

Background: The effect of GDF-15 on the regulation of cardiac remodeling is poorly understood.
Results: GDF-15 inhibits norepinephrine induced EGF receptor transactivation and associated hypertrophy and positively associates with myocardial hypertrophy of hypertension patients.
Conclusion: GDF-15 blocks norepinephrine-induced myocardial hypertrophy via a novel pathway involving inhibition of EGF receptor transactivation.
Significance: We indicate a negative feedback mechanism of GDF-15 in regulating catecholamine-induced cardiac remodeling.
Keywords: Cardiac Hypertrophy, Cell Signaling, Epidermal Growth Factor Receptor (EGFR), Growth Factors, Hypertension, Growth Differentiation Factor 15, Norepinephrine, Transactivation
Abstract
Accumulating evidence suggests that growth differentiation factor 15 (GDF-15) is associated with the severity and prognosis of various cardiovascular diseases. However, the effect of GDF-15 on the regulation of cardiac remodeling is still poorly understood. In this present study, we demonstrate that GDF-15 blocks norepinephrine (NE)-induced myocardial hypertrophy through a novel pathway involving inhibition of EGFR transactivation. Both in vivo and in vitro assay indicate that NE was able to stimulate the synthesis of GDF-15. The up-regulation of GDF-15 feedback inhibits NE-induced myocardial hypertrophy, including quantitation of [3H]leucine incorporation, protein/DNA ratio, cell surface area, and ANP mRNA level. Further research shows that GDF-15 could inhibit the phosphorylation of EGF receptor and downstream kinases (AKT and ERK1/2) induced by NE. Clinical research also shows that serum GDF-15 levels in hypertensive patients were significant higher than in healthy volunteers and were positively correlated with the thickness of the posterior wall of the left ventricle, interventricular septum, and left ventricular mass, as well as the serum level of norepinephrine. In conclusion, NE induces myocardial hypertrophy and up-regulates GDF-15, and this up-regulation of GDF-15 negatively regulates NE-induced myocardial hypertrophy by inhibiting EGF receptor transactivation following NE stimulation.
Introduction
Growth differentiation factors (GDFs)3 are a subfamily of proteins belonging to the transforming growth factor β (TGFβ) superfamily that have functions predominantly in development (1). Several members of this subfamily have been described and named GDF-1 through GDF-15. GDF-15, also known as MIC-1, NAG-1, and PLAB, has been identified as a biphasic regulator of cell survival and inflammation in several pathological processes including injury and tumorigenesis (2, 3).
In recent years, an increasing number of investigations have focused on the association between GDF-15 and cardiovascular diseases. Normally, GDF-15 is expressed at very low levels in healthy heart, but its expression can be significantly up-regulated in myocardial injury, ischemia, and cardiac remodeling. GDF-15 has proved valuable in the prognosis of coronary heart disease and heart failure (4–13). Recent studies showed that GDF-15 was involved in the process of cardiac remodeling, including hypertrophic cardiomyopathy, primary hypertension, and ischemic heart diseases (13–17). Our previous data also indicated that GDF-15 may be involved in the compensatory stage of the pathological process of heart failure (12). These results suggested that GDF-15 could play a role in the process of myocardial hypertrophy. So far, both pro- and anti-hypertrophic effects have been characterized (18, 19); however, whether GDF-15 acts as an injury factor or a protector, as well as its precise function and mechanism of action, remains unclear.
Sustained activation of the sympathetic nervous system leads to alterations in normal myocardial structure (20). Previous results indicated that the endogenous adrenergic agonist norepinephrine (NE) was responsible for the hypertrophic process in vivo and in vitro (21). The regulatory features of NE-induced hypertrophy were highly complex, however, and numerous substances have been implicated (22–24). Whether GDF-15 might be a novel regulatory factor in NE-induced myocardial hypertrophy is still unknown.
In this research, we analyzed the relationships between GDF-15 and clinical features of myocardial hypertrophy, as well as between GDF-15 and NE. To explain the clinical relationships observed, the study focused on elucidating (i) whether the GDF-15 level could be affected by NE stimulation and (ii) the potential role of GDF-15 in NE-induced myocardial hypertrophy and its molecular mechanisms.
EXPERIMENTAL PROCEDURES
Clinical Analysis
A cohort study was conducted between July 2009 and August 2011 in the Department of Cardiology of Peking University Third Hospital (Beijing, China). A total of 63 consecutive inpatients, who were diagnosed with primary hypertension based on the European Society of Cardiology guidelines for the management of arterial hypertension (32), and 30 healthy volunteers, were enrolled. The cohort was tested for a correlation of serum GDF-15 level with echocardiograph items and the serum level of NE, together with other clinical parameters, to investigate the relationship between GDF-15 and myocardial hypertrophy. Two-dimensional echocardiography was performed with an Vivid 7 cardiac ultrasound unit (GE, VINGMED). Left ventricular hypertrophy was assessed by measurement of the left ventricle mass and the thickness of the posterior wall of left ventricle (LVPW) and interventricular septal (IVS). Mitral inflow was assessed in the apical four-chamber view, using pulsed-wave Doppler echocardiography, with the Doppler beam aligned parallel to the direction of flow and the sample volume at the leaflet tips. From the mitral inflow profile, the E wave was measured. Doppler tissue imaging of the mitral annulus was obtained from the apical four-chamber view, using a 1–2-mm sample volume placed in the septal mitral valve annulus. Left ventricular mass index (LVMI) and body surface area (BSA) were calculated based on the following formulas,
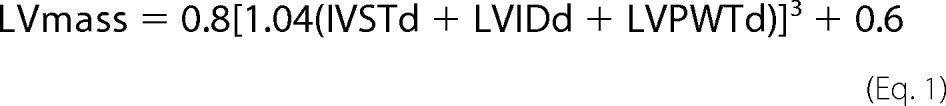 |
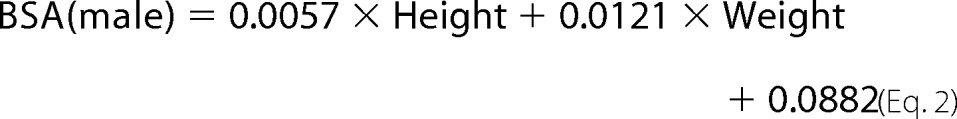 |
 |
where LVmass is left ventricular mass. All the blood samples were obtained at 8:00 on an empty stomach. Serum GDF-15 and NE levels were measured by ELISA (GDF-15: ELH-GDF15–001 RayBiotech, Inc.; NE: H21662; BioASSAY Co., Ltd. Hong Kong, China). Both GDF-15 and NE were measured according to the manufacturer's instructions.
Patients who developed cardiac shock or severe congestive heart failure (NYHA III-IV) were excluded from the study. Other exclusion criteria were acute myocardial infarction within 12 weeks, impairment of renal function, systemic inflammatory disease, infectious diseases, cancer, acute cerebral infarction, prostate diseases, pregnancy, or use of a steroid that might affect the serum GDF-15 levels.
This study was approved by the Medical Ethics Committee of Peking University, and all subjects provided written informed consent. The study was conducted in compliance with the Declaration of Helsinki.
Animals and Drug Treatment
The investigation was approved by the Biomedical Research Ethics Committee of Peking University (LA 2010-048) and adhered to the American Physiological Society's Guiding Principles in the Care and Use of Vertebrate Animals in Research and Training. One-day-old neonatal and 10-week-old adult Sprague-Dawley rats were provided by the Animal Department of Peking University Health Science Center (Beijing, China). All experimental protocols were approved by the Peking University Institutional Committee for Animal Care and Use. The rats were hypodermically injected with norepinephrine twice per day with a total dose of 2.4 mg/kg/day or ascorbic acid (control) for 7 days. The rats were weighed, and hearts were excised and then rinsed in PBS before use.
Preparation of Cardiomyocytes
Cardiomyocytes were isolated and cultured from 1-day-old neonatal Sprague-Dawley rats as described previously (28). In brief, neonatal rat cardiomyocytes (NRCMs) were grown in DMEM (Invitrogen) with 10% fetal bovine serum (Hyclone) and antibiotics (50 mg/ml streptomycin, 50 units/ml penicillin) at 37 °C in a 5% CO2 atmosphere. 5-BrdU (100 μmol/l) was added to the culture medium to prevent proliferation of non-myocardiocytes. NRCMs were starved overnight in DMEM medium without serum before use in experiments.
ELISA
GDF-15 protein levels in the serum and supernatant were measured using a commercially available ELISA kit according to the manufacturer's instructions. All samples were assayed in triplicate.
Western Blot
Cell lysates were subjected to SDS-PAGE and blotted on nitrocellulose membranes. Membranes were then incubated with antibodies directed against GDF-15, phospho-Akt (Ser-473), phospho-ERK1/2 (Thr-202/Tyr-204), and phospho-EGFR (Tyr-1173) and then against the total level of each protein, respectively (all the above antibodies were from Cell Signaling Technology Inc., Danvers, MA). Protein bands were visualized with the Gel Logic 212 imaging system (Kodak, Rochester, NY).
Immunohistochemistry
Hearts were excised and fixed with 4% paraformaldehyde overnight and embedded in paraffin. Tissue morphometry was evaluated in a blinded fashion using the Leica Q550 IW imaging work station (Leica Microsystems Imaging Solutions Ltd., Cambridge, UK). GDF-15 expression was revealed with diaminobenzidine staining (primary antibody: GDF-15 (E-19) antibody, sc-10607; Santa Cruz Biotechnology, Inc.). The number of positively stained (brown) cells was expressed as a percentage of total area.
Real Time PCR
Total RNA was extracted by using TRIzol (Invitrogen). One microgram of total RNA was then reverse transcribed into cDNA by use of the RT-PCR access kit (Invitrogen). One microliter of cDNA was used for PCR. Rat atrial natriuretic peptide (ANP), B-type natriuretic peptide (BNP), β myosin heavy chain (β-MHC), and GAPDH mRNA were amplified by PCR. The PCR mixture was incubated in a DNA Thermal Cycler (Stratagene, La Jolla, CA). The reactions were prepared in triplicate and heated to 95 °C for 5 min, followed by 40 cycles at 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 30 s. The resulting relative increase in reporter fluorescent dye (SYBR Green) emission was monitored with the use of the ABI PRISM 7700 sequence detector (Applied Biosystems).
Adenovirus Transfection
Recombinant adenovirus strains expressing EGFR (pAV-EGFR-eGFP), GDF-15 (pAV-GDF15-eGFP), β-arrestin1 (pAV-β-arrestin1-eGFP), or EGFP shRNA were generated by subcloning the cDNA encoding EGFR, GDF-15, or β-arrestin1 into the vector pAd/CMV/V5-DESTTM Gateway® and EGFR shRNA, GDF-15 shRNA, or β-arrestin1 shRNA into the pAV5′ U6-shRNA adenoviral vector. NRCMs were incubated with recombinant virus strains for 24 h before experimentation.
Immunofluorescence
NRCMs were treated with NE (10 μm) for 0, 5, or 15 min; then fixed with 4% paraformaldehyde; permeabilized with 0.2% Triton X-100 in PBS; and incubated with anti-EGFR (Tyr-1173) antibody (diluted 1:100) at 4 °C overnight and FITC-conjugated anti-rabbit IgG antibody (diluted 1:400, Santa Cruz Biotechnology) at 37 °C for 1 h. For actin staining, cardiomyocytes were also stained with phalloidin (5 μg/ml) at 37 °C for 30 min after permeabilization for cell size analysis. After washing with PBS, NRCMs were examined under an inverted fluorescence microscope (DM IRE2; Leica, Wetzlar, Germany). Cell size was measured by Image-Pro Plus 6.0 (Media Cybernetics Inc.).
[3H]Leucine Incorporation
After preincubation with or without GDF-15, cardiomyocytes were treated with NE (10 μm) for 48 h in serum-free medium and further incubated for 6 h with 1 μCi/ml [3H]leucine before harvesting. Protein was collected by 10% trichloroacetic acid precipitation, resuspended in 1 m sodium hydroxide, and analyzed using a liquid scintillation counter (LS 1801; Beckman Coulter Inc.). The radioactivity in each sample was normalized against protein concentration.
Statistics
All values are expressed as means ± S.D. The sample size in each group is denoted by n. An unpaired t test was used to assess the differences in GDF-15 levels between patients and healthy volunteers. Pearson's correlations and linear regressions were used to evaluate the relationship of GDF-15 to different clinical parameters and echocardiography measurements. One-way analysis of variance followed by the Newman-Keuls test was used for multiple comparisons. GraphPad Prism (GraphPad Software, San Diego, CA) software was used for statistical analysis.
RESULTS
NE Stimulates GDF-15 Secretion
To investigate the relationship between GDF-15 and NE in the induction of myocardial hypertrophy, NRCMs were stimulated with NE in the concentration range 1–100 μm. GDF-15 levels increased significantly in both NRCM supernatant (Fig. 1A) and cell lysate (Fig. 1B) in a dose-dependent manner after incubation with NE. GDF-15 concentration in the supernatant peaked at 24 h after NE (10 μm) stimulus and remained at high level over the next 24 h (Fig. 1C). GDF-15 concentration in cell lysate also peaked 24 h after NE(10 μm) stimulus and showed a slightly downward tendency in the next 24 h (Fig. 1D).
FIGURE 1.

NE induced GDF-15 expression in NRCMs. A, ELISA showing the GDF-15 levels in supernatant induced by NE in different concentrations for 24 h, the peak level of GDF-15 appeared when treated with NE (100 μm). B, Western blot showing the GDF-15 expression in cell lysate induced by NE in different concentrations for 24 h; the peak level of GDF-15 appeared when treated with NE (100 μm). C, ELISA showing the GDF-15 levels in supernatant induced at different time points after NE (10 μm) stimulation; the peak level of GDF-15 appeared 24 h after NE treatment. D, Western blot showing the GDF-15 level in cell lysate at different time points after NE (10 μm) stimulation; the level of GDF-15 increased significantly 24 h after NE treatment. Con, control; Vitc, vitamin C. *, p < 0.05; n = 5.
Similar results were observed in experiments with Sprague-Dawley rats. An ELISA revealed that serum GDF-15 levels increased significantly when rats were injected with NE at a concentration of either 1.2 or 2.4 mg/kg/day. The serum level of GDF-15 reached a maximum 24 h after NE injection and then declined to baseline after 3 days (Fig. 2, A and B). Western blot also revealed increased GDF-15 level in cardiac tissue after NE injection (Fig. 2C). Further, immunohistochemistry assay illustrated the maximum expression of GDF-15 in rat cardiac tissue at the time point of 24 h after the NE injection (Fig. 2D). These results suggested that the synthesis and secretion of GDF-15 could be stimulated by NE in either cardiomyocytes or at the whole animal level.
FIGURE 2.

NE induced GDF-15 expression in Sprague-Dawley rats. A, ELISA showing the GDF-15 levels in rat serum induced by NE in different doses. The level of serum GDF-15 elevated 3 h after NE injection, maintained a high level in 24 h, and declined to baseline in 72 h. B, ELISA revealed increased serum GDF-15 levels when treated with NE (both 1.2 and 2.4 mg/kg/day) for 12 h. C, Western blot revealed increased tissue GDF-15 levels when treated with NE (both 1.2 and 2.4 mg/kg/day) for 12 h. D, immunohistochemistry tissue section illustrating more GDF-15 expression in rat cardiomyocytes 24 h after NE (2.4 mg/kg/day) stimulation. Con or con, control, *, p < 0.05; n = 3.
GDF-15 Negatively Regulates NE-induced Myocardial Hypertrophy
To investigate the functional effects of increased GDF-15 in NE induced myocardial hypertrophy, NRCMs were stimulated with NE (10 μm) in the presence or absence of GDF-15 for 48 h. ANP, BNP, and β-MHC are known to be up-regulated during the process of myocardial hypertrophy (35). Our results revealed that GDF-15 (20 ng/ml) significantly reduced the effect of NE on ANP, BNP, and β-MHC mRNA levels (Fig. 3, A–C). Further, because it is considered the golden standard of myocardial hypertrophy, the level of cardiac myocyte protein synthesis was also analyzed by measurements of [3H]leucine incorporation and total protein/DNA ratio. Treatment in isolated cardiomyocytes with NE resulted in an increase in protein/DNA ratio (Fig. 3D) and [3H]leucine incorporation (Fig. 3E), both of which were inhibited by preincubation with GDF-15. NE stimulation resulted in more [3H]leucine incorporation in NRCMs with GDF-15 knockdown (Fig. 3F). In addition, the increased cell surface area of cardiomyocytes induced by NE was also inhibited by GDF-15 (20 ng/ml) (Fig. 3G). Taken together, these results indicate that GDF-15 negatively regulates NE-induced myocardial hypertrophy and therefore likely functions as a sympathetic regulator.
FIGURE 3.

GDF-15 inhibited NE-induced myocardial hypertrophy. A–C, RT-PCR showed that GDF-15 inhibited the increase of relative ratios of ANP (A), BNP (B), and β-MHC (C) mRNA levels induced by NE (10 μm). D, GDF-15 inhibited the increase of protein-DNA ratio induced by NE (10 μm). E, less [3H]leucine incorporation induced by NE was observed in GDF-15 preincubated NRCMs than in NRCMs without GDF-15 incubation. F, more [3H]leucine incorporation induced by NE was observed in GDF-15 knockdown NRCMs than in NRCMs without the knockdown of GDF-15. G, F-actin staining showed that GDF-15 inhibited the enlargement of cell surface area induced by NE (10 μm) with contributing analysis. Con, control; CPM, counts per minute. *, p < 0.05 n = 4.
GDF-15 Negatively Regulates NE-induced Transactivation of EGFR and Its Signaling Pathway
The kinases Akt and ERK have previously been shown to play an essential role in myocardial hypertrophy (33, 34). Western blot revealed the inhibitory effect of GDF-15 preincubation upon the phosphorylation of Akt and ERK induced by NE (Fig. 4, D–F). Data from our previous study and other recent studies indicated that adrenergic receptor (AR) agonists were capable of inducing transactivation of EGFR (28, 29). Considering that Akt and ERK both act downstream of EGFR, we hypothesized that NE transactivates EGFR and that GDF-15 probably inhibits the activation of EGFR. Our results revealed that NE significantly induced EGFR transactivation with immunofluorescence assay (Fig. 4A) and Western blot (Fig. 4, D–F) in cardiomyocytes, which could be inhibited by preincubation with GDF-15 (Fig. 4, D–F). Similar results were also observed in experiments with Sprague-Dawley rats. Immunochemistry assay and Western blot revealed that GDF15 (1 μg/kg) mediated inhibition of EGFR transactivation induced by NE treatment (Fig. 4, B and C). Furthermore, adenovirus-mediated knockdown of GDF-15 or EGFR were employed to confirm the inhibitory effect of GDF-15 upon NE-induced transactivation of EGFR. The phosphorylation of EGFR and its downstream signaling (Akt and ERK) induced by NE were significantly enhanced in NRCMs with GDF-15 knockdown alone (Fig. 4, G–I), whereas these enhancement could be abolished in NRCMs with co-knockdown of GDF-15 and EGFR (Fig. 4, J–L). Taken together, these results showed that GDF-15 inhibits the NE-induced phosphorylation of EGFR and its downstream kinases, Akt and ERK, thus implicating GDF-15 in a novel NE-induced EGFR transactivation pathway.
FIGURE 4.

GDF-15 inhibited the phosphorylation of EGFR induced by NE in NRCMs. A, immunofluorescence illustrating that NE (10 μm) was able to induced EGFR phosphorylation. B, immunochemistry assay revealed that intravenous injection of GDF-15 (1 μg/kg) significantly attenuated the EGFR phosphorylation induced by NE stimulation. C, Western blot revealed that GDF-15 inhibited the phosphorylation of EGFR induced by NE in tissue. D–F, less phosphorylation of Akt (D), ERK1/2 (E), and EGFR (F) induced by NE was observed in GDF-15 preincubated NRCMs than in NRCMs without GDF-15 incubation. G–I, more phosphorylation of Akt (G), ERK1/2 (H), and EGFR (I) induced by NE was observed in GDF-15 knockdown NRCMs than in NRCMs without the knockdown of GDF-15. J–L, less phosphorylation of Akt (J), ERK1/2 (K), and EGFR (L) induced by NE was observed in NRCMs with the knockdown of GDF-15 alone than in GDF-15/EGFR co-knockdown NRCMs. Con, control; Scr, scrambled; Vitc, vitamin C. *, p < 0.05 n = 3.
Adenovirus strains for EGFR overexpression was also constructed and introduced into NRCMs, to further elucidate the antagonistic effect of GDF-15 on NE-induced transactivation of EGFR. The phosphorylation of EGFR (Fig. 5A), ERK (Fig. 5B), and Akt (Fig. 5C) induced by NE in EGFR-overexpressing cardiomyocytes was significantly augmented compared with controls without EGFR overexpression (p < 0.05). In addition, [3H]leucine incorporation experiments showed that NE-induced protein incorporation was enhanced in EGFR-overexpressing cardiomyocytes (Fig. 5G) and was attenuated in EGFR knockdown cardiomyocytes (Fig. 5H). These results suggest that transactivation of EGFR is responsible for NE-induced myocardial hypertrophy. Moreover, GDF-15 blocked NE-stimulated EGFR transactivation (Fig. 5A) and further phosphorylation of the downstream kinases ERK (Fig. 5B) and Akt (Fig. 5C). GDF-15 also blocked EGFR transactivation-mediated myocardial hypertrophy ([3H]leucine incorporation) (Fig. 5G), and this effect was abolished in EGFR knockdown cardiomyocytes (Fig. 5H). In contrast, NE failed to induce phosphorylation of EGFR (Fig. 5D), ERK (Fig. 5E), and Akt (Fig. 5F) in EGFR knockdown cardiomyocytes. The blockage of EGFR transactivation by GDF-15 was also abolished in EGFR knockdown cardiomyocytes (Fig. 5, D–F). Our findings demonstrate that GDF-15 is a novel negative regulator of NE-induced myocardial hypertrophy through inhibition of NE-induced EGFR transactivation.
FIGURE 5.

GDF-15 inhibited NE induced transactivation of the EGFR signaling pathway and t myocardial hypertrophy. A–C, Western blot showed that the phosphorylation of EGFR (A), ERK1/2 (B), and Akt (C) induced by NE and the extent of inhibition by GDF-15 were more significant in NRCMs with EGFR overexpression than in NRCMs without overexpression. D–F, Western blots showing that the phosphorylation of EGFR (D), ERK1/2 (E), and Akt (F) induced by NE were reduced in EGFR knockdown NRCMs than in Scr, and no inhibition of phosphorylation by GDF-15 was observed. G, more [3H]leucine incorporation induced by NE was observed in EGFR-overexpressed NRCMs than in NRCMs without EGFR overexpression. The [3H]leucine incorporation of NRCMs with or without EGFR overexpression was inhibited by GDF-15 to a similar level. H, less [3H]leucine incorporation induced by NE was observed in EGFR knockdown NRCMs than in NRCMs without EGFR knockdown. GDF-15 could not inhibit NE-induced [3H]leucine incorporation in NRCMs with EGFR knockdown. Scr, scrambled; CPM, counts per minute. *, p < 0.05; n = 3.
Because β-arrestin1 was involved in NE induced EGFR transactivation and myocardial hypertrophy (28, 29), we assessed whether β-arrestin1 mediated the negative regulation of NE-induced EGFR transactivation by GDF-15. The results revealed that the knockdown of GDF-15 enhanced phosphorylation of EGFR, ERK, and Akt, which were abolished in β-arrestin1-deficient cardiomyocyte (Fig. 6, A–C). These data demonstrated that the inhibitory effect of GDF-15 upon the EGFR transactivation induced by NE was β-arrestin1-dependent.
FIGURE 6.
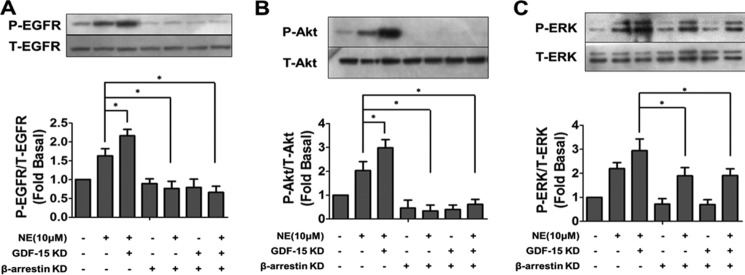
β-Arrestin1 mediated the negative regulation of NE-induced EGFR transactivation by GDF-15. Less phosphorylation of EGFR (A), Akt (B), and ERK1/2 (C) induced by NE was observed in NRCMs with GDF-15/β-arrestin1 co-knockdown than in NRCMs with GDF-15 knockdown alone. *, p < 0.05; n = 3.
GDF-15 Levels Positively Correlate with Echocardiograph Items and Norepinephrine Concentrations in Serum
To further analyze the clinical significance of GDF-15 in cardiac hypertrophy in hypertension, a total of 63 hypertensive patients and 30 volunteers joined our study. There was no age difference between patients and volunteers (62.79 ± 14.43 versus 58.26 ± 5.99, t = 1.649, p = 0.103). The serum GDF-15 levels in hypertensive patients ranged from 894.85 to 1505.20 pg/ml, with a mean value of 1193 ± 163.4 pg/ml, which was significantly higher than the GDF-15 levels of healthy volunteers (308.58 ± 239.371 pg/ml, p < 0.01) (detailed clinical and echocardiograph data not shown here). Patient GDF-15 levels significantly and positively correlated with parameters contributing to myocardial remodeling including BNP (r = 0.2786, p < 0.05) and echocardiograph parameters such as thickness of IVS (r = 0.4503, p < 0.01), LVPW (r = 0.7103, p < 0.01), left atrial pressure (r = 0.3906, p < 0.01), and mitral early diastolic flow versus early diastolic peak velocity (E/Em) (r = 0.3731, p < 0.01). Both serum levels of NE and GDF-15 were positively correlated with LVMI (Table 1 and Fig. 7). These results demonstrated a tight relation between GDF-15 and myocardial hypertrophy and the relation between myocardial hypertrophy and activation of the sympathetic nervous system. Further, a significant positive correlation between GDF-15 and serum levels of NE, an inducer of myocardial hypertrophy, was also demonstrated (r = 0.7334, p < 0.01), which indicated that GDF-15 could be involved in activation of the sympathetic nervous system.
TABLE 1.
GDF-15 levels of hypertension patients in relation to biochemical and echocardiography measures
The p value was calculated from the regression analysis with GDF-15 as the dependent variable. US-CRP, ultrasensitive-C reactive protein; E/Em, excitation/emission; LAP, left atrial pressure; Rho, Pearson's correlation coefficient.
| Value (n = 63) | Rho | p | |
|---|---|---|---|
| Biochemical parameters | |||
| BNP (pg/ml) | 541.0 ± 79.0 | 0.2786 | p < 0.05 |
| Triclycerides (mmol/liter) | 1.716 ± 0.8647 | −0.2730 | p < 0.05 |
| US-CRP (mmol/liter) | 4.106 ± 1.92 | 0.3045 | p < 0.05 |
| NE (pg/ml) | 135.9 ± 17.72 | 0.7334 | p < 0.01 |
| Echocardiography measures | |||
| IVS (mm) | 9.3 ± 2.1 | 0.4503 | p < 0.01 |
| LVPW (mm) | 8.5 ± 1.4 | 0.7103 | p < 0.01 |
| E/Em | 9.5 ± 4.9 | 0.3731 | p < 0.01 |
| LAP (mm Hg) | 13.0 ± 6.1 | 0.3906 | p < 0.01 |
| LVMI (g/m2) | 82.6 ± 25.3 | 0.5572 | p < 0.01 |
FIGURE 7.

A, positive correlation between GDF-15 and NE (r = 0. 7334, p < 0.01). B, positive correlation between GDF-15 and LVPW (r = 0.7103, p < 0.01). C, positive correlation between GDF-15 and IVS (r = 0.4503, p < 0.01). D, positive correlation between GDF-15 and LVMI (r = 0.5572, p < 0.01). E, positive correlation between NE and LVMI (r = 0.7264, p < 0.01).
DISCUSSION
Our previous studies indicated that serum GDF-15 concentration was elevated with increasing stage of heart failure and might have potential value in distinguishing stage B heart failure (12). The clinical value of GDF-15 as a biomarker in cardiovascular diseases was also demonstrated in other studies. GDF-15 was not only identified as a prognostic marker of coronary heart disease and heart failure, where higher serum GDF-15 levels are associated with worse outcomes (4–9), but was also valuable in guiding the selection of treatment (4). Further, GDF-15 was implicated in pathological processes such as the development of atherosclerosis and angiogenesis of lateral arteries (11). However, the relationship between GDF-15 and myocardial hypertrophy has been examined only infrequently. A recent study demonstrated that the serum level of GDF-15 elevated significantly in hypertension patients with left ventricular hypertrophy (36). Our current study revealed that GDF-15 positively correlated with IVS and LVPW at the prehypertrophy stage of hypertension, as well as left atrial pressure and excitation/emission, which indicated that GDF-15 was probably involved in the compensatory stage of myocardial hypertrophy. Further laboratory data indicated that GDF-15 negatively regulated NE-induced myocardial hypertrophy, which was consistent with the observation in a transverse abdominal aortic constriction model that GDF15 antagonized pressure overload-induced hypertrophy (18). These results are consistent with a role for GDF-15 as a protective factor in myocardial hypertrophy.
Chronic sympathetic activation is known to be a key factor in the progression of myocardial hypertrophy, with NE as an important endogenous agonist of the sympathetic system (20). Clinical data from our research demonstrated a positive correlation between serum GDF-15 and NE levels. Furthermore, our laboratory data indicated that this relationship was not just a concomitant phenomenon, but a cause and effect chain of events. Continuous treatment with NE was found to induce the synthesis and secretion of GDF-15 in myocardium, both at the cellular and whole animal levels. Previously, various exogenous agents such as cyclooxygenase inhibitors, dietary agents, PPAR agonists, and some anti-cancer drugs were shown to induce GDF-15 synthesis (3). Our work is the first to identify an endogenous stimulator of GDF-15 and to suggest a potential molecular network involving GDF-15 and catecholamines.
The data we present suggest a model whereby up-regulation of GDF-15 inhibits NE-induced myocardial hypertrophy by acting as a protective agent in a negative feedback loop with NE (Fig. 8). A similar protective effect has been reported in other conditions including ischemia/reperfusion injury and infarction, as well as pressure-induced hypertrophy (18, 25, 26). In previous studies, several endogenous agents have been identified as sympathetic regulators, including catestatin, spinophilin, syntrophins, and angiotensin II: syntrophin regulates α1-AR through a PDZ domain-mediated interaction (22), spinophilin stabilizes cell surface expression of α2-AR and blocks arrestin1 action on GPCR (23), and catestatin interferes with the level of catecholamine and the activation of phospholipase C (24). Here, we identified GDF-15 as a novel sympathetic regulator that protects against myocardial hypertrophy through repression of NE-induced transactivation of EGFR.
FIGURE 8.
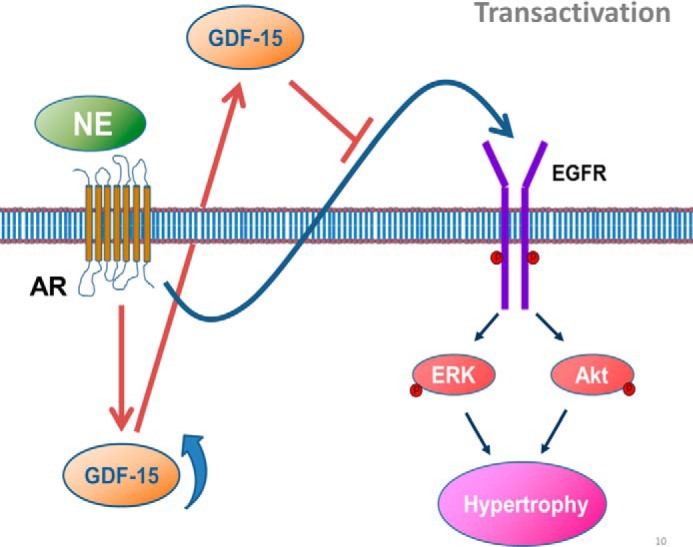
Working model of critical role for GDF-15 in NE-induced myocardial hypertrophy. GDF-15 synthesis and secretion could be stimulated by NE both in vivo and in vitro. Increased GDF-15 in turn inhibited NE-induced myocardial hypertrophy by inhibiting NE-induced transactivation of EGFR and the phosphorylation of downstream kinases Akt and ERK, which constructed a feedback manner.
Multiple pathways are involved in AR-related myocardial hypertrophy, with the transactivation of EGFR representing a novel pathway demonstrated in recent years (27). We showed previously that α-AR-induced transactivation of EGFR may lead to myocardial hypertrophy (28); β-AR-related EGFR transactivation was also demonstrated (29). As a major endogenous agonist of AR, NE simultaneously activates both the α- and β-AR and induces myocardial hypertrophy. The demonstration that NE can induce myocardial hypertrophy through transactivation of EGFR and its downstream kinases, Akt and ERK, and that GDF-15 can inhibit this process is the first report to our knowledge of a mechanism for the inhibition of the transactivation pathway of EGFR.
It has been demonstrated that β-arrestin1 was involved as a mediator in the process of transactivation. The β-arrestin1 recruitment was essential in adrenergic receptor-mediated EGFR transactivation (28, 29). Our results revealed that the knockdown of GDF-15 enhanced the phosphorylation of EGFR induced by NE, whereas co-knockdown of GDF-15 and β-arrestin1 abolished this enhancement, indicating that the inhibitory effect of GDF-15 upon the transactivation induced by NE was β-arrestin1-dependent.
Interestingly, the true effect of GDF-15 on myocardial hypertrophy remains controversial. GDF-15 was found to inhibit myocardial hypertrophy through a Smad2/3 pathway in a pressure-induced hypertrophy model (18), whereas directly treating cultured cardiomyocytes with GDF-15 results in pro-hypertrophic effect through a Smad1 pathway (19). It seems that the exact effect differs in different conditions. Previous studies mainly focused on the Smad-dependent mechanism, which was considered the canonical signaling pathway of GDF-15. However, the Smad pathway has not been proved to be so important in the hypertrophic model induced by sympathetic activation. Here, our data suggested that GDF-15 may protect the heart from NE-induced hypertrophy through a Smad-independent pathway.
In the recent guidelines for heart failure, several new pharmacotherapies including BNP, simendan, and ivabradine were suggested in clinical use (30). Although new pharmacotherapies have developed rapidly in recent years, β-blockers, together with angiotensin-converting enzyme inhibitors, were still considered the cornerstones for heart failure treatment and reversing cardiac remodeling. Our study demonstrated another potential role of β-blockers, and potentially also angiotensin-converting enzyme inhibitors, in the reversal of remodeling and hypertrophy by inhibition of the EGFR transactivation pathway. Finally, it seems clear that GDF-15, as a new modulator of sympathetic system, should not be considered purely as a biomarker, but also as a potential new pharmacotherapy for heart failure and cardiac remodeling.
CONCLUSION
Beyond its prognostic value of numerous cardiovascular diseases, GDF-15 secretion is also tightly correlated with the process of myocardial hypertrophy and chronic activation of the sympathetic nervous system in hypertensive patients. Investigations of function and molecular mechanisms demonstrated a feedback loop in which the synthesis of GDF-15 could be stimulated by NE in cardiomyocytes, and in turn, elevated GDF-15 could negatively regulate NE-induced hypertrophy. GDF-15 was identified as a new endogenous sympathetic regulator that restricts the transactivation of EGFR NE (Fig. 8). Our results suggested that GDF-15 could be a new therapeutic target for the treatment of myocardial hypertrophy.
Acknowledgment
We thank Alan Tunnacliffe for critical evaluation of the manuscript.
This work was supported by Grant PUCRP-004 from the Key Construction Program of the National 985 Project, National Natural Science Foundation of China Grants 30910103902, 81270157, 81070078, and 81121061, Grant from the 973 Program of the Major State Basic Research Development Program of China 2011CB503903, and Beijing Municipal Natural Science Foundation Grant 7102158.
- GDF
- growth differentiation factor
- NE
- norepinephrine
- EGFR
- EGF receptor
- LVPW
- left ventricle posterior wall
- IVS
- interventricular septum
- LVMI
- left ventricular mass index
- NRCM
- neonatal rat cardiomyocyte
- ANP
- atrial natriuretic peptide
- BNP
- type B natriuretic peptide
- β-MHC
- β myosin heavy chain
- AR
- adrenergic receptor.
REFERENCES
- 1. Lee S. J. (1990) Identification of a novel member (GDF-1) of the transforming growth factor-β superfamily. Mol. Endocrinol. 4, 1034–1040 [DOI] [PubMed] [Google Scholar]
- 2. Bootcov M. R., Bauskin A. R., Valenzuela S. M., Moore A. G., Bansal M., He X. Y., Zhang H. P., Donnellan M., Mahler S., Pryor K., Walsh B. J., Nicholson R. C., Fairlie W. D., Por S. B., Robbins J. M., Breit S. N. (1997) MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-β superfamily. Proc. Natl. Acad. Sci. U.S.A. 94, 11514–11519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xu X., Li Z., Gao W. (2011) Growth differentiation factor 15 in cardiovascular diseases: from bench to bedside. Biomarkers 16, 466–475 [DOI] [PubMed] [Google Scholar]
- 4. Wollert K. C., Kempf T., Peter T., Olofsson S., James S., Johnston N., Lindahl B., Horn-Wichmann R., Brabant G., Simoons M. L., Armstrong P. W., Califf R. M., Drexler H., Wallentin L. (2007) Prognostic value of growth-differentiation factor-15 in patients with non-ST-elevation acute coronary syndrome. Circulation 115, 962–971 [DOI] [PubMed] [Google Scholar]
- 5. Kempf T., von Haehling S., Peter T., Allhoff T., Cicoira M., Doehner W., Ponikowski P., Filippatos G. S., Rozentryt P., Drexler H., Anker S. D., Wollert K. C. (2007) Prognostic utility of growth differentiation factor-15 in patients with chronic heart failure. J. Am. Coll. Cardiol. 50, 1054–1060 [DOI] [PubMed] [Google Scholar]
- 6. Kempf T., Björklund E., Olofsson S., Lindahl B., Allhoff T., Peter T., Tongers J., Wollert K. C., Wallentin L. (2007) Growth-differentiation factor-15 improves risk stratification in ST-segment elevation myocardial infarction. Eur. Heart J. 28, 2858–2865 [DOI] [PubMed] [Google Scholar]
- 7. Khan S. Q., Ng K., Dhillon O., Kelly D., Quinn P., Squire I. B., Davies J. E., Ng L. L. (2009) Growth differentiation factor-15 as a prognostic marker in patients with acute myocardial infarction. Eur Heart J. 30, 1057–1065 [DOI] [PubMed] [Google Scholar]
- 8. Anand I. S., Kempf T., Rector T. S., Tapken H., Allhoff T., Jantzen F., Kuskowski M., Cohn J. N., Drexler H., Wollert K. C. (2010) Serial measurement of growth-differentiation factor-15 in heart failure: relation to disease severity and prognosis in the Valsartan Heart Failure Trial. Circulation 122, 1387–1395 [DOI] [PubMed] [Google Scholar]
- 9. Eggers K. M., Kempf T., Lagerqvist B., Lindahl B., Olofsson S., Jantzen F., Peter T., Allhoff T., Siegbahn A., Venge P., Wollert K. C., Wallentin L. (2010) Growth-differentiation factor-15 for long-term risk prediction in patients stabilized after an episode of non-ST-segment-elevation acute coronary syndrome. Circ Cardiovasc Genet. 3, 88–96 [DOI] [PubMed] [Google Scholar]
- 10. Kempf T., Horn-Wichmann R., Brabant G., Peter T., Allhoff T., Klein G., Drexler H., Johnston N., Wallentin L., Wollert K. C. (2007) Circulating concentrations of growth-differentiation factor 15 in apparently healthy elderly individuals and patients with chronic heart failure as assessed by a new immunoradiometric sandwich assay. Clin. Chem. 53, 284–291 [DOI] [PubMed] [Google Scholar]
- 11. Eggers K. M., Kempf T., Lind L., Sundström J., Wallentin L., Wollert K. C., Siegbahn A. (2012) Relations of growth-differentiation factor-15 to biomarkers reflecting vascular pathologies in a population-based sample of elderly subjects. Scand. J. Clin. Lab. Invest. 72, 45–51 [DOI] [PubMed] [Google Scholar]
- 12. Wang F., Guo Y., Yu H., Zheng L., Mi L., Gao W. (2010) Growth differentiation factor 15 in different stages of heart failure: potential screening implications. Biomarkers 15, 671–676 [DOI] [PubMed] [Google Scholar]
- 13. Dominguez-Rodriguez A., Abreu-Gonzalez P., Avanzas P. (2011) Relation of growth-differentiation factor 15 to left ventricular remodeling in ST-segment elevation myocardial infarction. Am. J. Cardiol. 108, 955–958 [DOI] [PubMed] [Google Scholar]
- 14. Lind L., Wallentin L., Kempf T., Tapken H., Quint A., Lindahl B., Olofsson S., Venge P., Larsson A., Hulthe J., Elmgren A., Wollert K. C. (2009) Growth-differentiation factor-15 is an independent marker of cardiovascular dysfunction and disease in the elderly: results from the Prospective Investigation of the Vasculature in Uppsala Seniors (PIVUS) Study. Eur. Heart J. 30, 2346–2353 [DOI] [PubMed] [Google Scholar]
- 15. Wang X., Yang X., Sun K., Chen J., Song X., Wang H., Liu Z., Wang C., Zhang C., Hui R. (2010) The haplotype of the growth-differentiation factor 15 gene is associated with left ventricular hypertrophy in human essential hypertension. Clin. Sci. 118, 137–145 [DOI] [PubMed] [Google Scholar]
- 16. Bjørnstad J. L., Neverdal N. O., Vengen O. A., Knudsen C. W., Husebye T., Pepper J., Lie M., Christensen G., Tønnessen T. (2008) Alterations in circulating activin, A., GDF-15, TGF-β3 and MMP-2, -3, and -9 during one year of left ventricular reverse remodeling in patients operated for severe aortic stenosis. Eur. J. Heart Fail. 10, 1201–1207 [DOI] [PubMed] [Google Scholar]
- 17. Montoro-García S., Hernández-Romero D., Jover E., García-Honrubia A., Vilchez J. A., Casas T., Martínez P., Climent V., Caballero L., Valdés M., Marín F. (2012) Growth differentiation factor-15, a novel biomarker related with disease severity in patients with hypertrophic cardiomyopathy. Eur. J. Intern Med. 23, 169–174 [DOI] [PubMed] [Google Scholar]
- 18. Xu J., Kimball T. R., Lorenz J. N., Brown D. A., Bauskin A. R., Klevitsky R., Hewett T. E., Breit S. N., Molkentin J. D. (2006) GDF15/MIC-1 functions as a protective and antihypertrophic factor released from the myocardium in association with SMAD protein activation. Circ. Res. 98, 342–350 [DOI] [PubMed] [Google Scholar]
- 19. Heger J., Schiegnitz E., von Waldthausen D., Anwar M. M., Piper H. M., Euler G. (2010) Growth differentiation factor 15 acts anti-apoptotic and pro-hypertrophic in adult cardiomyocytes. J. Cell. Physiol. 224, 120–126 [DOI] [PubMed] [Google Scholar]
- 20. Parati G., Esler M. (2012) The human sympathetic nervous system: its relevance in hypertension and heart failure. Eur. Heart J. 33, 1058–1066 [DOI] [PubMed] [Google Scholar]
- 21. Cacciapuoti F. (2011) Molecular mechanisms of left ventricular hypertrophy (LVH) in systemic hypertension (SH)-possible therapeutic perspectives. J. Am. Soc. Hypertens. 5, 449–455 [DOI] [PubMed] [Google Scholar]
- 22. Chen Z., Hague C., Hall R. A., Minneman K. P. (2006) Syntrophins regulate α1D-adrenergic receptors through a PDZ domain-mediated interaction. J. Biol. Chem. 281, 12414–12420 [DOI] [PubMed] [Google Scholar]
- 23. Petzhold D., da Costa-Goncalves A. C., Gross V., Morano I. (2011) Spinophilin is required for normal morphology, Ca2+ homeostasis and contraction but dispensable for β-adrenergic stimulation of adult cardiomyocytes. J. Muscle Res. Cell Motil. 32, 243–248 [DOI] [PubMed] [Google Scholar]
- 24. Schillaci G., De Vuono S., Pucci G. (2011) An endogenous brake on the sympathetic nervous system: the emerging role of catestatin in hypertension. J. Cardiovasc. Med. (Hagerstown) 12, 609–612 [DOI] [PubMed] [Google Scholar]
- 25. Kempf T., Eden M., Strelau J., Naguib M., Willenbockel C., Tongers J., Heineke J., Kotlarz D., Xu J., Molkentin J. D., Niessen H. W., Drexler H., Wollert K. C. (2006) The transforming growth factor-β superfamily member growth-differentiation factor-15 protects the heart from ischemia/reperfusion injury. Circ. Res. 98, 351–360 [DOI] [PubMed] [Google Scholar]
- 26. Kempf T., Zarbock A., Widera C., Butz S., Stadtmann A., Rossaint J., Bolomini-Vittori M., Korf-Klingebiel M., Napp L. C., Hansen B., Kanwischer A., Bavendiek U., Beutel G., Hapke M., Sauer M. G., Laudanna C., Hogg N., Vestweber D., Wollert K. C. (2011) GDF-15 is an inhibitor of leukocyte integrin activation required for survival after myocardial infarction in mice. Nat Med. 17, 581–588 [DOI] [PubMed] [Google Scholar]
- 27. Howes A. L., Miyamoto S., Adams J. W., Woodcock E. A., Brown J. H. (2006) Gαq expression activates EGFR and induces Akt mediated cardiomyocyte survival: dissociation from Gαq mediated hypertrophy. J. Mol. Cell. Cardiol. 40, 597–604 [DOI] [PubMed] [Google Scholar]
- 28. Li Y., Zhang H., Liao W., Song Y., Ma X., Chen C., Lu Z., Li Z., Zhang Y. (2011) Transactivated EGFR mediates α-AR-induced STAT3 activation and cardiac hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 301, H1941–H1951 [DOI] [PubMed] [Google Scholar]
- 29. Noma T., Lemaire A., Naga Prasad S. V., Barki-Harrington L., Tilley D. G., Chen J., Le Corvoisier P., Violin J. D., Wei H., Lefkowitz R. J., Rockman H. A. (2007) β-arrestin1-mediated β1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J. Clin. Invest. 117, 2445–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McMurray J. J., Adamopoulos S., Anker S. D., Auricchio A., Böhm M., Dickstein K., Falk V., Filippatos G., Fonseca C., Gomez-Sanchez M. A., Jaarsma T., Køber L., Lip G. Y., Maggioni A. P., Parkhomenko A., Pieske B. M., Popescu B. A., Rønnevik P. K., Rutten F. H., Schwitter J., Seferovic P., Stepinska J., Trindade P. T., Voors A. A., Zannad F., Zeiher A., and ESC Committee for Practice Guidelines (2012) ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Eur. Heart J. 33, 1787–1847 [DOI] [PubMed] [Google Scholar]
- 31. Deleted in proof. [Google Scholar]
- 32. Mancia G., De Backer G., Dominiczak A., Cifkova R., Fagard R., Germano G., Grassi G., Heagerty A. M., Kjeldsen S. E., Laurent S., Narkiewicz K., Ruilope L., Rynkiewicz A., Schmieder R. E., Struijker Boudier H. A., Zanchetti A., Vahanian A., Camm J., De Caterina R., Dean V., Dickstein K., Filippatos G., Funck-Brentano C., Hellemans I., Kristensen S. D., McGregor K., Sechtem U., Silber S., Tendera M., Widimsky P., Zamorano J. L., Kjeldsen S. E., Erdine S., Narkiewicz K., Kiowski W., Agabiti-Rosei E., Ambrosioni E., Cifkova R., Dominiczak A., Fagard R., Heagerty A. M., Laurent S., Lindholm L. H., Mancia G., Manolis A., Nilsson P. M., Redon J., Schmieder R. E., Struijker-Boudier H. A., Viigimaa M., Filippatos G., Adamopoulos S., Agabiti-Rosei E., Ambrosioni E., Bertomeu V., Clement D., Erdine S., Farsang C., Gaita D., Kiowski W., Lip G., Mallion J. M., Manolis A. J., Nilsson P. M., O'Brien E., Ponikowski P., Redon J., Ruschitzka F., Tamargo J., van Zwieten P., Viigimaa M., Waeber B., Williams B., Zamorano J. L., The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH), and of the European Society of Cardiology (ESC) (2007) 2007 Guidelines for the management of arterial hypertension. Eur. Heart J. 28, 1462–1536 [DOI] [PubMed] [Google Scholar]
- 33. Muslin A. J. (2008) MAPK signalling in cardiovascular health and disease: molecular mechanisms and therapeutic targets. Clin. Sci. 115, 203–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Muslin A. J., DeBosch B. (2006) Role of Akt in cardiac growth and metabolism. Novartis Found. Symp. 274, 118–131 [PubMed] [Google Scholar]
- 35. Ruskoaho H., Leppäluoto J. (1988) Immunoreactive atrial natriuretic peptide in ventricles, atria, hypothalamus, and plasma of genetically hypertensive rats. Circ. Res. 62, 384–394 [DOI] [PubMed] [Google Scholar]
- 36. Xue H., Fu Z., Chen Y., Xing Y., Liu J., Zhu H., Zhou X. (2012) The association of growth differentiation factor-15 with left ventricular hypertrophy in hypertensive patients. PLoS One 7, e46534. [DOI] [PMC free article] [PubMed] [Google Scholar]