

Background: In [16,16,16-d3]palmitic acid (M+3) perfused working hearts, M+3 stearoylcarnitine is formed.
Results: Rat hearts chain-elongate palmitate to stearate and arachidate. Furthermore, rat heart mitochondria catalyze the malonyl/acetyl-CoA-dependent chain elongation of palmitoyl-CoA.
Conclusion: The two-carbon units for fatty acid chain elongation are derived from mitochondrial fatty acid β-oxidation.
Significance: This pathway may represent the molecular basis for heart-specific fatty acid remodeling.
Keywords: Carnitine; Coenzyme A; Fatty Acid Oxidation; Heart; Mitochondria; Fatty Acid Chain Elongation, Malonyl-CoA, Carnitine Palmitoyltransferase, Acylcarnitines, Acyl-CoAs
Abstract
Rat hearts were perfused with [1,2,3,4-13C4]palmitic acid (M+4), and the isotopic patterns of myocardial acylcarnitines and acyl-CoAs were analyzed using ultra-HPLC-MS/MS. The 91.2% 13C enrichment in palmitoylcarnitine shows that little endogenous (M+0) palmitate contributed to its formation. The presence of M+2 myristoylcarnitine (95.7%) and M+2 acetylcarnitine (19.4%) is evidence for β-oxidation of perfused M+4 palmitic acid. Identical enrichment data were obtained in the respective acyl-CoAs. The relative 13C enrichment in M+4 (84.7%, 69.9%) and M+6 (16.2%, 17.8%) stearoyl- and arachidylcarnitine, respectively, clearly shows that the perfused palmitate is chain-elongated. The observed enrichment of 13C in acetylcarnitine (19%), M+6 stearoylcarnitine (16.2%), and M+6 arachidylcarnitine (17.8%) suggests that the majority of two-carbon units for chain elongation are derived from β-oxidation of [1,2,3,4-13C4]palmitic acid. These data are explained by conversion of the M+2 acetyl-CoA to M+2 malonyl-CoA, which serves as the acceptor for M+4 palmitoyl-CoA in chain elongation. Indeed, the 13C enrichment in mitochondrial acetyl-CoA (18.9%) and malonyl-CoA (19.9%) are identical. No 13C enrichment was found in acylcarnitine species with carbon chain lengths between 4 and 12, arguing against the simple reversal of fatty acid β-oxidation. Furthermore, isolated, intact rat heart mitochondria 1) synthesize malonyl-CoA with simultaneous inhibition of carnitine palmitoyltransferase 1b and 2) catalyze the palmitoyl-CoA-dependent incorporation of 14C from [2-14C]malonyl-CoA into lipid-soluble products. In conclusion, rat heart has the capability to chain-elongate fatty acids using mitochondria-derived two-carbon chain extenders. The data suggest that the chain elongation process is localized on the outer surface of the mitochondrial outer membrane.
Introduction
Fatty acids are the major source for ATP production in the heart. Under normoxic conditions, ∼60–70% of the heart's energy requirement is met by β-oxidation of fatty acids. In addition to ATP production, fatty acids are required for the synthesis of membrane phospholipids with tissue- and organelle-specific fatty acyl composition. The heart's fatty acid need for both processes, β-oxidation and phospholipid synthesis, is met by uptake of fatty acids from the circulation. Whereas ATP production by mitochondrial β-oxidation relies mostly on catabolism of saturated fatty acid with 16 and to a lesser extent with 18 carbons (i.e. palmitate and stearate), synthesis of phospholipids requires fatty acids with longer saturated and unsaturated carbon chains. However, it is unknown whether fatty acids for phospholipid synthesis are derived exclusively from blood or if the heart is capable of modifying fatty acids derived from circulation by chain elongation and desaturation (1–4). Synthesis of long-chain saturated and unsaturated fatty acids involves the malonyl-CoA-dependent elongation of fatty acids by enzymes, referred to as elongation of very long-chain fatty acids (ELOVL),3 and introduction of one or more double bounds into the chain-elongated fatty acids catalyzed by stearoyl-CoA and fatty acid desaturases. Both ELOVL and stearoyl-CoA/fatty acid desaturase enzymes are localized in microsomes (5–8).
Previously, we found that when rat hearts are perfused in the working mode with palmitate plus glucose (but not with glucose alone), there is a time-dependent accumulation of palmitoyl-, myristoyl-, and stearoylcarnitine. Our interpretation was that some of the perfused palmitate underwent chain elongation to stearate. Using the fatty acid-perfused working rat heart model in subsequent experiments, chain elongation of palmitate to stearate has been unequivocally shown by the formation of M+3 stearoylcarnitine from perfused [16,16,16-d3]palmitic acid (M+3) (9). These experiments demonstrated that the heart is able to catalyze fatty acid chain elongation. The nearly identical stable isotope enrichment in palmitoyl- and stearoylcarnitine suggested that the chain elongation of palmitate to stearate occurred in a compartment with carnitine palmitoyltransferase activity in mitochondria. This notion is supported by the finding that in rabbit heart perfused in the Langendorff mode, the conversion of perfused [9,10-3H]octadec-9′-enoic acid to [3H]eicosenoylcarnitine is confined to the acylcarnitine pool (4). Furthermore, the lack of detectable M+5 stearoylcarnitine in our studies with [16,16,16-d3]palmitic acid indicated that unlabeled malonyl-CoA served as the acyl acceptor in the chain elongation process. The formation of M+5 stearoylcarnitine is expected if the perfused [16,16,16-d3]palmitic acid (M+3) undergoes complete oxidation to acetyl-CoA (7 M+0 and 1 M+3) in mitochondria but not when palmitate is partially oxidized in peroxisomes and the two-carbon units of acetyl-CoA are used for fatty acid chain elongation. (The formation of M+2 malonyl-CoA from M+3 acetyl-CoA results in a loss of a proton from the methyl group of acetyl-CoA.) It has been shown that in the Langendorff-perfused heart, peroxisomal oxidation of fatty acids is a substantial contributor to malonyl-CoA synthesis (10).
The goal of the present study was to determine the source and metabolic origin of the two-carbon units for chain elongation of perfused palmitic acid. To this end, we perfused isolated rat hearts with [1,2,3,4-13C4]palmitic acid (M+4) and by UHPLC-MS/MS determined the relative enrichment of 13C in palmitoyl-, myristoyl-, and acetylcarnitine as well as in the respective CoA esters (intermediates and products of β-oxidation) and in stearoyl- and arachidylcarnitine and stearoyl-CoA, which are products of fatty acid chain elongation. Because malonyl-CoA, synthesized from acetyl-CoA by acetyl-CoA carboxylase (ACC), is the ultimate two-carbon donor in chain elongation, the relative 13C enrichment also was determined in acetyl-CoA and malonyl-CoA. Because limited data (3) suggested that fatty acid chain elongation is decreased with age, both 6-month-old adult and 24-month-old elderly Fisher rats were used. In addition, we assessed whether isolated intact mitochondria have the capacity to synthesize malonyl-CoA and to catalyze fatty acid chain elongation.
EXPERIMENTAL PROCEDURES
Chemicals
Unlabeled palmitic acid (≥99.0% by GC) was from Fluka, and [1,2,3,4-13C4]palmitic acid (M+4) (minimum 99 atom % 13C) was from Isotech. [1-14C]acetyl-CoA (54 mCi/mmol) and [2-14C]malonyl-CoA (55 mCi/mmol) were purchased from Moravek Biochemicals. Malate dehydrogenase (5 mg/ml, 1200 units/mg) and carnitine acetyltransferase (5 mg/ml, 80 units/mg) were from Roche Applied Science, and carnitine (inner salt) was a gift from Sigma-Tau (Rome, Italy). ATP-citrate lyase was isolated from livers of starved/high carbohydrate diet refed rats by ammonium sulfate precipitation and DEAE-cellulose chromatography (11), and the activity was determined as described (12). The isolated enzyme had a specific activity of 99.6 units (μmol/min/mg) of protein and was free of acetyl-CoA hydrolase and citrate synthase. The enzyme stored in the presence of 0.1 mm dithiothreitol at −60 °C is stable for at least 8 months. Trypsin and soybean trypsin inhibitor were from Worthington. All other chemicals were of the highest quality commercially available.
Heart Perfusion
6- and 24-month-old Fisher 344 rats were obtained from a colony maintained by the NIA, National Institutes of Health (Harlan Sprague-Dawley, Inc., Indianapolis, IN). The animals, housed in our animal care facility in a temperature- and humidity-controlled room with a 12-h light/dark cycle, had free access to food and water until the experiments. The perfusion protocol was the same as described previously (9, 13). Briefly, the animals were weighed, injected with 500 units of heparin (intraperitoneally), and euthanized with sodium pentobarbital (100 mg/kg body weight), and the hearts were canulated for perfusion. The perfusion protocol consisted of a 15 min of non-recirculating perfusion in the Langendorff mode with Krebs-Henseleit buffer containing 5.5 mm glucose and 0.1 units/liter insulin, followed by a 60-min perfusion in the working mode (left atrial flow 35–50 ml/min at a preload of 10–15 mm Hg) with 5.5 mm glucose, 0.1 units/liter insulin, and 1.2 mm [1,2,3,4-13C4]palmitic acid (M+4) complexed to 3% BSA. At the end of perfusion, the hearts were freeze-clamped and powdered under liquid nitrogen, and the powdered tissue was stored at −60 °C until analysis.
Isolation of Rat Heart Mitochondria
Rat heart subsarcolemmal (SSM) and interfibrillar mitochondria (IFM) were isolated as referenced (14), with the exception that nagarse was replaced by trypsin (5 mg/g wet heart weight); trypsin activity was stopped by soybean trypsin inhibitor (2.5 mg/g wet heart weight). The intactness of the mitochondrial outer and inner membrane was determined by measuring the activity of cytochrome oxidase as the first order rate constant using reduced cytochrome c before and after treatment of mitochondria with dodecyl maltoside.
Mitochondrial Malonyl-CoA Synthesis and Carnitine Palmitoyltranferase 1b (CPT1b) Activity
Rat heart mitochondria (2.0 mg/ml) were incubated at room temperature for 5, 10, and 15 min in the presence of 20 mm Hepes, 97 mm KCl, 5 mm MgCl2, 18 mm NaHCO3, 10 mm Na3-citrate, 2.2 mm ATP, 1.0 mm acetyl-CoA, pH 7.0 (complete reaction mixture), in a final volume of 1.0 ml. At the indicated time points, duplicate 0.1-ml aliquots were removed and immediately frozen in liquid nitrogen for later determination of malonyl-CoA. At the same time points, 10-μl aliquots were removed, and the activity of CPT1 was measured using the modified radiochemical forward assay as described (15) with the exception that the carnitine concentration was 5.0 mm. CPT1b activity is defined as the activity suppressed by 200 μm malonyl-CoA.
Determination of Fatty Acid Chain Elongation in Isolated Mitochondria
Fatty acid chain elongation with isolated intact mitochondria was assessed by measuring the palmitoyl-CoA-dependent incorporation of 14C from [2-14C]malonyl-CoA or [1-14C]acetyl-CoA into lipid-soluble products as described (16) with the exception that 20 mm Hepes, 130 mm KCl (pH 7.4) was used to replace 50 mm potassium phosphate (pH 7.0). The assay was linear with time and mitochondrial protein concentration, and was routinely carried out with 250 μg of protein for 5 or 15 min at 37 °C with acetyl-CoA and malonyl-CoA, respectively, as the substrates.
Analysis of 13C Enrichment in Cardiac Acylcarnitines and Acyl-CoAs
Acylcarnitines and acyl-CoAs were extracted from powdered heart tissue by homogenizing the tissue powder first with acetonitrile/isopropyl alcohol (3:1, v/v) followed by 0.1 m KH2PO4 (pH 6.7) and a second 30-s homogenization (17). Acylcarnitines were isolated by strong cation exchange solid phase extraction, derivatized with pentafluorophenacyl trifluoromethanesulfonate, and analyzed by UHPLC-MS/MS for content and for 13C enrichment as described in detail earlier (18, 19). Acyl-CoAs were isolated using solid-phase anion exchange columns (Supelco) as described (17). For acyl-CoAs, 500 μl of the tissue extract was applied to an SPE column containing 100 mg of 2-(2-pyridyl)ethyl-functionalized silica gel. On-line WAX trapping was used to trap all of the acyl-CoAs, followed by UHPLC reversed-phase chromatography and detection by positive ion multiple reaction monitoring (17, 20). The stable isotope enrichment was determined by deconvolution of the isotope patterns using the Isopat2 algorithm of Gruber et al. (21). This method of isotope deconvolution uses a comparison between a known unlabeled standard and the labeled analyte. Standard isotope abundances came from acylcarnitines and acyl-CoAs prepared along with the biological samples (18). The data are expressed as relative 13C enrichment. For stearoyl- and arachidylcarnitine, the values also are expressed after correction for the presence of unlabeled (M+0) species.
Analysis of 13C Enrichment of Mitochondrial (Citrate) Acetyl-CoA
The stable isotope enrichment of mitochondrial acetyl-CoA was determined essentially as described (10) with the modification that citrate was cleaved into acetyl-CoA and oxaloacetate, and the product acetyl-CoA was trapped in the form of acetylcarnitine in the presence of excess carnitine and carnitine acetyltransferase. Briefly, 400–500 mg of powdered heart was extracted with four volumes of cold 1 n HClO4, the supernatant (15 min, 16,000 × g) was neutralized with 10 n KOH, and the KClO4 was removed by centrifugation. To hydrolyze endogenous acetyl-CoA and acetylcarnitine, the pH of the neutralized supernatant was adjusted to ∼13 with KOH and left for 1 h at 50 °C. Following neutralization with 1 n HClO4 to pH 6–7, the KClO4 was removed, and the supernatant was used for converting the acetyl-moiety of citrate into acetylcarnitine as described briefly below. The incubation mixture contained (in a 1.0-ml final volume) 100 mm Tris, pH 8.7, 10 mm MgCl2, 5 mm ATP, 0.33 mm CoASH, 2 mm l-carnitine, 0.5 ml of base-treated and neutralized heart extract, 12 units (1 unit defined as 1 μmol/min/mg protein) of malate dehydrogenase, 0.8 unit of carnitine acetyltransferase, and 0.3 mm NADH. The reaction was initiated with 1.0 unit of ATP-citrate lyase and followed by measuring the decrease of absorbance at 340 nm. The reaction was complete in less than 8 min, after which excess N-ethylmaleimide (4.0 mm) is added to ensure that all acetyl-CoA is converted to acetylcarnitine. The pH of the reaction mixture was brought to ∼2–3 with 1 n HCl, the precipitated protein was removed, and the stable isotope enrichment in acetylcarnitine was determined as described above.
Statistical Analysis
Data are presented as means ± S.E. of at least three independent experiments. Differences between data sets were tested for statistical significance using Student's t test and analysis of variance (SigmaStat) with p < 0.05 considered statistically significant.
RESULTS
In contrast to the reported age-associated decrease in chain elongation of linoleic acid observed with isolated cardiomyocytes (3), no age-related differences with acyl-CoA and acylcarnitine stable isotope enrichment in data from the 6-month-old versus 24-month-old rats were observed in the present study. Therefore, the data obtained with 6- and 24-month-old animals were combined and discussed together.
Carnitine Status of Palmitate-perfused Working Heart
Previously, we have shown that in palmitate-perfused working rat heart, there is a time-dependent accumulation of palmitoyl-, myristoyl-, and stearoylcarnitine (9). In the present study, we first evaluated the effect of perfusion of rat heart in the working mode with unlabeled palmitic acid on the total and free carnitine and acylcarnitine content as well as on the amount of the individual short-, medium-, and long-chain acylcarnitines. As shown in Table 1, there is a large, although statistically not significant, loss of total and free carnitine and total acylcarnitines after a 60-min perfusion with unlabeled palmitic acid. Among the individual short-chain acylcarnitines, acetylcarnitine showed a statistically significant decrease, whereas there was only a trend for a decrease of all other short-chain acylcarnitines. These decreases occurred although there was a dramatic increase in palmitoylcarnitine content.
TABLE 1.
Effect of perfusion of rat heart with palmitate on cardiac acylcarnitine profile
Total, free, and individual short-, medium-, and long-chain acylcarnitines were quantified in rat hearts at baseline (no perfusion) and after 60-min perfusion in the working mode with unlabeled palmitic acid. The numbers represent the mean ± S.E. of six (baseline) and four (perfusion) separate experiments. ND, not determined.
| Acylcarnitine species | Heart acylcarnitines |
|
|---|---|---|
| Baseline (no perfusion) | Perfusion with palmitate | |
| nmol/g wet weight | nmol/g wet weight | |
| Total carnitine | 873.8 ± 150.3 | 579.9 ± 78.5 |
| Free carnitine | 563.2 ± 124.1 | 304 ± 15.8 |
| Acylcarnitines | 310.62 ± 64.4 | 276.0 ± 94.0 |
| Acetyl | 260.6 ± 63.6 | 62.7 ± 3.7a |
| Butyryl | 1.7 ± 0.8 | ND |
| Isobutyryl | 0.3 ± 0.4 | ND |
| (R)-3-Hydroxybutyryl | 2.3 ± 0.3 | ND |
| (S)-3-Hydroxybutyryl | 6.7 ± 2.5 | 1.6 ± 0.07 |
| 2-Hydroxyisovaleryl | 0.7 ± 0.5 | ND |
| 2-Methylbutyryl | 0.3 ± 0.4 | ND |
| Lauroyl | ND | 1.8 ± 1.2 |
| Myristoyl | 0.75 ± 0.67 | 15.4 ± 10.2a |
| (S)-3-hydroxymyristoyl | ND | 0.7 ± 0.85 |
| Palmitoyl | 2.81 ± 0.98 | 135.9 ± 40.7a |
| (S)-3-Hydroxypalmitoyl | 0.2 ± 0.25 | 6.0 ± 2.9a |
| Stearoyl | 1.11 ± 0.65 | 16.34 ± 9.8a |
| (S)-3-Hydroxystearoyl | ND | 0.6 ± 0.7 |
| Oleyl | 1.82 ± 0.8 | 1.5 ± 0.92 |
| Linoleyl | 2.08 ± 0.8 | 1.6 ± 0.99 |
| Malonyl | 3.3 ± 0.65 | 2.0 ± 0.35 |
| Succinyl | 7.2 ± 1.6 | 5.9 ± 1.06 |
a Carnitine species whose content showed a significant increase or decrease (p < 0.05, Student's t test) after perfusion.
Perfusion of rat hearts with palmitic acid also led to a statistically significant increase of myristoyl-, (S)-3-hydroxypalmitoyl-, and stearoylcarnitine, intermediates of β-oxidation and product of palmitate chain elongation, respectively. No changes were found in the cardiac content of unsaturated and dicarboxylyl carnitine derivatives.
Metabolism of [1,2,3,4-13C4]Palmitic Acid in Perfused Working Heart
Analysis of 13C Enrichment in Acylcarnitines
Acyl-CoAs produced in mitochondria and/or peroxisomes are in equilibrium with their respective acylcarnitines via carnitine palmitoyltransferase 1, carnitine palmitoyltransferase 2, and carnitine acetyltransferase (CPT 1/CPT 2/CAT) in mitochondria, and with carnitine octanoyltransferase and carnitine acetyltransferase (COT/CAT) in peroxisomes. Thus, acylcarnitines reflect both the content and isotope labeling of the acyl-CoA esters, and because of the higher carnitine concentration in heart, their content can be conveniently measured by UHPLC-MS/MS. Additionally, mass spectrometric analysis of 13C enrichment in acylcarnitines, as compared with acyl-CoAs, has the advantage of a lower background contribution (1.1% per carbon atom) even after derivatization with pentafluorophenacyl trifluoromethanesulfonate. The 13C enrichment of acylcarnitines is summarized in Table 2, and a representative chromatographic separation and mass isotopomer distribution of acylcarnitines along with their acyl-CoA counterparts formed by the heart during the 60-min perfusion is depicted in Figs. 1 and 2.
TABLE 2.
Relative percent 13C enrichment in acylcarnitine mass isotopomers produced from [1,2,3,4-13C4]palmitic acid (M+4) in perfused hearts
Rat heart extracts were analyzed for acylcarnitines by UHPLC-MS/MS, using multiple reaction monitoring transitions specific to the isotopic form of each acylcarnitine species. The deconvolution of the isotope patterns was performed using the “Isopat” algorithm of Gruber et al. (21). No 13C enrichment was found in acylcarnitines with a chain length ranging from 6 to 12 carbons. The corrected percent 13C enrichment values for M+4 and M+6 for stearoyl- and arachidylcarnitine are given in parenthesis. The data represent the mean ± S.E. of six or seven experiments. Boldface type identifies the mass isotopomers whose percent 13C enrichment is expected to be altered due to β-oxidation/chain elongation of perfused [1,2,3,4-13C]palmitate (M+2, M+4, M+6) or dilution by endogenous fatty acids (M+0).
| Palmitoyl-carnitine | Myristoyl-carnitine | Acetyl-carnitine | Stearoyl-carnitine | Arachidyl-carnitine | |
|---|---|---|---|---|---|
| M+0 | 4.5 ± 2.1 | 5.9 ± 1.4 | 80.4 ± 1.2 | 24.9 ± 6.7 | 27.6 ± 11.0 |
| M+1 | 0.2 ± 0.1 | 1.3 ± 0.1 | 0.1 ± 0.03 | 1.4 ± 0.4 | 0.0 |
| M+2 | 1.0 ± 0.3 | 95.7 ± 1.4a | 19.4 ± 1.2a | 2.4 ± 0.4 | 5.6 ± 2.9 |
| M+3 | 2.4 ± 0.1 | 0.0 | 0.0 ± | 1.5 ± 0.3 | 0.4 ± 2.4 |
| M+4 | 91.2 ± 2.1a | 0.0 | 0.5 ± 0.03 | 59.5 ± 1.2a | 45.5 ± 4.5a |
| (84.7 ± 1.2) | (69.9 ± 4.8) | ||||
| M+5 | 0.0 | 0.0 | 7.7 ± 2.1 | ||
| M+6 | 2.4 ± 0.2 | 11.6 ± 1.0a,b | 12.0 ± 2.3a,b | ||
| (16.2 ± 0.9a,b) | (17.8 ± 1.6a,b) | ||||
| M+7 | 0.0 | 7.4 ± 1.1 | |||
| M+8 | 0.0 | 0.0 |
a p < 0.05 versus M+0.
b p < 0.05, M+6 versus M+4 and M+0 (analysis of variance).
FIGURE 1.

Chromatographic separation and determination of mass isotopomer distribution of long-chain acylcarnitines and acyl-CoAs extracted from [1,2,3,4-13C4]palmitate-perfused working rat heart. Shown is a representative chromatogram showing the UHPLC separation and mass isotopomer distribution of the carnitine and CoA derivatives of myristic, palmitic, and stearic acid extracted from rat heart perfused for 60 min with [1,2,3,4-13C4]palmitic acid (M+4). In the insets, the most abundant isotopomer was set to 100%.
FIGURE 2.
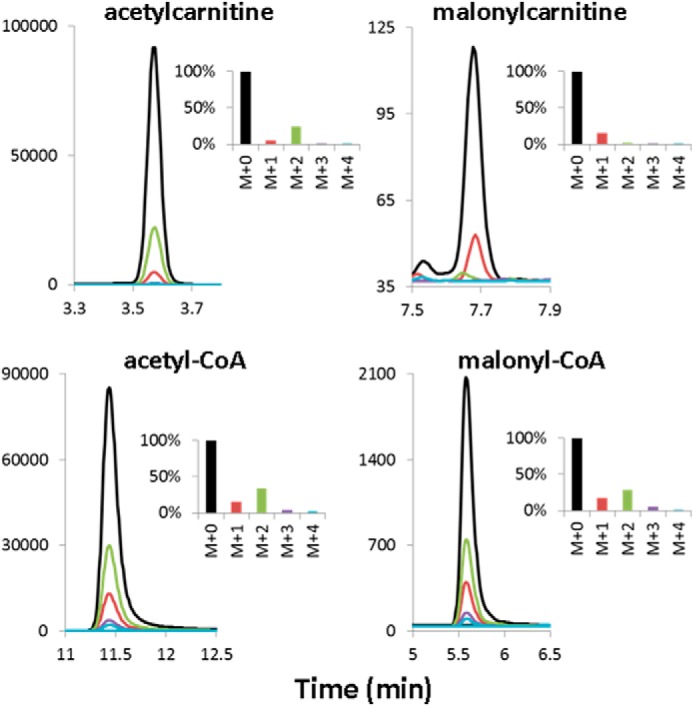
Chromatographic separation and determination of mass isotopomer distribution of short-chain acylcarnitines and acyl-CoAs extracted from [1,2,3,4-13C4]palmitate-perfused working rat heart. Shown is a representative chromatogram showing the UHPLC separation and mass isotopomer distribution of the acetyl- and malonylcarnitine and their respective CoA derivatives extracted from rat heart perfused for 60 min with [1,2,3,4-13C4]palmitic acid (M+4). In the insets, the most abundant isotopomer was set to 100%.
As shown by the formation of stable isotope-labeled M+4 palmitoylcarnitine, M+2 myristoylcarnitine, and M+2 acetylcarnitine (Table 2 and Figs. 1 and 2), rat hearts readily took up and β-oxidized palmitate. Because the perfused 13C-labeled palmitic acid was greater than 99 atom % 13C, the 4.5% relative amount of M+0 palmitoylcarnitine indicates dilution of the perfused palmitic acid by a negligible amount of unlabeled endogenous palmitic acid, probably generated by triglyceride and phospholipid hydrolysis during the perfusion period. In good agreement with this, M+4 palmitoylcarnitine represented 91.2% of all palmitoylcarnitine mass isotopomers (Table 2) without correction for the unlabeled (M+0) species. As expected from the isotopomer distribution of palmitoylcarnitine (M+4 much greater than M+0), if the perfused [1,2,3,4-13C4]palmitic acid undergoes β-oxidation, the predominant isotopomer of myristoylcarnitine will be M+2. Indeed, the relative amount of the M+2 myristoylcarnitine accounts for 95.7% (Table 2). Thus, the stable isotope enrichment in M+2 myristoylcarnitine is close to that found in M+4 palmitoylcarnitine. These results are consistent with a precursor-product relationship between M+4 palmitoylcarnitine(CoA) and M+2 myristoylcarnitine(CoA), regardless of whether the β-oxidation occurred either in mitochondria or peroxisomes (Tables 2 and 3 and Fig. 1). The mass isotopomer distribution of unlabeled (M+0) and labeled (M+2) acetylcarnitine was found to be 80.4 and 19.4%, respectively (Table 2). No 13C enrichment was observed in hexanoyl-, octanoyl-, decanoyl-, lauroyl-, oleyl-, and malonylcarnitine. For butyrylcarnitine, of seven perfused hearts, no enrichment was found in five samples, whereas in two samples, a 14.7 and 6.4% enrichment was observed in M+2 butyrylcarnitine isotopomer.
TABLE 3.
Relative percent 13C enrichment in acyl-CoA mass isotopomers produced from [1,2,3,4-13C4]palmitic acid (M+4) in perfused hearts
Rat heart extracts were analyzed for acyl-CoAs by UHPLC-MS/MS, using multiple reaction monitoring transitions specific to the isotopic form of each acyl-CoA species. The deconvolution of the isotope patterns was performed using the “Isopat” algorithm of Gruber et al. (21). The data represent the mean ± S.E. of six experiments. Boldface identifies the mass isotopomers whose percent 13C enrichment is expected to be altered due to β-oxidation/chain elongation of perfused [1,2,3,4-13C]palmitate (M+2, M+4, M+6) or by dilution by endogenous fatty acids (M+0).
| Palmitoyl-CoA | Myristoyl-CoA | Stearoyl-CoA | Mitochondrial acetyl-CoA (citrate) | Total Acetyl-CoA | Malonyl-CoA | |
|---|---|---|---|---|---|---|
| M+0 | 4.2 ± 0.8 | 8.2 ± 1.1 | 53.6 ± 11.5 | 81.1 ± 0.8 | 77.6 ± 0.9 | 80.0 ± 1.0 |
| M+1 | 0.0 | 1.2 ± 0.13 | 0.4 ± 0.53 | 0.2 ± 0.05 | 0.1 ± 0.05 | 1.3 ± 0.2 |
| M+2 | 0.3 ± 0.14 | 93.4 ± 1.2a | 2.5 ± 0.63 | 18.9 ± 0.8a | 22.4 ± 0.9a | 19.9 ± 1.1a |
| M+3 | 2.3 ± 0.03 | 0 | 0.8 ± 0.25 | 0.0 | 0.0 | 0.0 |
| M+4 | 93.2 ± 0.74a | 0 | 37.8 ± 10.1a | 0.0 | 0.6 ± 0.03 | 0.0 |
| M+5 | 0.0 | 0.1 ± 0.1 | 0.0 | |||
| M+6 | 3.8 ± 0.12 | 5.4 ± 1.6a,b | ||||
| M+7 | 0.0 | |||||
| M+8 | 0.0 |
a p < 0.05 versus M+0.
b p < 0.05 M+6 versus M+4 and M+0 (analysis of variance).
In addition to β-oxidation, a significant fraction of the perfused [1,2,3,4-13C4]palmitate was chain-elongated to C18 stearate and C20 arachidate, as shown by the presence of stable isotope-labeled M+4 and M+6 stearoylcarnitine and arachidylcarnitine species (Table 2 and Fig. 1). For stearoylcarnitine, the relative amount of the unlabeled (M+0) species is 24.9%, and that of the M+4 and M+6 species is 59.5 and 11.6%, respectively. Correcting for the contribution of the unlabeled species, the relative amount of the M+4 and M+6 is 84.7 and 16.2%, respectively. Similar 13C enrichment data were obtained for arachidylcarnitine (i.e. 27.6% M+0, 45.5% M+4, and 12% M+6 before and 69.0 and 17.8% for the M+4 and M+6, after correcting for the M+0 species) (Table 2). The isotope enrichment in M+6 species of stearoyl- and arachidylcarnitine is in close agreement with that found in M+2 acetylcarnitine (19.4%) and clearly shows that β-oxidation of [1,2,3,4-13C4]palmitate contributed the pool of two-carbon units used for chain elongation. The presence of 25–28% M+0 stearoyl- and arachidylcarnitine isotopomers indicates a contribution of these fatty acids from endogenous sources.
Analysis of 13C Enrichment in Acyl-CoA Esters
In order to test the notion that the two-carbon units used for fatty acid chain elongation are derived from mitochondrial fatty acid oxidation, we determined the 13C enrichment in cardiac long-chain (palmitoyl-, myristoyl-, stearoyl-CoA) (Fig. 1) and short-chain acyl-CoAs (malonyl-CoA and acetyl-CoA) (Fig. 2) with special emphasis on mitochondrial acetyl-CoA. These enrichment data are summarized in Table 3 and Figs. 1 and 2. The relative amounts of the M+2 isotopomers of malonyl-CoA and mitochondrial acetyl-CoA (as well as acetylcarnitine) are in good agreement, confirming that carbon atoms 1 and 2 of malonyl-CoA are derived from mitochondrial acetyl-CoA via β-oxidation of [1,2,3,4-13C4]palmitic acid. On the other hand, the enrichment in total cardiac M+2 acetyl-CoA is slightly higher than that of mitochondrial M+2 acetyl-CoA; however, this difference is statistically not significant. Comparison of the 13C enrichment in β-oxidation intermediate acyl-CoAs presented in Table 3 with that of the corresponding acylcarnitines in Table 2 shows an identical relative 13C enrichment in palmitoyl-, myristoyl-, and mitochondrial (citrate) acetyl-CoA and those of the respective acylcarnitines (Figs. 1 and 2). In contrast, in stearoyl-CoA, which is the product of fatty acid chain elongation, the contribution of M+0 is greater, and those of the M+4 and M+6 species are lower compared with the corresponding stearoylcarnitine mass isotopomers (Tables 2 and 3 and Fig. 1). Similarly, despite the significant 13C enrichment in malonyl-CoA, no stable isotope enrichment was observed in malonylcarnitine (Fig. 2). The lack of equilibrium in 13C enrichment between the stearoyl-CoA and stearoylcarnitine and the malonyl-CoA and malonylcarnitine suggests compartmentalization of stearyl-CoA and malonyl-CoA in rat heart. No 13C enrichment was detected in octanoyl-, decanoyl-, lauroyl-, and oleyl-CoA.
Mitochondrial Malonyl-CoA Synthesis and CPT1 Activity
The 13C enrichment data presented above suggest mitochondrial involvement in chain elongation of palmitate to stearate and arachidate. Because malonyl-CoA is synthesized from acetyl-CoA catalyzed by acetyl-CoA carboxylase 1 and 2 (ACC1 and ACC2), we determined whether isolated intact mitochondria, in the presence of ACC substrates, are able to synthesize malonyl-CoA. Because ACC2 is localized on the cytosolic side of the outer mitochondrial membrane (22), we determined whether the malonyl-CoA produced inhibits the activity of carnitine palmitoyltransferase 1b, also localized on the mitochondrial outer membrane (15, 23). To this end, freshly isolated mitochondrial populations (i.e. SSM and IFM), were incubated with ACC substrates for 5, 10, and 15 min, and at the indicated time points, aliquots were removed for determination of malonyl-CoA and CPT1 activity. The data presented in Fig. 3 show that freshly isolated intact rat heart mitochondria catalyzes the time-dependent formation of malonyl-CoA in the presence of ACC substrates. The rate of malonyl-CoA formation in both mitochondrial populations (SSM and IFM) was ∼150 pmol/min/mg protein. Under the experimental conditions, SSM and IFM synthesized sufficient malonyl-CoA to produce maximal inhibition of CPT1b. This inhibition was already achieved by 0.04 μm malonyl-CoA in the SSM and approximately at 0.06 μm malonyl-CoA in the IFM, concentrations lower than the reported IC50 values for malonyl-CoA on CPT1b (25–27). Both synthesis of malonyl-CoA and inhibition of CPT1b are absolutely dependent on the presence of acetyl-CoA, ATP, and bicarbonate but independent of citrate (data not shown).
FIGURE 3.
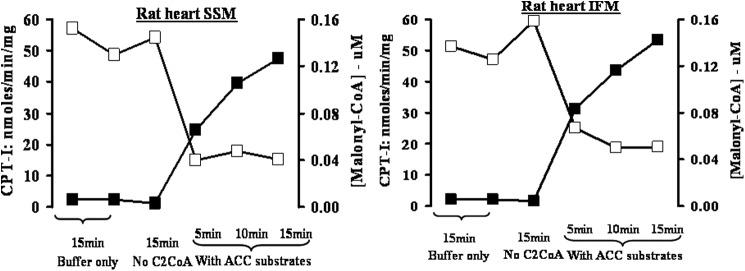
Mitochondrial synthesis of malonyl-CoA and its effect on CPT1b activity. Rat heart SSM and IFM were incubated for the indicated times in the presence of buffer only, buffer plus acetyl-CoA, and buffer containing all ACC substrates. At the indicated time points, malonyl-CoA content and CPT1b activity were determined as described under “Experimental Procedures.” Malonyl-CoA (closed symbols) is expressed as the micromolar concentration present in the CPT assay due to the addition of 10 μl of mitochondria preincubated with ACC substrates and CPT1b (open symbols) as nmol/min/mg mitochondrial protein. The values are the averages of three separate experiments.
Fatty Acid Chain Elongation with Isolated Intact Mitochondria
Freshly isolated mitochondria with membrane intactness greater than 97.2% (as assessed by cytochrome oxidase activity) were used to determine their capacity for fatty acid chain elongation. Chain elongation was determined by measuring the palmitoyl-CoA-dependent incorporation of 14C from [2-14C]malonyl-CoA or [1-14C]acetyl-CoA into lipid-soluble products (Table 4). The latter substrate was tested because of an earlier report that mitochondrial fatty acid chain elongation is acetyl-CoA- and NADH-dependent (28). As shown in Table 4, intact mitochondria incorporate 14C from either [2-14C]malonyl- or [1-14C]acetyl-CoA into lipid-soluble products with acetyl-CoA at nearly twice the rate as with malonyl-CoA. Furthermore, with both substrates, NADH is the preferred reducing agent over NADPH. When both reducing equivalents were simultaneously present, the 14C incorporation rates with malonyl- and acetyl-CoA were additive. Subjecting the mitochondria to three freeze/thaw cycles doubled the 14C incorporation rate with both substrates and co-substrates, suggesting a dual localization of fatty acid chain elongation in rat heart mitochondria. Omission of NADPH, NADH, or palmitoyl-CoA from the incubation mixture reduced the 14C incorporation from malonyl- and acetyl-CoA into lipid-soluble products by over 90%.
TABLE 4.
Chain elongation of palmitoyl-CoA by isolated intact rat heart mitochondria
Rat heart SSM were incubated at 37 °C in the presence of palmitoyl-CoA and [1-14C]acetyl-CoA or [2-14C]malonyl-CoA for 5 and 15 min, respectively, as described under “Experimental Procedures.” The values are expressed as picomol/min/mg mitochondrial protein and represent the mean ± S.E. of three separate experiments.
| Malonyl-CoA |
Acetyl-CoA |
|||
|---|---|---|---|---|
| Intact mitochondria | Frozen/thawed mitochondria | Intact mitochondria | Frozen/thawed mitochondria | |
| NADPH | 6.2 ± 4.2 | 14.4 ± 4.1 | 27.1 ± 5.0 | 43.7 ± 2.9 |
| NADH | 17.0 ± 9.2 | 41.0 ± 7.6 | 86.0 ± 12.5 | 169.9 ± 14.2 |
| NADPH + NADH | 21.3 ± 11.8 | 47.1 ± 17.9 | 106.2 ± 19.8 | 202.8 ± 20.2 |
DISCUSSION
Ex vivo perfusion of rat heart in the working mode with unlabeled palmitic acid led to dramatic increase in palmitoylcarnitine and a significant increase in myristoyl- and stearoylcarnitine with a concomitant decrease in acetylcarnitine. This change in cardiac acylcarnitine profile suggests that under the experimental conditions, the perfused palmitate underwent β-oxidation as well as chain elongation. Moreover, the striking increase in palmitoylcarnitine also suggests that CPT1b, the enzyme considered to be rate-controlling in mitochondrial fatty acid oxidation, is not limiting the entry of activated long-chain fatty acids into mitochondria in the fatty acid-perfused heart. Rather, the rate-limiting step resides downstream of CPT1b. The moderate accumulation of long-chain β-oxidation intermediates, such as 3-hydroxypalmitoyl and 3-hydroxymyristoyl derivatives of carnitine (Table 1), suggests that the limiting step of fatty acid oxidation is located at the step catalyzed by the trifunctional protein. The decrease in total carnitine content after the 60-min perfusion indicates leakage of carnitine from the heart. The increase in stearoylcarnitine content suggests chain elongation of palmitate either on the mitochondrial outer membrane or in the mitochondrial matrix.
The notion that perfused palmitic acid is β-oxidized and at the same time chain-elongated was unequivocally validated in experiments where hearts were perfused with [1,2,3,4-13C4]palmitate, and the 13C enrichment in fatty acid oxidation intermediates was determined. Measuring the stable isotope enrichment in either acylcarnitine or acyl-CoA β-oxidation intermediates revealed identical enrichment values, supporting the view that acylcarnitines and acyl-CoAs are in equilibrium and that the acylcarnitine content reflects that of acyl-CoAs (29). This seems to be true although in rat heart, ∼90% of carnitine and CoASH is in the cytosol and mitochondria, respectively (30). Lack of identical stable isotope enrichment in acyl-CoA and acylcarnitine derivatives, as observed for stearoyl and malonyl derivatives (see below), suggests metabolic compartmentalization of the respective acyl derivatives.
The mass isotopomer distribution of palmitoylcarnitine and palmitoyl-CoA (91 and 93.2% M+4), myristoylcarnitine and myristoyl-CoA (95 and 93% M+2), and acetylcarnitine and mitochondrial (citrate) acetyl-CoA (19.4 and 18.9% M+2) (Tables 2 and 3 and Figs. 2 and 3) shows that most, if not all, of the perfused [1,2,3,4-13C4]palmitate was oxidized in mitochondria. Because peroxisomal or cytosolic acetyl-CoA is not transferred to mitochondria (31), the slightly (but statistically not significantly) higher 13C enrichment in cardiac total M+2 acetyl-CoA (22.4%) compared with mitochondrial (citrate) M+2 acetyl-CoA (18.9%) (Table 3) could represent a minor contribution from peroxisomal β-oxidation. Based on malonyl-CoA turnover and 13C enrichment in malonyl-CoA and mitochondrial (citrate) acetyl-CoA in Langendorff-perfused rat heart, the estimated contribution of peroxisomal fatty acid oxidation was 2.5 nmol of acetyl units/min/g of wet weight (10). This rate is ∼100 times lower than the rate of acetyl-CoA production by mitochondrial oxidation of long-chain fatty acids using the Langendorff-perfused rat heart (32). In contrast to perfused heart, in heart homogenates, the contribution of peroxisomal β-oxidation to total fatty acid oxidation has been estimated to account for 19–27% in rat, depending on nutritional status (33), and 26–31% in pig (34). These values were arrived at by measuring the formation of 14CO2 and 14C-labeled acid-soluble intermediates from [1-14C]palmitate in heart homogenates in the absence and presence of inhibitors of mitochondrial respiration, antimycin and rotenone. Complete oxidation of [1,2,3,4-13C4]palmitate in mitochondria yields two [13C2]acetyl-CoA (M+2) and six M+0 acetyl-CoA molecules. Thus, with no contribution by oxidation of other unlabeled lipid and non-lipid substrates, the fraction of the M+2 acetyl-CoA isotopomer should theoretically be 25%. The experimentally found lower M+2 and higher M+0 acetyl-CoA isotopomer distribution probably represents dilution of M+2 acetyl-CoA by M+0 acetyl-CoA derived from oxidation of unlabeled glucose present in the perfusate as well as endogenous unlabeled substrates.
In addition to oxidation in mitochondria a fraction of the perfused [1,2,3,4-13C4]palmitate underwent chain elongation to the two- and four-carbon atom-longer M+4 and M+6 stearate and arachidate, respectively. The production of M+6 stearoyl- and arachidylcarnitine from perfused [1,2,3,4-13C4]palmitate can be explained only by recycling of the two-carbon units produced by β-oxidation of the perfused labeled fatty acid for chain elongation. Comparison of the 13C enrichment in M+6 isotopomers of stearoyl- and arachidylcarnitine (16.2 and 17.8%) with that of mitochondrial (citrate) M+2 acetyl-CoA (18.9%) and M+2 malonyl-CoA (19.9%) suggests that the majority of two-carbon chain extender units are derived from β-oxidation of [1,2,3,4-13C4]palmitate in mitochondria. The relative greater proportion of the M+0 stearoylcarnitine (24.9%) and arachidylcarnitine (27.6%) isotopomers compared with that of palmitoylcarnitine (4.5%) probably reflects a greater dilution of the smaller labeled pools by unlabeled stearic and arachidic acid released from endogenous sources. Stearic acid is the most abundant fatty acid in rat heart, and, compared with palmitate, it is in ∼2-fold molar excess both in phospholipids and total lipids (35, 36). Furthermore, the difference in isotopomer distribution between the carnitine and CoA derivatives of stearate is noteworthy. Among the CoA isotopomers of stearic acid, the M+0 species account for 53.6% of all stearoyl-CoA isotopomers, whereas among the carnitine derivatives, the M+0 stearoylcarnitine species account only for 24.9%. This ∼2-fold difference in M+0 stearoyl-CoA and M+0 stearoylcarnitine suggests compartmentalization of stearoyl-CoA with ∼25% not accessible to CPT1/CPT2 and therefore not localized in mitochondria.
Our findings raise the question about the nature of the chain extender (i.e. is it malonyl-CoA or acetyl-CoA?). Both microsomal fatty acid chain elongation (6, 8) and mitochondrial fatty acid synthesis (38–40) are malonyl-CoA-dependent. If the chain elongation of M+4 palmitate to M+4 and M+6 stearate and arachidate described in this paper is malonyl-CoA-dependent and intramitochondrially generated acetyl-CoA is the only source for the synthesis of malonyl-CoA, then the 13C enrichment in M+2 malonyl-CoA and M+2 mitochondrial (citrate) acetyl-CoA isotopomers should be the same. Indeed, the relative amounts of mitochondrial (citrate) M+2 acetyl- and cardiac M+2 malonyl-CoA isotopomers are identical (Table 3). Considering the identical 13C enrichment in the CoASH and carnitine derivatives of mitochondrial fatty acid oxidation intermediates (i.e. palmitoyl-, myristoyl-, and acetyl-CoA/carnitine (Table 2 and 3)), the lack of stable isotope enrichment in malonylcarnitine (Fig. 2) was unexpected. Malonylcarnitine has to come from malonyl-CoA via reversible transesterification, most likely catalyzed by CAT localized in the matrix or peroxisomes. A plausible explanation for the lack of stable isotope enrichment in malonylcarnitine is that malonyl-CoA is formed from mitochondrial (citrate) acetyl-CoA by ACC2 on the mitochondrial outer membrane and thus not accessible to CAT (or a carnitine acyltransferase specific for dicarboxylic acids yet to be identified) in the matrix. Then again, this still does not answer the question about the metabolic origin of unlabeled malonylcarnitine and its intracellular localization.
Our data presented herein clearly show that in the palmitate-perfused working heart, mitochondrial (citrate) acetyl-CoA is the precursor for malonyl-CoA synthesis. This is in contrast to our earlier finding that peroxisomal rather than mitochondrial fatty acid oxidation is a substantial source of the acetyl moiety of malonyl-CoA in the Langendorff-perfused heart (10). There are several possible reasons for the discrepancy between the two studies. In the present study, we used the working (ejecting) heart, which performs physiologically relevant mechanical work and is the model of choice for metabolic utilization studies (see Ref. 41 and references therein). In contrast, in the Langendorff-perfused heart used in our earlier study (10), the left ventricular chamber is empty, no perfusate is ejected from the heart, and the preparation is considered nonworking (see Ref. 41 and references therein). A second difference between the two studies known to profoundly affect fatty acid oxidation was the concentration of fatty acids in the perfusate (42). In the working heart (present study) the palmitate concentration was 1.2 mm, whereas in the earlier study with Langendorff-perfused hearts, the concentration of fatty acid was 0.2 mm. A further difference between the two studies concerns the analysis of stable isotope enrichment in malonyl-CoA. In the present work, the 13C enrichment was determined in malonyl-CoA isolated and specifically detected following solid-phase anion exchange extraction and HPLC separation (17, 43). In contrast, in our earlier work (10) the 13C enrichment of malonyl-CoA was determined indirectly as malonic acid by GC/MS following base hydrolysis of a fraction isolated using an oligonucleotide column. Any compound present in the eluate other than malonyl-CoA that, upon hydrolysis, gives rise to the formation of free malonic acid would lead to inaccuracy of determined 13C enrichment in malonyl-CoA.
Another question concerns the localization of the enzyme(s) catalyzing the synthesis of malonyl-CoA from intramitochondrially generated acetyl-CoA as well as of those catalyzing the chain elongation of [1,2,3,4-13C4]palmitate. Enzymes known to catalyze the synthesis of malonyl-CoA from acetyl-CoA are ACC1, confined to the cytosol, and ACC2, localized on the cytosolic surface of outer mitochondrial membrane (22), whereas enzymes catalyzing fatty acid chain elongation, known as Elovl1–7, are localized in endoplasmic reticulum (7, 45). It has been shown that of the seven Elovl enzymes, rat heart expresses Elovl1, Elovl3, and Elovl6 mRNAs (46), and in rat heart, the presence of Elovl6 also has been documented at the protein level (47). Thus, fatty acid chain elongation using intramitochondrial generated acetyl-CoA requires the export of acetyl-CoA into the cytosol and carboxylation of acetyl-CoA to malonyl-CoA, which serves as the chain extender for fatty acid chain elongation. Export of intramitochondrial acetyl-CoA in the form of acetylcarnitine is unlikely because CAT is absent from the cytosol (48). Alternatively, acetyl-CoA can be converted to citrate, exported to the cytosol, and reconverted to acetyl-CoA via ATP-citrate lyase. The enzyme is expressed in both mouse and rat heart (49, 50) and, as shown in the Langendorff-perfused rat heart, is indeed involved in export of acetyl units from mitochondria into the cytosol for malonyl-CoA synthesis (32, 51). The potential metabolic interrelationship between mitochondrial fatty acid oxidation in the matrix and fatty acid chain elongation on the mitochondrial outer membrane is depicted in Fig. 4. An alternative to Elovl6 on the mitochondrial outer membrane is the presence of mitochondria-associated membranes in the isolated heart mitochondria. In liver, Elovl6 is localized in the endoplasmic reticulum or microsomes. In heart, the localization of Elovl6 is unknown. Assuming that Elovl6 is localized in the sarcoplasmic reticulum and presumably in mitochondria-associated membranes would provide another possible explanation of our findings.
FIGURE 4.

Schematic of proposed mechanism of fatty acid chain elongation in working rat heart perfused with [1,2,3,4-13C4]palmitic acid (M+4) and unlabeled glucose. Palmitic acid is activated by long-chain acyl-CoA sythetase (ACS1) and transported into the matrix by the mitochondrial carnitine system CPT1b, CACTL, and CPT2 for β-oxidation. The end product acetyl-CoA is converted to citrate by citrate synthase and exported to the cytosol catalyzed by the tricarboxylate transporter (CT). In the cytosol, citrate is converted back to acetyl-CoA and oxaloacetate by ACL, and the acetyl-CoA is carboxylated to malonyl-CoA by ACC2 on the mitochondrial outer membrane. Malonyl-CoA is then used as the chain extender to chain-elongate [1,2,3,4-13C4]palmitic acid to M+4 and M+6 stearic acid either on outer mitochondrial membrane or in the endoplasmic reticulum, catalyzed by Elovl6.
The alternative to export of mitochondrial acetyl-CoA and synthesis of malonyl-CoA is the synthesis of malonyl-CoA in the mitochondrial matrix by propionyl-CoA carboxylase. If this was the case, then the stable isotope enrichment in malonylcarnitine should have been identical to that in malonyl-CoA. This expectation is based on increased excretion of succinylcarnitine and methylmalonylcarnitine in patients with succinate-CoA ligase and methylmalonyl-CoA mutase/epimerase deficiency, enzymes exclusively localized in the mitochondrial matrix (52–55). Therefore, mitochondria must have the capability to catalyze the reversible transesterification between dicarboxylyl-CoAs and carnitine, brought about either by CAT or an acylcarnitine transferase yet to be identified. However, no stable isotope enrichment was observed in malonylcarnitine, and therefore, we do not think malonyl-CoA is synthesized from acetyl-CoA via propionyl-CoA carboxylase in the mitochondrial matrix.
In the heart, ACC2 is the primary ACC isoform and is localized on the outer mitochondrial membrane (22). The function of ACC2 is thought to regulate mitochondrial fatty acid oxidation via inhibiting CPT1b, also localized in the outer membrane. As depicted in Fig. 3, isolated intact SSM and IFM synthesize malonyl-CoA in the presence of ACC substrates. Furthermore, the malonyl-CoA formed during the incubation is sufficient to inhibit CPT1b activity. These data confirm that malonyl-CoA is indeed produced on the outer mitochondrial membrane. Maximal inhibition of CPT1b was already observed at 0.04 and 0.06 μm malonyl-CoA in SSM and IFM, respectively. These inhibitor concentrations are lower than the reported IC50 values (25–27), suggesting that possible channeling of malonyl-CoA between ACC2 and CPT1b (56) and/or the malonyl-CoA formed during the preincubation induces a conformational change of CPT1b, locking the enzyme into a more inhibited state (57–60).
The finding that intact heart mitochondria have the capability to synthesize malonyl-CoA prompted us to test if they also catalyze fatty acid chain elongation. As shown in Table 4, intact rat heart mitochondria (SSM) catalyze the chain elongation of palmitoyl-CoA with malonyl- and acetyl-CoA as chain extenders in the presence of either NADPH or NADH. NADH seems to be the preferred reducing equivalent over NADPH with both malonyl- and acetyl-CoA; however, the simultaneous presence of both reducing equivalents is additive, suggesting that the two pathways are not identical. Furthermore, acetyl-CoA is preferred over malonyl-CoA. Because the membrane intactness of the isolated mitochondria was ∼98% for all three preparations, it is reasonable to propose that fatty acid chain elongation is localized to the outer mitochondrial membrane. Furthermore, the finding that disruption of mitochondrial membrane integrity by freezing/thawing doubles the rate of palmitoyl-CoA chain elongation with malonyl- and acetyl-CoA and with both reducing equivalents implies a dual mitochondrial localization (i.e. mitochondrial outer membrane and mitochondrial inner membrane or matrix). The mitochondrial localization of palmitate chain elongation also is supported by the lack of stable isotope enrichment in oleylcarnitine, the formation of which is catalyzed by stearoyl-CoA desaturase in the endoplasmic reticulum (61). Our findings are consistent with early reports on mitochondrial fatty acid chain elongation (62, 63). It has been shown that isolated guinea pig heart mitochondria catalyze the acetyl-CoA-dependent chain elongation of palmitoyl-CoA and palmitoylcarnitine with a preference for NADPH over NADH as the reducing equivalent (62). Malonyl-CoA as the chain extender has not been assessed in the referenced study. In rat liver mitochondria, fatty acid chain elongation localized in the outer and inner membrane were described by Colli et al. (63). Both systems had a preference of acetyl-CoA over malonyl-CoA and NADH over NADPH. The mitochondrial inner membrane system had a preference for medium chain fatty acids, whereas the chain elongation system localized in the outer membrane showed a predilection for palmitoyl-CoA as the primer.
The potential significance of cardiac fatty acid chain elongation is not readily apparent. Of the seven Elovls, the heart expresses Elovl1, Elovl5, and Elovl6 (7, 8, 46, 47). From the known substrate specificity of the three Elovls expressed in heart Elovl6, is most likely responsible for the observed chain elongation of palmitate to stearate (8, 16). The potential metabolic significance of this pathway is highlighted by the fact that the content of stearate, relative to palmitic acid, is nearly double in heart what it is in plasma (5). Consistent with the high cardiac stearate content reported previously (5), oleyl-CoA and stearoyl-CoA are the dominant long-chain acyl-CoAs in rat heart, independent of diet (47). Stearic acid is an important component of phospholipids, which in turn are important structural and functional components of membranes. In the heart, a tissue rich in mitochondria, mitochondrial phospholipids comprise a significant portion of total cellular phospholipids that fulfill diverse functions, such as electron transport, apoptosis, and protein and lipid import. Thus, alterations not only in phospholipid content but also in molecular composition of phospholipid fatty acids are critical for proper mitochondrial and cardiac function. Elovl6, by providing stearate for continuous remodeling of membrane phospholipid fatty acids, plays an important role in mitochondrial metabolism.
The significance of Elovl6-catalyzed fatty acid chain elongation is further highlighted by recent studies using Elovl6 knock-out mice. Mice with global deletion of Elovl6 exhibit partial embryonic lethality, underscoring the physiological significance of this enzyme. The surviving offspring are protected against development of diet-induced insulin resistance but not against development of obesity and hepatosteatosis (64).
This indicates the significance of tissue fatty acid composition in insulin sensitivity, especially the stearate/palmitate ratio, which is considered as the hallmark for Elovl6 activity. Accordingly, deletion of Elovl6 increased palmitate and palmitoleate (C16:1, n-7) and decreased stearate and oleate content that was associated with increased insulin sensitivity (37, 44, 64). The crucial role of palmitate conversion to stearate for the emergence of insulin resistance in liver raises the question of a similar effect in other tissues, such as the heart. If this exists, then cardiac Elovl6 could be a viable target to affect insulin resistance of the heart and impacts the heart in many ways (24).
In summary, using the fatty acid-perfused working rat heart in the present study, we have documented the chain elongation of perfused [1,2,3,4-13C4]palmitate (M+4) to M+4 and M+6 stearate and arachidate and that mitochondrial β-oxidation of the perfused [1,2,3,4-13C4]palmitate is the major contributor to the two-carbon acetyl units used as the chain extender. Furthermore, the data presented indicate that the described chain elongation pathway is localized on the mitochondrial outer membrane. The synthesis of malonyl-CoA on the mitochondrial outer membrane from acetyl-CoA, derived from mitochondrial β-oxidation in the mitochondrial matrix, provides malonyl-CoA for fatty acid chain elongation and represents a unique signaling mechanism by which excessive mitochondrial fatty acid oxidation is restrained via inhibition of carnitine palmitoyltransferase 1b by malonyl-CoA (feedback inhibition).
The novel finding that the heart, a non-lipogenic tissue, has the capability to chain-elongate fatty acids derived from the circulation raises questions about the metabolic significance of this pathway in the heart. Fatty acids are important constituents of membrane phospholipids that show tissue as well as organelle specificity and thus require remodeling of the free fatty acids derived from the circulation. This is especially true for the heart, an organ rich in mitochondria. We propose that in the heart, the function of the fatty acid chain elongation pathway described herein is to provide chain-elongated fatty acids for phospholipid remodeling. Further studies directed at the metabolic fate of the chain-elongated fatty acid products should provide new insight into the significance of cardiac mitochondrial fatty acid chain elongation.
Acknowledgments
We thank Sarah Stewart for performing the working heart perfusions, Maria Stoll for expert technical help in acylcarnitine analysis, Dr. Bernard Tandler and the Hoppel laboratory writing with style group for editorial assistance, and Dr. Jason Mears for help preparing Fig. 4.
This work was supported, in whole or in part, by National Institutes of Health Grant PO1 AG15885 Project 3 and Cores B and D.
- ELOVL
- elongation of very long-chain fatty acids
- UHPLC
- ultra-HPLC
- ACC
- acetyl-CoA carboxylase
- SSM
- subsarcolemmal mitochondria
- IFM
- interfibrillar mitochondria
- CAT
- carnitine acetyltransferase.
REFERENCES
- 1. Hagve T.-A., Sprecher H. (1989) Metabolism of long-chain polyunsaturated fatty acids in isolated cardiac myocytes. Biochim. Biophys. Acta 1001, 338–344 [DOI] [PubMed] [Google Scholar]
- 2. Hamilton C., Saggerson E. D. (2000) Malonyl-CoA metabolism in cardiac myocytes. Biochem. J. 350, 61–67 [PMC free article] [PubMed] [Google Scholar]
- 3. Lopez Jimenez J. A., Bordoni A., Lorenzini A., Rossi C. A., Biagi P. L., Hrelia S. (1997) Linoleic acid metabolism in primary cultures of adult rat cardiomyocytes is impaired by aging. Biochem. Biophys. Res. Commun. 237, 142–145 [DOI] [PubMed] [Google Scholar]
- 4. Ford D. A., Han X., Horner C. C., Gross R. W. (1996) Accumulation of unsaturated acylcarnitine molecular species during acute myocardial ischemia: metabolic compartmentalization of products of fatty acyl chain elongation in the acylcarnitine pool. Biochemistry 35, 7903–7909 [DOI] [PubMed] [Google Scholar]
- 5. Cinti D. L., Cook L., Nagi M. N., Suneja S. K. (1992) The fatty acid chain elongation system of mammalian endoplasmic reticulum. Prog. Lipid Res. 31, 1–51 [DOI] [PubMed] [Google Scholar]
- 6. Leonard A. E., Pereira S. L., Sprecher H., Huang Y.-S. (2004) Elongation of long-chain fatty acids. Prog. Lipid Res. 43, 36–54 [DOI] [PubMed] [Google Scholar]
- 7. Jakobsson A., Westerberg R., Jacobsson A. (2006) Fatty acid elongases in mammals: their regulation and roles in metabolism. Prog. Lipid Res. 45, 237–249 [DOI] [PubMed] [Google Scholar]
- 8. Guillou H., Zadravec D., Martin P. G., Jacobsson A. (2010) The key roles of elongases and desaturases in mammalian fatty acid metabolism: insight from transgenic mice. Prog. Lipid Res. 49, 186–199 [DOI] [PubMed] [Google Scholar]
- 9. Kerner J., Minkler P. E., Lesnefsky E. J., Hoppel C. L. (2007) Fatty acid chain-elongation in perfused rat heart: synthesis of palmitoylcarnitine from perfused palmitate. FEBS Lett. 581, 4491–4494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Reszko A. E., Kasumov T., David F., Jobbins K. A., Thomas K. R., Hoppel C. L., Brunengraber H., Des Rosiers C. (2004) Peroxisomal fatty acid oxidation is a substantial source of the acetyl moiety of malonyl-CoA in rat heart. J. Biol. Chem. 279, 19574–19579 [DOI] [PubMed] [Google Scholar]
- 11. Houston B., Nimmo H. G. (1984) Purification and some kinetic properties of rat liver ATP citrate lyase. Biochem. J. 224, 437–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Linn T. C., Srere P. A. (1979) Identification of ATP citrate lyase as a phosphoprotein. J. Biol. Chem. 254, 1691–1698 [PubMed] [Google Scholar]
- 13. Kudo N., Barr A. J., Barr R. L., Desai S., Lopaschuk G. D. (1995) High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl-CoA levels due to an increase in 5′-AMP-activated protein kinase inhibition of acetyl-CoA carboxylase. J. Biol. Chem. 270, 17513–17520 [DOI] [PubMed] [Google Scholar]
- 14. Palmer J. W., Tandler B., Hoppel C. L. (1977) Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J. Biol. Chem. 252, 8731–8739 [PubMed] [Google Scholar]
- 15. Hoppel C. L., Kerner J., Turkaly P., Turkaly J., Tandler B. (1998) The malonyl-CoA-sensitive form of carnitine palmitoyltransferase is not localized exclusively in the outer membrane of rat liver mitochondria. J. Biol. Chem. 273, 23495–23503 [DOI] [PubMed] [Google Scholar]
- 16. Moon Y.-A., Shah N. A., Mohapatra S., Warrington J. A., Horton J. D. (2001) Identification of a mammalian long chain fatty acyl elongase regulated by sterol regulatory element-binding proteins. J. Biol. Chem. 276, 45358–45366 [DOI] [PubMed] [Google Scholar]
- 17. Minkler P. E., Kerner J., Ingalls S. T., Hoppel C. L. (2008) Novel isolation procedure for short-, medium-, and long-chain acyl-coenzyme A esters from tissue. Anal. Biochem. 376, 275–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Minkler P. E., Stoll M. S., Ingalls S. T., Yang S., Kerner J., Hoppel C. L. (2008) Quantification of carnitine and acylcarnitines in biological matrices by HPLC electrospray ionization-mass spectrometry. Clin. Chem. 54, 1451–1462 [DOI] [PubMed] [Google Scholar]
- 19. Minkler P. E., Stoll M. S. K., Ingalls S. T., Hoppel C. L. (2012) Selective, accurate, and precise quantification of acylcarnitines by UHPLC-MS/MS for detailed analysis in metabolic disease research, Proceedings of the 60th ASMS Conference on Mass Spectrometry and Allied Topics, American Society for Mass Spectrometry, May 20–24, Vancouver, BC, Canada [Google Scholar]
- 20. Minkler P. E., Stoll M. S. K., Ingalls S. I., Hoppel C. L. (2013) Comprehensive, accurate, and precise quantification of acylcarnitines and acyl-CoAs in tissues using on-line ion-exchange trapping and UHPLC-MS/MS. Tenth Annual Ohio Mass Spectrometry Symposium, April 15, 2013, Columbus, OH Sponsored by Agilent Technologies, Bruker Daltonics, and Thermo Fisher Scientific [Google Scholar]
- 21. Gruber C. C., Oberdorfer G., Voss C. V., Kremsner J. M., Kappe C. O., Kroutil W. (2007) An algorithm for the deconvolution of mass spectrometric patterns in isotope labeling studies. Evaluation for the hydrogen-deuterium exchange reaction in ketones. J. Org. Chem. 72, 5778–5783 [DOI] [PubMed] [Google Scholar]
- 22. Abu-Elheiga L., Brinkley W. R., Zhong L., Chirala S. S., Woldegiorgis G., Wakil S. J. (2000) The subcellular localization of acetyl-CoA carboxylase 2. Proc. Natl. Acad. Sci. U.S.A. 97, 1444–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Murthy M. S., Pande S. V. (1987) Malonyl-CoA binding site and the overt carnitine palmitoyltransferase activity reside on opposite sides of the outer mitochondrial membrane. Proc. Natl. Acad. Sci. U.S.A. 84, 378–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abel E. D., O'Shea K. M., Ramasamy R. (2012) Insulin resistance: metabolic mechanism and consequences in the heart. Arterioscler. Thromb. Vasc. Biol. 32, 2068–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McGarry J. D., Mills S. E., Long C. S., Foster D. W. (1983) Observations on the affinity for carnitine, malonyl-CoA sensitivity, of carnitine palmitoyltransferase I in animal and human tissue. Biochem. J. 214, 21–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mills S. E., Foster D. W., McGarry J. D. (1983) Interaction of malonyl-CoA and related compounds with mitochondria from different rat tissues. Biochem. J. 214, 83–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Distler A. M., Kerner J., Hoppel C. L. (2009) Mass spectrometric demonstration of the presence of liver carnitine palmitoyltransferse-I (CPT-I) in rat heart mitochondria. Biochim. Biophys. Acta 1794, 431–437 [DOI] [PubMed] [Google Scholar]
- 28. Seubert W., Podack E. R. (1973) Mechanisms and physiological roles of fatty acid chain elongation in microsomes and mitochondria. Mol. Cell. Biochem. 1, 29–40 [DOI] [PubMed] [Google Scholar]
- 29. Pearson D. J., Tubbs P. K. (1967) Carnitine and derivatives in rat tissues. Biochem. J. 105, 953–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oram J. F., Wenger J. I., Neely J. R. (1975) Regulation of long chain fatty acid activation in heart muscle. J. Biol. Chem. 250, 73–78 [PubMed] [Google Scholar]
- 31. Bian F., Kasumov T., Thomas K. R., Jobbins K. A., David F., Minkler P. E., Hoppel C. L., Brunengraber H. (2005) Peroxisomal and mitochondrial oxidation of fatty acids in the heart, assessed from the 13C labeling of malonyl-CoA and the acetyl moiety of citrate. J. Biol. Chem. 280, 9265–9271 [DOI] [PubMed] [Google Scholar]
- 32. Vincent G., Comte B., Poirier M., Rosiers C. D. (2000) Citrate release by perfused rat hearts: a window on mitochondrial cataplerosis. Am. J. Physiol. Endocrinol. Metab. 278, E846–E856 [DOI] [PubMed] [Google Scholar]
- 33. Veerkamp J. H., van Moerkerk H. T. B. (1986) Peroxisomal fatty acid oxidation in rat and human tissues. Effect of nutritional state, clofibrate treatment and postnatal development in the rat. Biochim. Biophys. Acta 875, 301310. [DOI] [PubMed] [Google Scholar]
- 34. Yu X. X., Drackley J. K., Odle J. (1997) Rates on mitochondrial and peroxisomal β-oxidation of palmitate change during postnatal development and food deprivation in liver, kidney and heart of pigs. J. Nutr. 127, 1814–1821 [DOI] [PubMed] [Google Scholar]
- 35. Gómez-Tubio A., Carreras O., Tavares E., Delgado M. J. (1999) Effect of chronic ethanol consumption on fatty acid profile of heart tissue in rats. Alcohol Clin. Exp. Res. 23, 404–407 [PubMed] [Google Scholar]
- 36. Jones P. J., Toy B. R., Cha M. C. (1995) Differential fatty acid accretion in heart, liver and adipose tissues of rats fed beef tallow, fish oil, olive oil and safflower oils at three levels of energy intake. J. Nutr. 125, 1175–1182 [DOI] [PubMed] [Google Scholar]
- 37. Matsuzaka T., Shimano H. (2009) Elovl6: a new player in fatty acid metabolism and insulin sensitivity. J. Mol. Med. 87, 379–384 [DOI] [PubMed] [Google Scholar]
- 38. Hiltunen J. K., Chen Z., Haapalainen A. M., Wierenga R. K., Kastaniotis A. J. (2010) Mitochondrial fatty acid synthesis: an adopted set of enzymes making a pathway of major importance for cellular metabolism. Prog. Lipid Res. 49, 27–45 [DOI] [PubMed] [Google Scholar]
- 39. Zhang L., Joshi A. K., Smith S. (2003) Cloning, expression, characterization, and interaction of two components of a human mitochondrial fatty synthase. J. Biol. Chem. 41, 40067–40074 [DOI] [PubMed] [Google Scholar]
- 40. Witkowski A., Thweatt J., Smith S. (2011) Mammalian ACSF3 protein is a malonyl-CoA synthetase that supplies the chain extender units for mitochondrial fatty acid synthesis. J. Biol. Chem. 286, 33729–33736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liao R., Podesser B. K., Lim C. C. (2012) The continuing evolution of the Langendorff and ejecting murine heart: new advances in cardiac phenotyping. Am. J. Physiol. Heart Circ. Physiol. 303, H156–H167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Oram J. F., Bennetch S. L., Neely J. R. (1973) Regulation of fatty acid utilization in isolated perfused rat hearts. J. Biol. Chem. 248, 5299–5309 [PubMed] [Google Scholar]
- 43. Minkler P. E., Kerner J., Kasumov T., Parland W., Hoppel C. L. (2006) Quantification of malonyl-coenzyme A in tissue specimens by high-performance liquid chromatography/mass spectrometry. Anal. Biochem. 352, 24–32 [DOI] [PubMed] [Google Scholar]
- 44. Shimano H. (2012) Novel qualitative aspects of tissue fatty acids related to metabolic regulation: lessons from Elovl6 knockout. Prog. Lipid Res. 51, 267–271 [DOI] [PubMed] [Google Scholar]
- 45. Kihara A. (2012) Very long-chain fatty acids: elongation, physiology and related disorders. J. Biochem. 152, 387–395 [DOI] [PubMed] [Google Scholar]
- 46. Wang Y., Botolin D., Christian B., Busik J., Xu J., Jump D. B. (2005) Tissue-specific, nutritional, and developmental regulation of rat fatty acid elongases. J. Lipid Res. 46, 706–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Harmancey R., Wilson C. R., Wright N. R., Taegtmeyer H. (2010) Western diet changes cardiac acyl-CoA composition in obese rats: a potential role for hepatic lipogenesis. J. Lipid Res. 51, 1380–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Abbas A. S., Wu G., Schulz H. (1998) Carnitine acetyltransferase is not a cytosolic enzyme in rat heart and therefore cannot function in the energy-linked regulation of cardiac fatty acid oxidation. J. Mol. Cell. Cardiol. 30, 1305–1309 [DOI] [PubMed] [Google Scholar]
- 49. Berne C. (1975) Nicotinamide adenine dinucleotide phosphate-converting enzymes and adenosine triphosphate citrate lyase in some tissues and organs of New Zealand obese mice with special reference to the enzyme pattern of the pancreatic islets. J. Histochem. Cytochem. 23, 660–665 [DOI] [PubMed] [Google Scholar]
- 50. Lin G., Brownsey R. W., MacLeod K. M. (2009) Regulation of mitochondrial aconitase by phosphorylation in diabetic rat heart. Cell. Mol. Life Sci. 66, 919–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Poirier M., Vincent G., Reszko A. E., Bouchard B., Kelleher J. K., Brunengraber H., Des Rosiers C. (2002) Probing the link between citrate and malonyl-CoA in the perfused rat hearts. Am. J. Physiol. Heart Circ. Physiol. 283, H1379–H1386 [DOI] [PubMed] [Google Scholar]
- 52. Van Hove J. L., Saenz M. S., Thomas J. A., Gallagher R. C., Lovell M. A., Fenton L. Z., Shanske S., Myers S. M., Wanders R. J., Ruiter J., Turkenburg M., Waterham H. R. (2010) Succinyl-CoA ligase deficiency: a mitochondrial hepatoencephalomyopathy. Pediatr. Res. 68, 159–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jaberi E., Chitsazian F., Ali Shahidi G., Rohani M., Sina F., Safari I., Malakouti Nejad M., Houshmand M., Klotzle B., Elahi E. (2013) The novel mutation p.Asp251Asn in the β-subunit of succinate-CoA ligase causes encephalomyopathy and elevated succinylcarnitine. J. Hum. Genet. 58, 526–530 [DOI] [PubMed] [Google Scholar]
- 54. Yano S., Li L., Le T. P., Moseley K., Guedalia A., Lee J., Gonzalez I., Boles R. G. (2003) Infantile mitochondrial DNA-depletion syndrome associated with methylmalonic aciduria and 3-methylcrotonyl-CoA and propionyl-CoA carboxylase deficiencies in two unrelated patients: a new phenotype of mtDNA depletion syndrome. J. Inherit. Metab. Dis. 26, 481–488 [DOI] [PubMed] [Google Scholar]
- 55. Maeda Y., Ito T., Suzuki A., Kurono Y., Ueta A., Yokoi K., Sumi S., Togari H., Sugiyama N. (2007) Simultaneous quantification of acylcarnitine isomers containing dicarboxylic acylcarnitines in human serum and urine by high-performance liquid chromatography/electrospray ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom. 21, 799–806 [DOI] [PubMed] [Google Scholar]
- 56. Abu-Elheiga L., Matzuk M. M., Abo-Hashema K. A., Wakil S. J. (2001) Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science 291, 2613–2616 [DOI] [PubMed] [Google Scholar]
- 57. Zammit V. A. (1983) Reversible sensitization and desensitization of carnitine palmitoyltransferase I to inhibition by malonyl-CoA in isolated rat liver mitochondria. Significance for the mechanism of malonyl-CoA-induced sensitization. Biochem. J. 214, 1027–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cook G. A., Cox K. A. (1986) Hysteretic behavior of carnitine palmitoyltransferase. Biochem. J. 236, 917–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kerner J., Bieber L. L. (1994) Conversion of carnitine palmitoyltransferase of rat heart mitochondria into a high affinity, malonyl-CoA-inhibited state. Biochem. Arch. 10, 95–101 [Google Scholar]
- 60. Bieber L. L., Wagner M. (1996) Effect of pH and acyl-CoA chain length on the conversion of heart mitochondrial CPT-I/CPTo to a high affinity, malonyl-CoA-inhibited state. Biochim. Biophys. Acta 1290, 261–266 [DOI] [PubMed] [Google Scholar]
- 61. Hodson L., Fielding B. A. (2013) Stearoyl-CoA desaturase: rogue or innocent bystander. Prog. Lipid Res. 52, 15–42 [DOI] [PubMed] [Google Scholar]
- 62. Warshaw J. B., Kimura R. E. (1973) Palmitoyl-CoA and palmitoylcarnitine interactions in mitochondrial fatty acid elongation. Arch. Biochem. Biophys. 157, 44–49 [DOI] [PubMed] [Google Scholar]
- 63. Colli W., Hinkle P. C., Pullman M. E. (1969) Characterization of the fatty acid elongation system in soluble extracts and membrane preparations of rat liver mitochondria. J. Biol. Chem. 244, 6432–6443 [PubMed] [Google Scholar]
- 64. Matsuzaka T., Shimano H., Yahagi N., Kato T., Atsumi A., Yamamoto T., Inoue N., Ishikawa M., Okada S., Ishigaki N., Iwasaki H., Iwasaki Y., Karasawa T., Kumadaki S., Matsui T., Sekiya M., Ohashi K., Hasty A. H., Nakagawa Y., Takahashi A., Suzuki H., Yatoh S., Sone H., Toyoshima H., Osuga J., Yamada N. (2007) Crucial role of a long-chain fatty acid elongase, Elovl6, in obesity-induced insulin resistance. Nat. Med. 13, 1193–1202 [DOI] [PubMed] [Google Scholar]