] [PubMed] [Google Scholar]
- 39. Schindler U., Beckmann H., Cashmore A. R. (1993) HAT3.1, a novel Arabidopsis homeodomain protein containing a conserved cysteine-rich region. Plant J. 4, 137–150 [DOI] [PubMed] [Google Scholar]
- 40. Capili A. D., Schultz D. C., Rauscher F. J., Borden K. L. (2001) Solution structure of the PHD domain from the KAP-1 corepressor structural determinants for PHD, RING and LIM zinc-binding domains. EMBO J. 20, 165–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sanchez R., Zhou M. M. (2011) The PHD finger: a versatile epigenome reader. Trends Biochem. Sci. 36, 364–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li Y., Li H. (2012) Many keys to push: diversifying the ‘readership’ of plant homeodomain fingers. Acta Biochim. Biophys. Sin. 44, 28–39 [DOI] [PubMed] [Google Scholar]
- 43. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fiedler M., Sánchez-Barrena M. J., Nekrasov M., Mieszczanek J., Rybin V., Müller J., Evans P., Bienz M. (2008) Decoding of methylated histone H3 tail by the Pygo-BCL9 Wnt signaling complex. Mol. Cell 30, 507–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. He C., Li F., Zhang J., Wu J., Shi Y. (2013) The methyltransferase NSD3 has chromatin-binding motifs, PHD5-C5HCH, that are distinct from other NSD (nuclear receptor SET domain) family members in their histone H3 recognition. J. Biol. Chem. 288, 4692–4703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xi Q., Wang Z., Zaromytidou A. I., Zhang X. H., Chow-Tsang L. F., Liu J. X., Kim H., Barlas A., Manova-Todorova K., Kaartinen V., Studer L., Mark W., Patel D. J., Massagué J. (2011) A poised chromatin platform for TGF-β access to master regulators. Cell 147, 1511–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Peña P. V., Hom R. A., Hung T., Lin H., Kuo A. J., Wong R. P., Subach O. M., Champagne K. S., Zhao R., Verkhusha V. V., Li G., Gozani O., Kutateladze T. G. (2008) Histone H3K4me3 binding is required for the DNA repair and apoptotic activities of ING1 tumor suppressor. J. Mol. Biol. 380, 303–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lejon S., Thong S. Y., Murthy A., AlQarni S., Murzina N. V., Blobel G. A., Laue E. D., Mackay J. P. (2011) Insights into association of the NuRD complex with FOG-1 from the crystal structure of an RbAp48·FOG-1 complex. J. Biol. Chem. 286, 1196–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Linder B., Newman R., Jones L. K., Debernardi S., Young B. D., Freemont P., Verrijzer C. P., Saha V. (2000) Biochemical analyses of the AF10 protein the extended LAP PHD-finger mediates oligomerisation. J. Mol. Biol. 299, 369–378 [DOI] [PubMed] [Google Scholar]