

Background: The drug delivery vehicle 2-hydroxypropyl-β-cyclodextrin (HPβCD) prevents cholesterol storage.
Results: HPβCD treatment induces TFEB mediated activation of autophagy and clearance of the autophagic substrate ceroid lipopigment.
Conclusion: HPβCD administration results in enhancement of the innate autophagic clearance capacity.
Significance: Dissecting the cellular pathways impacted by HPβCD is crucial to design HPβCD-based therapeutic modalities.
Keywords: Apoptosis, Autophagy, Lipoprotein, Lysosomal Storage Disease, Lysosomes, Ceroid Lipopigment, Cyclodextrin, Neuronal Ceroid Lipofuscinosis, TFEB
Abstract
2-Hydroxypropyl-β-cyclodextrin (HPβCD) is a Food and Drug Administration-approved excipient used to improve the stability and bioavailability of drugs. Despite its wide use as a drug delivery vehicle and the recent approval of a clinical trial to evaluate its potential for the treatment of a cholesterol storage disorder, the cellular pathways involved in the adaptive response that is activated upon exposure to HPβCD are still poorly defined. Here, we show that cell treatment with HPβCD results in the activation of the transcription factor EB, a master regulator of lysosomal function and autophagy, and in enhancement of the cellular autophagic clearance capacity. HPβCD administration promotes transcription factor EB-mediated clearance of proteolipid aggregates that accumulate due to inefficient activity of the lysosome-autophagy system in cells derived from a patient with a lysosomal storage disorder. Interestingly, HPβCD-mediated activation of autophagy was found not to be associated with activation of apoptotic pathways. This study provides a mechanistic understanding of the cellular response to HPβCD treatment, which will inform the development of safe HPβCD-based therapeutic modalities and may enable engineering HPβCD as a platform technology to reduce the accumulation of lysosomal storage material.
Introduction
Cyclodextrins (CDs)3 comprise a family of cyclic oligosaccharides manufactured from starch degradation and used in a number of pharmaceutical applications. The three-dimensional ring-like structure of CDs presents unique chemical properties. Because of the large number of hydroxyl groups, CDs are water-soluble. In addition, they present hydrophilic exteriors, which can be easily manipulated to disrupt hydrogen bonding and increase water solubility, and apolar interiors, which create a hydrophobic microenvironment in their cavity. Most pharmaceutical applications of CDs are based on this dual structural nature and rely on the incorporation of lipophilic molecules in CD complexes in aqueous media (1). Particularly, 2-hydroxypropyl-β-cyclodextrin (HPβCD) is a Food and Drug Administration-approved drug delivery vehicle used in a variety of pharmaceutical applications (2).
The recent serendipitous finding that HPβCD alone could increase the life span of a mouse model of the cholesterol storage disorder (3, 4), Niemann-Pick type C disease (5, 6), raised significant questions regarding the inert nature of HPβCD. It was previously demonstrated that HPβCD can extract excess cholesterol from biological membranes by trapping it into its hydrophobic core (7, 8). This interaction provided a mechanistic hypothesis for the observed HPβCD-mediated clearance of cholesterol in the brain of Niemann-Pick type C mice and in fibroblasts derived from Niemann-Pick type C patients (3, 9–11). Not surprisingly, these findings have paved the way for a new therapeutic avenue for Niemann-Pick type C, culminating in the recently approved clinical trial in which HPβCD is the active agent (clinicaltrials.gov).
The dissection of the cellular pathways impacted by the administration of HPβCD, however, is still in its early infancy. Recent findings demonstrated that depletion of cholesterol using HPβCD triggers activation of autophagy, an important lysosomal pathway involved in cellular clearance (12). However, the molecular mechanisms that regulate activation of autophagy observed upon HPβCD treatment remain unclear. Moreover, whether HPβCD treatment may lead to depletion of additional cellular substrates that are normally cleared by autophagy is not known.
Autophagy is an evolutionarily conserved and highly regulated catabolic pathway that mediates bulk degradation of long-lived proteins, macromolecules, and organelles (13), thus providing an important survival mechanism to supply energy resources under nutrient deprivation (14–16). A number of studies have described the role of autophagy in the disposal of toxic cellular material (17), such as protein and proteolipid aggregates (18, 19), as well as in the defense against bacterial infections (20–22). Deregulation of autophagy in cancer progression and cell death is also a subject of intense investigation (23–26). Together, these findings reveal that autophagy is a key hub in the regulation of cell metabolism and that modulation of autophagy is likely to play a critical role in the treatment of a number of pathological conditions. Excessive activation of autophagic clearance, however, may also induce activation of cell death mechanisms (27, 28). Interestingly, several genes involved in autophagy and apoptosis are co-regulated (29, 30), suggesting that in addition to a pro-survival function, autophagy may also have a pro-death role. Although the molecular mechanism underlying activation of autophagy-associated apoptosis remains a subject of intense debate (31), a detailed characterization of the cellular response associated with HPβCD-mediated autophagy activation, including its relationship with cell death mechanisms, is critically needed to develop improved guidelines for using HPβCD in commercial products and therapeutic applications.
Autophagy initiates upon formation of autophagosomes, which are vesicles that sequester cytoplasmic material to be degraded, proceeds through the fusion of autophagosomes with lysosomes, which are organelles that contain a battery of hydrolytic enzymes capable of degrading any type of biomolecules, and culminates with the formation of autophagolysosomes, where degradation takes place (32). Evidence of integrated and co-regulated roles of lysosomes and autophagosomes emerged from the recent discovery of an overarching lysosomal regulatory gene network (CLEAR, Coordinated Lysosomal Expression and Regulation) and its master regulator, the transcription factor EB (TFEB). TFEB regulates the expression of genes encoding lysosomal proteins (33, 34), the processing of lysosomal proteins (35), and the expression of autophagy genes (36). This evidence points to the role of TFEB at the crossroad of the regulatory mechanisms that coordinates the autophagy and lysosomal pathways and, importantly, to the function of TFEB as a regulator of autophagic clearance (37, 38).
Inefficient autophagic activity has been linked to the development of a number of diseases characterized by aberrant accumulation of intracellular substrates. Neuronal ceroid lipofuscinoses (NCLs) are a group of more than 12 genetically distinct neurodegenerative lysosomal storage disorders affecting children and young adults (39, 40). The hallmark of NCLs is the aberrant intracellular accumulation of autofluorescent ceroid lipopigments due to mutations in genes encoding proteins involved in lysosomal biogenesis and function (41). NCL patient-derived cell lines exhibit slowed growth and increased propensity to undergo apoptosis (42). Interestingly, TFEB activation was shown to lower the accumulation of ceroid lipopigments in fibroblasts derived from a patient with juvenile NCL (43), suggesting a potential role of TFEB as a therapeutic target for the treatment of NCLs.
Because of the similarities between the biological effects of HPβCD administration and TFEB activation, including the ability to activate autophagy and promote cellular clearance, we hypothesized that TFEB mediates activation of autophagy observed upon HPβCD administration. In this study we investigated this hypothesis by testing the molecular mechanism of autophagy induction activated in response to cell treatment with HPβCD using an in vitro model system of TFEB activation, namely HeLa cells that overexpress TFEB (33). To investigate whether HPβCD treatment induces enhancement of autophagy-mediated clearance and whether enhancement of autophagic activity is accompanied by activation of apoptosis, we used fibroblasts derived from a patient with late infantile neuronal ceroid lipofuscinosis (LINCL). LINCL cells were used in this study because (i) they provide an in vitro model system of lysosomal storage that allows evaluating whether autophagy activation parallels enhanced clearance of storage material, and (ii) they are prone to activation of cell death pathways and thus enable detecting even basal activation of autophagy associated cell death. We found that HPβCD administration results in the activation of TFEB and up-regulation of genes involved in the lysosome-autophagy system. We observed dramatic reduction in autofluorescent ceroid lipopigment in LINCL fibroblasts treated with HPβCD. Our mechanistic studies reveal that TFEB activation mediates the observed clearance of autofluorescent material. We also found that activation of autophagy observed upon HPβCD administration, under conditions that result in activation of TFEB and clearance of autophagic material, is not associated with activation of apoptosis. In summary, this study demonstrates that HPβCD treatment results in enhancement of the cellular autophagic capacity and that this response is mediated by TFEB. These findings unveil the molecular pathway involved in the cellular response to HPβCD treatment and establish HPβCD as a platform technology to develop nanotherapeutics for the treatment of diseases characterized by accumulation of lysosomal storage material.
EXPERIMENTAL PROCEDURES
Reagents and Cell Cultures
2-Hydroxypropyl-β-cyclodextrin was purchased from Sigma, sucrose was from Calbiochem, bafilomycin was from Cayman Chemical, and DAPI nuclear stain was from Enzo Life Sciences. Cell culture media were from Lonza. TFEB small interfering RNAs (siRNA; catalog #SI00094969) and control siRNA (catalog #1027280) were from Qiagen. pBABEpuro GFP-LC3 plasmid was from Addgene.
HeLa cells stably transfected for the expression of TFEB-3×FLAG were generated as previously described (33). Fibroblasts derived from patients with LINCL were obtained from Coriell Cell Repositories. Direct sequencing of TPP1 coding sequences showed compound heterozygous variations: an Arg-208-to-Ter mutation and a G-to-C transversion of the consensus AG 3-prime splice acceptor site at exon 6, resulting in the retention of intron 5 in the spliced transcript. Cells were grown at 37 °C in 5% CO2 in minimal essential medium with Earle's salts supplemented with 10% heat-inactivated fetal bovine serum and 1% glutamine Pen-Strep. Medium was replaced every 2 or 3 days. Monolayers were passaged with TrypLE Express.
Enzymatic Assays
TPP1 activity assays were conducted as described previously (44). Briefly, fibroblasts derived from a patient with LINCL and from a healthy individual were plated and cultured overnight at 37 °C in 5% CO2. Cells were collected using TrypLE, washed with PBS, and incubated with lysis buffer (150 mm NaCl, 50 mm Tris, pH 8, 0.5% sodium deoxycholate, 1% Triton, 0.1% SDS) supplemented with 1% protease inhibitor for 1 h at 4 °C. Lysed cells were centrifuged, and the supernatant was collected for activity assays. The protein concentration was measured using BCA assay, and TPP1 activity was assayed as previously described (44) using 3.5 μg of total protein in the assay reaction.
Immunofluorescence Assays
Fibroblasts were seeded on glass coverslips, cultured in the presence of HPβCD, and fixed with 4% paraformaldehyde for 30 min. Cells were permeabilized with 0.1% Triton X for 5 min and incubated with 8% BSA for 1 h. After incubation for 1 h with primary antibodies (rabbit anti-3×FLAG (Sigma), rabbit anti-LC3 (MBL International), mouse anti-LAMP2 (BioLegend), or mouse anti-TFEB (Abcam)), cells were washed 3 times with 0.1% Tween 20, PBS and incubated with secondary antibodies for 1 h (Dylight 549 goat anti-mouse IgG or Dylight 633 goat anti-rabbit IgG (KPL)). Images were obtained using an Olympus IX81 confocal microscope. Co-localization was conducted using Fluoview software. Co-localization heatmap images and image quantifications were obtained using NIH ImageJ analysis software.
Images were obtained using an Olympus IX81 confocal microscope under a fixed voltage and saturation for each channel. All images were subjected to a fixed threshold to remove background noise and were reported without changing intensity or contrast.
Quantitative RT-PCR
RT-PCR was conducted as described previously (45). Briefly, cells were incubated with HPβCD for 24 or 72 h before total RNA was extracted using RNAGEM™ reagent (ZyGEM). cDNA was synthesized from total RNA using qScript™ cDNA SuperMix (Quanta Biosciences). Total cDNA amount was measured using a NanoDrop 2000 (Thermo Scientific). Quantitative PCR reactions were performed using cDNA, PerfeCTa™ SYBR Green FastMix™ (Quanta Biosciences), and the corresponding primers (Table 1 and Refs. 33, 45, and 46) in a CFX96™ Real-Time PCR detection system (Bio-Rad). Samples were heated for 2 min at 95 °C and amplified in 45 cycles of 1 s at 95 °C, 30 s at 60 °C, and 30 s at 72 °C. Analyses were conducted using CFX manager software (Bio-Rad), and the threshold cycle (CT) was extracted from the PCR amplification plot. The ΔCT values were calculated to evaluate the difference between the CT of a target gene and the CT of the housekeeping gene, GAPDH, as follows: ΔCT = CT (target gene) − CT (GAPDH). The relative mRNA expression level of a target gene in treated cells was normalized to that measured in untreated cells: relative mRNA expression level = 2 exp[−(ΔCT (treated cells) − ΔCT (untreated cells))]. Each data point was evaluated in triplicate and measured three times.
TABLE 1.
Primers
| Gene | Sequence |
|---|---|
| GBA | From Ref. 45 |
| HEXA | From Ref. 33 |
| LAMP1 | From Ref. 33 |
| ACTB | From Ref. 46 |
| MAPLC3B | Forward: 5′-GAGAAGCAGCTTCCTGTTCTGG-3′ |
| Reverse: 5′-GTGTCCGTTCACCAACAGGAAG-3′ | |
| SQSTM1 | Forward: 5′-GCACCCCAATGTGATCTGC-3′ |
| Reverse: 5′-CGCTACACAAGTCGTAGTCTGG-3′ | |
| BECN1 | Forward: 5′-GGCTGAGAGACTGGATCAGG-3′ |
| Reverse: 5′-CTGCGTCTGGGCATAACG-3′ | |
| GAPDH | From Ref. 45 |
Western Blot Analyses
Cells were lysed with the complete lysis-M buffer containing the protease inhibitor mixture (Roche Applied Science). Total protein concentrations were determined by Bradford assay, and each sample was diluted to the same protein concentration. Aliquots of cell lysates were separated by 15% SDS-PAGE gel, and Western blot analyses were performed using primary antibody (rabbit anti-LC3 (MBL) and rabbit anti-GAPDH (Santa Cruz Biotechnology)) and secondary antibody (HRP-conjugated goat anti-rabbit (Santa Cruz Biotechnology)). Blots were visualized using Luminata Forte Western HRP Substrate (Millipore) and quantified by NIH ImageJ analysis software.
Apoptosis Assays
CLN2 patient-derived cells were treated with HPβCD or Taxol for 24 h at 37 °C. Cells were collected and apoptosis-tested with the CytoGLOTM Annexin V-FITC Apoptosis Detection kit (IMGENEX) according to the manufacturer's instructions and analyzed by flow cytometry (FACSCantoTM II, BD Biosciences) with a 488-nm argon laser as previously described (47).
Cell Transfections
105 cells in 2 ml of growth medium were seeded in each well of a 6-well plate. 6 μl of jetPRIME™ reagent (Polyplus Transfection) was mixed with 0.6 μg of the plasmid pBABEpuro GFP-LC3 to form transfection complexes. When cells reached 70–80% confluency, transfection complexes were added to each well of the plate. After incubation for 20 h, the transfection medium was replaced with fresh medium containing HPβCD.
siRNA Transfections
siRNA transfections were performed using HiPerFect® transfection reagent as described in the manufacturer's manual (Qiagen). Briefly, each well of a 24-well plate was spotted with 75 ng of siRNA diluted in 18 μl of RNase-free water. 4.5 μl of HiPerFect reagent was resuspended in 145.5 μl of culture medium without serum and was added to the pre-spotted siRNA. The mixture was incubated for 10 min at room temperature to allow formation of transfection complexes. 3 × 104 cells in 1 ml of culture medium were seeded into each well on top of the transfection complexes and incubated for 2 days. Medium was replaced with fresh medium or medium containing HPβCD. Ceroid lipopigment autofluorescence intensity level and relative mRNA expression levels of representative TFEB or autophagy genes were evaluated after 3 days of HPβCD treatment.
Statistical Analyses
All data are presented as the mean ± S.D., and statistical significance was calculated using one-way analysis of variance analysis followed by post-hoc Tukey's test.
RESULTS
HPβCD Treatment Promotes TFEB Activation
Previous studies demonstrated that TFEB localizes predominantly in the cytoplasm in resting cells and translocates into the nucleus upon activation under conditions of lysosomal stress (33). Upon TFEB nuclear translocation, TFEB target genes are transcriptionally induced (33). To investigate the role of TFEB in regulating autophagy activation upon HPβCD treatment, we tested TFEB subcellular localization. Specifically, we monitored TFEB subcellular localization in a cellular model system of TFEB activation consisting of HeLa cells stably transfected with a TFEB-3×FLAG construct (HeLa/TFEB) (33). Cells were cultured in presence of HPβCD (0.1–10 mm; 24 h), and TFEB subcellular distribution was evaluated by confocal microscopy using DAPI nuclear staining and an anti-FLAG antibody (Fig. 1, A and B). The results showed that, as expected, TFEB localizes predominantly in the cytoplasm of untreated HeLa/TFEB cells. HPβCD treatment, however, resulted in increased translocation of TFEB into the nucleus (Fig. 1, A and B). The extent of TFEB nuclear accumulation correlated with HPβCD concentration, as it increased progressively in cells treated with HPβCD ranging from 0.1 to 10 mm HPβCD. Co-localization of TFEB and DAPI nuclear staining in cells treated with 1 mm HPβCD was comparable with that observed in cells treated with sucrose under conditions (100 mm; 24 h) previously reported to result in maximum activation of TFEB (33).
FIGURE 1.
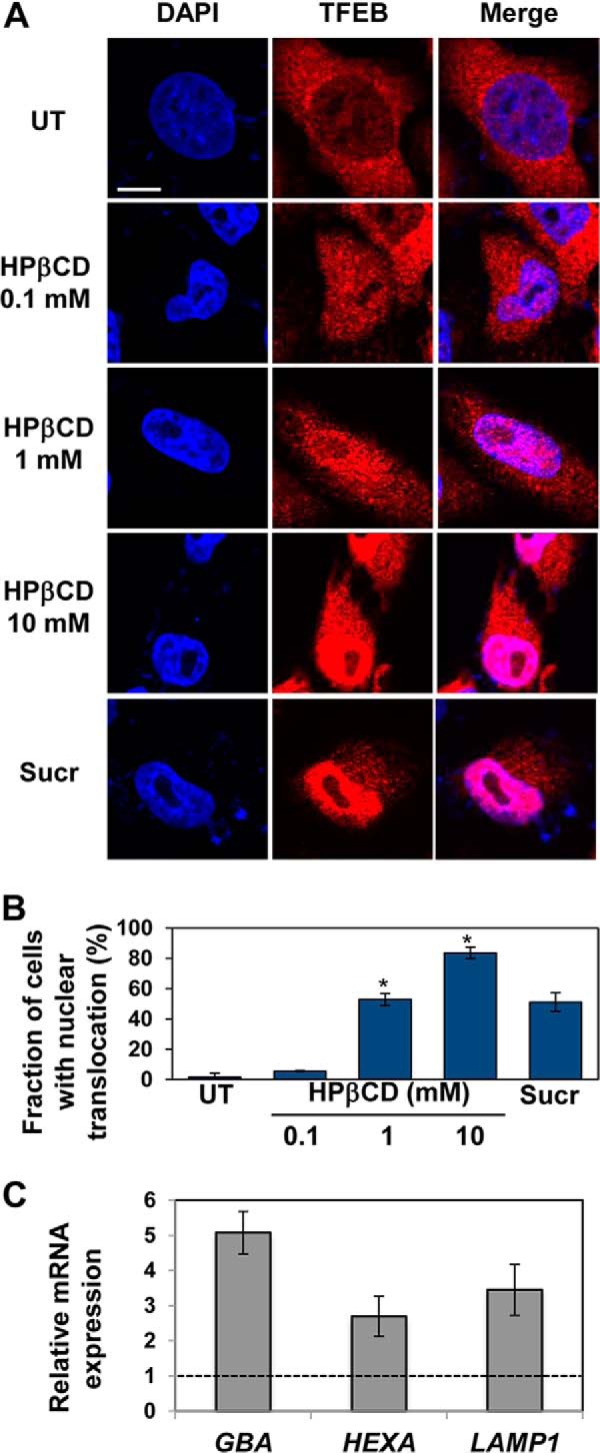
HPβCD treatment results in activation of TFEB and the CLEAR network. A, confocal microscopy analysis of TFEB subcellular localization in HeLa cells stably transfected with TFEB-FLAG treated with 0.1, 1, and 10 mm HPβCD or sucrose 100 mm for 1 day. Co-localization of DAPI (blue, first column) and an anti-FLAG antibody (red, first column) is shown in purple (third column). The scale bar is 10 μm. UT, untreated; Sucr, sucrose. B, percentage of cells presenting nuclear localization of TFEB-FLAG upon treatment with HPβCD (0.1, 1, and 10 mm) or sucrose (100 mm). Representative (∼30) fields containing ∼50 cells were analyzed (p < 0.05; *, p < 0.01). Data are reported as the mean ± S.D. C, relative mRNA expression levels of representative genes of the lysosome system in HeLa cells stably transfected with TFEB-FLAG treated with 1 mm HPβCD for 1 day. mRNA expression levels of GBA, HEXA, LAMP1, were obtained by qRT-PCR, corrected by the expression of the house-keeping gene GAPDH, and normalized to those of untreated cells (dashed line). Data are reported as the mean ± S.D. n ≥ 3; p < 0.05.
To investigate whether nuclear translocation of TFEB observed in cells treated with HPβCD results in activation of the CLEAR network, we tested the expression of representative genes of the CLEAR network that are involved in lysosomal function upon HPβCD administration. HeLa/TFEB cells were treated with HPβCD (1 mm), and the mRNA expression levels of TFEB targets were monitored by quantitative RT-PCR (Fig. 1C). We observed significant up-regulation of TFEB targets, namely, GBA (glucocerebrosidase; 5.1-fold, p < 0.05), HEXA (hexosaminidase A; 2.7-fold, p < 0.05), and LAMP1 (Lysosome-associated membrane glycoprotein 1; 3.5-fold, p < 0.05).
LC3 is a protein found on the membrane of autophagosomes (48) and widely used as a marker of autophagy activation (16). To investigate whether HPβCD treatment, under conditions that result in TFEB activation, induces activation of autophagy, we transfected HeLa/TFEB cells with a vector encoding the LC3 gene fused to GFP to facilitate visualization of LC3 structures. As expected, we observed a diffuse GFP signal in untreated cells, indicative of basal autophagic activity, and punctate GFP structures in cells treated with HPβCD, indicative of the formation of autophagic vesicles (Fig. 2A). Activation of autophagy upon HPβCD treatment was also confirmed by immunoblotting of LC3 isoforms. The increase in LC3-II in cells treated with HPβCD compared with untreated cells suggests increased formation of autophagic vesicles (Fig. 2B). The further increase in LC3-II levels observed in cells treated with HPβCD in the presence of the autophagy inhibitor bafilomycin (100 nm), compared with cells treated only with HPβCD (p < 0.01) and to cells treated only with bafilomycin (p < 0.05), is indicative of an increase of autophagic flux upon HPβCD treatment. A time-dependent analysis of LC3 conversion and consumption upon HPβCD was also conducted to confirm up-regulation of autophagy. The ratio of LC3-II over LC3-I levels was observed to increase with time of cell exposure to HPβCD (Fig. 2C; *, p < 0.05). These results confirm that HPβCD treatment induces activation of autophagy (49).
FIGURE 2.

HPβCD treatment results in activation of autophagy. A, confocal microscopy analysis of LC3 expression in HeLa/TFEB cells transfected for the expression of LC3-GFP for 20 h and treated with 1 mm HPβCD for additional 24 h. The scale bar is 10 μm. UT, untreated. B, Western blot analyses of LC3 isoforms and GAPDH (used as loading control) in HeLa/TFEB cells treated with 1 mm HPβCD and 100 nm bafilomycin for 24 h and quantification of LC3-II bands. Band intensities were quantified with ImageJ analysis software, corrected by GAPDH band intensities, and divided by the values obtained in untreated samples (p < 0.01; *, p < 0.05). Baf, bafilomycin. C, Western blot analyses of LC3 isoforms and GAPDH (used as loading control) in HeLa/TFEB cells treated with 1 mm HPβCD for 0, 12, 24, 48, and 72 h and quantification of LC3-II/LC3-I ratios at each time point. Band intensities were quantified as described in A (*, p < 0.05). D, relative mRNA expression levels of representative genes of the autophagy system in HeLa cells stably transfected with TFEB-FLAG treated with 1 mm HPβCD for days. mRNA expression levels of MAPLC3B, SQSTM1, and BECN1 were obtained as described in Fig. 1. Data are reported as the mean ± S.D. n ≥ 3; p < 0.01.
To further confirm that HPβCD treatment mediates activation of autophagy, we tested the expression of genes involved in different steps of the autophagic pathway. Cells were treated with HPβCD (1 mm), and mRNA levels were tested by quantitative RT-PCR (Fig. 2D). We detected up-regulation of MAPLC3 (microtubule-associated light chain protein 3 (LC3); 4.8-fold, p < 0.01), which is essential for the formation of autophagic vesicles, SQSTM1 (p62; 2.1-fold, p < 0.01), which is essential for cargo recognition, and BECN1 (Beclin-1; 3.0-fold, p < 0.01), which is required for the formation of autophagosomes. Interestingly, MAPLC3B and SQSTM1 are known to be direct targets of TFEB (34, 36). Taken together these results demonstrate that HPβCD treatment results in activation of TFEB, transcriptional up-regulation of genes involved in the lysosome-autophagy system, and up-regulation of the autophagic flux.
HPβCD Treatment Results in Clearance of Ceroid Lipopigment in LINCL Fibroblasts
Previous studies associated autophagy activation with HPβCD-induced cholesterol depletion in cultured human fibroblasts (12). To investigate whether the link between HPβCD-induced clearance and autophagy is specific for cholesterol storage or is a more general cellular response activated upon uptake of HPβCD, we focused on a different model of lysosomal storage, namely neuronal ceroid lipofuscinoses (NCLs). Cells derived from NCL patients are characterized by accumulation of ceroid lipopigment, a lipofuscin-like autofluorescent material that is readily visible in microscopic analyses (41). Specifically, we used fibroblasts derived from a patient with LINCL, a disease caused by deficiency of tripeptidyl peptidase (TPP1) activity. Previous studies showed that cells derived from patients with LINCL or other NCLs have an increased tendency to undergo apoptosis (42). Thus, apoptosis-sensitive LINCL fibroblasts were selected for this study to investigate whether HPβCD treatment affects autophagic clearance of ceroid lipofuscin and whether HPβCD-induced modulation of autophagy also activates cell death mechanisms (see below). We used cells carrying two heterozygous compound mutations in the TTP1 gene: a missense mutation (Arg-208 → Ter) and a splicing mutation resulting in the retention of intron 5 in the spliced transcript (50). Enzymatic activity assays confirmed that this cell line has null TPP1 enzyme activity compared with fibroblasts derived from a non-affected individual (Fig. 3).
FIGURE 3.
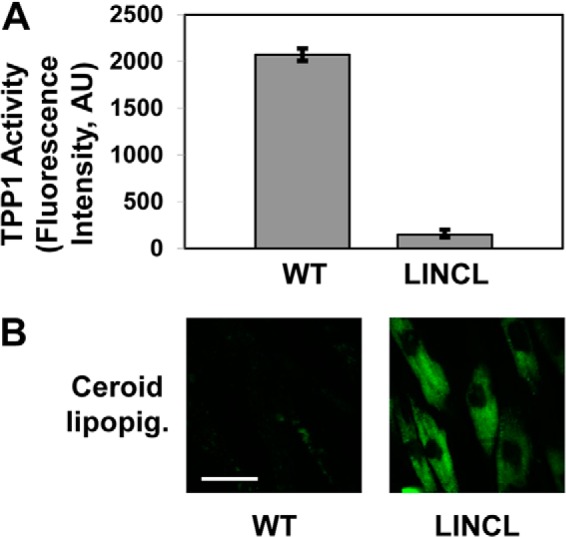
LINCL fibroblasts present null TPP1 enzyme activity and accumulation of ceroid lipopigment. A, TPP1 enzymatic activity in fibroblasts derived from a healthy individual (WT) and from a patient affected by LINCL conducted as described under “Experimental Procedures.” n ≥ 3; p < 0.01. B, confocal microscopy analysis of ceroid lipopigment in fibroblasts derived from a healthy individual (WT) and from a patient affected by LINCL. The scale bar is 50 μm. AU, absorbance units.
To test the effects of HPβCD treatment on the storage of the autofluorescent material in LINCL fibroblasts, we supplemented the culture medium with a range of HPβCD concentrations and incubated the cells for 3 days. Confocal microscopy analyses showed that HPβCD treatment resulted in clearance of ceroid lipopigment as observed by monitoring the loss of autofluorescence (Fig. 4, A and B). Notably, ceroid lipopigment-associated autofluorescence decreased with increasing concentrations of HPβCD, and maximum clearance occurred upon treatment with 1–10 mm HPβCD (Fig. 4, A and B), which was previously reported to activate autophagy (12).
FIGURE 4.

HPβCD treatment results in reduced storage of ceroid lipopigment. A, confocal microscopy analysis of ceroid lipopigment (top, green) and TFEB (bottom, red) in LINCL patient-derived fibroblasts treated with 0.1, 1, and 10 mm HPβCD evaluated by detecting green autofluorescence and binding of an anti-TFEB antibody, respectively. The scale bar is 20 μm. UT, untreated. B, quantification of ceroid lipopigment autofluorescence in LINCL patient-derived fibroblasts treated as described in A. Representative fields containing ∼50 cells were analyzed. The scale bar is 20 μm. UT, untreated. Data are reported as the mean ± S.D. (p < 0.05; *, p < 0.01). C, confocal microscopy analysis of ceroid lipopigment (top, green) and TFEB (bottom, red) in LINCL fibroblasts treated with 1 mm HPβCD for 1, 3, and 7 days and evaluated as described in a. D and E, relative mRNA expression levels of representative genes of the lysosome-autophagy system in LINCL fibroblasts treated with 1 mm HPβCD for 3 days. GBA, HEXA, LAMP1, MAPLC3B, SQSTM1, and BECN1 mRNA expression levels were obtained as described in Fig. 1 (p < 0.01).
To test whether clearance of ceroid lipopigment parallels activation of TFEB in cells treated with HPβCD, we evaluated TFEB subcellular localization in LINCL cells treated with a range of HPβCD concentrations. Confocal microscopy analyses revealed that TFEB preferentially accumulates in the nucleus in LINCL fibroblasts treated with HPβCD and that the extent of TFEB nuclear translocation increases with increasing concentrations of HPβCD in the culture medium (Fig. 4A). Partial nuclear translocation of TFEB was observed in untreated LINCL fibroblasts, as expected, due to storage-induced lysosomal stress (33).
To confirm that the clearance of ceroid lipopigment in LINCL fibroblasts depends on HPβCD treatment, we monitored autofluorescence in LINCL fibroblasts treated with a fixed concentration of HPβCD (1 mm) for up to 7 days. Confocal microscopy analyses showed that both ceroid lipopigment clearance and TFEB nuclear translocation increased with increasing time of incubation with HPβCD (Fig. 4C).
To investigate whether the nuclear translocation of TFEB observed in LINCL fibroblasts treated with HPβCD results in the activation of the CLEAR network, we measured the expression of representative genes of the CLEAR network upon HPβCD administration. LINCL cells were treated with HPβCD (1 mm), and the mRNA expression levels of TFEB targets were monitored by quantitative RT-PCR (Fig. 4D). We found that HPβCD treatment resulted in transcriptional up-regulation of all genes of the lysosomal system tested: GBA (2.7-fold; p < 0.01), HEXA (2.8-fold, p < 0.01), and LAMP1 (1.6-fold, p < 0.01). To test whether the autophagy system was also transcriptionally activated, we measured the mRNA expression levels of representative genes involved in different steps of the autophagy pathway as described above. We detected significant up-regulation of MAPLC3 (3.2-fold, p < 0.01), SQSTM1 (2.5-fold, p < 0.01), and BECN1 (2.5-fold, p < 0.01) (Fig. 4E). Taken together, these results demonstrate that the reduced deposition of ceroid lipopigment observed upon HPβCD administration parallels activation of TFEB and transcriptional up-regulation of genes involved in the lysosome-autophagy system. These results, therefore, suggest that the clearance of ceroid lipopigment correlates with the activation of TFEB.
TFEB Mediates the Autophagic Clearance Observed upon HPβCD Treatment
To determine whether clearance of ceroid lipopigment observed in HPβCD-treated LINCL fibroblasts depends on TFEB activation, we silenced TFEB expression using specific siRNA. We observed an increase in ceroid lipopigment accumulation in cells treated with TFEB siRNA compared with cells treated with a control siRNA (Fig. 5, A, top panels, and B), suggesting that TFEB is involved in the clearance of lipopigment deposits. TFEB silencing resulted in a 60% reduction in TFEB expression levels compared with cells transfected with a control siRNA, as evaluated by quantitative RT-PCR (Fig. 5C, solid yellow and solid blue bars). Interestingly, the clearance of autofluorescent storage material upon HPβCD treatment was decreased by TFEB silencing (Fig. 5, A, bottom panels, and B). The decrease in ceroid lipopigment storage observed upon administration of HPβCD to TFEB-silenced cells was likely due to HPβCD-mediated activation of residual TFEB that is present upon partial silencing of TFEB expression (Fig. 5C). Accordingly, the residual amount of TFEB detected in silenced cells was found to localize preferentially in the cytoplasm in untreated cells and in the nucleus in cells treated with HPβCD (Fig. 5A).
FIGURE 5.

HPβCD-induced activation of clearance is regulated by TFEB. A, confocal microscopy analysis of ceroid lipopigment (green) and TFEB (red) in LINCL fibroblasts treated with control siRNA or TFEB siRNA and with 1 mm HPβCD, evaluated by detecting green autofluorescence and binding of an anti-TFEB antibody, respectively. The scale bar is 20 μm. UT, untreated. B, quantification of ceroid lipopigment autofluorescence in LINCL patient-derived fibroblasts treated as described in A. Representative fields containing ∼50 cells were analyzed. siCtrl, control siRNA; siTFEB, TFEB siRNA. Data are reported as the mean ± S.D. (p < 0.05, *, p < 0.01). C, relative mRNA expression levels of representative genes of the lysosome-autophagy system in LINCL fibroblasts treated with control siRNA or TFEB siRNA for 2 days and with 1 mm HPβCD for 3 days. TFEB, HEXA, MAPLC3B, and BECN1 mRNA expression levels were obtained as described in Fig. 1 (n ≥ 3; p < 0.01).
To further investigate the effect of TFEB silencing in LINCL fibroblasts treated with HPβCD, we also measured the expression of genes that encode proteins involved in the lysosome-autophagy system. LINCL fibroblasts were incubated with TFEB siRNA for 2 days and with HPβCD for 3 additional days, and mRNA expression levels were measured by quantitative RT-PCR (Fig. 5C). As mentioned above, TFEB silencing reduced TFEB transcription to 40% that measured in control LINCL cells. Interestingly, HPβCD treatment resulted in a 6.9-fold increase (p < 0.01) in TFEB transcription in non-silenced LINCL cells. Similar results were obtained upon TFEB silencing: HPβCD treatment resulted in a 5.6-fold increase (p < 0.01) in TFEB transcription compared with untreated (silenced) cells, suggesting that administration of HPβCD causes up-regulation of TFEB. Representative genes of the lysosomal system, GBA and HEXA, were found to be up-regulated in cells treated with HPβCD and control siRNA (3.4- and 2.7-fold, respectively, confirming the results reported in Fig. 4D; p < 0.01), down-regulated in cells treated with TFEB siRNA (0.65- and 0.6-fold, respectively; p < 0.01), and up-regulated in cells treated with TFEB siRNA and HPβCD (1.7- and 1.4-fold, respectively; p < 0.01) compared with cells treated with control siRNA. The increase in expression levels of TFEB target genes observed in cells treated with HPβCD in both control and silenced cells suggests that HPβCD treatment has a dual effect and results in transcriptional up-regulation of TFEB as well as TFEB protein activation. In summary, we found that HPβCD treatment causes an increase in expression of TFEB target genes that is subsequent to TFEB nuclear translocation, confirming that LINCL cells respond to HPβCD treatment by activating TFEB and the CLEAR network.
To investigate the role of TFEB in the activation of autophagy observed upon HPβCD treatment, we also measured the expression of representative genes involved in the autophagy pathway, namely MAPLC3B and BECN1. As observed for genes involved in lysosomal function, MAPLC3B and BECN1 were found to be up-regulated in LINCL fibroblasts treated with HPβCD and control siRNA (2.4- and 3.2-fold; p < 0.01, respectively), down-regulated in cells treated with TFEB siRNA (0.5-fold; p < 0.01), and up-regulated in cells treated with TFEB siRNA and HPβCD (1.6- and 1.3-fold; p < 0.01, respectively) compared with cells treated with control siRNA (Fig. 5C).
Taken together these results demonstrate that HPβCD treatment results in coordinated up-regulation of lysosome biogenesis and autophagy and enhanced clearance of autophagic material. Importantly, these data also demonstrate that TFEB plays a key role in mediating autophagy activation observed upon HPβCD administration and that the up-regulation of genes involved in the lysosome-autophagy system and the reduction of ceroid lipopigment accumulation correlate with TFEB expression levels and parallel TFEB activation.
HPβCD Treatment Results in Activation of Autophagy without Inducing Apoptosis
To investigate whether clearance of ceroid lipopigment in LINCL cells treated with HPβCD depends on activation of autophagy, we analyzed the expression of LC3 by confocal microscopy. Endogenous LC3 was visualized as a diffuse cytoplasmic pool in untreated cells, but it appeared as punctate structures that primarily represent autophagosomes and autophagolysosomes (49) in cells treated with HPβCD (1 mm, 3 days). Interestingly, the increase in expression of LC3 and punctate appearance, indicative of an increase in autophagosome formation, paralleled a reduction in autofluorescence of storage material (Fig. 6A, left panels).
FIGURE 6.
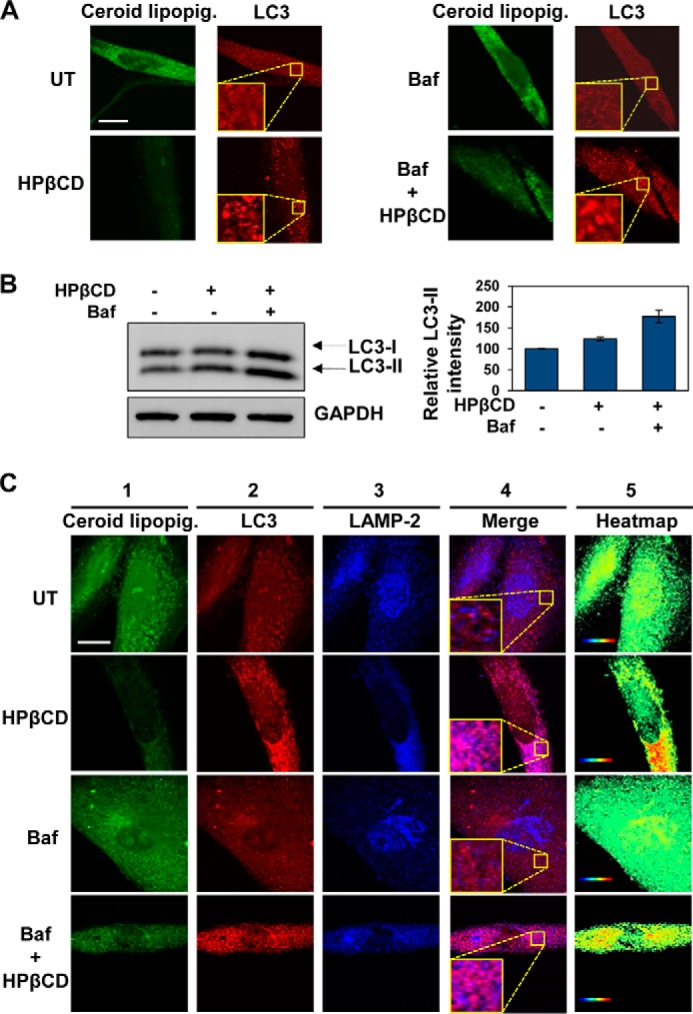
HPβCD treatment activates autophagic clearance. A, confocal microscopy analysis of ceroid lipopigment (green) and LC3 (red) in LINCL fibroblasts treated with 1 mm HPβCD and 100 nm bafilomycin for 3 days, evaluated by detecting green autofluorescence and binding of an anti-LC3 antibody, respectively. The scale bar is 20 μm. UT, untreated; Baf, bafilomycin. B, Western blot analyses of LC3 isoforms and GAPDH (used as loading control) in LINCL fibroblasts treated with 1 mm HPβCD and 100 nm bafilomycin for 24 h and quantification of LC3-II bands. Band intensities were quantified with ImageJ analysis software, corrected by GAPDH band intensities, and divided by the values obtained in untreated samples (p < 0.05). C, confocal microscopy analysis of ceroid lipopigment (green, first column), LC3 (red, second column), and LAMP-2 (blue, third column) in LINCL fibroblasts treated with 1 mm HPβCD and 100 nm bafilomycin for 3 days, evaluated by detecting green autofluorescence, binding of anti-LC3 antibody, and binding of anti-LAMP-2 antibody, respectively. Co-localization of LC3 and LAMP-2 is shown in purple (fourth column). Heatmaps of co-localization images were obtained with ImageJ analysis software (fifth column). Hot colors represent positive correlation (co-localization), whereas cold colors represent negative correlation (exclusion). The scale bar is 20 μm.
To confirm that that clearance of ceroid lipopigment observed upon HPβCD treatment depends on activation of autophagy, we treated LINCL fibroblasts with bafilomycin (100 nm). Bafilomycin, a specific inhibitor of vacuolar H+ ATPase (V-ATPase) that prevents fusion of autophagosomes with lysosomes, thereby impairing autophagic flux (51), did not result in a significant increase in lipopigment autofluorescence, confirming that LINCL cells have a defective autophagy (52). However, lipopigment autofluorescence was partially reduced by the addition of HPβCD (1 mm). HPβCD treatment in cells cultured in the presence of bafilomycin was found not only to reduce accumulation of ceroid lipopigment but also to enhance the appearance of punctate LC3 structures compared with cells only treated with bafilomycin (Fig. 6A, right panels).
Activation of autophagy upon treatment of LINCL cells with HPβCD was also confirmed by immunoblotting of LC3 isoforms. HPβCD treatment was observed to increase the amount of LC3-II, which is indicative of autophagy induction (Fig. 6B). The increase in LC3-II levels observed in cells treated with HPβCD in the presence of bafilomycin compared with cells treated only with HPβCD confirmed that HPβCD treatment results in an increase in autophagic flux.
To verify activation of the autophagic flux, we tested the extent to which HPβCD treatment resulted in fusion of autophagosomes with lysosomes and subsequent formation of autophagolysosomes. To this end we evaluated the co-localization of LC3 with LAMP-2, a protein that resides on the lysosomal membrane. LINCL fibroblasts were treated with HPβCD (1 mm), bafilomycin (100 nm), or a combination thereof for 3 days. Accumulation of ceroid lipopigment was evaluated under all conditions tested in this experiment by monitoring autofluorescence by confocal microscopy (Fig. 6B, first column). The overlap of LC3 (Fig. 6B, second column) and LAMP-2 (Fig. 6B, third column) signals was evaluated using the ImageJ script Colocalization Colormap (Fig. 6B, fourth and fifth columns). HPβCD treatment resulted in enhanced formation of autophagolysosomes as indicated by the punctate LC3 appearance and by the hot colors in the co-localization heatmap. As expected, the formation of autophagolysosomes decreased in cells treated with bafilomycin and was partially restored upon the addition of HPβCD.
To confirm that the HPβCD treatment of LINCL fibroblasts results in clearance of ceroid lipopigment specifically through the autophagy pathway, we silenced ATG7 expression using specific siRNAs. ATG7 is an essential autophagy gene required for basal as well starvation-induced autophagy (53). We observed an increase in ceroid lipopigment accumulation in cells treated with ATG7 siRNA compared with cells treated with a control siRNA (Fig. 7, A, top panels, and B), confirming that ATG7 is required for clearance of lipopigment deposits. Interestingly, ATG7 silencing decreased clearance of autofluorescent storage material even upon HPβCD treatment (Fig. 7, A, bottom panels, and B). As expected, ATG7 silencing did not alter HPβCD-induced activation of TFEB. These results suggest a model in which HPβCD treatment results in TFEB-induced activation of the autophagy system, but blockage of downstream steps of the autophagic flux (e.g. blockage of ATG7 expression) prevents clearance of ceroid lipopigment.
FIGURE 7.
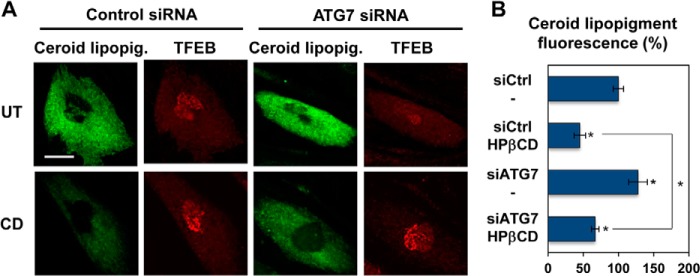
HPβCD-induced activation of autophagy is reduced upon genetic inhibition of the autophagic flux. A, confocal microscopy analysis of ceroid lipopigment (green) and TFEB (red) in LINCL fibroblasts treated with control siRNA or ATG7 siRNA and with 1 mm HPβCD, evaluated by detecting green autofluorescence and binding of an anti-TFEB antibody, respectively. The scale bar is 20 μm. UT, untreated. B, quantification of ceroid lipopigment autofluorescence in LINCL fibroblasts treated as described in A. Representative fields containing ∼50 cells were analyzed. siCtrl, control siRNA; siTFEB, TFEB siRNA. Data are reported as the mean ± S.D. *, p < 0.01.
Activation of the apoptotic pathway is often observed in association with excessive autophagic activity (27, 28). To investigate whether administration of HPβCD increases apoptosis under the conditions used in this study, we used LINCL fibroblasts, which present higher propensity to undergo apoptosis compared with wild type fibroblast (42). We monitored induction of early and late apoptosis by measuring membrane rearrangement, characteristic of early apoptosis (annexin V binding), and membrane fragmentation, characteristic of late apoptosis (propidium iodide binding), using the Cyto-GLOTM annexin V-FITC apoptosis detection kit as previously described (47). HPβCD (0.1–10 mm, 3 days) did not cause significant changes in activation of early or late apoptosis compared with untreated cells (FITC binding affinity, p < 0.01; propidium iodide binding population, p < 0.01; respectively). Taxol (50 nm) was used as a positive control in this study because it is known to stabilize microtubules leading to cell cycle arrest and apoptosis induction (54) (Fig. 8). These data suggest that LINCL fibroblasts respond to treatment with HPβCD by activating the pro-survival autophagy pathway, which under the conditions used in this study results in extensive clearance of ceroid lipopigment deposits.
FIGURE 8.
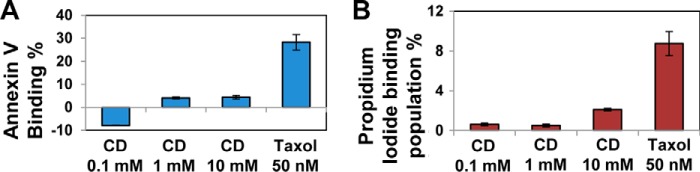
HPβCD treatment does not induce activation of apoptosis. A, annexin V binding affinity change (%) in NCL2 patient-derived fibroblasts treated with HPβCD (0.1, 1, and 10 mm) or taxol (50 nm) for 1 day normalized to untreated cells (analysis of variance, p < 0.01). B, propidium iodide binding population change (%) of NCL2 patient-derived fibroblasts treated with HPβCD (0.1, 1, and 10 mm) or taxol (50 nm) for 1 day normalized to untreated cells (analysis of variance, p < 0.01). The number of total counted cells was 10,000. The data are reported as the mean ± S.D. (n ≥ 3).
In summary, these results demonstrate that (i) HPβCD treatment results in clearance of ceroid lipopigment deposits in a model of neuronal ceroid lipofuscinosis, (ii) clearance of ceroid lipopigment observed in cells treated with HPβCD is dependent on the activation of the autophagy pathway, and (iii) induction of autophagy observed in cells treated with HPβCD does not parallel activation of apoptosis.
DISCUSSION
Exposure to natural and anthropogenic nanosized particles induces activation of a series of compensatory mechanisms to maintain cellular homeostasis. Engineered nanomaterials are typically perceived by the cell as foreign or toxic, such as virus and pathogens (20–22), and may stimulate the reaction of cellular clearance mechanisms. In this study we investigated the autophagic response that is activated upon exposure to HPβCD. We provide evidence for the first time that cell treatment with HPβCD induces a series of adaptive changes mediated by TFEB and that culminate in enhancement of the innate cellular clearance capacity.
Although cyclodextrins were first described over a century ago and have been widely used for a variety of industrial and pharmaceutical applications, their potential use for applications beyond the solubilization and stabilization of small molecules was only recently recognized. It has long been known that cyclodextrins can trap cholesterol into their hydrophobic core (7, 8). However, the molecular mechanism involved in HPβCD-mediated reduction of cholesterol accumulation in cells is unclear. We report here that HPβCD administration induces a series of adaptive changes in the lysosome-autophagy systems that are mediated by activation of TFEB, a master regulator of lysosomal biogenesis and autophagy. Importantly, we found that HPβCD treatment results in TFEB-mediated clearance of ceroid lipopigment deposits in disease cells that have a defective lysosomal system. We propose a model in which cellular uptake of HPβCD and accumulation into late endosomes and lysosomes (10, 55) results in activation of the cellular clearance response mediated by the autophagy pathway, which in turn results in degradation of the autophagic substrate ceroid lipopigment (Fig. 9). Importantly, activation of autophagy observed upon HPβCD administration, under conditions that result in activation of TFEB and clearance of autophagic material, is not associated with activation of apoptosis, suggesting that cells respond to HPβCD treatment by activating the prosurvival autophagic pathway.
FIGURE 9.
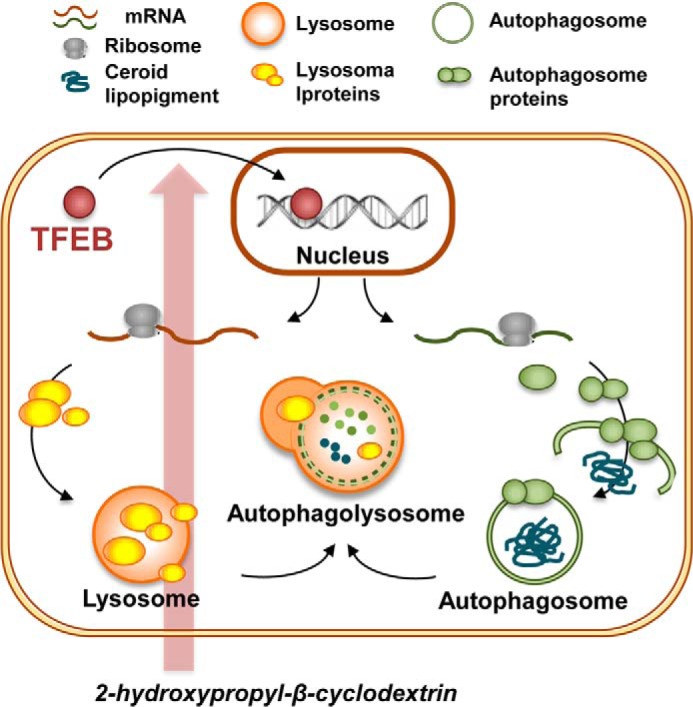
Schematic representation of the proposed adaptive cellular response to HPβCD treatment. Administration of HPβCD results in activation of TFEB. Upon translocation from the cytoplasm to the nucleus, TFEB regulates the expression of genes involved in biogenesis and fusion of lysosomes and autophagosomes. As a result, HPβCD administration results in enhanced clearance of the autophagic substrate ceroid lipopigment.
This study provides for the first time a detailed mechanistic understanding of the changes induced by HPβCD administration on this important catabolic pathway, which plays a key role in the regulation of cellular metabolism with implications ranging from cell survival under nutrient deprivation to defense against virus and parasites infections. On the one hand, these results provide a solid mechanistic groundwork to reevaluate the use of HPβCD as a drug delivery vector in conditions that may be negatively impacted by deregulation of lysosomal pathways and autophagy. On the other hand, our findings will extend the capability of designing therapeutic solutions based on the use of HPβCD for the treatment of diseases characterized by inefficient autophagic clearance and accumulation of storage material.
It was recently appreciated that impairment or deregulation of autophagy is linked to the development and progression of a number of human diseases ranging from neurodegenerative diseases to cancer. For instance, the accumulation of lysosomal substrates typically observed in affected cells from patients with lysosomal storage disorders was shown to impair fusion of lysosomes with autophagosomes and, ultimately, lower the autophagic clearance capacity (56). Generally speaking, lysosomal storage disorders include more than 50 inherited metabolic diseases caused by defective lysosomal functions and consequent accumulation of metabolites, such as lipids and glycoproteins (57). Among lysosomal storage disorders, NCLs are among the most devastating inherited disorders of childhood and the most common cause of neurodegeneration in children in the United States. In light of these findings, it is important to reevaluate the design of therapeutic strategies based on the use of HPβCD as the drug delivery vehicle or as the active agent.
Acknowledgment
We thank Dr. Junghae Suh for critical reading of the manuscript.
This work was supported by the Welch Foundation (C-1824; to L. S.), the National Science Foundation (CBET 1254318; to L. S.), and the Beyond Batten Disease Foundation (to M. S.).
- CD
- cyclodextrin
- HPβCD
- 2-hydroxypropyl-β-cyclodextrin
- TFEB
- transcription factor EB
- NCL
- neuronal ceroid lipofuscinose
- LINCL
- late infantile neuronal ceroid lipofuscinosis
- CT
- threshold cycle
- GBA
- glucocerebrosidase
- HEXA
- hexosaminidase A
- LAMP1
- lysosome-associated membrane glycoprotein 1
- MAPLC3
- microtubule-associated light chain protein 3 (LC3)
- BECN1
- Beclin-1
- SQSTM1
- Sequestosome 1
- NCL
- neuronal ceroid lipofuscinose
- CLEAR
- Coordinated Lysosomal Expression and Regulation
- Ter
- termination or stop codon.
REFERENCES
- 1. Davis M. E., Brewster M. E. (2004) Cyclodextrin-based pharmaceutics. Past, present, and future. Nat. Rev. Drug Discov. 3, 1023–1035 [DOI] [PubMed] [Google Scholar]
- 2. Rajewski R. A., Stella V. J. (1996) Pharmaceutical applications of cyclodextrins. 2. In vivo drug delivery. J. Pharm. Sci. 85, 1142–1169 [DOI] [PubMed] [Google Scholar]
- 3. Davidson C. D., Ali N. F., Micsenyi M. C., Stephney G., Renault S., Dobrenis K., Ory D. S., Vanier M. T., Walkley S. U. (2009) Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS ONE 4, e6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu B., Turley S. D., Burns D. K., Miller A. M., Repa J. J., Dietschy J. M. (2009) Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1(−/−) mouse. Proc. Natl. Acad. Sci. U.S.A. 106, 2377–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu B., Li H., Repa J. J., Turley S. D., Dietschy J. M. (2008) Genetic variations and treatments that affect the lifespan of the NPC1 mouse. J. Lipid Res. 49, 663–669 [DOI] [PubMed] [Google Scholar]
- 6. Mukherjee S., Maxfield F. R. (2004) Lipid and cholesterol trafficking in NPC. Biochim. Biophys. Acta 1685, 28–37 [DOI] [PubMed] [Google Scholar]
- 7. Atger V. M., de la Llera Moya M., Stoudt G. W., Rodrigueza W. V., Phillips M. C., Rothblat G. H. (1997) Cyclodextrins as catalysts for the removal of cholesterol from macrophage foam cells. J. Clin. Invest. 99, 773–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Christian A. E., Haynes M. P., Phillips M. C., Rothblat G. H. (1997) Use of cyclodextrins for manipulating cellular cholesterol content. J. Lipid Res. 38, 2264–2272 [PubMed] [Google Scholar]
- 9. Abi-Mosleh L., Infante R. E., Radhakrishnan A., Goldstein J. L., Brown M. S. (2009) Cyclodextrin overcomes deficient lysosome-to-endoplasmic reticulum transport of cholesterol in Niemann-Pick type C cells. Proc. Natl. Acad. Sci. U.S.A. 106, 19316–19321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rosenbaum A. I., Zhang G., Warren J. D., Maxfield F. R. (2010) Endocytosis of β-cyclodextrins is responsible for cholesterol reduction in Niemann-Pick type C mutant cells. Proc. Natl. Acad. Sci. U.S.A. 107, 5477–5482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Loftus S. K., Morris J. A., Carstea E. D., Gu J. Z., Cummings C., Brown A., Ellison J., Ohno K., Rosenfeld M. A., Tagle D. A., Pentchev P. G., Pavan W. J. (1997) Murine model of Niemann-Pick C disease. Mutation in a cholesterol homeostasis gene. Science 277, 232–235 [DOI] [PubMed] [Google Scholar]
- 12. Cheng J., Ohsaki Y., Tauchi-Sato K., Fujita A., Fujimoto T. (2006) Cholesterol depletion induces autophagy. Biochem. Biophys. Res. Commun. 351, 246–252 [DOI] [PubMed] [Google Scholar]
- 13. Wang C. W., Klionsky D. J. (2003) The molecular mechanism of autophagy. Mol. Med. 9, 65–76 [PMC free article] [PubMed] [Google Scholar]
- 14. Kundu M., Thompson C. B. (2008) Autophagy. Basic principles and relevance to disease. Annu Rev. Pathol 3, 427–455 [DOI] [PubMed] [Google Scholar]
- 15. Levine B., Kroemer G. (2008) Autophagy in the pathogenesis of disease. Cell 132, 27–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mizushima N., Yamamoto A., Matsui M., Yoshimori T., Ohsumi Y. (2004) In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 15, 1101–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Glick D., Barth S., Macleod K. F. (2010) Autophagy. Cellular and molecular mechanisms. J. Pathol. 221, 3–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hara T., Nakamura K., Matsui M., Yamamoto A., Nakahara Y., Suzuki-Migishima R., Yokoyama M., Mishima K., Saito I., Okano H., Mizushima N. (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889 [DOI] [PubMed] [Google Scholar]
- 19. Komatsu M., Waguri S., Chiba T., Murata S., Iwata J., Tanida I., Ueno T., Koike M., Uchiyama Y., Kominami E., Tanaka K. (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884 [DOI] [PubMed] [Google Scholar]
- 20. Nakagawa I., Amano A., Mizushima N., Yamamoto A., Yamaguchi H., Kamimoto T., Nara A., Funao J., Nakata M., Tsuda K., Hamada S., Yoshimori T. (2004) Autophagy defends cells against invading group a Streptococcus. Science 306, 1037–1040 [DOI] [PubMed] [Google Scholar]
- 21. Oh J. E., Lee H. K. (2012) Autophagy in Innate Recognition of Pathogens and Adaptive Immunity. Yonsei Med. J. 53, 241–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Delgado M., Singh S., De Haro S., Master S., Ponpuak M., Dinkins C., Ornatowski W., Vergne I., Deretic V. (2009) Autophagy and pattern recognition receptors in innate immunity. Immunol. Rev. 227, 189–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kondo Y., Kanzawa T., Sawaya R., Kondo S. (2005) The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 5, 726–734 [DOI] [PubMed] [Google Scholar]
- 24. Tsujimoto Y., Shimizu S. (2005) Another way to die. Autophagic programmed cell death. Cell Death Differ. 12, 1528–1534 [DOI] [PubMed] [Google Scholar]
- 25. Hait W. N., Jin S., Yang J. M. (2006) A matter of life or death (or both). Understanding autophagy in cancer. Clin. Cancer Res. 12, 1961–1965 [DOI] [PubMed] [Google Scholar]
- 26. Ogier-Denis E., Codogno P. (2003) Autophagy: a barrier or an adaptive response to cancer. Biochim. Biophys. Acta 1603, 113–128 [DOI] [PubMed] [Google Scholar]
- 27. Debnath J., Baehrecke E. H., Kroemer G. (2005) Does autophagy contribute to cell death? Autophagy 1, 66–74 [DOI] [PubMed] [Google Scholar]
- 28. Eisenberg-Lerner A., Bialik S., Simon H. U., Kimchi A. (2009) Life and death partners. Apoptosis, autophagy, and the cross-talk between them. Cell Death Differ 16, 966–975 [DOI] [PubMed] [Google Scholar]
- 29. Gorski S. M., Chittaranjan S., Pleasance E. D., Freeman J. D., Anderson C. L., Varhol R. J., Coughlin S. M., Zuyderduyn S. D., Jones S. J., Marra M. A. (2003) A SAGE approach to discovery of genes involved in autophagic cell death. Curr. Biol. 13, 358–363 [DOI] [PubMed] [Google Scholar]
- 30. Lee C. Y., Clough E. A., Yellon P., Teslovich T. M., Stephan D. A., Baehrecke E. H. (2003) Genome-wide analyses of steroid- and radiation-triggered programmed cell death in Drosophila. Curr. Biol. 13, 350–357 [DOI] [PubMed] [Google Scholar]
- 31. Kroemer G., Levine B. (2008) Autophagic cell death. The story of a misnomer. Nat. Rev. Mol. Cell Biol. 9, 1004–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mizushima N., Komatsu M. (2011) Autophagy. Renovation of cells and tissues. Cell 147, 728–741 [DOI] [PubMed] [Google Scholar]
- 33. Sardiello M., Palmieri M., di Ronza A., Medina D. L., Valenza M., Gennarino V. A., Di Malta C., Donaudy F., Embrione V., Polishchuk R. S., Banfi S., Parenti G., Cattaneo E., Ballabio A. (2009) A gene network regulating lysosomal biogenesis and function. Science 325, 473–477 [DOI] [PubMed] [Google Scholar]
- 34. Palmieri M., Impey S., Kang H., di Ronza A., Pelz C., Sardiello M., Ballabio A. (2011) Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 20, 3852–3866 [DOI] [PubMed] [Google Scholar]
- 35. Song W., Wang F., Savini M., Ake A., di Ronza A., Sardiello M., Segatori L. (2013) TFEB regulates lysosomal proteostasis. Hum. Mol. Genet. 22, 1994–2009 [DOI] [PubMed] [Google Scholar]
- 36. Settembre C., Di Malta C., Polito V. A., Garcia-Arencibia M., Vetrini F., Erdin S., Erdin S. U., Huynh T., Medina D., Colella P., Sardiello M., Rubinsztein D. C., Ballabio A. (2011) TFEB Links Autophagy to Lysosomal Biogenesis. Science 332, 1429–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Settembre C., Ballabio A. (2011) TFEB regulates autophagy An integrated coordination of cellular degradation and recycling processes. Autophagy 7, 1379–1381 [DOI] [PubMed] [Google Scholar]
- 38. Sardiello M., Ballabio A. (2009) Lysosomal enhancement A CLEAR answer to cellular degradative needs. Cell Cycle 8, 4021–4022 [DOI] [PubMed] [Google Scholar]
- 39. Pierret C., Morrison J. A., Kirk M. D. (2008) Treatment of lysosomal storage disorders: focus on the neuronal ceroid-lipofuscinoses. Acta Neurobiol. Exp. 68, 429–442 [DOI] [PubMed] [Google Scholar]
- 40. Williams R. E., Mole S. E. (2012) New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. Neurology 79, 183–191 [DOI] [PubMed] [Google Scholar]
- 41. Kohlschütter A., Schulz A. (2009) Towards understanding the neuronal ceroid lipofuscinoses. Brain Dev. 31, 499–502 [DOI] [PubMed] [Google Scholar]
- 42. Persaud-Sawin D. A., Mousallem T., Wang C., Zucker A., Kominami E., Boustany R. M. (2007) Neuronal ceroid lipofuscinosis. A common pathway? Pediatr. Res. 61, 146–152 [DOI] [PubMed] [Google Scholar]
- 43. Medina D. L., Fraldi A., Bouche V., Annunziata F., Mansueto G., Spampanato C., Puri C., Pignata A., Martina J. A., Sardiello M., Palmieri M., Polishchuk R., Puertollano R., Ballabio A. (2011) Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell 21, 421–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ghosh A., Corbett G. T., Gonzalez F. J., Pahan K. (2012) Gemfibrozil and fenofibrate, Food and Drug Administration-approved lipid-lowering drugs, up-regulate tripeptidyl-peptidase 1 in brain cells via peroxisome proliferator-activated receptor α. Implications for late infantile batten disease therapy. J. Biol. Chem. 287, 38922–38935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang F., Song W., Brancati G., Segatori L. (2011) Inhibition of endoplasmic reticulum-associated degradation rescues native folding in loss of function protein misfolding diseases. J. Biol. Chem. 286, 43454–43464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kondo S., Kubota S., Mukudai Y., Moritani N., Nishida T., Matsushita H., Matsumoto S., Sugahara T., Takigawa M. (2006) Hypoxic regulation of stability of connective tissue growth factor/CCN2 mRNA by 3′-untranslated region interacting with a cellular protein in human chondrosarcoma cells. Oncogene 25, 1099–1110 [DOI] [PubMed] [Google Scholar]
- 47. Wang F., Chou A., Segatori L. (2011) Lacidipine remodels protein folding and Ca2+ homeostasis in Gaucher's disease fibroblasts. A mechanism to rescue mutant glucocerebrosidase. Chem. Biol. 18, 766–776 [DOI] [PubMed] [Google Scholar]
- 48. Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mizushima N., Yoshimori T., Levine B. (2010) Methods in mammalian autophagy research. Cell 140, 313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sleat D. E., Donnelly R. J., Lackland H., Liu C. G., Sohar I., Pullarkat R. K., Lobel P. (1997) Association of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis. Science 277, 1802–1805 [DOI] [PubMed] [Google Scholar]
- 51. Boya P., González-Polo R. A., Casares N., Perfettini J. L., Dessen P., Larochette N., Métivier D., Meley D., Souquere S., Yoshimori T., Pierron G., Codogno P., Kroemer G. (2005) Inhibition of macroautophagy triggers apoptosis. Mol. Cell. Biol. 25, 1025–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cao Y., Espinola J. A., Fossale E., Massey A. C., Cuervo A. M., MacDonald M. E., Cotman S. L. (2006) Autophagy is disrupted in a knock-in mouse model of juvenile neuronal ceroid lipofuscinosis. J. Biol. Chem. 281, 20483–20493 [DOI] [PubMed] [Google Scholar]
- 53. Komatsu M., Waguri S., Ueno T., Iwata J., Murata S., Tanida I., Ezaki J., Mizushima N., Ohsumi Y., Uchiyama Y., Kominami E., Tanaka K., Chiba T. (2005) Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 169, 425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bacus S. S., Gudkov A. V., Lowe M., Lyass L., Yung Y., Komarov A. P., Keyomarsi K., Yarden Y., Seger R. (2001) Taxol-induced apoptosis depends on MAP kinase pathways (ERK and p38) and is independent of p53. Oncogene 20, 147–155 [DOI] [PubMed] [Google Scholar]
- 55. Maxfield F. R., van Meer G. (2010) Cholesterol, the central lipid of mammalian cells. Curr. Opin. Cell Biol. 22, 422–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Settembre C., Fraldi A., Rubinsztein D. C., Ballabio A. (2008) Lysosomal storage diseases as disorders of autophagy. Autophagy 4, 113–114 [DOI] [PubMed] [Google Scholar]
- 57. Futerman A. H., van Meer G. (2004) The cell biology of lysosomal storage disorders. Nat. Rev. Mol. Cell Biol. 5, 554–565 [DOI] [PubMed] [Google Scholar]