

Background: Tissue transglutaminase (tTG) promotes various aspects of oncogenesis, including cell survival.
Results: Ectopically expressed tTG in non-transformed cells triggers a survival response that involves c-Src and PI3-kinase.
Conclusion: tTG promotes survival by activating PI3-kinase through a c-Src-dependent mechanism.
Significance: These findings demonstrate that tTG has an intrinsic capability to promote cell survival and explains how it contributes to oncogenesis.
Keywords: Akt, Apoptosis, PI3-kinase (PI3K), Src, Transformation, Cell Survival, p70 S6-kinase, Tissue Transglutaminase
Abstract
Tissue transglutaminase (tTG) functions as a GTPase and an acyl transferase that catalyzes the formation of protein cross-links. tTG expression is frequently up-regulated in human cancer, where it has been implicated in various aspects of cancer progression, including cell survival and chemo-resistance. However, the extent to which tTG cooperates with other proteins within the context of a cancer cell, versus its intrinsic ability to confer transformed characteristics to cells, is poorly understood. To address this question, we asked what effect the ectopic expression of tTG in a non-transformed cellular background would have on the behavior of the cells. Using NIH3T3 fibroblasts stably expressing a Myc-tagged form of tTG, we found that tTG strongly protected these cells from serum starvation-induced apoptosis and triggered the activation of the PI3-kinase/mTOR Complex 1 (mTORC1)/p70 S6-kinase pathway. We determined that tTG forms a complex with the non-receptor tyrosine kinase c-Src and PI3-kinase, and that treating cells with inhibitors to block tTG function (monodansylcadaverine; MDC) or c-Src kinase activity (PP2) disrupted the formation of this complex, and prevented tTG from activating the PI3-kinase pathway. Moreover, treatment of fibroblasts over-expressing tTG with PP2, or with inhibitors that inactivate components of the PI3-kinase pathway, including PI3-kinase (LY294002) and mTORC1 (rapamycin), ablated the tTG-promoted survival of the cells. These findings demonstrate that tTG has an intrinsic capability to stimulate cell survival through a novel mechanism that activates PI3-kinase signaling events, thus highlighting tTG as a potential target for the treatment of human cancer.
Introduction
Tissue transglutaminase (tTG)3 is a protein that is capable of multiple catalytic activities. In particular, tTG can bind and hydrolyze GTP-like members of the large and small families of GTPases (i.e. Rho, Rac, Cdc42, and Ras) (1–3). It also exhibits a calcium-dependent acyl transferase activity (transamidation) that catalyzes the formation of an amide bond between the γ-carboximide group of a glutamine residue within one protein and the primary amino groups or the ϵ-amino group of a lysine residue within another protein (4, 5). Because its transamidation activity requires millimolar concentrations of calcium, it seems likely that this activity becomes most relevant when tTG is secreted from cells. tTG has been implicated in the regulation of a wide array of cellular processes, ranging from the maintenance of the extracellular matrix and cell adhesion to the induction of cellular differentiation and apoptosis (6–10). However, tTG has also been suggested to play crucial roles in the progression of a number of human disease states. In particular, during the past decade, several laboratories, including our own, have shown that increases in tTG expression are hallmarks of various types of human cancer including breast, brain, ovarian, and pancreatic cancers (11–16). In many of these same studies, it was also shown that knocking-down tTG expression by siRNA in cancer cell lines where it was aberrantly expressed, or treating the cells with the small molecule MDC, which binds as a competitive inhibitor/substrate at the transamidation active site of tTG, either ablated the growth of the cancer cells or made them more sensitive to chemotherapy and other types of apoptotic-inducing cellular stress (11–13, 16).
The indications that the overexpression of tTG contributes to tumor progression and metastasis raise an important question, namely to what extent are the contributions of tTG to cancer progression shaped by the cancer cell context and the various signaling proteins present within transformed cells, versus the intrinsic ability of tTG to alter normal cellular behavior. Indeed, various studies have suggested that tTG can work together with different signaling proteins in the background of a cancer cell (17–20). One example from studies performed in our laboratory involves the ability of tTG to influence the transformed characteristics of human breast cancer cells. In particular, we discovered, when using the human SKBR3 breast cancer cell line as a model, that tTG expression and activation were strongly up-regulated in an epidermal growth factor (EGF)-dependent manner. Moreover, tTG was essential for the EGF-stimulated growth of these cancer cells in monolayer, as well as for their anchorage-independent growth and importantly, their survival in the face of stress conditions and apoptotic challenges such as chemotherapeutic agents (20). We then demonstrated that a key element in the transformed characteristics of these breast cancer cells, as imparted by tTG, was its ability to form a complex with the non-receptor tyrosine kinase and proto-oncogene c-Src.
Here, we have set out to determine whether tTG has the ability to alter the behavior of non-transformed cells, as a means of obtaining insights into the capability of this protein in the absence of a cancer cell context to induce characteristics necessary for malignant transformation. To address this important question, we have examined the biological consequences of ectopically expressing tTG in NIH3T3 cells, a fibroblast cell line. Interestingly, we found that tTG strongly promoted NIH3T3 cell survival by enhancing the activation of the canonical PI3-kinase/mTORC1/p70 S6-kinase pathway. We then went on to demonstrate that the ability of tTG to activate this signaling pathway was through the assembly of a complex consisting of tTG, c-Src, and PI3-kinase. Importantly, treating the cells with either the Src inhibitor, PP2, or MDC, disrupted the interaction between c-Src and tTG, as well as blocked the ability of tTG to stimulate PI3-kinase-mediated signaling events. Thus, these findings point to tTG as being a key participant in a c-Src-PI3-kinase signaling pathway and that it is able to associate with these signaling proteins even in a non-transformed cellular context. This capability is likely to have important consequences for enabling cancer cells to survive various apoptotic challenges including treatment with chemotherapeutic drugs.
EXPERIMENTAL PROCEDURES
Materials
All cell culture reagents (unless mentioned otherwise), Lipofectamine, EGF, and protein G-agarose beads were from Invitrogen. Monodansylcadaverine (MDC) and 6-diamidino-2-phenylindole (DAPI) were obtained from Sigma, while PP2, LY294002, and rapamycin were from Calbiochem. Biotinylated pentylamine (BPA) was obtained from Pierce, and the Myc and HA antibodies were from Covance. The Ras, pan-p70 S6-kinase, and p85 antibodies were from Millipore, the tTG antibody was from Zedira, the p110 antibody was from Santa Cruz Biotechnology, and the actin antibody was from Neomarkers. The anti-phosphotyrosine, cleaved caspase-3, and cleaved PARP antibodies, as well as the antibodies that recognize the total, activated, and/or phosphorylated forms of ERK (Thr-202/Tyr-204), c-Jun (Ser-63/Ser-73), AKT (Thr-308 and Ser-473), mTOR (S2448), p70 S6-kinase (Thr-389), p85 (Tyr-458), and PTEN (Ser-380/Thr-382/Thr-383) were from Cell Signaling.
Cell Culture
Parental NIH3T3 cells were grown in DMEM containing 10% calf serum (CS), while HEK293T cells were grown in DMEM containing 10% fetal bovine serum (FBS). The pcDNA3 constructs encoding the Myc-tagged forms of wild-type tTG or tTG C277V and the HA-tagged forms of H-Ras G12V, c-Src, v-Src, p85, or p110α, were transfected into cells using Lipofectamine. Clones of NIH3T3 mouse fibroblasts stably expressing the vector alone or a Myc-tagged form of wild-type tTG were selected by culturing the cells in DMEM containing 10% CS and 2 μg/ml puromycin. Once individual clones expressing either the vector alone or the Myc-tagged form of tTG were obtained, the cells were then maintained in the same growth medium supplemented with 0.5 μg/ml puromycin. Where indicated, cells were treated with 50 μm MDC, 0.1 μg/ml EGF, 10 μm PP2, 10 μm LY294002, or 50 nm rapamycin. Cells were lysed with cell lysis buffer (25 mm Tris, 100 mm NaCl, 1% Triton X-100, 1 mm EDTA, 1 mm DTT, 1 mm NaVO4, 1 mm β-glycerol phosphate, 1 μg/ml aprotinin, 1 μg/ml leupeptin). The Bio-Rad DC protein assay was used to determine the protein concentrations of the cell lysates.
Transamidation Activity Assays
Fifteen micrograms of whole cell extracts collected from parental NIH3T3 cells or NIH3T3 cells stably expressing the vector alone or a Myc-tagged form of tTG, were incubated in a buffer containing 10 mm dithiothreitol, 10 mm CaCl2, and 50 μm BPA for 10 min followed by the addition of Laemmli sample buffer. The samples were boiled, subjected to SDS-PAGE, transferred to polyvinylidene fluoride (PVDF) membranes, and blocked for 1 day in BBST (100 mm boric acid, 20 mm sodium borate, 0.01% SDS, 0.01% Tween 20, 80 mm NaCl) containing 10% bovine serum albumin (BSA). The PVDF membranes were then incubated with horseradish-peroxidase-conjugated streptavidin at a dilution of 1:2000 in BBST containing 5% BSA for 1 h, followed by extensive washing with BBST. The membranes were then exposed to ECL reagent, and the proteins that had incorporated BPA were visualized using x-ray film.
Focus Formation Assays
Nearly confluent cultures of fibroblasts were transfected without (Mock) or with expression plasmids encoding a Myc-tagged form of tTG or an HA-tagged form of activated H-Ras (H-Ras G12V) and were maintained in DMEM containing 10% CS for 10 days. The cells were then fixed with 3.7% formaldehyde and stained with 0.4% crystal violet to visualize any resulting foci that formed.
Cell Migration (Scratch) Assays
Multiple sets of NIH3T3 cells stably expressing either the vector alone or a Myc-tagged form of tTG were grown to confluence, at which time a wound was struck down the center of each plate using a pipette tip and the cultures rinsed with phosphate-buffered saline to remove the detached cells. One set of cells was immediately fixed with 3.7% formaldehyde after striking the wound to indicate the size of the initial wound. The remaining sets of cells were then placed in DMEM containing 0.1% CS for 1 day before being fixed. The ability of the cells to migrate into the wound was visualized by light microscopy and photographed.
Cell Growth Assays
Multiple sets of NIH3T3 cells stably expressing the vector alone or a Myc-tagged form of tTG were plated in 6-well dishes at a density of 2 × 104 cells/dish and maintained in DMEM containing 0.1% CS. One set of the cells was counted every 2 days, while the medium on the remaining sets of cells was replenished. The growth assays were carried-out over a span of 6 days.
Apoptotic Assays
NIH3T3 cells stably expressing either the vector alone or a Myc-tagged form of tTG were seeded in 6-well dishes and then maintained in serum-free medium without or with PP2, LY294002, MDC or rapamycin for the indicated lengths of time. The cells (both floating and attached) were then collected and stained with DAPI (2 μg/ml) for viewing by fluorescence microscopy. Apoptotic cells were identified by condensed and/or blebbed nuclei.
PI3-kinase Activity Assays
Serum-starved cultures of NIH3T3 cells stably expressing the vector alone or a Myc-tagged form of tTG were treated without or with EGF for 10 min and lysed. Immunoprecipitations using a p85 antibody were performed on the cell lysates, and then the immunocomplexes were subjected to kinase reactions through the addition of ATP and PI(4,5)P2. The resulting production of PI(3,4,5)P3 was quantified using an ELISA-based assay kit obtained from Echelon Biosciences. All procedures were carried out according to the manufacturer's instructions.
Immunoprecipitations
Cell lysates (typically 1.2 mg) were initially pre-cleared using protein G-agarose beads. The pre-cleared lysates were then incubated with an antibody for 1.5 h, followed by the addition of protein G-agarose beads for another 1.5 h. After extensive washing of the beads with cell lysis buffer, Laemmli sample buffer was added to the beads and the samples were boiled.
Immunoblot Analysis
Whole cell lysates (60 μg of each) and the resulting immunoprecipitates were resolved by SDS-PAGE, and the proteins were transferred to PVDF membranes. The membranes were incubated in primary antibodies prepared in 20 mm Tris, 135 mm NaCl, and 0.02% Tween 20. Horseradish-peroxidase conjugated secondary antibodies were used to detect the primary antibodies, followed by exposure to ECL reagent.
Statistical Analysis
All assays/experiments were performed a minimum of three separate times and many of the results were presented as bar graphs. In these cases, the error bars indicate standard deviation. Statistical analyses of the results were done using Excel to perform Student's t-tests. p values <0.05 were considered statistically significant and were indicated with asterisks.
RESULTS
Ectopic Expression of tTG in Fibroblasts Promotes Cell Survival
Increases in tTG expression and activation occur in several different types of human cancer resulting in a wide range of potential interactions and cross-talk with other proteins that drive transformation (12, 14, 20–22). This has contributed to some confusion in the field regarding the many possible roles that tTG plays in cancer progression. In particular, it has been extremely difficult to identify the inherent and fundamental actions of tTG that contribute to the development of the malignant state. To determine the intrinsic capability of tTG to mediate actions relevant to cancer progression, we examined the effects of overexpressing this protein in a non-transformed cell type. The NIH3T3 mouse fibroblast cell line was chosen as our model system for this study, as it is commonly used to read-out various types of cellular outcomes (23–25). Fig. 1A (top panel, first lane) shows that these cells express low, but detectable, levels of tTG. We then generated two different NIH3T3 stable cell lines; one expressing the vector alone and the other expressing a Myc-tagged form of wild-type tTG (Fig. 1A, second panel from the top, second and third lanes). The tTG ectopically expressed in the fibroblasts is functionally active, as indicated by its ability to catalyze the calcium-dependent incorporation of biotinylated pentylamine (BPA) into lysate proteins, as compared with parental or vector alone-expressing fibroblasts, which exhibit little detectable activity in this assay (Fig. 1A, bottom panel).
FIGURE 1.

Ectopic expression of tTG in NIH3T3 fibroblasts promotes cell growth and survival. A, whole cell lysates of parental NIH3T3 cells, or NIH3T3 cells stably expressing the vector alone or a Myc-tagged form of tTG, were immunoblotted with tTG, Myc, and actin antibodies. The same cell lysates were also assayed for transamidation (cross-linking) activity by determining the incorporation of BPA into lysate proteins as described in “Experimental Procedures.” B, focus formation assays were carried-out on parental fibroblasts that were transiently transfected without (Mock), or with expression plasmids encoding either Myc-tagged tTG (tTG), or an HA-tagged activated form of Ras (H-Ras G12V). The cells were maintained in DMEM supplemented with 10% CS for 10 days, at which time they were fixed and stained with crystal violet. Shown are representative images of the resulting foci that formed for each condition. C, cell migration (scratch) assays were performed on NIH3T3 cells stably expressing the vector alone or a Myc-tagged form of tTG. Twenty-four hours after striking the wound, the cells were fixed and then visualized by light microscopy to determine the extent of wound closure. One set of vector alone-expressing fibroblasts was fixed immediately after striking the wound (Control 0 h.) to indicate the width of the initial wound (indicated by dashed lines). D, cultures of the NIH3T3 cells stably expressing the vector alone or a Myc-tagged form of tTG were placed in serum-free medium for the indicated lengths of time, at which point they were collected and stained with DAPI to identify condensed and/or blebbed nuclei. Percent apoptosis was determined by calculating the ratio of apoptotic to non-apoptotic cells. The experiments were performed in triplicate, and the results were averaged. The error bars indicate standard deviation, and the p values determined for the different conditions are as follows; *, p < 0.05 and **, p < 0.01. E, stable cell lines were cultured in serum free medium for the indicated lengths of time and lysed. The extracts were then immunoblotted with Myc, actin, cleaved caspase-3, and cleaved PARP antibodies. F, growth in low serum assays were performed on NIH3T3 cells stably expressing the vector alone or Myc-tagged tTG by plating them at a density of 2 × 104 cells/dish in 6-well dishes and then placing them in DMEM containing 0.1% CS. Every other day for 6 days, one set of cells was counted, while on the remaining sets of cells the medium was replenished. The experiments were performed in triplicate, and the results were averaged together and graphed. The error bars indicate standard deviation, and the p values determined for the different conditions are as follows; *, p < 0.05 and **, p < 0.01.
Previous work from our laboratory had suggested that over-expressing tTG in non-transformed cell lines was not sufficient to induce transformation, as indicated by their inability to exhibit anchorage-independent growth (i.e. as assayed by colony formation in soft agar) (26). Here, we followed-up on these findings by performing focus formation assays on NIH3T3 cells ectopically expressing tTG. The ability of cells to form foci (i.e. distinct areas of high cell density) represents another indicator of cellular transformation that measures the ability of cells to overcome the contact inhibition exhibited by non-transformed cells when grown in monolayer. For these experiments, cultures of parental fibroblasts were transiently transfected without (Mock), or with expression plasmids encoding either tTG (tTG) or an oncogenic form of Ras (H-Ras G12V), and then were maintained in normal growth medium (DMEM containing 10% CS) for 10 days. The resulting cell cultures were then fixed and stained with crystal violet to highlight any differences in cell densities (i.e. foci) that might have occurred as an outcome of expressing tTG or activated H-Ras in the cells. As anticipated, fibroblasts expressing oncogenic Ras (H-Ras G12V) formed numerous foci (Fig. 1B, bottom panel), while neither the control fibroblasts (Mock) (Fig. 1B, top panel), nor the NIH3T3 cells transfected with the tTG plasmid (Fig. 1B, middle panel), showed detectable foci.
Given that tTG has been shown to localize to the leading edges of actively migrating cancer cells where it promotes the EGF-stimulated migration of the HeLa cervical carcinoma cell line, as well as the constitutive migration exhibited by the MDAMB231 breast cancer cell line (17, 27), we next examined whether the over-expression of tTG in NIH3T3 cells would enhance their ability to migrate. The NIH3T3 cells stably expressing the vector alone or a Myc-tagged form of tTG were subjected to wound healing (scratch) assays to determine whether there was any difference in the rate at which these cell lines migrated. Fig. 1C shows that the extent of cell migration into the wound by fibroblasts expressing the vector alone, versus Myc-tagged tTG, was similar, suggesting that tTG is not important for promoting the general migration of these cells.
We then examined the ability of tTG to impact cell survival. Serum starvation is a stress that induces a cell death response in a number of cell types, including NIH3T3 fibroblasts (26, 28). Thus, we took cultures of NIH3T3 cells stably expressing the vector alone or Myc-tagged tTG and placed them in serum-free medium for increasing lengths of time ranging from 0 to 48 h. The cells were then collected, and the extent of cell death was determined by staining the cells with DAPI and examining them for the appearance of condensed and/or blebbed nuclei, an indicator of apoptosis. Fig. 1D shows that the vector alone-expressing cells undergo a time-dependent apoptotic response, with ∼75% of the cells dying after being cultured in medium lacking serum for 48 h. In contrast, ∼20% of the tTG-expressing fibroblasts were apoptotic under the same culturing conditions, suggesting tTG strongly promotes the survival of the fibroblasts. To further substantiate these findings, the stable cell lines, after being subjected to a similar serum starvation time course, were collected and analyzed by Western blot analysis for the presence of the apoptotic markers cleaved caspase-3 and cleaved PARP. Fig. 1E shows that cleaved forms of caspase-3 (second panel from the top) and PARP (third panel from the top) start to be detectable in the fibroblasts expressing the vector alone by 24 h of serum deprivation, and are near maximal at 36 h. In contrast, the amounts of cleaved caspase-3 and PARP detected in the cells expressing tTG remained low throughout the duration of the experiment.
The growth rates of the stable cell lines maintained in medium containing 0.1% serum (low serum conditions) over the course of 6 days were also assessed. The results in Fig. 1F show that tTG promoted the growth of fibroblasts cultured under low serum conditions, as evidenced by the continued growth of the cells stably expressing tTG, whereas, the growth rate of the cells expressing the vector alone was stunted. These data suggest that tTG expression in a normal non-transformed cell type is sufficient to promote some aspects of cellular transformation, in particular, cell survival, and growth in low serum.
tTG Activates PI3-kinase
To further explore how tTG promotes cell survival, the NIH3T3 stable cell lines were maintained in serum-free medium for 24 h and lysed. The whole cell lysates were then subjected to Western blot analysis using antibodies which detect the activated or phosphorylated forms of several traditional signaling proteins known to promote cell survival, with the expectation being that if tTG promotes cell viability by activating certain signaling proteins, then we should be able to detect an increase in the activation/phosphorylation of these proteins in fibroblasts expressing Myc-tagged tTG, compared with cells expressing the vector alone. The proteins whose activities were examined included members of the mitogen-activated protein (MAP) kinase family, specifically, extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK), as well as components of the canonical PI3-kinase signaling cascade, namely, AKT, mammalian target of rapamycin (mTOR), and p70 S6-kinase (p70S6K). Fig. 2A shows that when using antibodies recognizing the activated form of ERK, as well as the phosphorylation of the transcription factor c-Jun at Ser-63 and Ser-73 (i.e. two known JNK phosphorylation sites), we found that neither of these MAP kinase family members were activated by tTG overexpression (third and fourth panels from the top, respectively). However, components of the PI3-kinase pathway were significantly affected. Specifically, fibroblasts ectopically expressing tTG showed higher levels of AKT (fifth and sixth panels from the top), mTOR (eighth panel from the top), and p70 S6-kinase phosphorylation (tenth panel from the top) compared with control cells, while the overall expression levels of each of these signaling proteins remained constant (seventh, ninth, and eleventh panels from the top, respectively).
FIGURE 2.

tTG promotes activation of the PI3-kinase/mTOR/p70 S6-kinase pathway. A, NIH3T3 cells stably expressing the vector alone or a Myc-tagged form of tTG were placed in serum-free medium for 24 h, at which time they were lysed and subjected to Western blot analysis using the indicated antibodies. B, serum-starved cultures of the same stable cell lines were left untreated or were treated with EGF for 10 min prior to being lysed. Immunoprecipitations using a p85 antibody (IP: p85) were carried-out on the cell extracts (1.2 mg), as was a beads only (no antibody) immunoprecipitation performed on lysates collected from EGF-stimulated NIH3T3 cells expressing the vector alone (the negative control for these experiments). The resulting immunocomplexes and 60 μg of each whole cell lysate (WCL) were subjected to Western blot analysis using the indicated antibodies (left panels). The immunocomplexes were also subjected to kinase reactions (right panel). The amount of PI(3,4,5)P3 generated by the samples was read out by ELISA. The assays were performed in triplicate, and the results were averaged together and graphed. The error bars indicate standard deviation, and the p values determined for the different conditions are as follows; *, p < 0.05. C, stable cell lines were cultured in serum-free medium supplemented without (not treated; NT) or with MDC for 24 h at which time they were lysed. The whole cell lysates were then subjected to Western blot analysis using the indicated antibodies.
We examined whether PI3-kinase activity is up-regulated in the cells expressing tTG. Serum-starved cultures of NIH3T3 cells stably expressing the vector alone or a Myc-tagged form of tTG were either left untreated or were stimulated with EGF for 10 min prior to being lysed. Immunoprecipitations using a p85 antibody were carried out on the cell extracts, and then the resulting immunocomplexes, together with the whole cell extracts, were immunoblotted with an antibody that detects p85 and another that recognizes the different isoforms of p110. Fig. 2B (left panels) shows that the levels of p85 and p110 isoforms detected in the stable cell lines (WCL) were similar (third and fourth panels from the top), as was the amount of p85 that was immunoprecipitated from each of the lysates (top panel). The immunocomplexes were then subjected to kinase reactions, where the amount of PI(3,4,5)P3 generated by each sample was determined using an ELISA-based detection assay. Fig. 2B (right panel) shows that PI3-kinase activity in the cells expressing the vector alone is low and is comparable to the background levels of activity seen in the negative control (i.e. immunoprecipitations without p85 antibody performed on lysates from EGF-stimulated NIH3T3 cells expressing the vector alone). However, the cells expressing tTG exhibited an ∼4-fold increase in PI3-kinase activity. In fact, the level of PI3-kinase activity detected in these cells was similar to that obtained in the positive control, where cells expressing the vector alone were treated with EGF. Thus, these results demonstrate that PI3-kinase activity is increased in the cells overexpressing tTG, and is consistent with the increases in the phosphorylations of AKT, mTOR, and p70 S6-kinase detected in these cells (Fig. 2A).
To further confirm that these results were due to the ectopic expression of tTG, we treated the stable cell lines without or with the tTG inhibitor, MDC, which binds at the transamidation active site. Fig. 2C shows that MDC treatment blocked the increases in AKT, mTOR, and p70 S6-kinase activities observed in fibroblasts over-expressing tTG (third, fourth, sixth, and eighth panels from the top, respectively), again without altering the expression levels of each of these proteins (fifth, seventh, and ninth panels from the top, respectively).
The Ability of tTG to Activate the PI3-kinase Pathway Requires Src Kinase Activity
We wanted to learn more about how the overexpression of tTG in NIH3T3 cells leads to increased activation of PI3-kinase and several of its downstream effectors, namely AKT, mTOR, and p70 S6-kinase. Based on a recent suggestion that tTG can down-regulate the expression of the lipid phosphatase and major negative regulator of PI3-kinase signaling, PTEN (for phosphatase and tensin homologue deleted on chromosome ten), in pancreatic cancer cells (22), we first examined whether tTG affected PTEN expression in a manner that might promote PI3-kinase signaling activity in NIH3T3 cells. However, this was ruled out when we compared the levels of PTEN in the lysates collected from the NIH3T3 fibroblasts expressing the vector alone, to those expressing Myc-tagged tTG, and found that they were similar (Fig. 2A, bottom panel). Moreover, we also determined whether the phosphorylation of PTEN was increased in fibroblasts overexpressing tTG, as the phosphorylation of PTEN in its C-terminal tail has been shown to inhibit its phosphatase activity, as well as enhance its stability (29, 30). However, the relative amounts of phosphorylated PTEN detected in these same whole cell lysates were nearly identical (Fig. 2A, second panel from the bottom), further suggesting that tTG is not stimulating the PI3-kinase pathway by inhibiting PTEN.
The non-receptor tyrosine kinase c-Src, as well as other members of the Src family, can bind and phosphorylate the p85 regulatory subunit of PI3-kinase (31–33). This phosphorylation event is believed to induce a conformational change in p85 that allows the p110 catalytic subunit of PI3-kinase to become activated and signal to its downstream effectors (i.e. AKT, mTOR, and p70 S6-kinase) (34, 35). Moreover, it has been shown that the ability of Src to stimulate PI3-kinase activity is critical for Src-mediated cellular transformation, highlighting that these two proteins can participate in a common signaling pathway important for malignant transformation (36, 37). Interestingly, our laboratory recently reported that tTG can bind and activate c-Src in human SKBR3 breast cancer cells stimulated with EGF (20). Since c-Src interacts with both PI3-kinase and tTG, and the overexpression of tTG in fibroblasts enhances the activation of the PI3-kinase pathway, we asked whether Src activity was important for the ability of tTG to stimulate PI3-kinase/mTOR/p70 S6-kinase signaling. To address this question, serum-starved fibroblasts stably expressing the vector alone or Myc-tagged tTG were incubated without or with the Src kinase inhibitor PP2, before being lysed and then immunoblotted to read-out the effects of this treatment on p70 S6-kinase activity. Fig. 3A shows that inhibiting Src in cells expressing Myc-tagged tTG reduced the levels of p70 S6-kinase activity (third panel from the top) to those observed in control cells expressing the vector alone. Thus, c-Src kinase activity is indeed necessary for tTG to activate the PI3-kinase pathway.
FIGURE 3.

Src activity is required for tTG-stimulated PI3-kinase activation. A, NIH3T3 cells stably expressing the vector alone or Myc-tagged tTG were placed in serum-free medium for 12 h, at which time they were treated without (not treated; NT) or with PP2 for an additional 12 h. The cells were then lysed, and the cell extracts were subjected to Western blot analysis using the indicated antibodies. B, stable cell lines were transfected without (Mock) or with an HA-tagged constitutively active form of Src (v-Src) and then were maintained in serum-free medium for 12 h. The cells were lysed, and the extracts subjected to Western blot analysis using the indicated antibodies.
This finding prompted us to then determine whether the activation of c-Src was sufficient to mimic the effects of tTG and stimulate PI3-kinase-mediated signaling events. NIH3T3 cells stably expressing the vector alone or a Myc-tagged form of tTG were transiently transfected without (Mock) or with a plasmid encoding HA-tagged viral Src that lacks a critical C-terminal inhibitory phosphorylation site, making it constitutively active (HA-v-Src) (38). The transfectants were maintained in serum-free medium for 12 h, before being lysed and subjected to Western blot analysis. Using p70 S6-kinase activation as the read-out for activation of the PI3-kinase pathway, we found that despite the relatively high expression of HA-tagged v-Src in the vector alone-expressing cells (Fig. 3B, second panel from the top), only a modest increase in p70 S6-kinase activity (fourth panel from the top) was detected, that was significantly less than the activity observed in fibroblasts overexpressing tTG. It is also worth noting that the ectopic expression of HA-tagged v-Src in cells stably expressing tTG did not enhance p70 S6-kinase activity beyond that observed in cells stably expressing tTG alone. Taken together, these results suggest that while c-Src kinase activity is necessary for tTG to activate PI3-kinase, it is not sufficient to completely mimic the effects of tTG.
tTG Forms a Complex with Src and PI3-kinase
How then does tTG cooperate with c-Src to activate the PI3-kinase pathway? Since Src was previously shown to bind and phosphorylate the p85 regulatory subunit of PI3-kinase, which induces the activation of its p110 catalytic subunit, we examined the possibility that tTG stimulates PI3-kinase activity by binding to c-Src and enhancing its ability to phosphorylate p85. Whole cell lysates generated from HEK293T cells that had been transiently transfected with a Myc-tagged tTG construct, alone, or together with an HA-tagged c-Src construct (Fig. 4A, left panels), were subjected to immunoprecipitations using an HA antibody. Fig. 4A (right panels) shows that, indeed, tTG co-immunoprecipitates with c-Src, similar to what our laboratory has shown in the past (20). Surprisingly, we also found that tTG is capable of interacting with PI3-kinase. For these experiments, rather than transiently expressing Myc-tagged tTG together with HA-tagged c-Src, we instead co-expressed Myc-tagged tTG together with one of the two subunits of PI3-kinase; the p110α catalytic subunit (HA-tagged p110) or the p85 regulatory subunit (HA-tagged p85). Fig. 4B (left panels) shows that the ectopic expression of these constructs in cells was similar. Immunoprecipitations using an HA antibody performed on the whole cell lysates prepared from these transfectants showed that tTG co-immunoprecipitates with both of the PI3-kinase subunits (Fig. 4B, right panels).
FIGURE 4.

tTG binds Src and PI3-kinase. A, immunoprecipitations were performed using an HA antibody on whole cell lysates (1.2 mg) collected from HEK293T cells ectopically expressing Myc-tagged tTG, or Myc-tagged tTG and HA-tagged c-Src. A beads only control was included to confirm the specificity of the interaction. The resulting immunocomplexes (IP:HA) and 60 μg of each whole cell lysate (WCL) were blotted with Myc and HA antibodies. B, immunoprecipitations were performed using an HA antibody on extracts (1.2 mg) collected from HEK293T cells ectopically expressing Myc-tagged tTG, or Myc-tagged tTG together with either the HA-tagged form of the p110 catalytic subunit of PI3-kinase (HA-p110) or the p85 regulatory subunit of PI3-kinase (HA-p85). A beads-only control was included to confirm the specificity of the interactions. The resulting immunocomplexes (IP:HA) and 60 μg of each whole cell lysate (WCL) were blotted with Myc and HA antibodies. C, immunoprecipitations using a Myc antibody were performed on extracts (1.2 mg) collected from NIH3T3 cells stably expressing a Myc-tagged form of tTG that were further transfected with HA-tagged forms of the p110 catalytic subunit of PI3-kinase (HA-p110), the p85 regulatory subunit of PI3-kinase (HA-p85), or c-Src (HA-c-Src). Similar immunoprecipitations were performed on extracts (1.2 mg) collected from NIH3T3 cells expressing the vector alone that were further transfected with the same constructs to confirm the specificity of the interactions. The resulting immunocomplexes (IP:Myc) and 60 μg of each whole cell lysate (WCL) were blotted with Myc and HA antibodies.
These same protein interactions also occur in NIH3T3 cells. HA-tagged forms of c-Src, p85, and p110 were transiently transfected into the fibroblasts stably expressing either Myc-tagged tTG or the vector alone (Fig. 4C, WCL panels) and the cell lysates generated from these cell cultures were subjected to immunoprecipitation using a Myc antibody. Fig. 4C (IP: Myc panels) shows that c-Src, as well as the PI3-kinase subunits, p85 and p110, immunoprecipitates with Myc-tagged tTG in NIH3T3 mouse fibroblasts.
Is the formation of a complex between tTG, Src, and PI3-kinase responsible for the enhanced PI3-kinase signaling observed in the tTG-expressing fibroblasts? Given that the tTG inhibitor MDC and the Src inhibitor PP2 can abrogate the enhanced PI3-kinase signaling observed in fibroblasts stably expressing tTG, we reasoned that if the formation of a trimeric complex was important for this outcome, MDC and PP2 might then inhibit its formation. To test this, tTG was expressed alone, or together with c-Src or p85, in HEK293T cells and then the cells were treated without or with either PP2 or MDC for 6 h prior to being lysed. The cell extracts were then incubated with an HA antibody to immunoprecipitate either the HA-tagged forms of c-Src or p85 expressed in the cell lysates (Fig. 5A, second and third panels from the top, respectively). The resulting immunocomplexes were also immunoblotted with a Myc antibody to detect the extent that tTG interacted with c-Src or p85 under conditions where Src or tTG activity was inhibited. Fig. 5A (top panel) shows that the interaction between tTG and p85 was insensitive to treatment with either PP2 or MDC, suggesting that the ability of these to proteins to interact with one another does not require c-Src kinase activity, nor access to the transamidation active site of tTG (i.e. that would be blocked by the competitive inhibitor MDC). On the other hand, PP2 and MDC blocked the ability of tTG to co-immunoprecipitate with Src. Importantly, Fig. 5B (right side) shows that a transamidation (crosslinking)-defective form of tTG generated by mutating the essential active site cysteine to valine (Myc-tagged tTG C277V) is still capable of co-immunoprecipitating with HA-tagged c-Src in HEK293T cells, suggesting that the transamidation activity of tTG, per se, is not required for this interaction to occur. Rather, it appears that the interaction of MDC with tTG in some way (e.g. sterically) blocks its ability to form a stable complex with c-Src.
FIGURE 5.

Inhibiting tTG or Src activity blocks the ability of tTG to bind Src and the ability of Src to phosphorylate p85. A, HEK293T cells ectopically expressing Myc-tagged tTG (Myc-tTG), Myc-tagged tTG and HA-tagged c-Src (Myc-tTG/HA-c-Src), or Myc-tagged tTG and the HA-tagged p85 regulatory subunit of PI3-kinase (Myc-tTG/HA-p85), were treated without (not treated; NT) or with PP2 or MDC for 6 h and then lysed. The cell lysates (1.2 mg) were subjected to immunoprecipitations using an HA antibody. A beads-only control was included to confirm the specificity of the interactions. The resulting immunocomplexes (IP:HA) and 60 μg of each whole cell lysate (WCL) were blotted using Myc and HA antibodies. B, immunoprecipitations were performed using an HA antibody on whole cell lysates (1.2 mg) collected from HEK293T cells ectopically expressing various combinations of Myc-tagged tTG wild-type (WT), Myc-tagged tTG C277V, and HA-tagged c-Src. A beads-only control was included to confirm the specificity of the interaction. The resulting immunocomplexes (IP:HA) and 60 μg of each whole cell lysate (WCL) were blotted with Myc and HA antibodies. C, HEK293T cells co-expressing Myc-tagged tTG and HA-tagged p85 (Myc-tTG/HA-p85) were treated without (not treated; NT) or with PP2 or MDC for 6 h and then lysed. The lysates (1.2 mg) were subjected to immunoprecipitations using an HA antibody. 60 μg of each whole cell lysate (WCL) was blotted with Myc and HA antibodies. The resulting immunocomplexes (IP:HA) were first blotted with a phosphotyrosine antibody. The blot was then stripped and re-probed with an HA antibody to confirm that an equal amount of p85 was immunoprecipitated for each condition. D, experiment shown in C was performed in triplicate and the extent of HA-tagged p85 tyrosine phosphorylation detected by Western blot analysis was quantified for each condition and averaged and graphed. The error bars represent standard deviation, and the p values determined for the different conditions are as follows; *, p < 0.05 and **, p < 0.01. E, NIH3T3 cells stably overexpressing Myc-tagged tTG were treated without (not treated; NT) or with PP2 or MDC for 12 h and then lysed. The extracts were subjected to Western blot analysis using an antibody that recognizes a tyrosine-phosphorylated form of p85 (p-p85 (Y458)), as well as ones that detect total p85 (pan-p85), Myc-tTG, and actin.
Src has been shown to phosphorylate p85 and to stimulate the signaling capability of PI3-kinase (31). This, coupled with the fact that we find tTG is able to form a complex with both PI3-kinase and c-Src, raised the interesting possibility that tTG functions to help mediate the activation of PI3 kinase by c-Src. To test this idea, Myc-tagged tTG was transiently expressed along with HA-tagged p85 in HEK293T cells, and then the cells were incubated without or with PP2 or MDC. The cells were lysed and the HA-tagged p85 was immunoprecipitated from the extracts using an HA antibody. The resulting immunocomplexes were first immunoblotted with a phosphotyrosine antibody to detect whether p85 was phosphorylated. Fig. 5C (top panel) shows that p85 is indeed phosphorylated in the control lysates (not treated; NT), whereas the tyrosine phosphorylation is decreased by ∼80% with PP2 treatment and ∼60% with MDC. The blot was then stripped and re-probed with HA antibody to confirm that an equivalent amount of p85 was immunoprecipitated for each condition (Fig. 5C, second panel from the top). These experiments were performed multiple times and the results from each experiment were quantified and graphed (Fig. 5D).
The ability of tTG to promote the c-Src-dependent phosphorylation of p85 was also seen in the NIH3T3 fibroblasts stably expressing Myc-tTG. For these experiments, the cells were treated without or with PP2 or MDC, similar to what we had done in the HEK293T cells, and the effects of the inhibitors on the phosphorylation of endogenously expressed p85 was read-out using an antibody that recognizes p85 when it is phosphorylated on tyrosine 458, a known c-Src phosphorylation site (31). Fig. 5E (second panel from the top) shows that PP2 and MDC treatment reduced the extent of p85 phosphorylation in NIH3T3 cells overexpressing tTG, as compared with cells not-treated with inhibitors (NT).
Inhibiting tTG, Src, and Components of the PI3-kinase Pathway Eliminate tTG-enhanced Cell Survival
The overexpression of tTG in NIH3T3 cells protects them from serum-starvation-induced apoptosis, as well as increases the activities of AKT, mTORC1, and p70 S6-kinase, leading us to propose that tTG-promoted cell survival is dependent on its ability to activate the PI3-kinase pathway. Thus, we examined whether treating serum-starved fibroblasts expressing tTG with inhibitors of different components of this pathway, including PP2 (to inhibit Src activity), MDC (to block the transamidation active site of tTG), LY294002 (to inhibit PI3-kinase activity), and rapamycin (which specifically inhibits mTORC1), eliminated the protective effects of tTG and re-sensitized the cells to serum-starvation-induced apoptosis. Fig. 6A shows again that tTG overexpression strongly prevented the cells deprived of serum from undergoing cell death. However, the protective effect of tTG was completely ablated when the cells were treated with PP2. Likewise, MDC, LY294002, and rapamycin treatment blocked the ability of tTG to promote cell survival (Fig. 6B), indicating that this tTG-dependent effect requires its ability to activate the PI3-kinase signaling pathway, thus leading to the model shown in Fig. 7. tTG binds to c-Src and PI3-kinase to help facilitate the c-Src-dependent phosphorylation of the p85 regulatory subunit of PI3-kinase. This gives rise to the activation of the p110 catalytic subunit and its downstream effectors, AKT, mTORC1, and p70 S6-kinase, which ultimately results in enhanced cell survival.
FIGURE 6.

Inhibition of Src, PI3-kinase, or mTORC1 blocks the tTG-enhanced cell survival. A and B, NIH3T3 cells stably expressing the vector alone or Myc-tagged tTG were placed in serum-free medium supplemented without (not treated; NT) or with PP2, LY294002, MDC, or rapamycin for 36 h, at which time the cells were collected and stained with DAPI to identify condensed and/or blebbed nuclei. Percent apoptosis was determined by calculating the ratio of apoptotic to non-apoptotic cells. The experiments were performed in triplicate, and the results were averaged. The error bars indicate standard deviation, and the p values determined for the different conditions are as follows; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
FIGURE 7.
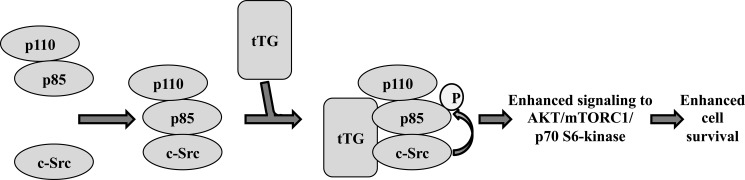
Model depicting how tTG promotes c-Src-mediated PI3-kinase signaling and cell survival. Our results suggest that tTG functions to promote cell survival by potentiating the activation of PI3-kinase. Specifically, tTG forms a complex with c-Src and PI3-kinase, an outcome that promotes a c-Src-dependent phosphorylation of the p85 regulatory subunit of PI3-kinase causing it to adopt a conformation that allows the p110 catalytic subunit of PI3-kinase to become activated and signal to its downstream effectors.
DISCUSSION
There have been an increasing number of studies implicating tTG in cancer progression. tTG expression is frequently up-regulated in a number of types of human cancer, particularly those that are highly aggressive, metastatic, and chemo-resistant (11–16, 21, 39). Importantly, several of these studies have shown that knocking-down tTG expression in cancer cells with siRNAs, or treating cells with compounds that bind at the transamidation active site of tTG, often sensitizes these cancer cells to chemotherapeutic agents and reduces many of their transformed features (11–16). While these findings point to tTG as being an important player in cancer progression, exactly how tTG contributes to the malignant state and whether it represents a valid target for cancer therapy, remains to be determined.
In this study, our goal was to use a simplified model system to better understand the intrinsic capability of tTG to confer cellular changes that might shed important light on the role that it plays in cancer progression. Thus, we sought to examine the actions of tTG in a non-transformed cellular context, specifically NIH3T3 fibroblasts, so as to avoid the aberrant oncogenic signaling inputs characteristic of transformed/cancer cells that might complicate the picture. In fact, we found that the overexpression of tTG in NIH3T3 fibroblasts did not cause the cells to lose their normal contact inhibition and form foci when grown in monolayer, nor did it enhance their migration. However, interestingly, we discovered that tTG provided a marked survival advantage to the cells challenged with serum starvation, and that treatment of the cells with the tTG inhibitor MDC, ablated this effect. This finding was consistent with our earlier observations that tTG confers cancer cells with a strong protective effect against apoptotic stresses, such as those elicited by treatment of cells with chemotherapeutic agents, and thus potentially would explain the advantage provided by the overexpression of tTG in high grade and highly aggressive forms of human cancer (6, 39).
So how does the overexpression of tTG in non-transformed fibroblasts enhance their survival capability? We obtained an interesting clue when we analyzed the activities and/or phosphorylation levels of components of the MAP kinase and PI3-kinase signaling pathways and found that significant increases in the phosphorylation of AKT, mTOR, and p70 S6-kinase occurred in the tTG-expressing fibroblasts, compared with those expressing the vector alone. We then took this one step further by directly showing that PI3-kinase activity was increased in the cells ectopically expressing tTG. These results led us to suspect that tTG promotes cell survival by enhancing the activation of the PI3-kinase/mTOR/p70 S6-kinase pathway.
This raised an important question, namely, how is tTG able to activate the PI3-kinase pathway? While there have been suggestions in the literature that tTG may regulate the levels of PTEN, and in turn, enhance AKT activation (22), we assessed the levels of PTEN in the NIH3T3 stable cell lines and found that they were unaffected by tTG. This led us to examine whether c-Src is contributing to the activation of PI3-kinase in tTG-expressing fibroblasts. c-Src binds PI3-kinase and requires this interaction to promote cellular transformation (31–33, 36, 37). Given our recent findings in the human breast cancer cell line SKBR3, that tTG is able to bind and increase c-Src activity in response to EGF treatment (20), we wondered whether a similar mechanism might be operating in fibroblasts over-expressing tTG. Indeed, we found that the activation of the PI3-kinase pathway that signals to mTOR and p70 S6-kinase, in NIH3T3 cells overexpressing tTG, is sensitive to Src inhibition.
Because we had shown previously that Src co-immunoprecipitates with tTG, and it has been reported that Src can bind to and activate PI3-kinase, we examined the possibility that Src, tTG, and PI3-kinase form a signaling complex. Indeed, we discovered that in addition to binding Src, tTG can be co-immunoprecipitated with both the p110 catalytic subunit and the p85 regulatory subunit of PI3-kinase. We reasoned that if the assembly of a complex that includes tTG, Src, and PI3-kinase is essential for sending a survival signal, then we would expect that inhibitors like MDC and PP2, which block these signals would have a corresponding inhibitory effect on complex formation. Interestingly, while inhibiting tTG and Src activity had no effect on the ability of tTG to be co-immunoprecipitated with p85, we found that these inhibitors blocked the interaction between tTG and Src. Thus, apparently Src and p85 have different requirements for binding to tTG. Importantly, however, disrupting the ability of tTG to associate with Src eliminates the Src-mediated tyrosine phosphorylation of p85, which in turn reduces the activation of the PI3-kinase signaling pathway. We have recently found that the tTG C277V mutant adopts a different conformation (i.e. an open conformational state) than wild-type tTG (which exists in cells as a closed conformational state), based on biochemical studies and small angle x-ray scattering measurements (40). Thus, the tTG C277V mutant adopts an open conformational state that apparently can still allow complex formation with c-Src and PI3 kinase. MDC, which also induces an open conformational state within tTG (40), as well as binds to its transamidation active site, apparently is able to interfere with the binding of c-Src to tTG. Thus, we suspect that the ability of tTG to bind to c-Src and p85 in a manner that leads to the activation of PI3 kinase activity is more likely an outcome of tTG adopting the proper conformational state, rather than being dependent upon its protein crosslinking (transamidation) activity. Indeed, we have thus far failed to find any involvement of protein crosslinking in these interactions.
Collectively, these findings point to a novel mechanism by which tTG is capable of triggering signals essential for cell survival (Fig. 7). Given the established link between Src and PI3-kinase, it is clear that these two proteins can work together to promote cellular transformation, particularly in cells expressing oncogenic Src mutants. However, we now find that tTG, when overexpressed even in a non-transformed cellular setting, is capable of forming a complex with c-Src and PI3-kinase that facilitates the activation of PI3-kinase, thereby providing a survival advantage to cells. These findings also offer important insights into how the overexpression of tTG in cancer cells can have important consequences that enable their survival in the face of stress and apoptotic signals such as chemotherapeutic drugs, and thus highlight tTG as a potentially an important therapeutic target in human cancer.
Acknowledgment
We thank Cindy Westmiller for assistance in the preparation of this manuscript.
Footnotes
- tTG
- tissue transglutaminase
- BPA
- biotinylated pentylamine
- CS
- calf serum
- DAPI
- 6-diamidino-2-phenylindole
- EGF
- epidermal growth factor
- MAP kinase
- mitogen-activated protein kinase
- MDC
- monodansylcadaverine
- mTOR
- mammalian target of rapamycin
- mTORC1
- mTOR complex 1
- PVDF
- polyvinylidene fluoride
- ERK
- extracellular signal-regulated kinase
- JNK
- c-Jun-N-terminal kinase
- p70 S6-kinase
- ribosomal protein S6 kinase
- c-Src
- cellular sarcoma
- PTEN
- phosphatase and tensin homologue deleted on chromosome ten.
REFERENCES
- 1. Singh U. S., Erickson J. W., Cerione R. A. (1995) Identification and biochemical characterization of an 80 kilodalton GTP-binding/transglutaminase from rabbit liver nuclei. Biochemistry 34, 15863–15871 [DOI] [PubMed] [Google Scholar]
- 2. Nakaoka H., Perez D. M., Baek K. J., Das T., Husain A., Misono K., Im M. J., Graham R. M. (1994) Gh: a GTP-binding protein with transglutaminase activity and receptor signaling function. Science 264, 1593–1596 [DOI] [PubMed] [Google Scholar]
- 3. Jaffe A. B., Hall A. (2005) Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21, 247–269 [DOI] [PubMed] [Google Scholar]
- 4. Folk J. E. (1980) Transglutaminases. Annu. Rev. Biochem. 49, 517–531 [DOI] [PubMed] [Google Scholar]
- 5. Greenberg C. S., Birckbichler P. J., Rice R. H. (1991) Transglutaminases: multifunctional cross-linking enzymes that stabilize tissues. Faseb J 5, 3071–3077 [DOI] [PubMed] [Google Scholar]
- 6. Antonyak M. A., Singh U. S., Lee D. A., Boehm J. E., Combs C., Zgola M. M., Page R. L., Cerione R. A. (2001) Effects of tissue transglutaminase on retinoic acid-induced cellular differentiation and protection against apoptosis. J. Biol. Chem. 276, 33582–33587 [DOI] [PubMed] [Google Scholar]
- 7. Aeschlimann D., Wetterwald A., Fleisch H., Paulsson M. (1993) Expression of tissue transglutaminase in skeletal tissues correlates with events of terminal differentiation of chondrocytes. J. Cell Biol. 120, 1461–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mehta K., Lopez-Berestein G. (1986) Expression of tissue transglutaminase in cultured monocytic leukemia (THP-1) cells during differentiation. Cancer Res. 46, 1388–1394 [PubMed] [Google Scholar]
- 9. Telci D., Griffin M. (2006) Tissue transglutaminase (TG2)–a wound response enzyme. Front Biosci. 11, 867–882 [DOI] [PubMed] [Google Scholar]
- 10. Verderio E. A., Johnson T., Griffin M. (2004) Tissue transglutaminase in normal and abnormal wound healing: review article. Amino Acids 26, 387–404 [DOI] [PubMed] [Google Scholar]
- 11. Yuan L., Siegel M., Choi K., Khosla C., Miller C. R., Jackson E. N., Piwnica-Worms D., Rich K. M. (2007) Transglutaminase 2 inhibitor, KCC009, disrupts fibronectin assembly in the extracellular matrix and sensitizes orthotopic glioblastomas to chemotherapy. Oncogene 26, 2563–2573 [DOI] [PubMed] [Google Scholar]
- 12. Kim D. S., Park S. S., Nam B. H., Kim I. H., Kim S. Y. (2006) Reversal of drug resistance in breast cancer cells by transglutaminase 2 inhibition and nuclear factor-kappaB inactivation. Cancer Res. 66, 10936–10943 [DOI] [PubMed] [Google Scholar]
- 13. Hwang J. Y., Mangala L. S., Fok J. Y., Lin Y. G., Merritt W. M., Spannuth W. A., Nick A. M., Fiterman D. J., Vivas-Mejia P. E., Deavers M. T., Coleman R. L., Lopez-Berestein G., Mehta K., Sood A. K. (2008) Clinical and biological significance of tissue transglutaminase in ovarian carcinoma. Cancer Res. 68, 5849–5858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Verma A., Wang H., Manavathi B., Fok J. Y., Mann A. P., Kumar R., Mehta K. (2006) Increased expression of tissue transglutaminase in pancreatic ductal adenocarcinoma and its implications in drug resistance and metastasis. Cancer Res. 66, 10525–10533 [DOI] [PubMed] [Google Scholar]
- 15. Satpathy M., Cao L., Pincheira R., Emerson R., Bigsby R., Nakshatri H., Matei D. (2007) Enhanced peritoneal ovarian tumor dissemination by tissue transglutaminase. Cancer Res. 67, 7194–7202 [DOI] [PubMed] [Google Scholar]
- 16. Mangala L. S., Fok J. Y., Zorrilla-Calancha I. R., Verma A., Mehta K. (2007) Tissue transglutaminase expression promotes cell attachment, invasion and survival in breast cancer cells. Oncogene 26, 2459–2470 [DOI] [PubMed] [Google Scholar]
- 17. Antonyak M. A., Li B., Regan A. D., Feng Q., Dusaban S. S., Cerione R. A. (2009) Tissue transglutaminase is an essential participant in the epidermal growth factor-stimulated signaling pathway leading to cancer cell migration and invasion. J. Biol. Chem. 284, 17914–17925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mann A. P., Verma A., Sethi G., Manavathi B., Wang H., Fok J. Y., Kunnumakkara A. B., Kumar R., Aggarwal B. B., Mehta K. (2006) Overexpression of tissue transglutaminase leads to constitutive activation of nuclear factor-kappaB in cancer cells: delineation of a novel pathway. Cancer Res. 66, 8788–8795 [DOI] [PubMed] [Google Scholar]
- 19. Zhang R., Tremblay T. L., McDermid A., Thibault P., Stanimirovic D. (2003) Identification of differentially expressed proteins in human glioblastoma cell lines and tumors. Glia 42, 194–208 [DOI] [PubMed] [Google Scholar]
- 20. Li B., Antonyak M. A., Druso J. E., Cheng L., Nikitin A. Y., Cerione R. A. (2010) EGF potentiated oncogenesis requires a tissue transglutaminase-dependent signaling pathway leading to Src activation. Proc. Natl. Acad. Sci. U.S.A. 107, 1408–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cao L., Petrusca D. N., Satpathy M., Nakshatri H., Petrache I., Matei D. (2008) Tissue transglutaminase protects epithelial ovarian cancer cells from cisplatin-induced apoptosis by promoting cell survival signaling. Carcinogenesis 29, 1893–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Verma A., Guha S., Wang H., Fok J. Y., Koul D., Abbruzzese J., Mehta K. (2008) Tissue transglutaminase regulates focal adhesion kinase/AKT activation by modulating PTEN expression in pancreatic cancer cells. Clin Cancer Res. 14, 1997–2005 [DOI] [PubMed] [Google Scholar]
- 23. Wang J. B., Erickson J. W., Fuji R., Ramachandran S., Gao P., Dinavahi R., Wilson K. F., Ambrosio A. L., Dias S. M., Dang C. V., Cerione R. A. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 18, 207–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Khosravi-Far R., White M. A., Westwick J. K., Solski P. A., Chrzanowska-Wodnicka M., Van Aelst L., Wigler M. H., Der C. J. (1996) Oncogenic Ras activation of Raf/mitogen-activated protein kinase-independent pathways is sufficient to cause tumorigenic transformation. Mol. Cell. Biol. 16, 3923–3933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Magdalena J., Millard T. H., Machesky L. M. (2003) Microtubule involvement in NIH 3T3 Golgi and MTOC polarity establishment. J. Cell Sci. 116, 743–756 [DOI] [PubMed] [Google Scholar]
- 26. Antonyak M. A., Li B., Boroughs L. K., Johnson J. L., Druso J. E., Bryant K. L., Holowka D. A., Cerione R. A. (2011) Cancer cell-derived microvesicles induce transformation by transferring tissue transglutaminase and fibronectin to recipient cells. Proc. Natl. Acad. Sci. U.S.A. 108, 4852–4857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Boroughs L. K., Antonyak M. A., Johnson J. L., Cerione R. A. (2011) A unique role for heat shock protein 70 and its binding partner tissue transglutaminase in cancer cell migration. J. Biol. Chem. 286, 37094–37107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Antonyak M. A., McNeill C. J., Wakshlag J. J., Boehm J. E., Cerione R. A. (2003) Activation of the Ras-ERK pathway inhibits retinoic acid-induced stimulation of tissue transglutaminase expression in NIH3T3 cells. J. Biol. Chem. 278, 15859–15866 [DOI] [PubMed] [Google Scholar]
- 29. Leslie N. R., Batty I. H., Maccario H., Davidson L., Downes C. P. (2008) Understanding PTEN regulation: PIP2, polarity and protein stability. Oncogene 27, 5464–5476 [DOI] [PubMed] [Google Scholar]
- 30. Tamguney T., Stokoe D. (2007) New insights into PTEN. J. Cell Sci. 120, 4071–4079 [DOI] [PubMed] [Google Scholar]
- 31. Auger K. R., Carpenter C. L., Shoelson S. E., Piwnica-Worms H., Cantley L. C. (1992) Polyoma virus middle T antigen-pp60c-src complex associates with purified phosphatidylinositol 3-kinase in vitro. J. Biol. Chem. 267, 5408–5415 [PubMed] [Google Scholar]
- 32. Liu X., Marengere L. E., Koch C. A., Pawson T. (1993) The v-Src SH3 domain binds phosphatidylinositol 3′-kinase. Mol. Cell. Biol. 13, 5225–5232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haefner B., Baxter R., Fincham V. J., Downes C. P., Frame M. C. (1995) Cooperation of Src homology domains in the regulated binding of phosphatidylinositol 3-kinase. A role for the Src homology 2 domain. J. Biol. Chem. 270, 7937–7943 [DOI] [PubMed] [Google Scholar]
- 34. von Willebrand M., Williams S., Saxena M., Gilman J., Tailor P., Jascur T., Amarante-Mendes G. P., Green D. R., Mustelin T. (1998) Modification of phosphatidylinositol 3-kinase SH2 domain binding properties by Abl- or Lck-mediated tyrosine phosphorylation at Tyr-688. J. Biol. Chem. 273, 3994–4000 [DOI] [PubMed] [Google Scholar]
- 35. Cuevas B. D., Lu Y., Mao M., Zhang J., LaPushin R., Siminovitch K., Mills G. B. (2001) Tyrosine phosphorylation of p85 relieves its inhibitory activity on phosphatidylinositol 3-kinase. J. Biol. Chem. 276, 27455–27461 [DOI] [PubMed] [Google Scholar]
- 36. Ling L. E., Druker B. J., Cantley L. C., Roberts T. M. (1992) Transformation-defective mutants of polyomavirus middle T antigen associate with phosphatidylinositol 3-kinase (PI3-kinase) but are unable to maintain wild-type levels of PI3-kinase products in intact cells. J. Virol. 66, 1702–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Serunian L. A., Auger K. R., Roberts T. M., Cantley L. C. (1990) Production of novel polyphosphoinositides in vivo is linked to cell transformation by polyomavirus middle T antigen. J. Virol. 64, 4718–4725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Martin G. S. (2004) The road to Src. Oncogene 23, 7910–7917 [DOI] [PubMed] [Google Scholar]
- 39. Antonyak M. A., Miller A. M., Jansen J. M., Boehm J. E., Balkman C. E., Wakshlag J. J., Page R. L., Cerione R. A. (2004) Augmentation of tissue transglutaminase expression and activation by epidermal growth factor inhibit doxorubicin-induced apoptosis in human breast cancer cells. J. Biol. Chem. 279, 41461–41467 [DOI] [PubMed] [Google Scholar]
- 40. Zhang J., Antonyak M. A., Singh G., Cerione R. A. (2013) A mechanism for the upregulation of EGF receptor levels in glioblastoma. Cell Rep 3, 2008–2020 [DOI] [PMC free article] [PubMed] [Google Scholar]