

Background: Neph1 is a podocyte protein that is critical for maintaining renal function.
Results: Inhibiting Neph1 signaling preserves podocyte structure and function in response to glomerular injury-inducing agents.
Conclusion: Maintaining a robust expression of Neph1 at the podocyte cell membrane protects podocytes from renal injury.
Significance: This is the first report demonstrating that Neph1 signaling is a therapeutic target for preventing podocyte damage.
Keywords: Actin, Cell Signaling, Kidney, Phosphorylation, Podocytes, Neph1, Renal Injury
Abstract
Podocytes are specialized epithelial cells that are critical components of the glomerular filtration barrier, and their dysfunction leads to proteinuria and renal failure. Therefore, preserving podocyte function is therapeutically significant. In this study, we identified Neph1 signaling as a therapeutic target that upon inhibition prevented podocyte damage from a glomerular injury-inducing agent puromycin aminonucleoside (PAN). To specifically inhibit Neph1 signaling, we used a protein transduction approach, where the cytoplasmic domain of Neph1 (Neph1CD) tagged with a protein transduction domain trans-activator of transcription was transduced in cultured podocytes prior to treatment with PAN. The PAN-induced Neph1 phosphorylation was significantly reduced in Neph1CD-transduced cells; in addition, these cells were resistant to PAN-induced cytoskeletal damage. The biochemical analysis using subfractionation studies showed that unlike control cells Neph1 was retained in the lipid raft fractions in the transduced cells following treatment with PAN, indicating that transduction of Neph1CD in podocytes prevented PAN-induced mislocalization of Neph1. In accordance, the immunofluorescence analysis further suggested that Neph1CD-transduced cells had increased ability to retain endogenous Neph1 at the membrane in response to PAN-induced injury. Similar results were obtained when angiotensin was used as an injury-inducing agent. Consistent with these observations, maintaining high levels of Neph1 at the membrane using a podocyte cell line overexpressing chimeric Neph1 increased the ability of podocytes to resist PAN-induced injury and PAN-induced albumin leakage. Using a zebrafish in vivo PAN and adriamycin injury models, we further demonstrated the ability of transduced Neph1CD to preserve glomerular function. Collectively, these results support the conclusion that inhibiting Neph1 signaling is therapeutically significant in preventing podocyte damage from glomerular injury.
Introduction
The structural and functional alterations to the filtration barrier of kidney observed during nephrotic syndromes or other glomerular disorders are the primary causes of proteinuria (1–3). This filtration barrier is primarily composed of three major cellular layers as follows: fenestrated endothelium, glomerular basement membrane, and podocytes (2, 3). The podocytes are highly specialized cells that are characterized by the presence of numerous interdigitating foot processes that arise from the cell body and surround the glomerular basement membrane (3, 4). These foot processes are inter-connected via a thin membranous structure that is 40 nm wide and commonly known as the filtration slit or “slit diaphragm.” Because of their critical involvement in maintaining the renal function, podocytes have been the subject of intense investigation by several investigators worldwide (1–3). In addition, podocytes are being recognized as a therapeutic target for discovering therapies directed toward the prevention of loss of glomerular function observed in various genetic and acquired renal disorders (1–3). Several studies indicate that the podocyte proteins Neph1 and nephrin are critical for maintaining the structural integrity of slit diaphragm and therefore the filtration function of the glomerulus (3–6). Studies in recent years have highlighted the significance of tyrosine phosphorylation in these proteins that plays a central role in regulating the function of these proteins particularly in the actin remodeling of podocytes that may occur during development and in response to glomerular injury (5–11). The cytoplasmic domains of Neph1 and nephrin are tyrosine-phosphorylated by Src family tyrosine kinases, namely Fyn (11), leading to the recruitment of adapter proteins Grb2 and Nck that are required for actin cytoskeleton rearrangement (5, 7, 10, 11).
Previous studies from our group have shown that the cytoplasmic domain of Neph1 plays a key role in actin cytoskeleton remodeling events that are directly associated with the tyrosine phosphorylation of this domain (6, 12). This phosphorylation event that is primarily mediated by the phosphorylation of residues 637 and 638 in Neph1 was shown to recruit several proteins, including Grb2, Csk tyrosine kinase, and ZO-1 (10, 12). Although the recruitment of Grb2 in cultured podocytes induced actin cytoskeleton reorganization at the cytoplasmic tail of Neph1 (6), the phosphorylation-dependent increased association between Neph1 and ZO-1 played a key role in defining the tight junction formation in podocytes (12).
In the various in vivo models of glomerular injury that have been shown to induce podocyte damage and podocyte effacement, an increased Neph1 tyrosine phosphorylation has been reported, indicating Neph1 phosphorylation as an important signaling event that takes place in response to an injury (5, 6, 12). Through what mechanism the injury-induced phosphorylation of Neph1 participates in actin cytoskeletal organization is not clear. It is possible that different injury models induce separate mechanisms for generating a cellular response that leads to changes in the actin cytoskeleton and alteration of the podocyte structure and function. One of the most common podocyte injury-inducing agents that has been used under both in vitro and in vivo conditions is PAN.3 Treatment of PAN in cultured podocytes has been shown to induce the disassembly of cell-cell junctions, actin cytoskeleton rearrangement, and the retraction of cellular processes (13–17). In the rat model, PAN treatment has been shown to induce podocyte effacement, a characteristic of actin reorganization, and loss of podocyte function associated with proteinuria (12, 18). More importantly, these changes in cultured podocytes and rat PAN model were associated with rapid tyrosine phosphorylation of Neph1 (10). Apart from PAN, the tyrosine phosphorylation of Neph1 was also noted in the ischemia reperfusion model of podocyte injury (12). Therefore, it is possible that phosphorylation of Neph1 is the primary event that induces a signaling cascade leading to the activation of downstream signaling events.
Because podocyte injury is a common denominator in many glomerular diseases associated with proteinuria, it is likely that multiple approaches and targets need to be identified to develop a viable therapy for preventing podocyte damage. Cultured podocytes have been widely used and are excellent tools for understanding the molecular mechanisms of cellular signaling in podocytes (11, 12, 19). In addition, these cells have been used in the high throughput drug discovery experiments (20) and protein transduction studies where delivery of cell-penetrating peptides in these cells was reported (21). Many pharmacological agents, including corticosteroids, antagonists of the renin-angiotensin system, vitamin derivatives, and mycophenolate, have been shown to possess the ability to prevent podocyte damage in response to various glomerular injury-inducing agents (22–27). Despite these studies, there is a greater need to identify additional targets and agents with therapeutic values for preventing podocyte damage given their central role regulating the filtration function of the glomerulus.
Protein transduction domains, including TAT (also known as cell-penetrating peptides), are short peptides that can rapidly translocate proteins independent of their size and shape across the biological membranes (28, 29). Protein transduction by TAT is a fast developing scientific tool whose emerging functions include probing the function of a particular protein and understanding its role in the molecular and cellular mechanism of a disease and the diagnosis and treatment of a variety of human diseases (30, 31). In this study, we report that TAT-mediated delivery of the cytoplasmic domain of Neph1 in podocytes protects them from PAN-induced injury. We further demonstrate that inhibition of Neph1 phosphorylation and preventing the loss of Neph1 expression at the podocyte cell membrane contributes toward the mechanism for this protection.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
Myo1c monoclonal antibody and polyclonal antibodies to Neph1, phospho-Neph1, and Neph1 extracellular domain have been previously described (12, 19). Other antibodies, including the His monoclonal antibody (Santa Cruz Biotechnology), ZO-1 monoclonal antibody (Invitrogen), caveolin 1 polyclonal antibody (Abcam), transferrin receptor monoclonal antibody (Abcam), and actin monoclonal antibody (Cell Signaling) were commercially purchased. The fluorophore-labeled secondary antibodies Alexa Fluor 488 goat anti-rabbit IgG(H+L), Alexa Fluor 568 goat anti-mouse IgG(H+L), Alexa Fluor 647 goat anti-mouse IgG(H+L), Alexa Fluor phalloidins, and Vybrantô DiI cell-labeling dye were obtained from Invitrogen. All the chemical reagents used were obtained commercially from Sigma and Calbiochem.
Cell Culture
The human podocyte cell line (19, 32) was cultured in RPMI 1640-based medium supplemented with 10% fetal bovine serum (FBS) (Invitrogen), 2 g/liter sodium bicarbonate (NaHCO3), insulin/transferrin/selenium supplement (33) (Sigma), and 200 units/ml penicillin and streptomycin (Roche Applied Science), at 33 °C and 5% CO2 as described previously (12, 19). The retroviruses overexpressing mCherry-Neph1 were generated by the transfection of mCherry Neph1 cDNA into Phoenix cells according to the manufacturer's instructions as described earlier (19). The target subconfluent podocytes were then infected with viral particles three times over a period of 36 h as described previously (19). The stably transfected cells overexpressing mCherry Neph1 were selected using puromycin as a selection marker.
Plasmids
The bacterial expression plasmids of TAT-Neph1 cytoplasmic domain (TAT-Neph1CD) and TAT-green fluorescence protein (TAT-GFP) were cloned in pTAT vector. The pTAT vector was a generous gift from Dr. Mark Payne (34). Neph1 cytoplasmic domain and the green fluorescence protein (GFP) were cloned in pTAT vector at the XhoI and EcoRI site with an N-terminal His tag as described earlier (12, 34, 35). The mCherry fluorescence tag was inserted between residues Val-575 and Asn-576of Neph1, which is immediately after the transmembrane domain and before any potential tyrosine phosphorylation sites in the cytoplasmic domain of Neph1 (Fig. 8A). This site was carefully chosen because insertions at the other sites such as at the C terminus (residue 789) drastically affected the localization of Neph1 (data not shown). A standard PCR cloning technique was used to generate the mammalian expression plasmid encoding mCherry fluorescent-tagged full-length Neph1 in the pBABE-puro vector. The PCR amplified mCherry-Neph1 was cloned at the EcoRI and SalI sites in the pBABE-puro vector. All the constructs were validated by complete sequencing of the clone and expression in the cultured podocytes. The mCherry-Neph1 protein correctly localized at the membrane in a fashion similar to the endogenous Neph1 (Fig. 8B).
FIGURE 8.

Podocytes overexpressing Neph1 are resistant to PAN-induced injury. A, schematic representation of the construction of Cherry-Neph1 construct (where Cherry was introduced after the transmembrane domain and before any potential tyrosine phosphorylation sites in Neph1) is shown. B, expression pattern of Cherry-Neph1 was similar to endogenous Neph1 in podocytes, where both proteins localized at the cell-cell junctions (arrows). C, podocytes without and with stable expression of Cherry-Neph1 were created and subjected to PAN treatment for 48 h. Unlike the control cells transfected with empty vector, the Cherry-Neph1-transfected cells were resistant to PAN-induced cytoskeletal damage, and their cell-cell junctions were well maintained. D, BSA permeability assay was performed with vector-transfected (control) and Cherry-Neph1-overexpressing podocytes that were cultured as a monolayer on Transwell filters and treated with PAN. The passage of albumin across the podocytes monolayer was assessed by a paracellular albumin flux assay using Texas red-labeled albumin. The Cherry-Neph1-overexpressing cells showed increased resistance to PAN-induced albumin leakage as compared with control podocytes. ns, nonsignificant. Scale bar, 20 μm (B); 10 μm (C).
Expression and Purification of TAT Proteins
The detailed procedures of expression and purification of TAT-Neph1CD and TAT-GFP proteins have been described previously (12, 35). Briefly, the relevant plasmids were transformed in Escherichia coli BL21 (DE3) strain, and cultures were grown at 37 °C in LB (Luria-Bertani) medium (Merck), and the protein expression was induced with 0.4 mm isopropyl β-d-thiogalactopyranoside. The bacterial pellet was obtained and suspended in 50 mm Tris-HCl buffer (pH 8.0) containing protease inhibitor (Roche Applied Science), and the cells were lysed by sonication followed by centrifugation. The supernatant was incubated overnight with the Ni-NTA column (Sigma), and the bound proteins were eluted with 250 mm imidazole. The eluates were further purified by dialysis using dye Slide-A-Lyzer dialysis cassettes (Thermo Scientific). The purified protein was concentrated using membrane concentrators (3 kDa cutoff; Millipore), and the protein quality and concentration were confirmed by SDS-PAGE and spectrophotometry. For the removal of endotoxins from purified protein preparations for our in vivo experiments, the purified proteins were processed through Pierce high capacity endotoxin removal resin (Thermo Scientific) according to the manufacturer's protocol. Briefly, the resin was regenerated by adding 0.2 n NaOH overnight at 4 °C and equilibrated with Tris-HCl buffer containing 0.2 m NaCl (pH 7.4). After washing with endotoxin-free water, the proteins were added and incubated for 1 h at 4 °C, and the endotoxin-free proteins were collected in the flow-through.
Protein Transduction in Cultured Human Podocytes
The human podocyte cell line cultured in RPMI 1640-based medium supplemented with 10% fetal bovine serum (FBS) was allowed to form a monolayer. Following overnight serum starvation, the TAT protein was added in different amounts (5–10 μg/ml) in serum-free medium for a period of 30 min. The transduced cells were washed once with 1× PBS followed by an acid wash (0.5% acetic acid and 0.5 m NaCl (pH 3.0)) to remove any nonspecific cell surface-bound protein, followed by washing with 1× PBS (three times). The transduced cells either continued to grow in normal RPMI 1640 medium or were lysed in RIPA buffer as indicated.
Protein Labeling
The TAT-Neph1CD protein was labeled with Texas red using Alexa Fluor 594 protein labeling kit (Molecular Probes) according to the manufacturer's protocol. Texas-red TAT-Neph1CD was transduced into the serum-starved cultured podocytes as described above. The transduced podocytes were later washed and analyzed for the transduction of labeled protein using fluorescence microscopy.
TUNEL Assay
A terminal deoxynucleotidyltransferase dUTP-mediated nick-end labeling (TUNEL) assay was performed to evaluate apoptosis in TAT-Neph1CD-transduced cultured podocytes. The TUNEL staining was performed using an in situ cell death detection kit (Roche Diagnostics) using the manufacturer's protocol. Briefly, the transduced cells were washed with 1× PBS and fixed in freshly prepared 4% paraformaldehyde (in 1× PBS, pH 7.4), followed by permeabilization with 0.5% Triton X-100 for 10 min on ice. The cells were washed with 1× PBS (twice) and incubated with the TUNEL reaction mixture for 60 min at 37 °C in the dark. Finally, the cells were washed three times with 1× PBS, mounted, and analyzed using a fluorescence microscope.
Pulldown
The cell lysate from cells transduced with TAT-Neph1CD was prepared in RIPA buffer as described above. The TAT-Neph1CD and ZO-1 complex was separated by pulldown using Ni-NTA beads, and the beads were washed with PBS containing 0.1% Tween 20. The protein complexes were eluted with SDS sample buffer and resolved by SDS-PAGE, followed by immunoblotting with the indicated ZO-1 antibody.
PAN Treatment
The cultured podocytes were grown to 80–90% confluence, serum-starved overnight, and treated with PAN (100 μg/ml) for time periods ranging from 5 to 48 h. After incubation, the cells were fixed with 4% PFA (in 1× PBS) and analyzed by immunofluorescence. For subfractionation studies, the podocytes were lysed in RIPA buffer after 48 h of PAN treatment and processed for the isolation of lipid raft fractionations as described below.
Angiotensin II (Ang II) Treatment
The culture human podocytes (at 90% confluency) were treated with Ang II (100 nm) for a period of 48 h to induce injury (36). After treatment, the podocytes were fixed with 4% PFA and labeled with the desired antibodies and analyzed by immunofluorescence.
Lipid Raft Preparation
The lipid rafts were prepared from podocyte cells as described previously (19). Briefly, podocytes were lysed in cold Triton X-100 buffer containing 0.25% Triton X-100, 150 mm NaCl, 1 mm EGTA, and 25 mm Tris-HCl (pH 7.4) with protease and phosphatase inhibitors. A solution of density gradient was prepared using the Optiprep density gradient medium (Sigma; catalogue number D1556). In an ultracentrifuge tube, 0.67 ml of the cell lysate and 1.33 ml of 60% Optiprep were mixed by pipetting to obtain a final density of 40%; a total of 2.0 ml of 30% gradient Optiprep solution was layered at the top of this solution followed by the addition of 1.0 ml of 5% gradient Optiprep solution, thus making a total of three layers. The ultracentrifugation was performed at 45,000 rpm using a Beckman SW 55Ti rotor (Sorvall Discovery 90SE; Hitachi) at 4 °C for a period of 3.5–4.0 h. The Triton X-100-insoluble lipid raft layer was formed at the interface of 5 and 30% Optiprep solutions. One milliliter of fractions was sequentially collected from top-to-bottom in separate tubes and analyzed by SDS-PAGE followed by Western blotting using the indicated antibodies.
Cellular Imaging
Podocytes were grown on coverslips to 80–90% confluency and transduced with TAT-Neph1CD or TAT-GFP for 30 min. The podocyte cells stably expressing mCherry Neph1 protein were also cultured in a similar fashion. Following transduction and treatment with PAN, the cells were washed with 1× PBS and fixed with 4% paraformaldehyde (in 1× PBS) followed by permeabilization with 0.1% SDS and immunostained with Neph1 antibodies (raised against the extracellular or intracellular domain, as described previously (19). Alexa Fluor 488, 594, or 350 phalloidins were used to label actin cytoskeleton, and the coverslips were mounted with GelMount containing DAPI to label nuclei. Podocyte membrane was labeled using Vybrantô DiI cell-labeling dye (Invitrogen catalogue number V-22885). Epifluorescence microscopy was performed using a Zeiss inverted microscope (Zeiss Axio Observer D1m) fitted with ×63 and ×25 oil immersion objectives. Confocal microscopy was performed at the Molecular Pathology and Imaging Core, University of Pennsylvania, on a Nikon Eclipse Ti-U spinning disk confocal microscope fitted with a ×60 immersion objective. Metamorph software was used for the acquisition and analysis of the microscopic images. All parameters were maintained constant throughout the acquisition of images, including the exposure time. All images were recorded in 16-bit format, and ImageJ software was used for quantitative analysis. The three-dimensional reconstructions were made using FIJI (FIJI Is Just ImageJ, version 1.48p). Representative images from three independent experiments are provided for each experiment. Student's t tests were performed to compare two different groups, and p < 0.05 was considered as statistically significant.
Measurement of Albumin Flux (the Permeability Assay)
The transepithelial permeability of podocytes for BSA was assessed by measuring the passage of Texas Red-labeled BSA across the podocyte monolayer cultured in Transwell plates (0.4-μm pore; Corning Glass). Three days post-confluence, the cells were washed with 1× PBS, and the medium in wells (lower chamber) and inserts (upper chamber) was replaced with serum-free medium. Texas Red-labeled BSA at a final concentration of 50 μg/ml was added to the wells of the Transwell plate. Aliquots were removed from the inserts at regular intervals to measure the flow of fluorescence using 590 nm excitation and emission at 625 nm. A standard curve, derived from the measurement of serial dilutions of Texas Red-labeled BSA, was used to calculate the amount of BSA moved from the wells to inserts (19).
Zebrafish Stock and Injection
Wild-type zebrafish embryos were obtained from the Zebrafish core faculty of the university, where they are maintained and bred for experimental purposes. The egg medium (E3) was used to grow the zebrafish at 28.5 °C (37). The in vivo zebrafish kidney injury models that were used in this study have been described earlier (38, 39). Common cardinal vein injections (37) were performed for PAN injections, where PAN (25 mg/ml) was co-injected with either TAT-GFP or TAT-Neph1CD (0.25 mg/ml) at 72 hpf. The injected zebrafish were further placed in the culture medium and observed for the development of pericardial edema at 96 hpf. For adriamycin-induced podocyte injury model (39), the one-cell stage fish embryos were injected with 0.25 mg/ml either TAT-GFP or TAT-Neph1CD. The zebrafish were then grown in the E3 medium containing adriamycin at a final concentration of 30.3 mg/liter. The embryos were maintained in this medium throughout the entire growth period up to 96 hpf, and the edema was noted (39, 40). For histopathological analysis, the 72-hpf wild-type zebrafish were dechorionated, anesthetized, fixed with 4% paraformaldehyde, paraffin-embedded, and sectioned (5 μm thick). The sectioned fish were subjected to hematoxylin and eosin staining to analyze the glomerulus and pronephric tubules. A transgenic zebrafish expressing vitamin D-binding protein-GFP protein was used for the analysis of glomerular function, which is determined by estimating the leakage of VDBP-GFP in the medium (37, 41). 50 embryos of zebrafish injected with TAT-Neph1CD and uninjected were incubated with adriamycin (30.3 mg/liter) for 96 h in a 6-well plate. After 96 h, the adriamycin-containing medium was replaced with the normal medium. After 24 h (at 120 hpf), the media were concentrated using the Amicon Ultracentrifugal filters (Millipore), and the release of VDBP-GFP in the medium was estimated by GFP fluorescence intensity measurement at fluorescein channel using an ELISA plate reader (Beckman coulter). The protein concentration was calculated by comparing the sample values with the values drawn from a standard curve plotted using a purified GFP protein.
Statistical Analysis
All examined data are presented as mean ± S.E. Statistical analysis was performed using Microsoft Excel and Origin Software (OriginLab Corp.). Comparison between two groups was performed by using one-way analysis of variance, followed by the Student-Newman-Keuls test. p < 0.05 was considered as statistically significant.
RESULTS
TAT-Neph1CD Was Transduced in Podocytes Where It Retained Its Functionality
We have previously shown that the extracellular engagement of Neph1 induces its activation that includes Neph1 phosphorylation, its internalization, and subsequent downstream signaling mediated by the cytoplasmic domain of Neph1 (12, 42). Therefore, we hypothesized that if we selectively target the cytoplasmic domain of Neph1 by competitive inhibition, it may result in the inhibition of Neph1 phosphorylation and therefore Neph1 internalization. Because Neph1 has a tendency to dimerize and its cytoplasmic domain has been shown to interact with the native protein (43) and in the absence of any known molecular inhibitors for Neph1, we devised a strategy whereby we could compete the endogenous Neph1 (and the intracellular events mediated by its cytoplasmic domain) through increased expression of the Neph1CD. However, for efficient competition, relatively higher levels of expression of the Neph1 cytoplasmic domain was required. Moreover, we wanted to devise a molecular approach that can be later exploited commercially. Therefore, we chose to tranduce the cytoplasmic domain of Neph1 that was tagged with a protein transduction domain TAT. This approach allowed us to introduce sufficiently high levels of Neph1CD inside cultured podocytes with relative ease. Therefore, recombinant TAT-Neph1CD protein was purified, and protein quality was validated by the SDS-PAGE and Western blotting (Fig. 1A). TAT-Neph1CD protein was transduced in cultured podocytes by incubating the cells with 5 μg/ml protein in serum-free medium for the indicated periods ranging from 30 min to 24 h; after washing with PBS, the cells were lysed, and the lysate was subjected to Western blotting with Neph1 antibody. The results presented in Fig. 1B suggest that the protein rapidly transduced inside the cells within 30 min and was stably present inside the cells after 24 h. Transduction of the TAT-Neph1CD protein had no effect on the expression of Neph1-interacting protein ZO-1. Transduction of the His-TAT-GFP protein was used as a control to further validate the ability of podocyte cells as efficient transducers (Fig. 1C). To further confirm the intracellular presence of TAT-Neph1CD protein, the TAT-Neph1CD-transduced cells were analyzed by immunofluorescence using His antibody (Fig. 1D). The transduced protein was observed as a punctate staining in the perinuclear and the cytoplasmic regions (Fig. 1D). The quantitative analysis of the transduced podocytes suggested that more than 90% of cells were transduced (Fig. 1F). To further confirm the internalization of transduced protein, the TAT-Neph1CD protein was labeled with Texas red dye and transduced in human podocytes for a period of 30 min. Podocytes were washed, and the immunofluorescence imaging analysis of the live cells was performed. A significant amount of fluorescence was observed in the transduced cells (arrows) indicating that the labeled TAT-Neph1CD was present inside the podocytes (more than 95% transduction when compared with the untransduced podocytes) (Fig. 1, E and G). To evaluate if the transduced TAT-Neph1CD induces apoptosis in cells, TUNEL assay was performed. The results from TUNEL assay suggested that even at a higher concentration the TAT-Neph1CD (50 μg/ml) does not induce apoptosis in cells. It is to be noted that in all the subsequent transduction studies, 5 μg/ml concentration of TAT-Neph1CD was used (Fig. 1H). We further investigated whether the transduced TAT-Neph1CD was functional, and therefore, the cells transduced with His-TAT-Neph1CD podocytes were lysed, and a pulldown was performed using Ni-NTA beads. The pulldown complex was analyzed by Western blotting for the presence of Neph1 binding protein ZO-1 (12). Results presented in Fig. 1I demonstrate that TAT-Neph1CD retains its ability to interact with the endogenous ZO-1 following transduction in the podocytes.
FIGURE 1.

TAT-Neph1-CD is transduced in podocytes and remains functional. A, recombinantly expressed TAT-Neph1CD was purified, and its purity and identity were assessed by staining with Coomassie and Western blotting with Neph1 antibody, respectively. B, TAT-Neph1CD protein (5 μg/ml) was transduced in cultured podocytes for the indicated time periods ranging from 30 min to 24 h and subjected to Western blotting (WB) with Neph1 antibody. The protein rapidly transduced inside the cells within 30 min and was detected even at 24 h post-transduction. C, in a similar fashion, TAT-GFP was transduced for indicated times and was used as a control. D, TAT-Neph1CD was transduced in cultured human podocytes for a period of 30 min, washed, and fixed with 4% PFA (1× PBS). His primary antibody and Alexa Fluor 594 secondary antibodies were used to label the transduced TAT-Neph1CD (arrows), and the immunofluorescence imaging analysis was performed using a fluorescent microscope (×60 magnification). Additionally, the cells were visualized by differential interference contrast (DIC) microscopy. E, TAT-Neph1CD protein was labeled with Texas red dye and transduced in cultured human podocytes for 30 min. The cells were then washed and fixed with 4% PFA (in 1× PBS) and analyzed by immunofluorescence imaging that showed significant accumulation of fluorescent TAT-Neph1CD inside the transduced podocytes as compared with the untransduced control podocytes. F, statistical analysis of the TAT-Neph1CD transduction from D suggested that 94.6 ± 1.5% of the podocytes were transduced with this protein. G, statistical analysis of the Texas red TAT-Neph1CD transduction suggested that 96.3 ± 1.5% of podocytes were transduced with this protein. H, TUNEL assay was performed on transduced and untransduced podocytes. No apoptosis could be seen in the podocytes that were transduced with 10 times (50 μg/ml) higher than the usual dose (5 μg/ml) of TAT-Neph1CD in podocytes. Bright field and DAPI images were also obtained to visualize the overall cell population. I, ability of transduced TAT-Neph1CD to interact with endogenous ZO-1 was examined in a pulldown experiment using Ni-NTA beads. Podocytes transduced with TAT-Neph1CD were lysed, and the Neph1-ZO-1 complex was pulled down using nickel beads, separated on SDS-PAGE, and analyzed by Western blotting to detect the presence of ZO-1 in the complex. Scale bars, 10 μm (D), 20 μm (E); 20 μm (H).
Transduction of TAT-Neph1CD Inhibits Tyrosine Phosphorylation of Endogenous Neph1
Because previous studies have shown that the recombinant Neph1 can dimerize with the endogenous Neph1 (43), we hypothesized that the transduction of this domain will prevent the access of endogenous Neph1 to kinases such as Fyn, thereby affecting Neph1 phosphorylation, which is a strong indicator of Neph1 signaling. To test this hypothesis, we tested the ability of the transduced TAT-Neph1CD to inhibit PAN-induced phosphorylation of the endogenous Neph1. Therefore, cells transduced with either His-TAT-GFP or His-TAT-Neph1CD were treated with PAN, and the cell lysates were analyzed for the presence of phosphorylated Neph1 by Western blotting with phospho-Neph1 (p-Neph1) and Neph1 antibodies. The results presented in Fig. 2A show that the transduction of TAT-Neph1CD in podocytes significantly reduced PAN-induced endogenous Neph1 phosphorylation when compared with the control transduced cells. Further densitometric analysis of the transduced and untransduced cells revealed a 60–70% decrease in the PAN-induced endogenous Neph1 phosphorylation in the TAT-Neph1CD-transduced cells (Fig. 2B).
FIGURE 2.
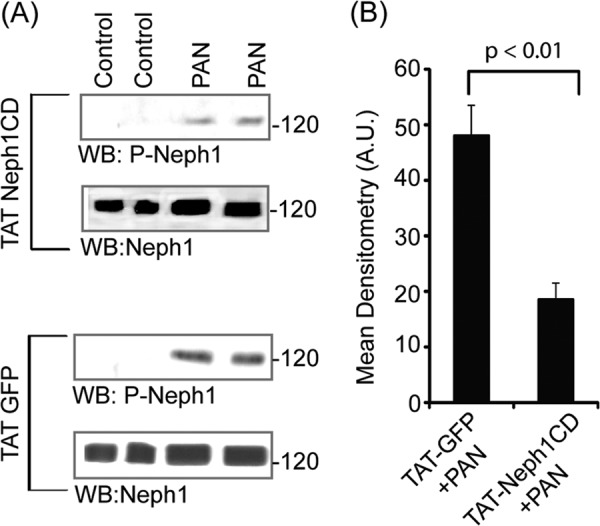
Transduced TAT-Neph1CD inhibited tyrosine phosphorylation of endogenous Neph1. A, podocytes transduced with either TAT-GFP or TAT-Neph1CD were treated with PAN for a period of 48 h, and the cell lysates were analyzed for the presence of phosphorylated Neph1 by Western blotting (WB) with phospho-Neph1 (p-Neph1) and Neph1 antibodies. The transduction of TAT-Neph1CD in podocytes significantly reduced PAN-induced endogenous Neph1 phosphorylation as compared with the control. B, further densitometric analysis revealed a 60–70% decrease of the PAN-induced endogenous Neph1 phosphorylation in cells transduced with TAT-Neph1CD as compared with the TAT-GFP-transduced cells.
Transduction of TAT-Neph1CD in Podocytes Prevented Redistribution of Endogenous Neph1 from Lipid Rafts to the Cytosol in Response to PAN Treatment
Using subfractionation studies, we previously demonstrated that Neph1 is primarily localized in the lipid raft fractions of the podocytes (19). Our previous results also showed that PAN induces a change in the distribution of Neph1 from podocyte cell membrane to the cytoplasm (12). Because TAT-Neph1CD transduction inhibited PAN-induced Neph1 phosphorylation, we hypothesized that it also affects the distribution of Neph1 in response to PAN. To test this hypothesis, the TAT-GFP- and TAT-Neph1CD-transduced cells were treated with PAN for 48 h followed by cell lysis. To evaluate the cellular distribution of endogenous Neph1, the cell lysates were subjected to fractionation (19), and each isolated fraction was analyzed for the presence of Neph1 and markers of subcellular compartments, including caveolin, Myo1c (lipid rafts), and transferrin (cytosolic fractions) (Fig. 3, A–D). As shown in Fig. 3B, treatment with PAN redistributed Neph1, and a significant reduction (p < 0.05) in the amount of Neph1 in lipid rafts was noticed (the densitometric analysis suggested a loss of 60–70% from the lipid rafts (Fig. 3E)). In contrast, the majority of Neph1 was retained in the lipid raft fraction in PAN-treated cells that were transduced with TAT-Neph1CD (Fig. 3D). It is to be noted that the treatment of TAT-Neph1CD alone had no effect on the Neph1 distribution (Fig. 3C). These results indicate that the transduction of TAT-Neph1CD prevents PAN-induced redistribution of Neph1 from the podocyte cell membrane to the cytoplasm. To further confirm these observations, the TAT-GFP- and TAT-Neph1CD-transduced cells were treated with PAN and labeled with a Neph1 antibody (without permeabilization) directed toward the extracellular domain of Neph1 (19), and confocal imaging was performed. Results presented in Fig. 4A and the quantitative analysis shown in Fig. 4B suggest that although PAN treatment significantly reduced (p < 0.05) the localization of Neph1 at the membrane in TAT-GFP-transduced cells, the transduction of TAT-Neph1CD maintained a robust localization of Neph1 at the podocyte cell membrane and was unaffected by the PAN treatment. Quantitative analysis of the single plane images (16 bits) was performed using ImageJ software where different regions of interest were selected. Images from three independent experiments were analyzed for the quantitation. Collectively, these results suggest that the transduction of TAT-Neph1CD in podocyte cells prevents PAN-induced Neph1 mis-localization.
FIGURE 3.

Redistribution of endogenous Neph1 to non-lipid raft fractions in response to PAN was inhibited by TAT-Neph1CD transduction. A–D, TAT-GFP- and TAT-Neph1CD-transduced podocytes were treated with or without PAN and subjected to subfractionation using Optiprep gradient. The lysates were run on an SDS-PAGE, transferred to nitrocellulose, and Western blotted (WB) using Neph1, Myo1c, caveolin 1, transferrin receptor, and actin antibodies. PAN-induced redistribution of endogenous Neph1 to non-lipid raft fractions (5–8) is shown in B, whereas treatment with TAT-Neph1CD (D) retained Neph1 in the lipid raft fraction (3). E, densitometric analysis of PAN-treated cells suggested a 70% loss of Neph1 from the lipid rafts (marked in dotted box), which was restored by the transduction of TAT-Neph1CD.
FIGURE 4.

Transduction of TAT-Neph1CD prevents PAN-induced loss of Neph1 from podocyte cell membrane. A, transduced cells were analyzed by immunofluorescence using antibodies directed against actin and the extracellular domain of Neph1 (Neph1-Ex). Confocal images were collected at ×60 magnification and presented after deconvolution in XY and XZ orientation. B, analysis of mean pixel intensity at the membrane of transduced cells suggests that PAN treatment significantly reduced Neph1 localization at the membrane (p < 0.05) in control cells (TAT-GFP+PAN), whereas the cells transduced with TAT-Neph1CD maintained a robust localization of Neph1 at the podocyte cell membrane and were unaffected by treatment with PAN. ns, nonsignificant. Scale bar, A, 10 μm.
Transduction of TAT-Neph1CD Protects Podocytes from PAN- and Ang II-induced Cytoskeletal Damage
In addition to affecting Neph1 phosphorylation and distribution, the treatment of PAN in podocytes has been shown to induce severe cytoskeletal damage and loss of cell-cell junctions (Fig. 5A). The cytoskeletal damage is often characterized by a gradual increase in stress fibers and retraction of cellular processes with PAN treatment (13, 15, 44). Eventually in a time-dependent manner, the stress actin fibers started moving toward the cell periphery, and the cell-cell contacts were lost (Figs. 5A and 6A). Additionally, it was noted (as described previously) (13, 15) that the distribution of Neph1 at the junctions was significantly altered with PAN treatment (Fig. 5A). The presence of Neph1 at the junctions is marked with arrows (Fig. 5). Because the transduction of TAT-Neph1CD in podocytes prevented loss of Neph1 from the membrane in PAN-treated cells, we asked if this also results in the prevention of PAN-induced cytoskeletal damage. Consistent with the above observations, the transduction of TAT-Neph1CD significantly reduced the PAN-induced cytoskeletal damage in podocytes (Fig. 5A) (please note that the increased cytoplasmic Neph1 staining observed in the TAT-Neph1CD cells is due to the Neph1 antibody that is directed toward the cytoplasmic domain of Neph1 and thus stains the transduced Neph1 along with the endogenous Neph1). Additionally, a comparative analysis was performed using the intensity profile that was plotted using a line spanning the junction between two cells (Fig. 5B). This analysis suggested that there was a significant loss of Neph1 at the cell-cell junctions in cells treated with PAN and transduced with control protein TAT-GFP; in contrast, cells transduced with TAT-Neph1 and treated with PAN showed minimal loss of Neph1 at the junctions (marked with arrows) (p < 0.05) (Fig. 5, A and B). It is to be noted that the TAT-GFP-transduced cells treated with PAN (48 h) failed to make efficient cell-cell contacts as noted previously (16, 17) and also demonstrated by the staining of podocyte cell membrane using Vybrantô DiI cell-labeling dye (Invitrogen catalog number V-22885). To quantitatively assess the ability of cells to form cell-cell junctions, we estimated the number of cells involved in making cell-cell contacts in treated and untreated cells. As shown in Fig. 6, the number of cells involved in making cell-cell contacts in PAN-treated TAT-GFP-transduced cells was significantly reduced, whereas the number of cells involved in cell-cell contacts in TAT-Neph1CD-transduced cells treated with PAN was similar to the 0-h time point (Fig. 6B). As shown previously (16), there was no indication of overall cell death at the 48-h time point in PAN-treated cells (data not shown). Multiple images from three different experiments were used for the statistical analysis. Overall, these results suggested that maintaining a robust expression of Neph1 at the podocyte cell-cell junctions may increase the ability of podocytes to resist PAN-induced cytoskeletal damage.
FIGURE 5.

TAT-Neph1CD-transduced podocytes are resistant to PAN-induced cytoskeletal damage. A and B, TAT-GFP- and TAT-Neph1CD-transduced podocytes were treated with PAN for indicated time periods (0–48 h) and immunostained with phalloidin (Alexa 488) and Neph1 (Alexa 594), and mounted with DAPI. Confocal imaging showed that PAN treatment resulted in drastic changes to the actin cytoskeleton in TAT-GFP-transduced cells. Additionally, the distribution of junctional proteins, including Neph1, was significantly altered in these cells. In contrast, the PAN treatment of podocytes transduced with TAT-Neph1CD showed minimal alteration to the actin cytoskeleton with increased localization of Neph1 at the junctions. Confocal images were collected at ×60 magnification and presented after deconvolution in XY and XZ orientation. B, pixel intensity profile was created by placing a line spanning the cell-cell junction and analyzed using ImageJ software. This analysis suggested that the intensity of Neph1 at the cell-cell junctions was maintained in TAT-Neph1CD-transduced cells treated with PAN (p < 0.05). Scale bar, 10 μm (A).
FIGURE 6.

Cell-cell contacts were preserved in TAT-Neph1CD-transduced podocytes treated with PAN. A and B, podocytes were labeled with Vybrantô DiI cell membrane labeling dye for easier visualization of cell boundaries. Number of podocyte cells that were engaged in cell-cell contacts were identified and analyzed. The cell-cell contacts were calculated by analyzing 100 cells in each group (in triplicate), and the cells with more than 50% of their surface (defined manually using actin and membrane DiI staining images) in contact with the neighboring cells were considered positive. The podocytes transduced with TAT-GFP showed significant reduction in the cell-cell contacts, and thus the number of cells involved in contact formation were significantly reduced in response to 48 h of PAN treatment; in contrast, the cell-cell contacts were well preserved in the TAT-Neph1CD-transduced cells treated with PAN. Scale bar, 10 μm (A).
To further validate our findings, another podocyte cell culture injury model was used that utilized Ang II as the injury-inducing agent (36). Untransduced or transduced podocytes with TAT-Neph1CD were subjected to Ang II (100 nm) treatment in a time-dependent fashion (Fig. 7, A and B). Cytoskeleton changes similar to PAN, such as increase in stress fibers close to the cell periphery (16, 36) and gradual loss of Neph1 from the junctions, were noted in the Ang II-treated cells. In contrast, the distribution of Neph1 at the junctions was significantly higher (p < 0.05) in TAT-Neph1CD-transduced podocytes as compared with the untransduced cells (Fig. 7A). The intensity plot of Neph1 at the junctions further suggested an increased presence of Neph1 at the cell-cell junctions (Fig. 7B).
FIGURE 7.

TAT-Neph1CD-transduced podocytes are resistant to cytoskeletal damage induced by Ang II. A and B, TAT-GFP- and TAT-Neph1CD-transduced podocytes were treated with Ang II for the indicated time periods (0–48 h) and immunostained with phalloidin (Alexa 488) and Neph1 (Alexa 594) and mounted with DAPI containing mounting media. Confocal imaging revealed that Ang II treatment resulted in changes to the actin cytoskeleton (where increased stress fibers were noted) along with loss of Neph1 at the cell-cell junctions (A). Notably, the transduction of TAT-Neph1CD prevented these changes (see Fig. 5A). Confocal images were collected at ×60 magnification and constructed after deconvolution in XY and XZ orientation. B, quantitation using pixel intensity profile further demonstrated that similar to PAN, the loss of Neph1 at the cell-cell junctions was minimal in TAT-Neph1CD transduced cells treated with Ang II when compared with the TAT-GFP transduced cells treated with Ang II (p < 0.05). Scale bar, 10 μm (A).
Increased Expression of Neph1 at the Podocyte Cell Membrane Protected Podocytes from PAN-induced Injury
Collectively, the above mentioned studies provide preliminary evidence that maintaining Neph1 localization at the podocyte membrane is protective in nature. Therefore, to further validate the results obtained from transduction studies, we investigated if the overexpression of Neph1 at the podocyte cell membrane provides similar protection from PAN-induced injury as observed in our transduction experiments. To test this, we constructed a chimeric Neph1 in which a fluorescent protein domain (Cherry) was inserted between the transmembrane and the cytoplasmic domain of Neph1 (schematic of the insertion site is shown in Fig. 8A). The insertion site was carefully chosen to avoid any potential tyrosine phosphorylation sites in the cytoplasmic domain of Neph1 and the functionally important C-terminal “THV” residues (9, 10). It is to be noted that tagging Neph1 at the C-terminal end with any fluorescent tag significantly decreased its ability to localize at the membrane (data not shown). The cells stably expressing mCherry-Neph1 were created using this construct, and the expression pattern of the overexpressed Neph1 closely matched that of endogenous Neph1 (both the proteins were correctly localized at the cell-cell junctions, Fig. 8B). These cells were subjected to PAN treatment in a fashion similar to the TAT-Neph1CD-transduced cells. The results presented in Fig. 8C show that these cells were resistant to PAN-induced cytoskeletal damage. Accordingly, the PAN-induced loss of Neph1 from the podocyte cell membrane was also not observed in the mCherry-Neph1-overexpressed cells (Fig. 8C).
We have previously shown that in addition to the loss of Neph1 from the cell membrane, PAN induces loss of tight junctions resulting in an increased BSA permeability in podocytes (19). However, if the tight junctions are preserved in the mCherry-Neph1-overexpressing cells, it is likely that these cells will be resistant to BSA permeability in response to PAN. Therefore, to test this hypothesis, a BSA permeability assay was performed (19). The passage of albumin across the monolayer of podocytes overexpressing mCherry-Neph1 or control cells treated with PAN was assessed by a paracellular albumin flux assay using Texas Red-labeled albumin. Results shown in Fig. 8D demonstrate that the Cherry-Neph1-overexpressing cells were resistant to PAN-induced albumin leakage. Interestingly, the mere overexpression of Cherry-Neph1 increased the overall resistance of podocyte cells to the passage of albumin (Fig. 8D). Collectively, these results provide preliminary evidence that the events that result in the robust maintenance of Neph1 at the membrane provide podocytes with an increased ability to maintain tight junctions and resistance from damage by injury-inducing agents.
Transduction of TAT-Neph1CD Prevented PAN and Adriamycin-induced Injury in a Zebrafish Renal Injury Model
Zebrafish is a fast developing in vivo model system to study renal diseases; in addition, many of the renal injury-inducing agents, including PAN and adriamycin, have been shown to induce phenotypes related to renal disorders (38, 40). Therefore, to further validate our in vitro studies, and to evaluate the in vivo ability of TAT-Neph1CD in preventing the PAN-induced injury, we used zebrafish as an in vivo model system (38). Therefore, 25 mg/ml PAN (3 nl) was injected through the common cardinal vein along with either TAT-GFP or TAT-Neph1CD (10 nm in 1.5 nl, each) at 72 hpf, and the injected zebrafish were developed for 96 hpf to observe the phenotype. As shown in Fig. 9A, a significant increase in the number of zebrafish with pericardial edema and curved body shape (a strong indicator of impaired renal function) was observed in embryos co-injected with TAT-GFP and PAN. In contrast, the phenotype was relatively mild in the zebrafish co-injected with TAT-Neph1CD and PAN (Fig. 9A). Quantitative analysis from three independent experiments suggested a 40% reduction in the PAN-induced phenotype in TAT-Neph1CD injected zebrafish when compared with the TAT-GFP-injected zebrafish (Fig. 9B). These results further support the conclusion that the transduction of TAT-Neph1CD can induce renal protection in vivo.
FIGURE 9.

Zebrafish injected with TAT-Neph1CD were resistant to PAN- and Ang II-induced injury. A, PAN (25 mg/ml) was co-injected with 0.25 mg/ml of either TAT-GFP (control) or TAT-Neph1CD (test) in 72-hpf zebrafish via the common cardinal vein, and the development of pericardial edema was monitored at 96 hpf. Significant increase in pericardial edema and curved body shape was noted in the control, whereas the test zebrafish had minimal or no edema, and their body shapes were grossly normal. B, quantitative analysis from three independent experiments suggested about 40% reduction of PAN-induced edema in test zebrafish as compared with the control. C and D, embryos either injected with 0.25 mg/ml of either TAT-GFP (control) or TAT-Neph1CD (test) were grown in the E3 medium containing adriamycin (30.3 mg/liter). The phenotype (pericardial edema an indication of renal abnormality) was observed at 96 hpf (more than 90% embryos develop edema in this model). However, the number of zebrafish embryos with pericardial edema was significantly reduced with TAT-Neph1CD injection. D, quantitative analysis further suggested a 25% decrease in pericardial edema in embryos injected with TAT-Neph1CD when compared with the embryos injected with TAT-GFP (p < 0.05). E, histological analysis of sections from 72 hpf of TAT-GFP-injected embryos treated with adriamycin and stained with hematoxylin and eosin showed prominent dilation of the pronephric tubules, whereas this phenotype was absent in TAT-Neph1CD-injected embryos treated with adriamycin. F, schematic representation of the in vivo filtration assay using transgenic zebrafish expressing GFP-VDBP from liver. Zebrafish injected with TAT-Neph1CD and uninjected were treated with adriamycin to induce glomerular injury, and the excreted GFP-VDBP was estimated in the medium. G, zebrafish embryos injected with TAT-Neph1CD showed a measurable reduction (∼30% with a p value of <0.05) in the excreted GFP-VDBP in the medium when compared with the control embryos.
To further validate these results, another in vivo zebrafish injury model using adriamycin as an injury agent (39) was used. In this model, the embryos were grown in the E3 medium containing adriamycin (30.3 mg/liter) (39), and the pericardial edema was observed at 96 hpf (39, 40) in more than 90% embryos (Fig. 9, C and D). In contrast, the transduction of TAT-Neph1CD decreased the pericardial edema in about 25% of embryos when compared with TAT-GFP-transduced embryos (Fig. 9D). Collectively, these results provide in vivo validation of this approach. To provide further confirmation of our results, the same zebrafish were histologically analyzed to confirm a renal phenotype. The zebrafish embryos transduced with TAT-GFP and treated with adriamycin showed significant pronephric tubular dilation (a hallmark of renal injury) as compared with the TAT-Neph1CD-transduced zebrafish that were treated with adriamycin (Fig. 9E). To further demonstrate the ability of TAT-Neph1CD to preserve the glomerular filtration function in response to a glomerular injury, we performed an in vivo filtration assay (Fig. 9F) using a transgenic zebrafish model that constitutively expressed VDBP-GFP protein. It is to be noted that in this model, under proteinuric conditions, the glomerulus is unable to retain this protein in the blood plasma, and therefore, this protein is leaked out and detected in the fish medium. Thus, zebrafish expressing the VDBP-GFP transgene (50 embryos) were injected with TAT-Neph1CD at the one-cell stage and treated with adriamycin to induce glomerular damage. The excretion of VDBP-GFP in the medium was measured. As shown in Fig. 9G, introduction of TAT-Neph1CD in zebrafish offered significant (∼30% with p < 0.05) protection from the adriamycin-induced loss of glomerular function.
DISCUSSION
Neph1 is a membrane protein that is localized at the podocyte cell membrane where it has been shown to participate in maintaining the structural and functional integrity of the slit diaphragm. Previous studies have shown that glomerular injury induces rapid tyrosine phosphorylation of the Neph1 cytoplasmic domain and the onset of subsequent signaling events resulting in actin cytoskeletal reorganization (9, 10, 12). This further suggests that Neph1 signaling initiated by its cytoplasmic domain is an important event that may define the response of podocytes to injury. Therefore, we hypothesized that inhibiting the Neph1 phosphorylation will inhibit the subsequent cellular response and diminish the response of podocytes to injury. Indeed, in this study, we demonstrate that Neph1 phosphorylation was inhibited by transducing the cytoplasmic domain of Neph1 in podocytes, and this resulted in the protection of podocytes from PAN-induced injury.
Several lines of evidence indicate that tyrosine phosphorylation of Neph1 plays a key role in regulating the actin cytoskeleton reorganization of podocytes (5, 45). Our previous study suggested that in addition to Neph1 phosphorylation, the extracellular engagement of Neph1 also induces its internalization and subsequent downstream signaling from the cytoplasmic domain of Neph1 (12, 42). This led us to hypothesize that selectively targeting the cytoplasmic domain of Neph1 will affect Neph1 phosphorylation and its ability to internalize.
The ability to transduce proteins rapidly inside cells and in vivo has gained increased attention over the last few years due to the discovery of protein transduction domains (29, 30). One of the most common protein transduction domains widely used in the in vivo and in vitro studies is TAT that has been used to deliver proteins intracellularly aimed at understanding the signaling mechanisms of the target proteins and identifying potential therapeutic targets (30, 46–48). In this study we tagged the cytoplasmic domain of Neph1 with TAT, to introduce Neph1CD inside the cultured podocytes at sufficiently high levels with relative ease. Indeed, the TAT-Neph1CD protein transduced quickly inside cultured podocytes and was detected by immunofluorescence and Western blotting analysis (Fig. 1). More importantly, the transduced Neph1CD retained its functionality as determined by its ability to interact with the endogenous ZO-1 (Fig. 1).
In this study, we showed that the transduction of TAT-Neph1CD in podocytes significantly reduced (∼60–70%) PAN-induced endogenous Neph1 phosphorylation, when compared with the control cells (Fig. 2B). The increased Neph1 tyrosine phosphorylation has been shown to associate with podocyte injury that is defined by the loss of actin cytoskeleton organization, internalization of the slit diaphragm proteins, including Neph1, nephrin, and podocin, and leading to the foot process effacement (13–15). Additionally, PAN treatment has been shown to alter the distribution of Neph1 from plasma membrane to the cytoplasmic region in the cultured podocytes (12). Interestingly, the transduction of TAT-Neph1CD prevented the movement of endogenous Neph1 from the membrane to cytoplasm. The subfractionation studies showed that 60–70% of the endogenous Neph1 was retained in the lipid raft fraction in TAT-NephCD-transduced podocytes that is consistent with the presence of Neph1 at the cell membrane (Fig. 3). The immunofluorescence analysis of the extracellular Neph1 in the TAT-Neph1CD-transduced podocytes further confirmed these observations. Collectively, these results presented evidence that the transduced TAT-Neph1CD induces resistance in podocytes toward PAN-induced redistribution of Neph1 from the podocyte cell membrane to the cytoplasm.
Changes in the actin cytoskeleton of podocytes in response to injury defines the morphological alterations to the podocyte architecture that is associated with the loss of glomerular filtration function (13, 15). The PAN-induced podocyte injury leads to actin cytoskeleton damage that is characterized by loss of fine F-actin stress fibers and the appearance of thick bundles of cortical actin filaments in podocytes (13, 44). In vivo PAN treatment has been shown to induce foot process effacement along with the decreased expression and abnormal distribution of the slit diaphragm proteins Neph1, nephrin, and podocin (14, 15, 50). Similarly in this study, we find that the cytoskeletal damage was induced by PAN treatment of podocytes; however, transduction of the TAT-Neph1CD protein protected podocytes from PAN-induced injury and preserved the cell-cell junctions (Figs. 5A and 6A) (13, 15, 51). Additionally, the transduced TAT-Neph1CD prevented redistribution of Neph1 from the junctions and loss of Neph1 from the membrane in response to PAN treatment (Fig. 5, A and B). We further report that this protection is not specific to injury induced by PAN, because similar protection was observed when Ang II was used as an injury-inducing agent. These cell culture studies highlight the mechanism through which transduced TAT-Neph1CD imparts protection in podocytes toward PAN-induced injury. The cellular loss of a protein can often be compensated by the transient overexpression of the protein through transfection of specific cDNA inside cells (19). Therefore, to compensate for the loss of Neph1 from the podocyte cell membrane in response to PAN, we constructed a stable cell line overexpressing chimeric Neph1 with the fluorescent domain that was targeted to the podocyte cell membrane. Results from the experiments using this cell line provided additional evidence that the podocytes with increased expression of Neph1 at the cell membrane are resistant to PAN-induced damage. Because these conclusions were based on the evidence provided from cell culture studies, it was necessary to test the validity of this approach in an in vivo model system. Therefore, we took advantage of a fast developing approach that utilizes zebrafish as a model system, where in vivo screening studies can be performed with relative ease (38, 40). In this model system, pericardial edema has commonly been used as a measure of renal dysfunction that is induced by the loss of several slit diaphragm proteins, including Neph1, nephrin, and podocin (37, 52, 53). Our in vivo experiments utilizing zebrafish as a model system further confirmed the protective nature of TAT-Neph1CD, where embryos injected with the TAT-Neph1CD protein showed increased protection from pericardial edema induced by either PAN or adriamycin. Furthermore, histological analysis of TAT-GFP-injected embryos showed significant dilation of pronephric tubules as compared with fishes transduced with TAT-Neph1CD protein. The dilation of renal proximal tubules has been used as a marker of renal dysfunction in the genetic knock-out mouse models of nephrin (54–56) and in zebrafish models where specific knockdown of slit diaphragm proteins, including nephrin, Neph1, podocin, and Myo1c, was performed (37). It has been suggested that the loss of slit diaphragm proteins alters glomerular functionality leading to a leaky filter in these morphants, thus increasing luminal pressure resulting in tubular distension (52). Additionally, the ability of TAT-Neph1CD to prevent the loss of glomerular filtration function in response to a glomerular injury-inducing agent was also demonstrated.
Because Neph1 is expressed in multiple cell types (49), it is possible that the transduced TAT-Neph1CD has extrarenal affects; however, identifying and understanding these affects will require further investigations, possibly using additional in vivo model systems, including mice.
Overall, these results provide evidence that chemically induced podocyte damage can be prevented by maintaining a robust expression of Neph1 at the podocyte cell membrane. In addition, the results suggest that inhibiting the Neph1 tyrosine phosphorylation and its internalization (necessary events for preserving Neph1 expression at the membrane) serve as a potential mechanism for this protection (Fig. 10). It is possible that these events directly affect Neph1 trafficking, especially its endocytosis; however, Neph1 trafficking has never been explored and will be the subject of our future investigations. Because a number of other proteins, including nephrin, podocin, and synaptopodin, have been shown to contribute toward podocyte maintenance and function, it remains to be known if inhibiting the signaling from these proteins has similar protective effects on podocytes. Additionally, whether this approach is applicable to the protection from other podocyte injury-inducing agents remains to be determined. Nevertheless, the results presented in this study are consistent with the role of Neph1 phosphorylation and signaling as a critical event that can be targeted to develop therapies for preserving podocyte structure/function in the event of glomerular disease. Because we recently determined the solution structure of the Neph1CD, we now have the ability to either design or identify small molecules with the ability to modulate Neph1 signaling and demonstrate their therapeutic value (35).
FIGURE 10.

Schematic presentation of the proposed TAT-Neph1CD action. The tyrosine phosphorylation of endogenous Neph1 is increased in response to PAN that induces downstream signaling leading to cytoskeleton damage and kidney malfunction. Transduction of TAT-Neph1CD competes for endogenous Neph1 interactions, thereby blocking Neph1 phosphorylation and inhibiting the downstream signaling. Thus, the transduced cells are unable to respond to injury and are therefore protected from the PAN-induced podocyte damage.
Acknowledgment
The imaging core was supported by Center Grant P30 DK050306 from the National Institutes of Health.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 1R01DK087956 (to D. N.) and DK080751 (to L. B. H.) from NIDDK. This work was also supported by the NephCure Foundation, NephCure Postdoctoral Grant 2012-RFP-001 (to E. A.), and the United States-India Educational Foundation (to Y. S. R.).
- PAN
- puromycin aminonucleoside
- TAT
- trans-activator of transcription
- Neph1CD
- cytoplasmic domain of Neph1
- Ni-NTA
- nickel-nitrilotriacetic acid
- Ang II
- angiotensin II
- hpf
- hours post-fertilization
- PFA
- paraformaldehyde
- DiI
- 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate.
REFERENCES
- 1. Haraldsson B., Nyström J., Deen W. M. (2008) Properties of the glomerular barrier and mechanisms of proteinuria. Physiol. Rev. 88, 451–487 [DOI] [PubMed] [Google Scholar]
- 2. Pavenstädt H., Kriz W., Kretzler M. (2003) Cell biology of the glomerular podocyte. Physiol. Rev. 83, 253–307 [DOI] [PubMed] [Google Scholar]
- 3. Tryggvason K., Patrakka J., Wartiovaara J. (2006) Hereditary proteinuria syndromes and mechanisms of proteinuria. N. Engl. J. Med. 354, 1387–1401 [DOI] [PubMed] [Google Scholar]
- 4. Tryggvason K., Pikkarainen T., Patrakka J. (2006) Nck links nephrin to actin in kidney podocytes. Cell 125, 221–224 [DOI] [PubMed] [Google Scholar]
- 5. Verma R., Kovari I., Soofi A., Nihalani D., Patrie K., Holzman L. B. (2006) Nephrin ectodomain engagement results in Src kinase activation, nephrin phosphorylation, Nck recruitment, and actin polymerization. J. Clin. Invest. 116, 1346–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Garg P., Verma R., Nihalani D., Johnstone D. B., Holzman L. B. (2007) Neph1 cooperates with nephrin to transduce a signal that induces actin polymerization. Mol. Cell. Biol. 27, 8698–8712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Verma R., Wharram B., Kovari I., Kunkel R., Nihalani D., Wary K. K., Wiggins R. C., Killen P., Holzman L. B. (2003) Fyn binds to and phosphorylates the kidney slit diaphragm component Nephrin. J. Biol. Chem. 278, 20716–20723 [DOI] [PubMed] [Google Scholar]
- 8. Zhu J., Sun N., Aoudjit L., Li H., Kawachi H., Lemay S., Takano T. (2008) Nephrin mediates actin reorganization via phosphoinositide 3-kinase in podocytes. Kidney Int. 73, 556–566 [DOI] [PubMed] [Google Scholar]
- 9. Huber T. B., Schmidts M., Gerke P., Schermer B., Zahn A., Hartleben B., Sellin L., Walz G., Benzing T. (2003) The carboxyl terminus of Neph family members binds to the PDZ domain protein zonula occludens-1. J. Biol. Chem. 278, 13417–13421 [DOI] [PubMed] [Google Scholar]
- 10. Harita Y., Kurihara H., Kosako H., Tezuka T., Sekine T., Igarashi T., Hattori S. (2008) Neph1, a component of the kidney slit diaphragm, is tyrosine-phosphorylated by the Src family tyrosine kinase and modulates intracellular signaling by binding to Grb2. J. Biol. Chem. 283, 9177–9186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harita Y., Kurihara H., Kosako H., Tezuka T., Sekine T., Igarashi T., Ohsawa I., Ohta S., Hattori S. (2009) Phosphorylation of nephrin triggers Ca2+ signaling by recruitment and activation of phospholipase C-γ1. J. Biol. Chem. 284, 8951–8962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wagner M. C., Rhodes G., Wang E., Pruthi V., Arif E., Saleem M. A., Wean S. E., Garg P., Verma R., Holzman L. B., Gattone V., Molitoris B. A., Nihalani D. (2008) Ischemic injury to kidney induces glomerular podocyte effacement and dissociation of slit diaphragm proteins Neph1 and ZO-1. J. Biol. Chem. 283, 35579–35589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zheng C. X., Chen Z. H., Zeng C. H., Qin W. S., Li L. S., Liu Z. H. (2008) Triptolide protects podocytes from puromycin aminonucleoside induced injury in vivo and in vitro. Kidney Int. 74, 596–612 [DOI] [PubMed] [Google Scholar]
- 14. Guan N., Ding J., Deng J., Zhang J., Yang J. (2004) Key molecular events in puromycin aminonucleoside nephrosis rats. Pathol. Int. 54, 703–711 [DOI] [PubMed] [Google Scholar]
- 15. Mundel P., Reiser J., Zúñiga Mejía Borja A., Pavenstädt H., Davidson G. R., Kriz W., Zeller R. (1997) Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp. Cell Res. 236, 248–258 [DOI] [PubMed] [Google Scholar]
- 16. Vega-Warner V., Ransom R. F., Vincent A. M., Brosius F. C., Smoyer W. E. (2004) Induction of antioxidant enzymes in murine podocytes precedes injury by puromycin aminonucleoside. Kidney Int. 66, 1881–1889 [DOI] [PubMed] [Google Scholar]
- 17. Rico M., Mukherjee A., Konieczkowski M., Bruggeman L. A., Miller R. T., Khan S., Schelling J. R., Sedor J. R. (2005) WT1-interacting protein and ZO-1 translocate into podocyte nuclei after puromycin aminonucleoside treatment. Am. J. Physiol. Renal Physiol. 289, F431–F441 [DOI] [PubMed] [Google Scholar]
- 18. Hagiwara M., Yamagata K., Capaldi R. A., Koyama A. (2006) Mitochondrial dysfunction in focal segmental glomerulosclerosis of puromycin aminonucleoside nephrosis. Kidney Int. 69, 1146–1152 [DOI] [PubMed] [Google Scholar]
- 19. Arif E., Wagner M. C., Johnstone D. B., Wong H. N., George B., Pruthi P. A., Lazzara M. J., Nihalani D. (2011) Motor protein Myo1c is a podocyte protein that facilitates the transport of slit diaphragm protein Neph1 to the podocyte membrane. Mol. Cell. Biol. 31, 2134–2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Reiser J., Gupta V., Kistler A. D. (2010) Toward the development of podocyte-specific drugs. Kidney Int. 77, 662–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Morris M. C., Deshayes S., Heitz F., Divita G. (2008) Cell-penetrating peptides: from molecular mechanisms to therapeutics. Biol. Cell 100, 201–217 [DOI] [PubMed] [Google Scholar]
- 22. Matsusaka T., Asano T., Niimura F., Kinomura M., Shimizu A., Shintani A., Pastan I., Fogo A. B., Ichikawa I. (2010) Angiotensin receptor blocker protection against podocyte-induced sclerosis is podocyte angiotensin II type 1 receptor-independent. Hypertension 55, 967–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Durvasula R. V., Shankland S. J. (2006) Podocyte injury and targeting therapy: an update. Curr. Opin. Nephrol. Hypertens. 15, 1–7 [DOI] [PubMed] [Google Scholar]
- 24. Liu H. F., Guo L. Q., Huang Y. Y., Chen K., Tao J. L., Li S. M., Chen X. W. (2010) Thiazolidinedione attenuate proteinuria and glomerulosclerosis in adriamycin-induced nephropathy rats via slit diaphragm protection. Nephrology 15, 75–83 [DOI] [PubMed] [Google Scholar]
- 25. Panchapakesan U., Chen X. M., Pollock C. A. (2005) Drug insight: Thiazolidinediones and diabetic nephropathy–relevance to renoprotection. Nat. Clin. Pract. Nephrol. 1, 33–43 [DOI] [PubMed] [Google Scholar]
- 26. Picard F., Auwerx J. (2002) PPARγ and glucose homeostasis. Annu. Rev. Nutr. 22, 167–197 [DOI] [PubMed] [Google Scholar]
- 27. Eto N., Wada T., Inagi R., Takano H., Shimizu A., Kato H., Kurihara H., Kawachi H., Shankland S. J., Fujita T., Nangaku M. (2007) Podocyte protection by darbepoetin: preservation of the cytoskeleton and nephrin expression. Kidney Int. 72, 455–463 [DOI] [PubMed] [Google Scholar]
- 28. Albarran B., To R., Stayton P. S. (2005) A TAT-streptavidin fusion protein directs uptake of biotinylated cargo into mammalian cells. Protein Eng. Des. Sel. 18, 147–152 [DOI] [PubMed] [Google Scholar]
- 29. Tünnemann G., Martin R. M., Haupt S., Patsch C., Edenhofer F., Cardoso M. C. (2006) Cargo-dependent mode of uptake and bioavailability of TAT-containing proteins and peptides in living cells. FASEB J. 20, 1775–1784 [DOI] [PubMed] [Google Scholar]
- 30. Vázquez J., Sun C., Du J., Fuentes L., Sumners C., Raizada M. K. (2003) Transduction of a functional domain of the AT1 receptor in neurons by HIV-Tat PTD. Hypertension 41, 751–756 [DOI] [PubMed] [Google Scholar]
- 31. Donnini S., Solito R., Monti M., Balduini W., Carloni S., Cimino M., Bampton E. T., Pinon L. G., Nicotera P., Thorpe P. E., Ziche M. (2009) Prevention of ischemic brain injury by treatment with the membrane penetrating apoptosis inhibitor, TAT-BH4. Cell Cycle 8, 1271–1278 [DOI] [PubMed] [Google Scholar]
- 32. Saleem M. A., O'Hare M. J., Reiser J., Coward R. J., Inward C. D., Farren T., Xing C. Y., Ni L., Mathieson P. W., Mundel P. (2002) A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J. Am. Soc. Nephrol. 13, 630–638 [DOI] [PubMed] [Google Scholar]
- 33. Shih N. Y., Li J., Karpitskii V., Nguyen A., Dustin M. L., Kanagawa O., Miner J. H., Shaw A. S. (1999) Congenital nephrotic syndrome in mice lacking CD2-associated protein. Science 286, 312–315 [DOI] [PubMed] [Google Scholar]
- 34. Del Gaizo V., Payne R. M. (2003) A novel TAT-mitochondrial signal sequence fusion protein is processed, stays in mitochondria, and crosses the placenta. Mol. Ther. 7, 720–730 [DOI] [PubMed] [Google Scholar]
- 35. Mallik L., Arif E., Sharma P., Rathore Y. S., Wong H. N., Holzman L. B., Ashish, Nihalani D. (2012) Solution structure analysis of cytoplasmic domain of podocyte protein Neph1 using small/wide angle x-ray scattering (SWAXS) J. Biol. Chem. 287, 9441–9453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hsu H. H., Hoffmann S., Endlich N., Velic A., Schwab A., Weide T., Schlatter E., Pavenstädt H. (2008) Mechanisms of angiotensin II signaling on cytoskeleton of podocytes. J. Mol. Med. 86, 1379–1394 [DOI] [PubMed] [Google Scholar]
- 37. Arif E., Kumari B., Wagner M. C., Zhou W., Holzman L. B., Nihalani D. (2013) Myo1c is an unconventional myosin required for zebrafish glomerular development. Kidney Int. 84, 1154–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hentschel D. M., Mengel M., Boehme L., Liebsch F., Albertin C., Bonventre J. V., Haller H., Schiffer M. (2007) Rapid screening of glomerular slit diaphragm integrity in larval zebrafish. Am. J. Physiol. Renal Physiol. 293, F1746–F1750 [DOI] [PubMed] [Google Scholar]
- 39. Zhang C., Willett C., Fremgen T. (2003) Zebrafish: an animal model for toxicological studies Curr. Protoc. Toxicol. Chapter 1, Unit 1.7, 10.1002/0471140856.tx0107s17 [DOI] [PubMed] [Google Scholar]
- 40. Kari G., Rodeck U., Dicker A. P. (2007) Zebrafish: an emerging model system for human disease and drug discovery. Clin. Pharmacol. Ther. 82, 70–80 [DOI] [PubMed] [Google Scholar]
- 41. Zhou W., Hildebrandt F. (2012) Inducible podocyte injury and proteinuria in transgenic zebrafish. J. Am. Soc. Nephrol. 23, 1039–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Otaki Y., Miyauchi N., Higa M., Takada A., Kuroda T., Gejyo F., Shimizu F., Kawachi H. (2008) Dissociation of NEPH1 from nephrin is involved in development of a rat model of focal segmental glomerulosclerosis. Am. J. Physiol. Renal Physiol. 295, F1376–F1387 [DOI] [PubMed] [Google Scholar]
- 43. Liu G., Kaw B., Kurfis J., Rahmanuddin S., Kanwar Y. S., Chugh S. S. (2003) Neph1 and nephrin interaction in the slit diaphragm is an important determinant of glomerular permeability. J. Clin. Invest. 112, 209–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Srivastava T., Sharma M., Yew K. H., Sharma R., Duncan R. S., Saleem M. A., McCarthy E. T., Kats A., Cudmore P. A., Alon U. S., Harrison C. J. (2013) LPS and PAN-induced podocyte injury in an in vitro model of minimal change disease: changes in TLR profile. J. Cell Commun. Signal. 7, 49–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jones N., Blasutig I. M., Eremina V., Ruston J. M., Bladt F., Li H., Huang H., Larose L., Li S. S., Takano T., Quaggin S. E., Pawson T. (2006) Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature 440, 818–823 [DOI] [PubMed] [Google Scholar]
- 46. Sawant R., Torchilin V. (2010) Intracellular transduction using cell-penetrating peptides. Mol. Biosyst. 6, 628–640 [DOI] [PubMed] [Google Scholar]
- 47. Hotchkiss R. S., McConnell K. W., Bullok K., Davis C. G., Chang K. C., Schwulst S. J., Dunne J. C., Dietz G. P., Bähr M., McDunn J. E., Karl I. E., Wagner T. H., Cobb J. P., Coopersmith C. M., Piwnica-Worms D. (2006) TAT-BH4 and TAT-Bcl-xL peptides protect against sepsis-induced lymphocyte apoptosis in vivo. J. Immunol. 176, 5471–5477 [DOI] [PubMed] [Google Scholar]
- 48. Cao G., Pei W., Ge H., Liang Q., Luo Y., Sharp F. R., Lu A., Ran R., Graham S. H., Chen J. (2002) In vivo delivery of a Bcl-xL fusion protein containing the TAT protein transduction domain protects against ischemic brain injury and neuronal apoptosis. J. Neurosci. 22, 5423–5431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Donoviel D. B., Freed D. D., Vogel H., Potter D. G., Hawkins E., Barrish J. P., Mathur B. N., Turner C. A., Geske R., Montgomery C. A., Starbuck M., Brandt M., Gupta A., Ramirez-Solis R., Zambrowicz B. P., Powell D. R. (2001) Proteinuria and perinatal lethality in mice lacking NEPH1, a novel protein with homology to NEPHRIN. Mol. Cell. Biol. 21, 4829–4836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Oh J., Reiser J., Mundel P. (2004) Dynamic (re)organization of the podocyte actin cytoskeleton in the nephrotic syndrome. Pediatr. Nephrol. 19, 130–137 [DOI] [PubMed] [Google Scholar]
- 51. Guo J. K., Menke A. L., Gubler M. C., Clarke A. R., Harrison D., Hammes A., Hastie N. D., Schedl A. (2002) WT1 is a key regulator of podocyte function: Reduced expression levels cause crescentic glomerulonephritis and mesangial sclerosis. Hum. Mol. Genet. 11, 651–659 [DOI] [PubMed] [Google Scholar]
- 52. Kramer-Zucker A. G., Wiessner S., Jensen A. M., Drummond I. A. (2005) Organization of the pronephric filtration apparatus in zebrafish requires Nephrin, Podocin and the FERM domain protein Mosaic eyes. Dev. Biol. 285, 316–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Neumann-Haefelin E., Kramer-Zucker A., Slanchev K., Hartleben B., Noutsou F., Martin K., Wanner N., Ritter A., Gödel M., Pagel P., Fu X., Müller A., Baumeister R., Walz G., Huber T. B. (2010) A model organism approach: defining the role of Neph proteins as regulators of neuron and kidney morphogenesis. Hum. Mol. Genet. 19, 2347–2359 [DOI] [PubMed] [Google Scholar]
- 54. Putaala H., Sainio K., Sariola H., Tryggvason K. (2000) Primary structure of mouse and rat nephrin cDNA and structure and expression of the mouse gene. J. Am. Soc. Nephrol. 11, 991–1001 [DOI] [PubMed] [Google Scholar]
- 55. Rantanen M., Palmén T., Pätäri A., Ahola H., Lehtonen S., Aström E., Floss T., Vauti F., Wurst W., Ruiz P., Kerjaschki D., Holthöfer H. (2002) Nephrin TRAP mice lack slit diaphragms and show fibrotic glomeruli and cystic tubular lesions. J. Am. Soc. Nephrol. 13, 1586–1594 [DOI] [PubMed] [Google Scholar]
- 56. Hamano Y., Grunkemeyer J. A., Sudhakar A., Zeisberg M., Cosgrove D., Morello R., Lee B., Sugimoto H., Kalluri R. (2002) Determinants of vascular permeability in the kidney glomerulus. J. Biol. Chem. 277, 31154–31162 [DOI] [PubMed] [Google Scholar]