

Background: Ras nanoclusters contain 6–8 Ras proteins on the plasma membrane and serve as indispensable signaling platforms for Ras-MAPK signaling.
Results: Ras membrane conformer mutants impart specific galectin-1-dependent nanoclustering responses.
Conclusion: Mutations in Ras can affect its nanoclustering response and thus allosterically effector recruitment and downstream signaling.
Significance: Disease-associated mutations that perturb Ras membrane conformers may alter signaling through nanoclustering.
Keywords: Cancer, Fluorescence Correlation Spectroscopy, Fluorescence Resonance Energy Transfer (FRET), Ras, Signaling, Fluorescence Recovery after Photobleaching (FRAP), Fluorescence-lifetime Microscopy Imaging (FLIM), Stimulated Emission Depletion (STED), Membrane, Nanocluster
Abstract
Solution structures and biochemical data have provided a wealth of mechanistic insight into Ras GTPases. However, information on how much the membrane organization of these lipid-modified proteins impacts on their signaling is still scarce. Ras proteins are organized into membrane nanoclusters, which are necessary for Ras-MAPK signaling. Using quantitative conventional and super-resolution fluorescence methods, as well as mathematical modeling, we investigated nanoclustering of H-ras helix α4 and hypervariable region mutants that have different bona fide conformations on the membrane. By following the emergence of conformer-specific nanoclusters in the plasma membrane of mammalian cells, we found that conformers impart distinct nanoclustering responses depending on the cytoplasmic levels of the nanocluster scaffold galectin-1. Computational modeling revealed that complexes containing H-ras conformers and galectin-1 affect both the number and lifetime of nanoclusters and thus determine the specific Raf effector recruitment. Our results show that mutations in Ras can affect its nanoclustering response and thus allosterically effector recruitment and downstream signaling. We postulate that cancer- and developmental disease-linked mutations that are associated with the Ras membrane conformation may exhibit so far unrecognized Ras nanoclustering and therefore signaling alterations.
Introduction
The Ras-MAPK pathway is spatiotemporally regulated on many levels (1, 2). Its central switch Ras toggles between GTP-on- and GDP-off-states, which are characterized by marked conformational differences of the so-called switch I and II regions. A wealth of structural and biochemical data of Ras have been accumulated, which describe the thermodynamics of its interactions and explain Ras activity, however, only of the soluble portion of the protein (3). Thus, the mechanistic basis for the most frequent oncogenic Ras mutations in codons 12, 13, and 61, as well as mutations in its regulators, guanine nucleotide exchange factors, which activate Ras or GTPase-activating proteins, that critically accelerate deactivation were understood (4). However, Ras proteins are dynamically membrane anchored by a farnesyl group in conjunction with palmitoyl groups or a stretch of basic amino acids that are all located at its C terminus (2, 5). Relatively little is known about the Ras reaction system in its native membranous environment. In the plasma membrane, ∼40% of Ras proteins laterally segregate into distinct nanoscopic proteolipid domains, the so-called nanoclusters. Nanoclusters are only 6–20 nm in dimension and contain 6–8 Ras proteins (6–8). Current evidence suggests that nanoclusters are immobile with lifetimes broadly estimated to be as short as microseconds or to reach up to 0.5 s (7, 9, 10).
Although nanoclustering is an intrinsic property of membrane-bound Ras polypeptides (9), its stability may be regulated by nanocluster scaffolding proteins such as galectin-1 (Gal-1)3 and -3 (10–13). Structural modeling and experimental data suggest that Gal-1 can accommodate the farnesyl moiety on the C terminus of H-ras, yet it must recognize the G-domain directly or indirectly, as it binds only to active GTP-loaded H-ras (14, 15). The exact structural mechanism, stoichiometry, and complex composition of H-ras- and Gal-1-containing nanoclusters are, however, unknown. Overexpression of Gal-1 enhances the stability of H-ras nanoclusters, which can be followed by an increased immobilization of Ras and downstream MAPK signaling (10, 11, 14), whereas its down-modulation decreases H-ras nanoclustering (12). Importantly, nanoclustering does not only lend the Ras reaction system new properties, such as signaling robustness and noise resistance, but is required for proper signaling (16).
We previously described a novel mechanism of how Ras operates on the membrane (17–19). Computational simulations of membrane-bound H-ras in combination with Förster/Fluorescence resonance energy transfer (FRET) experiments in intact mammalian cells suggested that H-ras exists in a nucleotide-dependent conformational equilibrium on the membrane that is guided by the novel switch III. Switch III is formed by the β2-β3 loop (also known as interswitch in Rab and Arf proteins (20)) in conjunction with helix α5 and exhibits less overt conformational changes than the classical switch I and II regions (21, 22). When structural simulations were carried out with GTP-bound H-ras, an unexpected conformer with a reoriented G-domain was found, which was stabilized by membrane contacts of Arg-128/Arg-135 on helix α4 (Fig. 1A, right). Conversely, when simulated with GDP-H-ras, a conformer with a more conventional membrane anchorage was observed, which was specifically stabilized by membrane contacts of residues Arg-169/Lys-170 in the hypervariable region (hvr) (Fig. 1A, left). These results suggested a GTP-dependent conformational equilibrium of H-ras on the membrane.
FIGURE 1.
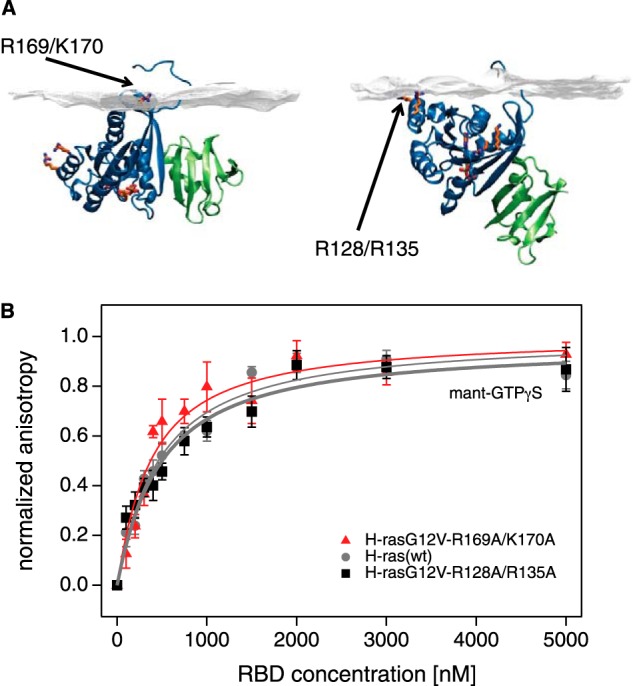
Identical binding of H-ras orientation mutants to the RBD in solution. A, structures of the c-Raf RBD (green) bound to the computationally generated models of membrane-bound H-ras (blue) in the GDP (left) and GTP (right) conformations. These conformers are stabilized by residues Arg-169/Lys-170 in the hvr and Arg-128/Arg-135 of the helix α4, respectively. The structure 1GUA was overlaid onto the previously described computational models of membrane-bound H-ras (19). Note that mutations of these residues modulate effective RBD interaction in cells, but they are distant from the effector binding site. B, fluorescence anisotropy binding assay of the RBD and H-ras wild type (residues 2–189) (gray circles), H-ras-R169A/K170A (red triangles), or H-ras-R128A/R135A (black squares) loaded with mant-GTPγS. The latter two orientation mutants, where membrane-contacting residues were neutralized, represent the two conformers shown (A, right and left, respectively). Binding curves for the mant-GTPγS condition were obtained using the global fitting function reported under “Experimental Procedures.” Data for mant-GDP (data not shown) did not suggest any significant interaction. Error bars represent the mean ± S.E. (n ≥ 4).
We subsequently showed that the Ras isoform/paralog-specific variations of the helix α4 and the hvr have a systematic impact on effector recruitment (18), suggesting that the conformational equilibrium that is modulated by the properties of helix α4 and the hvr is important for Ras paralog-specific activity. Recent work by others supported G-domain membrane contacts (23, 24) and nucleotide-dependent G-domain reorientation of membrane-bound Ras (25, 26).
Here, we investigated the mechanism by which the H-ras membrane conformers realize differential effector recruitment. Using in vitro binding of H-ras membrane conformers and the RBD, we confirmed that outside of the membrane, binding is identical. We then showed that differences in the Gal-1-dependent nanoclustering response determine effector recruitment. Using STED-FCS, FRAP, and FLIM-FRET, we followed the emergence, manifestation, and the activity of the mutant-specific H-ras signaling nanocluster. Our results significantly extend the previous model by revealing a complex, allosteric coupling between H-ras membrane conformers, Gal-1, and nanoclustering that dictates effector recruitment and signaling.
EXPERIMENTAL PROCEDURES
DNA Constructs and Molecular Cloning
Different expression plasmids for in vitro and cellular experiments were used. For protein expression in Escherichia coli, the pQE-A1 plasmid was generated from pQE-30 Xa (Qiagen, Hilden, Germany) with the following modifications; the sequence coding for the PreScission protease (GE Healthcare) recognition site and a sequence coding for the 12-amino acid A1 tag followed by a BglII restriction site were introduced between the original factor Xa recognition site and the multiple cloning site to allow for in-frame BgIII/KpnI cloning of H-ras mutant cDNAs. The PreScission protease recognition site was introduced for an efficient removal of the His tag. The A1 tag (GDSLDMLEWSLM) is an alternative acyl carrier protein tag (New England Biolabs, Ipswich, MA) and is used for selective fluorescent protein labeling in vitro (27). To construct the pQE-A1 plasmid, the sequence of N-terminally HA-tagged H-ras wild type (WT) was amplified from the pmGFP-H-ras(WT) plasmid (17) by two consecutive PCRs. First, the A1 tag (27) and the BglII restriction site were added with a forward primer, 5′-ATCGAGGGAAGGCCTCTGGAAGTTCTGTTTCAGGGTCCTGGCGATAGCCTGATATGCTGGAATG-3′ (Sigma), and next the StuI restriction and PreScission protease recognition sites were added using a second forward primer 5′-GGCGATAGCCTGGATATGCTGGAATGGAGCCTGATGGGAGATCTCGAGCTCACCACCATGTACC-3′. For both PCRs the same reverse primer, 5′-CATTTTATGTTTCAGGTTC AGGG-3′, was used. PCR products were purified and subcloned into pCRTM II-Blunt-TOPO (Invitrogen). From there, the fragment was cloned into the pQE-30 Xa vector using StuI and KpnI restriction sites to produce pQE-A1-H-ras (WT). A1-tagged H-ras-R169A/K170A and H-ras-R128A/R135A plasmids were generated from corresponding pmGFP plasmids (17) by cloning via BglII and KpnI restriction sites into the pQE-A1 vector. For the RBD of c-Raf expression plasmid, the open reading frame of the RBD was amplified by PCR with addition of appropriate restriction sites from the pmRFP-RBD plasmid (17), and the insert was cloned in-frame with the A1 tag (27) into the BglII and KpnI sites of the pQE-A1 vector. All final constructs were verified by DNA sequencing (GATC, Cologne, Germany). pOPINE2-Gal-1-His prokaryotic expression plasmid for galectin-1 was constructed in the protein expression facility of the University of Queensland, Brisbane, Australia, via in vivo ligation (28). Mammalian expression plasmids encoding N-terminally mGFP-tagged H-ras orientation mutants, pmGFP-H-rasG12V-R169A/K170A, pmGFP-H-rasG12V-R128A/R135A, and pmGFP-H-rasG12V, as well as mRFP-tagged RBD of c-Raf (pmRFP-RBD) and galectin-1 (pmRFP-Gal-1) that were used for cellular experiments, have been described previously (17, 18). Plasmids used for either knockdown (pcDNA3-asGal-1) or overexpression of untagged galectin-1 (pcDNA3-Gal-1) were described elsewhere (14). To generate SNAP-tagged orientation mutants for fluorescence correlation spectroscopy enhanced with stimulated emission depletion (STED-FCS) experiments, H-rasG12V, H-rasG12V-R169A/K170A, and H-rasG12V-R128A/R135A sequence fragments from pmGFP-H-rasG12V, pmGFP-H-rasG12V-R169A/K170A, and pmGFP-H-rasG12V-R128A/R135A were subcloned into the XhoI and PstI restriction sites of the pSNAP vector. The backbone of pSNAP vector is a pEGFP-C1 vector (Clontech) in which enhanced GFP was replaced by the SNAP tag (New England Biolabs, Ipswich, MA) between NheI and BsrGI restriction sites, while preserving the original reading frame.
Protein Expression and Purification
All A1-tagged proteins used in this study were expressed in the E. coli strain M15 (pREP4) (Qiagen, Hilden, Germany). Competent E. coli M15 (pREP4) cells were transformed with corresponding plasmids, and cultures were supplemented with ampicillin (100 μg/ml) and kanamycin (50 μg/ml) and induced with 100–500 μm isopropyl β-d-thiogalactopyranoside at an A600 of 0.6–0.8. Cells were cultured for 3 h at 37 °C, collected by centrifugation at 6000 × g for 20 min, and resuspended in 20 mm sodium phosphate, 5 mm MgCl2, 5 mm imidazole, 150 mm NaCl, pH 8.0. The bacteria were disrupted by freezing/thawing three times followed by sonication. Nondisrupted cells and large debris were removed by centrifugation at 49,000 × g for 60 min at 4 °C. The His-tagged proteins were further purified using a HiTrap nickel-nitrilotriacetic acid column (GE Healthcare) and eluted with 150–400 mm imidazole. The His tag was then removed by proteolysis with PreScission protease (GE Healthcare), according to the manufacturer's instructions (2 units of protease per 100 μg of protein overnight), and the final protein was further separated from uncleaved, His-tagged protein by nickel-nitrilotriacetic acid chromatography. The removal of salt was carried out by dialysis against 25 mm HEPES, 40 mm NaCl, 3 mm MgCl2, pH 7.2, for 16 h at 4 °C. Nucleotide exchange on the H-ras molecule was performed according to Tucker et al. (29) with minor modifications. In brief, the bound GDP present in purified samples of H-ras could be exchanged against other nucleotides (GTP and mant-GTP/GDP) in the presence of EDTA. The mant-GTPγS-loaded H-ras was obtained by titration of 100 nm mant-GTPγS (or 500 nm mant-GDP) with purified H-ras in the presence of 5 mm EDTA. The reaction was monitored by following the fluorescence anisotropy of the mant-nucleotide. Once the binding was saturated, the titration was stopped by addition of 20 mm MgCl2. Mant-GTPγS-loaded H-ras was immediately used to study the affinity between the RBD and H-ras mutants in solution. No further purifications were necessary as the nucleotide-free H-ras has a much lower affinity for the RBD (30, 31). The change in anisotropy due to a loss of mant-GTPγS from H-ras was negligible for the duration of a typical experiment (less than 1% in 2 h), as established in control experiments in the absence of the RBD (koff ∼1.5 × 10−4 s−1; data not shown).
Fluorescence Anisotropy Measurements
Fluorescence anisotropy was used to study the affinity between the RBD and H-ras mutants in solution. Typically, 100 nm mant-GTPγS or 500 nm mant-GDP-loaded H-ras were used for each assay. Steady-state anisotropy measurements were performed with a Synergy H1 hybrid fluorescence plate reader (BioTek, Winooski, VT) equipped with a polarization filter cube. Fluorescence measurements were done using a bandpass 340/30-nm excitation filter and by monitoring the emission using a bandpass 485/20-nm filter. Parallel and perpendicular excitations were used, and the emission intensities were acquired sequentially. Fluorescence anisotropies were calculated from the measured fluorescence intensities, according to Equation 1,
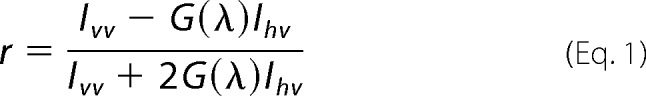 |
where r is the fluorescence anisotropy; Ivv is the fluorescence emission intensity detected with vertically polarized excitation and vertically polarized emission; Ihv is the fluorescence emission intensity detected with horizontally polarized excitation and vertically polarized emission, and G(λ) is the correction factor (G(λ) = 1 for λ between 300 and 700 nm, BioTek communication) for the Synergy H1 hybrid reader (32). Measurements were conducted at room temperature in buffer containing 25 mm HEPES, pH 7.2, 100 mm NaCl, and 3 mm MgCl2. Data processing was done using Gen5 software (version 2.01, BioTek), and Kd values were determined using GraphPad Prism 6 software (GraphPad Software Inc., La Jolla, CA). The global fitting of anisotropy data were done taking the RBD depletion into account as shown in Equation 2,
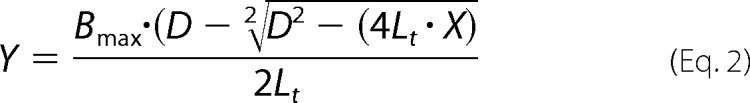 |
where Y is the change in anisotropy after the addition of the chosen concentration of RBD; Bmax is the maximum change in anisotropy due to the specific binding, and D is calculated as shown in Equation 3,
where Lt is the concentration of the fluorescently labeled H-ras (100 nm); Kd is the dissociation constant of the complex (fluorescently labeled H-ras · RBD), and X is the concentration of RBD (33, 34).
Cell Culture
BHK cells and Gal-1 knock-out mouse embryonic fibroblasts (Gal-1−/− MEFs) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS, l-glutamine, penicillin (100 units/ml), and streptomycin (100 μg/ml). They were grown to a confluency of 80% (8 × 107 cells/ml) and subcultured every 2–3 days. Gal-1−/− MEFs were a kind gift from the laboratory of Prof. Yoel Kloog (35). For Western blotting analysis and fluorescence recovery after photobleaching (FRAP) experiments, 200,000 cells were seeded in a 6-well plate (Cellstar catalog no. 657 160, Greiner Bio-one, Stonehouse, Gloucestershire, UK), and after 24 h, they were transfected using JetPRIME transfection reagent (Polyplus Transfection, New York) with the following plasmids: pmGFP-H-rasG12V, pmGFP-H-rasG12V-R169/K170A, or pmGFP-H-rasG12V-R128/135A. Plasmids were transfected alone or together with galectin-1 cDNA containing plasmids pcDNA3-asGal-1 in the case of depletion or with plasmids pmRFP-Gal-1 or pcDNA3-Gal-1 in the case of overexpression. For FLIM experiments, 100,000 cells were seeded on a 6-well plate on glass coverslips, and transfection with corresponding plasmids was done after 24 h using FuGENE 6 transfection reagent (Promega, Fitchburg, WI).
FLIM
FLIM-FRET experiments were done using a lifetime fluorescence imaging attachment (Lambert Instruments, Leutingwolde, The Netherlands) on an inverted microscope (Zeiss AXIO Observer.D1, Jena, Germany). 48 h post-transfection, cells expressing the H-ras mutants pmGFP-H-rasG12V, pmGFP-H-rasG12V-R169A/K170A, and pmGFP-H-rasG12V-R128A/R135A alone or with pmRFP-RBD, pcDNA3-Gal-1, or pmRFP-Gal-1 were fixed in 4% paraformaldehyde/PBS for 20 min and washed in PBS, and coverslips with cells were then mounted with Mowiol 4-88 (81381, Sigma) on microscope slides. Samples were excited with sinusoidally modulated (40 MHz) epi-illumination at 3 watts and 470 nm, using a temperature-stabilized multi-LED system (Lambert Instruments). Cells were imaged with a ×63 NA 1.4 oil objective using an appropriate GFP filter set (excitation, bandpass 470/40; beam splitter, FT 495; emission, bandpass 525/50). The phase and modulation fluorescence lifetimes were determined per pixel from images acquired at 12 phase settings using the manufacturer's software. Fluorescein at 0.01 mm, pH 9, was used as a lifetime reference standard (lifetime 4.0 ns). The phase lifetime of the donor (mGFP constructs) was determined for regions of interest containing 1–5 cells, coexpressing the indicated constructs. The percentage of the apparent FRET efficiency (Eapp) was calculated using the measured lifetimes of each donor-acceptor pair (τDA) and the average lifetime of the donor only (τD) samples. The formula employed for this was taken from (36) Equation 4,
 |
Confocal FLIM (TCSPC-FLIM)
Fluorescence lifetime imaging with subcellular resolution was done on a confocal microscope by means of time-correlated single photon counting (TCSPC). FLIM was performed on an inverted confocal laser-scanning microscope (Leica TCS STED, Leica Microsystems GmbH, Wetzlar, Germany) using a ×100 silicone oil immersion objective (NA 1.4). Samples were excited using a tunable Titanium:Sapphire Mai Tai laser (Spectra-Physics, Santa Clara, CA) delivering femtosecond pulses at a rate of 80 MHz with an output power of 1.9 watts for the selected wavelength of 900 nm. This wavelength was found to be optimal for the two-photon excitation of mGFP in mGFP/mRFP FRET pairs. Collection of the fluorescence was done by employing an avalanche photodiode detector and a bandpass filter to select the signal in the range 500–530 nm. Emitted photons from the donor fluorophore were collected and counted using the TimeHarp 200 (PicoQuant, Berlin, Germany) time-correlated single photon counting board. Laser power was adjusted to achieve a photon collection count of 150 photons/s. Images were acquired during 7 min in a 256 × 256 format, and the fluorescence lifetime was calculated for each pixel. Data acquisition and processing was done using the SymPhoTime software (PicoQuant).
STED-FCS Measurements in Cells
A combination of STED and FCS was used to assess the diffusion dynamics of H-ras. The STED-FCS setup used and its calibration procedure include minor changes from the setup and calibration described in detail elsewhere (37). BHK-21 cells were seeded on microscopy grade glass coverslips (diameter 18 mm, number 1 thickness) to a confluency of about 90% and grown at 37 °C in a water-saturated atmosphere of 5% CO2 in air. SNAP-tagged H-rasG12V proteins expressed in those cells were fluorescently labeled with the silicon-containing rhodamine (SiR) dye (38). The SNAP tag is a 20-kDa mutant of the DNA repair protein O6-alkylguanine-DNA alkyltransferase. O6-Alkylguanine-DNA alkyltransferase binds irreversibly and covalently benzylguanine derivatives. In the presence of a benzylguanine derivative that is bound to a synthetic probe, such as the fluorophore SiR, the SNAP-tagged protein becomes labeled with that synthetic probe (39).
During measurements cells were kept at 23 °C in Dulbecco's modified Eagle's medium (DMEM) without phenol red and buffered with 10 mm HEPES (HDMEM). We assessed the H-ras dynamics by placing the co-centered excitation and STED beams on random positions in the plasma membrane adherent to the surface and completed all measurements before any significant morphological changes of the cell could occur. Acquisition times were 10 s, which were thus 2 orders of magnitude longer than the typical transient times of H-ras proteins through the confocal observation area. The apparent diffusion coefficients for each H-ras orientation mutant were calculated from at least 7 and up to 30 measurements per given observation diameter. Repeated measurements were performed on the same cell, as well as on different cells. Results are given as the average ± S.E. of measurements acquired under identical treatment conditions.
Western Blot Analysis
Twenty four hours after transfection, cells were harvested using a buffer containing 50 mm dithiothreitol, 2% SDS, 10% glycerol, 0.1 m bromphenol blue, and 10 mm Tris-HCl, pH 6.8. Proteins from 20 μl of cell extract were first separated using SDS-PAGE (14%) and then electroblotted on a nitrocellulose membrane (GE Healthcare). After blocking with 5% milk powder in TBS + 0.1% Tween 20, membranes were probed with primary antibody for galectin-1 (sc-28248 Santa Cruz Biotechnology, Dallas, TX) diluted 1:3000 and then with secondary peroxidase-conjugated bovine anti-rabbit IgG antibody (sc-2370 Santa Cruz Biotechnology) diluted 1:3000. β-Actin or GAPDH was used for normalization. Membranes were first probed with primary antibody for β-actin (A1978 Sigma) or GAPDH (SAB1405848 Sigma) diluted 1:500,000 and then secondary peroxidase-conjugated bovine anti-mouse IgG antibody (Santa Cruz Biotechnology, sc-2954) diluted 1:3000. Secondary antibodies were detected using enhanced chemiluminescence (ECL Prime Western Blotting Detection Reagent, GE Healthcare). Quantification of the signal intensities was performed using Quantity One software (Bio-Rad). Endogenous levels of Gal-1 in BHK cells were determined from the Western blot standard curve of purified Gal-1 that was expressed from the pOPINE2-Gal-1-His plasmid.
Fluorescence Recovery after Photobleaching
FRAP was used to monitor diffusion properties of H-rasG12V mutants, under three different conditions: endogenous cellular Gal-1, mRFP-Gal-1 overexpression, and Gal-1 depletion (see under “DNA Constructs and Molecular Cloning” for details). Experiments were performed 24 h after transfection. Cells were kept in Ringer's buffer (10 mm HEPES, 10 mm glucose, 2 mm NaH2PO4·H2O, 1 mm MgCl2·6H2O, 2 mm CaCl2, 5 mm KCl, 155 mm NaCl, pH 7.2) at 22 °C. All data were acquired on a Leica TCS SP5 STED microscope (Leica Microsystems GmbH, Wetzlar, Germany) using the FRAP wizard from the manufacturer. Cells were imaged using confocal microscopy (excitation, 488 nm; detection, 497–568 nm) with bidirectional scanning at a 700 Hz frequency, 512 × 512 resolution, and ×15 zoom (pixel size 0.0201 μm). Under these conditions, we obtained a time between frames of 1.514 s. The first five frames were taken with only 10% of the laser intensity (laser nominal power is 65 milliwatts) and were used as a reference for normalization. In the next 20 frames, a square region of interest of 2.5 × 2.5 μm2 was bleached using the full laser power to reduce the fluorescence signal in the region to 30–50% of the initial intensity. To monitor the recovery, an additional 75 frames were taken with 10% of the laser intensity. Fluorescence signals were quantified in ImageJ 1.47g (National Institutes of Health, Bethesda), and recovery curves were normalized using the fluorescence intensity obtained in the first five frames, before photobleaching. The apparent characteristic half-time of recovery and the immobile fraction were determined using curve fitting analysis in IGOR Pro 6 (WaveMetrics, Tigard, OR) according to Equation 5 by Feder et al. (40).
 |
In this equation, F(t) represents the normalized fluorescence intensity; t is the time; F∞ is the normalized intensity after an infinite time; t½ is the half-time of recovery, and F0 is the fluorescence intensity at t = 0, immediately after bleaching. Immobile fractions Q were calculated using Q = (1 − F∞)/(1 − F0). For each sample ∼10 experiments were performed.
Statistical Analysis
Statistical differences between the different samples studied by FLIM, FRAP, and fluorescence anisotropy were established using analysis of variance. The analysis of variance test was complemented by Tukey's honestly significant difference test to establish which pairs of samples were significantly different. Analysis was performed using the software R version 2.15.2 (R Development Core Team, Vienna, Austria).
Simulations of Nanocluster Formation
Simulations of nanocluster formation were performed according to the schematic representation of the different reaction paths presented in Fig. 8, and using the variables and equations introduced in supplemental Tables S1 and S2. In our nanocluster model system, we had 1000 H-ras-GTP (in the following short H) molecules and observed the changes in the number of nanoclusters, depending on the number of Gal-1 molecules (in the following short G) and most importantly depending on the orientation mutant-specific HG complex stability, as defined by its lifetime. The concentration changes of the individual complexes were described using ordinary differential equations, which were solved by numerical simulation using the software R version 2.15.2 (R Development Core Team) and the ordinary differential equation solver package “deSolve” created by Karline Soetaert, Thomas Petzoldt, and R. Woodrow Setzer. Simulations were run until stability of the concentrations of all individual complexes was reached, typically 100 steps of 0.1 s. Each of these concentrations at stability was recorded as the final concentrations for the particular set of initial conditions. The initial concentration (number of molecules) for H-ras was always the same (H(t = 0) = 1000)), whereas different initial concentrations of Gal-1 (G(t = 0)) were evaluated between 0 and 15,000 with 100-unit increments. Initial concentrations for any other individual complex was always inputted as zero.
FIGURE 8.
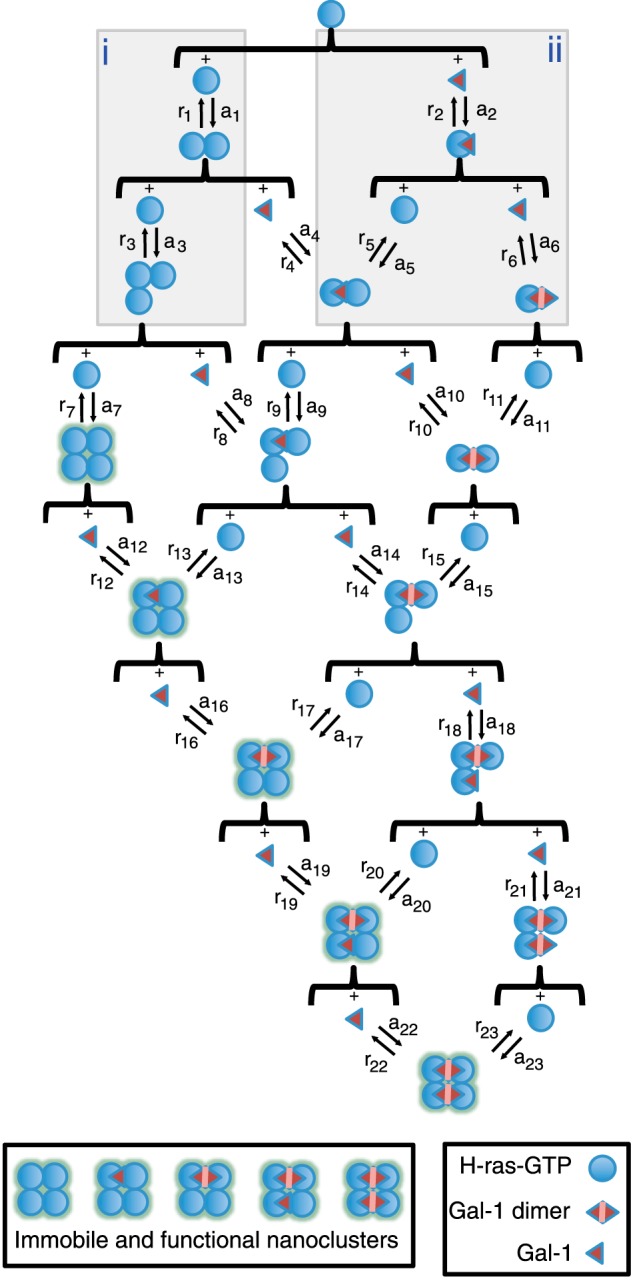
Schematic representation of the different reaction paths that were used to computationally describe nanocluster formation. Starting from single H-ras-GTP and galectin-1 (Gal-1) molecules, we present the different paths studied with our simulation leading to nanoclustering. An immobile and functional nanocluster is assumed to be reached once there are four H-ras-GTP molecules bound together, independent of the number of bound galectin-1 molecules. The presence of galectin-1 on a nanocluster increases its stability. Buildup of nanoclusters takes place in a step-by-step fashion, where single molecules are added one at a time depending on the association and dissociation rates (a# and b#, respectively) of the respective complexation reaction. Not displayed here are collapse rates (c#) for every multimer. Association, dissociation, and collapse rates vary systematically if additional molecules of H-ras-GTP (box i) or galectin-1 (box ii) are present in a given multimer. See supplemental Tables S1 and S2 and under “Experimental Procedures” for details.
To find the specific conditions for the model to reproduce the experimental data, eight fitting parameters were used as follows: dissociation rates r1 and r2; association rates a1 and a2; collapse rate c1; and parameters RasOn, GalOn, and CollGal that describe the changes of those rates as higher n-mer clusters are reached. Two of these parameters have constraints. One is due to the fact that r1 and a1 are linked through the dissociation constant of two Ras proteins Kd1, and the other one is due to the fact that r2 and a2 are linked through the dissociation constant of the HG complex Kd2. This left a total of six unconstrained parameters. Final fitting parameters are presented and described in detail in supplemental Tables S1 and S2.
Fits have been performed first for H-rasG12V using as starting point for the association, dissociation, and collapse rates, the experimentally derived formation time of a nanocluster (<1 s) (41) or the lifetime of nanoclusters (<0.5 s) (7, 41, 42). For RasOn, GalOn, and CollGal, initial values were chosen as 1, meaning identical conditions of binding at all levels of clustering (monomer, dimer, trimer, etc.). For GalOn and CollGal, values higher than 1 establish the positive cooperative effect present when more molecules of Gal-1 are involved (Fig. 8, box i). GalOn leads to higher association rates (both for binding of additional Gal-1 or H-ras), and CollGal decreases the collapse rate of higher n-mer clusters. In the case of RasOn, values higher than 1 represent negative cooperativity as RasOn decreases the association rates of H-ras at higher levels of organization in the nanocluster formation process (Fig. 8, box ii). This negative cooperativity is necessary to prevent all H-ras from being bound into nanoclusters.
To obtain the fits for mutants H-rasG12V-R169A/K170A and H-rasG12V-R128A/R135A, parameter results obtained from H-rasG12V were used as starting values. Most importantly, these were adjusted to reflect the experimentally determined relative affinities between H-ras and Gal-1, which increase in the order H-rasG12V-R169A/K170A > H-rasG12V ≥ H-rasG12V-R128A/R135A (18). Affinities between H-ras and H-ras were found in the model to be unequal and increase in the order H-rasG12V-R128A/R135A ≫ H-rasG12V ≥ H-rasG12V-R169A/K170A. Although this assumption cannot be verified experimentally, it is in line with a recent coarse grain structural computational simulation of the two H-ras conformers that are represented by our orientation mutants, showing that the two conformers interacted differently in their clusters, which is consistent with the modeled different affinities (43).
An immobile and functional nanocluster is formed, once four H molecules have assembled. This H-ras-GTP 4-mer represents the smallest nanocluster unit size found by both structural and Monte-Carlo computational simulations of Ras polypeptides (44, 45) and agrees with the minimal number of Ras found experimentally in a nanocluster (6). To lower computational efforts, we had to exclude higher cluster oligomers. Any immobile (4-mer) H-ras-GTP nanocluster will recruit RBD molecules. This incorporates evidence from single molecular data that suggest that the effector Raf is recruited only to immobilized Ras-GTP (7, 41, 42). For this recruitment step, which we do not explicitly model, we assume that the average time for an RBD to find an H-ras-GTP in a nanocluster is smaller than the lifetime of a nanocluster. Thus, the RBD can “sense” the different lifetimes of nanoclusters. Also, the lifetime of an H-ras-GTP·RBD complex has to be of similar magnitude as that of the nanocluster, to prevent that the RBD remains bound to H-ras-GTP after the nanocluster has collapsed. Both assumptions are based on published data, such as the relation of the lifetime of a nanocluster (<0.5 s) (7, 41, 42), compared with the binding time and the lifetime of the highly dynamic H-ras-GTP/RBD binding process (binding time 0.007–0.05 s; lifetime 0.1–0.5 s). These values were calculated from literature values of kon (30–35 μm−1 s−1) and koff (2–6 s−1), respectively (46–48). In the case of the binding time, we are assuming an equilibrium concentration of the H-ras-GTP·RBD complex in the range 0.5–5 μm.
RESULTS
H-ras Orientation Mutants Interact Identically with the Ras Binding Domain of the Effector c-Raf in Solution
Our previous structure of focused computational and cell biological work established different Raf effector recruitment and MAPK signaling strengths for H-rasG12V mutants that represent different conformational states of Ras on the membrane (Fig. 1A) (17, 19). H-rasG12V with mutations R169A/K170A in the hvr increases, whereas mutations R128A/R135A on helix α4 decreases effector recruitment and MAPK signaling. Because of their bona fide impact on the conformation or orientation of H-ras on the membrane, we here refer to these mutations as “orientation mutations.” H-ras orientation-mutations are distant from the effector interaction surface (Fig. 1A); therefore, the structural basis for their specific Raf effector recruitment levels and ensuing signaling changes remained unresolved.
To understand how they exhibit their specific biological activity, we first verified our previous assumption that effector binding to nonmembrane-bound H-ras is thermodynamically unaffected by the orientation mutations on helix α4 or the hvr. We therefore studied the interaction of purified H-ras orientation mutants with the Ras binding domain of c-Raf (in the following short: RBD) in solution, using a fluorescence anisotropy binding assay. Both H-ras orientation mutants showed RBD binding identical to that of WT H-ras in vitro (Fig. 1B and Table 1).
TABLE 1.
Dissociation constants of the interaction of H-ras orientation mutants with the Ras binding domain of C-Raf
In vitro dissociation constants (Kd) of the C-Raf-RBD and mant-GTPγS-bound H-ras (residues 2–189; 100 nm) with or without orientation mutations determined from fluorescence anisotropy measurements. Kd values ± S.E. are listed (n ≥ 4). Statistical analysis did not reveal any significant difference between the three mant-GTPγS-bound H-ras orientation mutants; see under “Experimental Procedures” for details about statistical analysis.
| H-ras, WT | H-ras- R169A/K170A | H-ras- R128A/R135A | |
|---|---|---|---|
| Mant-GTPγS Kd (nm) | 412 ± 71 | 327 ± 72 | 422 ± 76 |
We next wanted to confirm that this was also true in cells, by analyzing these interactions using FRET between mGFP-labeled H-ras (mGFP-H-rasG12V, donor) and mRFP-labeled RBD (mRFP-RBD, acceptor). Confocal FLIM-FRET images showed increased FRET mainly at the cell periphery consistent with the recruitment of the RBD to plasma membrane-bound active Ras (Fig. 2A, middle). We then analyzed cells that were treated with compactin, a HMG-CoA reductase inhibitor that efficiently blocks prenylation and thus membrane anchorage of Ras (49, 50). This led to a cytoplasmic redistribution of the Ras·RBD complex (Fig. 2A, right), which to our surprise was accompanied by a reduction of FRET throughout the cytoplasm. Therefore, compactin treatment allowed us to study the interaction of the H-ras orientation mutants with the RBD in solution, while remaining in the intact cellular environment that contained any putative binding modulators.
FIGURE 2.

Cytoplasmic relocalization by compactin abrogates FRET differences of H-ras orientation mutant·RBD complexes. A, confocal FLIM-FRET revealed that compactin treatment relocalized the H-rasG12V·RBD complex to the cytoplasm and led to a decrease in FRET. Imaging examples of mGFP-H-rasG12V-R169A/K170A only (left) + mRFP-RBD (center) and + mRFP-RBD and compactin treatment at a 5 μm concentration (4 h after transfection for an additional 24 h; right) are shown. Image color look-up table on the left shows fluorescence lifetimes. Schematics on the top depict the interaction situations in each experiment with the black line representing the membrane. B, interactions of pmGFP-H-rasG12V-R169A/K170A, pmGFP-H-rasG12V, or pmGFP-H-rasG12V-R128A/R135A and the acceptor mRFP-RBD in BHK cells were measured using FLIM-FRET. Treatment with 5 μm compactin was done 4 h after transfection and cells were fixed 24 h later. The apparent FRET efficiency was calculated from obtained fluorescence lifetimes. Blue vertical band represents the average ± 1 S.D. of the FRET efficiencies of conditions with compactin treatment. It would represent the baseline where there is typically no Ras signaling activity observed. Error bars correspond to the mean ± S.E., and numbers inside the bars correspond to the total number of cells imaged in each case. Three independent biological repeats were analyzed. Statistical significance was determined for the difference between compactin and non-compactin-treated samples. See under “Experimental Procedures” for details about statistical analysis (NS = nonsignificant, *, p < 0.05; **, p < 0.01; ***, p < 0.001).
We next used this assay to establish quantitative differences between the orientation mutants, using wide-field FLIM-FRET measurements. In the absence of compactin, the FRET levels of the hyperactive H-rasG12V-R169A/K170A and H-rasG12V or H-rasG12V-R128A/R135A were significantly different, as reported previously (17). Consistent with our in vitro data, compactin treatment abolished these differences (Fig. 2B), confirming that different RBD recruitment arises only if H-ras orientation mutants are membrane-bound. As already observed in the confocal FLIM data (Fig. 2A, right), the apparent FRET efficiency dropped significantly, while still being high enough to suggest that a complex containing Ras and the RBD was relocalized to the cytoplasm after compactin treatment. In conclusion, outside of the membrane H-ras orientation mutants bind identically to the c-Raf-RBD, consistent with our model in which we propose different conformational states on the membrane that dictate the downstream activity of H-ras by an unknown mechanism.
STED-FCS Experiments Reveal Specific Gal-1-dependent Transient Nanoclustering of H-ras Orientation Mutants
We hypothesized that the higher FRET efficiency of membrane-anchored H-ras orientation mutants with the RBD, as compared with the cytoplasmically redistributed complex, was due to an increase in the recruitment efficiency of Ras up-concentrated in the membrane. In the plasma membrane, Ras is organized in nanoclusters (8). We showed previously that the nanocluster scaffolding protein galectin-1 (Gal-1) interacts differently with the H-ras orientation mutants (18). Gal-1 can increase H-rasG12V nanoclustering and effector recruitment (11). This led us to investigate in detail whether orientation mutant-dependent differences in Gal-1 interaction could give rise to specific changes in H-ras nanoclustering and subsequent effector recruitment.
We reasoned that this interaction would lead to specific incorporation probabilities of H-ras into an immobile nanocluster (Fig. 3A). To assess specific membrane diffusion dynamics of different H-ras orientation mutants due to transient nanoclustering, we employed STED-FCS. Conventional FCS can be applied to study membrane dynamics by giving insight into diffusion characteristics of membrane proteins and lipids (51, 52). Nevertheless, being limited by the diffraction of light, this technique cannot resolve dynamics at the nanoscale. By combining it with STED microscopy, a method that breaks the diffraction limit of light in fluorescence microscopy, STED-FCS is able to resolve membrane dynamics with unprecedented resolution (53). In STED-FCS, the diffusion coefficient of a target molecule is measured at different scales. How the apparent diffusion coefficient varies with the area of observation defines the diffusion characteristics of the target molecule. For example, free diffusion is characterized by a constant diffusion coefficient, which is independent of the scale of observation. Transient clustering, however, is characterized by an apparent diffusion coefficient that decreases as the observation area is decreased (51, 53). Transient clustering occurs because as the observation area is reduced it gets closer and closer to the characteristic cluster size, making anomalous diffusion more and more evident.
FIGURE 3.
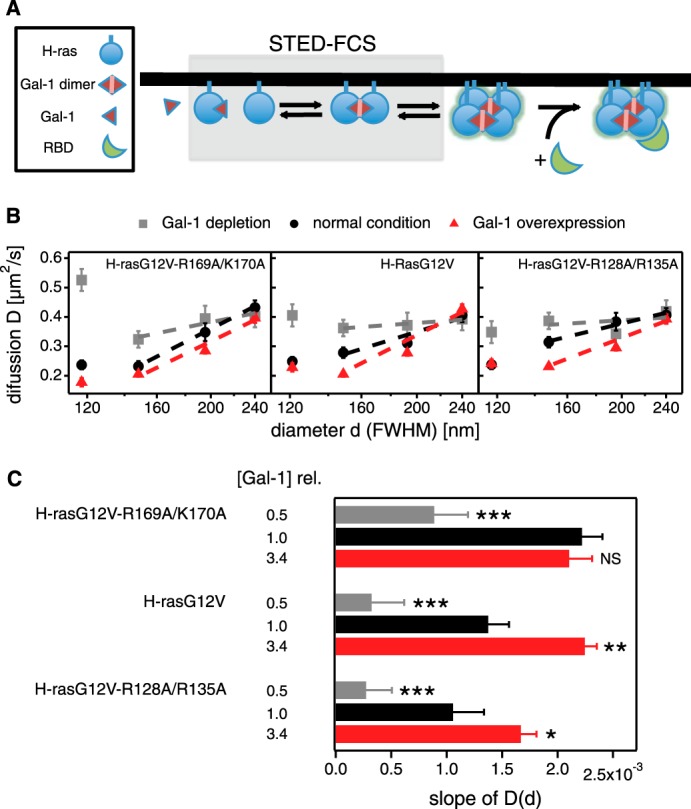
STED-FCS experiments reveal specific hindrance of H-ras orientation mutant diffusion in response to galectin-1 levels. A, schematic representation of nanocluster formation and effector recruitment. Gray box highlights the events that are studied by STED-FCS. B, diffusion of H-ras orientation mutants was studied using STED-FCS to determine their degree of hindered diffusion. The apparent diffusion coefficient D of H-rasG12V-R169A/K170A (left), H-rasG12V (center), and H-rasG12V-R128A/R135A (right) was determined in cells with Gal-1 depletion (gray squares), normal endogenous Gal-1 levels (black circles), and Gal-1 overexpression (red triangles). The values for D are displayed against the diameter d (full width at half-maximum) of the observation area to identify anomalous diffusion, which is apparent by deviations from a constant D(d) (i.e. a flat line). For each value of D, a minimum of seven and up to 30 STED-FCS autocorrelation curves were analyzed. Linear fits to D(d) for d between 150 and 240 nm are plotted. Error bars represent the mean ± S.E. C, slopes of linear fits to data shown in B. The greater the slope the more hindered the diffusion. The cellular total Gal-1 concentration relative to endogenous galectin-1 in control BHK cells ([Gal-1]rel) is displayed to the left of the data. Colors of data bars match those in B. Error bars represent 1 S.D. of the linear fits. The coefficients of determination (R2) from top to bottom are as follows: 0.84, 0.99, 0.99, 0.96, 0.94, 0.97, 0.17 (contains basically one outlier), 0.93, and 0.98. Statistical differences between treated and untreated cells were tested. See under “Experimental Procedures” for details about statistical analysis (NS, nonsignificant, *, p < 0.05; **, p < 0.01; ***, p < 0.001).
In live BHK cells, we labeled H-ras orientation mutants N-terminally with the silicon-containing rhodamine dye SiR, using SNAP tag technology (38). Then we determined H-ras mutant diffusion coefficients at various diameters of the observation area under three different conditions (Fig. 3B) as follows: normal/endogenous Gal-1 levels (10.0 ± 0.9 μm; Fig. 4, A and B); antisense-mediated Gal-1 depletion (reduction of endogenous levels to ∼50%; Fig. 4, C and D), and Gal-1 overexpression (here ∼2-fold over endogenous; Fig. 4, E–G, and Table 2).
FIGURE 4.
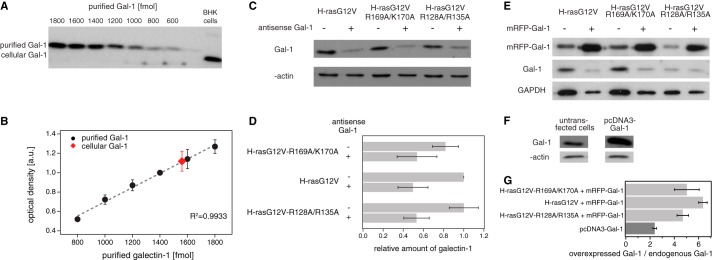
Galectin-1 level calibration in BHK cells. A, representative Western blot images displaying protein bands of serial dilutions of purified Gal-1 and of cellular Gal-1. B, standard curve showing the optical density of the band as a function of the amount of Gal-1 (n = 3). Displayed in red is the cellular Gal-1. The amount of Gal-1 in 105 untransfected BHK cells was established to be 1560.01 fmol, and the volume of an average cell was calculated to be 1564.06 μm3 (radiuses of trypsinized cells, r = 7.20 ± 0.16 μm, n = 33). Thus, we determined that the Gal-1 concentration in an average BHK cell is ∼10.0 ± 0.9 μm. C, representative Western blot of Gal-1 knockdown experiments. Indicated samples were probed with antibodies as shown on the left. D, relative amounts of Gal-1 in BHK cells expressing indicated constructs. Raw intensities were first normalized against intensities of β-actin and then plotted relative to the normalized signal of galectin-1 in the control sample (H-rasG12V). E, representative Western blots for co-overexpression using mRFP-Gal-1. Labeling shows expressed constructs on top and probed molecular species on the left. Endogenous and overexpressed Gal-1 have different molecular weights due to the mRFP label in the overexpressed Gal-1. The unspecific signal from the galectin-1-antibody that is present when mRFP-Gal-1 is overexpressed was subtracted in our quantification. F, representative Western blots for overexpression using pcDNA3-Gal-1. G, ratio between the overexpressed Gal-1 (pcDNA3-Gal-1 or pmRFP-Gal-1) and the endogenous Gal-1. In the case of overexpressed pmRFP-Gal-1, calculations were done independently for cells expressing indicated H-ras mutants. In the case of pcDNA3-Gal-1, levels were determined on BHK cells with endogenous Ras (see Table 2). Results are averaged from three independent experiments.
TABLE 2.
Quantification of overexpressed Gal-1 relative to endogenous Gal-1 in BHK cells
| H-ras mutation | Overexpressed Gal-1 | Ratio of overexpressed Gal-1/endogenous Gal-1 ± S.E. |
|---|---|---|
| H-rasG12V-R169A/K170A | mRFP-Gal-1 | 5.03 ± 1.02 |
| H-rasG12V | mRFP-Gal-1 | 6.36 ± 0.35 |
| H-rasG12V-R128A/R135A | mRFP-Gal-1 | 4.69 ± 0.47 |
| Averagea | mRFP-Gal-1 | 5.44 ± 0.45 |
| Endogenous Ras | pcDNA3-Gal-1 | 2.38 ± 0.16 |
a Average corresponds to the combination of the three mutants coexpressed with mRFP-Gal-1. ANOVA test showed that differences between mutants were statistically not significant (p = 0.33, n = 3).
With normal/endogenous Gal-1, all of the mutants had decreasing apparent diffusion coefficients (D) with decreasing observation spot size (diameter d), indicating transient nanoclustering (Fig. 3B). Overexpression of Gal-1 further enhanced the decrease of D with smaller d, while decreasing Gal-1 levels render the D(d) dependences for all orientation mutants more consistent with free diffusion, i.e. very low levels of Gal-1 do not affect transient nanoclustering sufficiently; therefore, the diffusion of Ras proteins resembles more free Brownian motion (Fig. 3, B and C).
We quantified the hindered diffusion using the slopes of the D(d) curves in the approximate linear regime with d ranging between 150 and 240 nm (Fig. 3C). Thus, we obtained three different slope profiles for the three different orientation mutants, which act like fingerprints of the three different molecular (conformational) states. H-rasG12V-R169A/K170A showed the highest slope already at normal Gal-1 levels, although this was reached by H-rasG12V only when Gal-1 was overexpressed. However, even in this Gal-1 overexpression condition, H-rasG12V-R128A/R135A did not reach the highest slope. This evolution of increasing Gal-1-dependent hindered diffusion in the order H-rasG12V-R169A/K170A > H-rasG12V > H-rasG12V-R128A/R135A is consistent with the order of membrane-dependent Gal-1 binding of these mutants (18) and their biological activity profiles (17, 19). Therefore, our STED-FCS data suggested different Gal-1-dependent nanoclustering propensities of H-ras orientation mutants.
H-rasG12V Orientation Mutants Show Specific and Gal-1 Dose-dependent Immobilization Responses in FRAP Experiments
After we had shown that Gal-1 mutant-specifically affects the emergence of nanoclusters, we next addressed whether this is also true in established nanoclusters (Fig. 5A). Single molecule data showed that active Ras is immobilized in nanoclusters (10, 54, 55), which is why the immobile fraction determined in FRAP experiments corresponds to the nanoclustered fraction of Ras (10). Hence we used a FRAP assay to detect Gal-1-dependent differences in nanoclustering of H-ras orientation mutants (Fig. 5, B and C).
FIGURE 5.
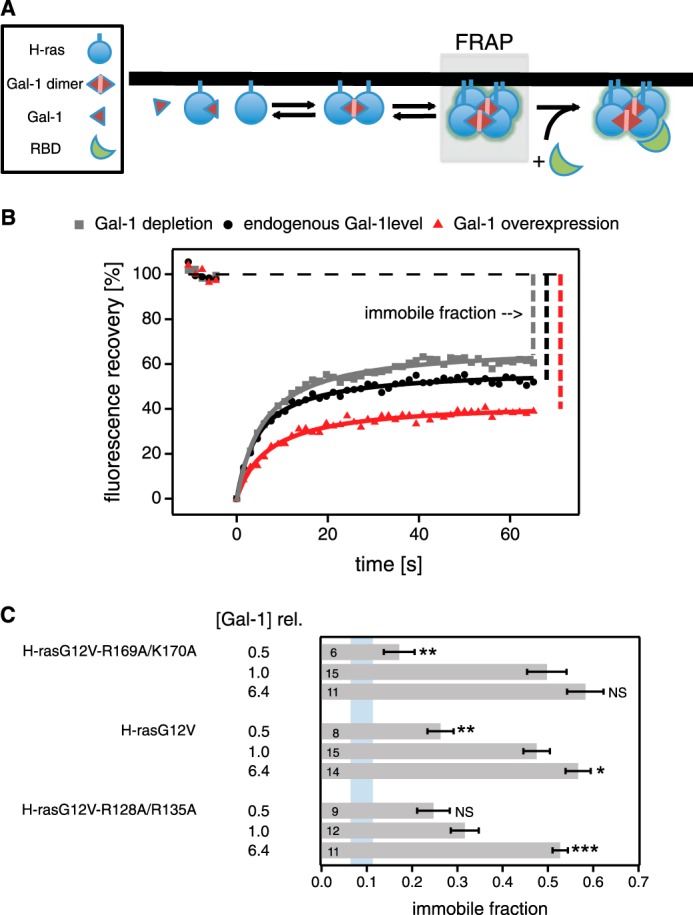
H-ras orientation mutants display different galectin-1-dependent immobilization responses in FRAP experiments. A, schematic representation of nanocluster formation and effector recruitment. Gray box highlights the events that are studied by FRAP analysis. The fraction of H-ras present in (immobile) nanoclusters is identified by the immobile fraction of the fluorescence recovery. B, FRAP analysis was performed on BHK cells transiently expressing pmGFP-H-rasG12V-R169A/K170A, pmGFP-H-rasG12V, or pmGFP-H-rasG12V-R128A/R135A. For each mutant, measurements with endogenous Gal-1 levels were compared with those with Gal-1 depletion or mRFP-Gal-1 overexpression. An example of fluorescence recovery traces for H-rasG12V-R128A/R135A is shown. Immobile fractions are indicated for illustration. Data were fitted as described under “Experimental Procedures.” C, average values with mean ± S.E. of the immobile fractions for each mutant under three different Gal-1 conditions are shown. The cellular total Gal-1 concentration relative to endogenous galectin-1 in control BHK cells ([Gal-1]rel) is displayed to the left of the data. For comparison, the blue vertical band represents the immobile fraction ± S.D. for pmGFP-H-rasG12V in Gal-1−/− MEF cells (immobile fraction = 0.08 ± 0.04; n = 9). Numbers inside the bars correspond to the total number of cells studied in each case. Statistical differences are evaluated for all mutants independently, looking for differences in the means of the immobile fractions of samples with endogenous Gal-1 levels and those either depleted of Gal-1 or overexpressing mRFP-Gal-1. See under “Experimental Procedures” for details about statistical analysis (NS, nonsignificant, *, p < 0.05; **, p < 0.01; ***, p < 0.001).
With normal Gal-1 levels, both H-rasG12V and the more active H-rasG12V-R169A/K170A have a high immobile fraction of ∼0.5 (Fig. 5C). Conversely, the less active mutant H-rasG12V-R128A/R135A was significantly (p < 0.1) less immobile. Interestingly, with high overexpression of Gal-1 (this time ∼5-fold over endogenous; Fig. 4, E and G, and Table 2) we could only achieve a maximum of ∼60% immobile H-ras, irrespective of which mutant we studied (Fig. 5B). With Gal-1 depletion, the immobile fraction of H-rasG12V and H-rasG12V-R128A/R135A significantly decreased to ∼0.26, whereas that of H-rasG12V-R169A/K170A decreased a little further down to ∼0.17 (Fig. 5C). For comparison, the immobile fraction of H-rasG12V in Gal-1 knock-out MEFs was ∼0.08. These data are in line with our STED-FCS data, which indicated that the nanoclustering behavior of H-rasG12V-R169A/K170A is more sensitive to Gal-1 levels than that of H-rasG12V or H-rasG12V-R128A/R135A. In summary, FRAP data revealed a mutation-specific dependence of the immobilization (nanoclustering) response of our H-ras mutants on cellular Gal-1 levels.
c-Raf-RBD Recruitment Is Mutant-specifically Modulated by Gal-1 Levels
If changes in nanoclustering underlie the differential recruitment of the effector fragment RBD to H-ras orientation mutants, then Gal-1 levels should also modulate this process (Fig. 6A). We therefore again performed FLIM-FRET experiments in BHK cells and measured FRET of mGFP-tagged H-ras orientation mutants when coexpressed with the mRFP-tagged RBD at increasing Gal-1 levels (Fig. 6, B and C).
FIGURE 6.

Effector recruitment to H-ras orientation mutants correlates with Gal-1-dependent nanoclustering. A, schematic representation of nanocluster formation and effector recruitment. Gray box highlights the interaction between H-ras orientation mutants in nanoclusters and the c-Raf Ras binding domain (RBD) that is studied here. B, BHK cells transiently expressing pmGFP-H-rasG12V-R169A/K170A, pmGFP-H-rasG12V, or pmGFP-H-rasG12V-R128A/R135A and the acceptor mRFP-RBD were analyzed. Three different levels of galectin-1 were tested as follows: Gal-1 depletion, with or without 5 μm compactin treatment, normal/endogenous cellular Gal-1 level, and Gal-1 overexpression. Examples of FLIM-FRET images of cells, expressing H-rasG12V-R128A/R135A under the above-described conditions. Image color look-up table on the right shows fluorescence lifetimes. C, apparent FRET efficiency was calculated from obtained fluorescence lifetimes. Blue vertical band represents the average ± S.D. of the FRET efficiencies of our three mutants with Gal-1 depletion combined with compactin treatment. The cellular total Gal-1 concentration relative to endogenous galectin-1 in control BHK cells ([Gal-1]rel) is displayed to the left of the data. Error bars correspond to the mean ± S.E., and numbers inside the bars correspond to the total number of cells imaged in each case. Three independent biological repeats were analyzed. Statistical differences between treated and untreated cells were tested; see under “Experimental Procedures” for details about statistical analysis (NS, nonsignificant, *, p < 0.05; **, p < 0.01; ***, p < 0.001).
As compared with the normal Gal-1 condition, ∼2-fold overexpression of Gal-1 (Fig. 4, F and G) led to a small but significant increase of FRET for all mutants, indicating increased recruitment of the RBD. Importantly, the relative difference between the orientation mutants remained the same, i.e. the hyperactive mutant H-rasG12V-R169A/K170A showed the highest FRET (Fig. 6C). However, knockdown of Gal-1 drastically decreased the FRET to an extent that there were no more significant differences between the H-ras orientation mutants. Additional treatment with compactin did not further decrease FRET. Thus, three Gal-1-dependent RBD recruitment responses were observed. Similar to the FRAP and STED-FCS data, we found that H-rasG12V-R169A/K170A displayed the largest change in FRET, when moving from low to normal Gal-1 levels. In addition, H-rasG12V FRET changes were smaller and identical to those of H-rasG12V-R128A/R135A. In conclusion, these FRET data confirmed our hypothesis that Gal-1-dependent differences in nanoclustering direct the H-ras orientation mutant-specific effector recruitment from the cytosol.
Computational Modeling Reveals That RBD Recruitment Depends on the Fraction and the Lifetime of H-ras Nanocluster
It was previously observed that the nanoclustered fraction of K-ras is similar to the fraction of the RBD recruited to Ras on the plasma membrane, consistent with recruitment of the RBD only to nanoclustered Ras (16). However, in addition to the fraction of Ras in nanoclusters, also the stability or lifetime of nanoclusters would affect how many Raf molecules can be recruited. For H-ras it is known that Gal-1 strongly regulates these parameters (10, 11), and it is plausible that they vary depending on the H-ras-GTP·Gal-1 complexation (18).
Because of the complexity of the nanocluster-reaction system with three interaction partners and many unknown molecular mechanistic details, we developed a computational model, which would help to understand the quantitative relationship between the fraction of nanoclustered H-ras mutants (Fig. 5C) and that of the recruited c-Raf-RBD (Fig. 6C). We incorporated current knowledge on Ras nanoclustering in our model system, which we otherwise tried to keep as simple as possible (Fig. 8 and supplemental Tables S1 and S2). In brief, we used a deterministic set of ordinary differential equations and solved them by numerical simulation. In our mathematical nanoclustering model, we assumed that an immobile nanocluster is formed step-by-step and contains four H-ras-GTP molecules (Fig. 8). The rate of nanocluster formation depends on the number of proteins of the nanocluster scaffold Gal-1 and most importantly on the known H-ras-GTP orientation mutant·Gal-1 complexation tendencies (18). This was basically observed in the STED-FCS experiments (Fig. 3), which showed that Gal-1-dependent incorporation of our H-ras variants into nanocluster follows the order H-rasG12V-R169A/K170A > H-rasG12V ≥ H-rasG12V-R128A/R135A. Finally, any immobile nanocluster was assumed to be capable of recruiting the RBD for the duration of its lifetime.
With our model, we could reproduce several important experimental observations. We could capture the ratio between H-rasG12V and endogenous Gal-1 (1:2.5 experimentally and 1:2.3 by modeling) that was required to realize the same immobile fraction (Fig. 7A). The experimentally observed lower and upper limits in the immobile (nanoclustered) fraction were also found in the simulations. For all mutants, the simulations showed an immobile fraction obeying 0 < immobile fraction < 0.1 in the absence of Gal-1. This is supported by our experimental FRAP data in Gal-1 knock-out MEFs, where we found an immobile fraction of 0.08 ± 0.04 (Fig. 5C). Also, the observed maximal immobile fraction of ∼0.6 was properly reflected in our model. Most importantly, our model evidenced that small or even no differences in the nanoclustered fraction could lead to large differences in RBD recruitment (Fig. 7, A and B).
FIGURE 7.
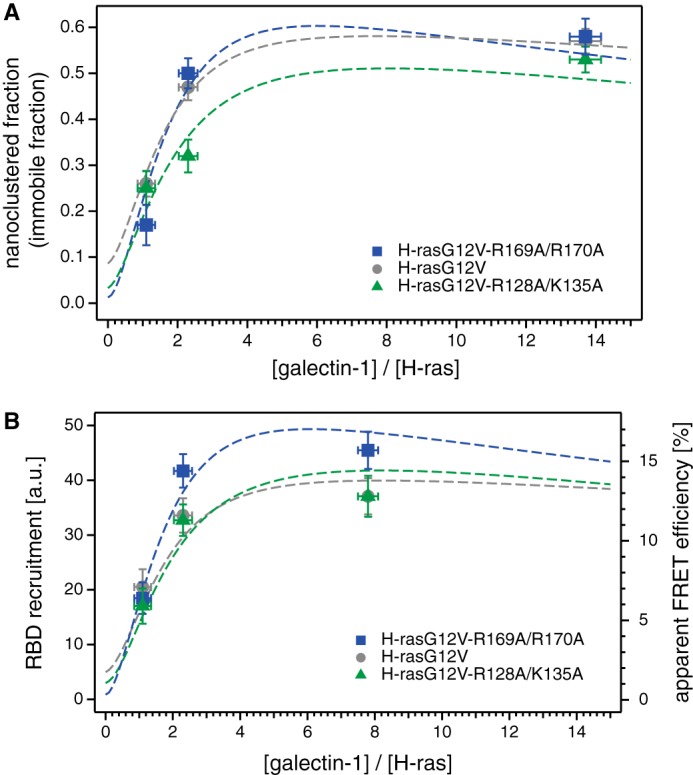
Computational modeling explains that the nanoclustered fraction and the nanocluster lifetime dictate specific RBD recruitment levels. We developed a mathematical model of nanocluster formation, which incorporates essential published experimental parameters. Thus, we could show that the RBD recruitment is determined by the nanoclustered fraction in conjunction with the nanocluster lifetime, which both depend on the H-ras orientation mutant·Gal-1 complexation. Our previous results suggest that RBD recruitment (input into the MAPK-pathway) correlates exactly with MAPK signaling output. A, nanoclustered (immobile) fractions of H-rasG12V-R169A/K170A (blue), H-rasG12V (gray), and H-rasG12V-R128A/R135A (green) displayed as a function of the ratio between the Gal-1 concentration and the H-ras concentration. Symbols correspond to the experimental data already presented in Fig. 5, and dotted lines to the simulation results. Error bars on the experimental data correspond to the mean ± S.E. of the nanocluster fraction (vertical) and of the Gal-1 concentration (horizontal). B, RBD recruitment (simulation, left axis) and apparent FRET efficiency (experimental, right axis) are displayed together as a function of the ratio between Gal-1 concentration and H-ras concentration. Symbols correspond to the experimental data already presented in Fig. 6, and dotted lines to the simulation results. Error bars on the experimental data correspond to ±S.E. of the apparent FRET efficiency (vertical) and of the Gal-1 concentration (horizontal).
This explains our observation of the up- and down-modulated nanoclustering of the orientation mutants as compared with H-rasG12V. This can be seen especially when focusing on results obtained at low Gal-1 concentrations; H-rasG12V-R128A/R135A has low binding rates with Gal-1, as compared with its rate of association with another Ras into a growing nanocluster. This results in preferential incorporation of Gal-1 not into growing nanoclusters (<4 H-ras-GTP; Fig. 8) but into intermediate, less stable (Gal-1 devoid or poor) four H-ras-GTP-containing nanocluster configurations (H4Gn, n <4; Figs. 8 and 9A). Thus, relatively few fully stabilized and few less stable intermediate nanoclusters are formed by H-rasG12V-R128A/R135A under low Gal-1 conditions (Fig. 9B). Conversely, high Gal-1 binding rates favor binding of Gal-1 to free H-rasG12V-R169A/K170A and allow for the formation of many new nanoclusters (Fig. 9B) that include Gal-1 from early steps (Fig. 8, box ii). This H-ras mutant therefore effectively bypasses intermediate nanocluster configurations and goes straight to fully stabilized (H4G4) nanoclusters (Fig. 9B). At higher Gal-1 concentrations our simulation was allowed to follow the development of intermediate nanocluster number and stability, both of which decrease due to the formation of more and more fully stabilized nanoclusters (Fig. 9, C and D).
FIGURE 9.

Individual contributions of the intermediate nanocluster configurations to the total immobile fraction and RBD recruitment. In our model, we assume that the number of Gal-1 proteins bound to a nanocluster affects its stability. The specific H-rasG12V mutant·Gal-1 complex formation tendency thus significantly determines the rate of nanocluster formation and the stability of nanoclusters. Both the fraction of H-ras in nanoclusters and the nanocluster stability (measured by the lifetime) affect the recruitment of the effector (here the Ras-binding domain of c-Raf, RBD). A, schematic representation of the five nanocluster configurations, where we define those with <4 Gal-1 as intermediate, as they do not yet possess the final number of four Gal-1 on four H-ras-GTP. Arrows pointing out of the nanoclusters correspond to rates at which a specific nanocluster configuration will cease to exist or actually change configuration. These arrows are therefore contributing to the lifetime of the particular configuration. Red arrows correspond to association rates with free Gal-1 that lead to nanoclusters with more Gal-1 incorporated. The sizes of the red arrows have been adjusted to show the increasingly higher association rates as the number of Gal-1 in a nanocluster increases. Arrows pointing into the formation of each nanocluster configuration have not been displayed, as they do not have any effect on the nanocluster lifetime. See Fig. 8 for a complete scheme of reactions. The same legend as in Fig. 8 applies. Detailed results of our nanocluster simulation for (B) depleted, (C) normal, and (D) overexpressed Gal-1 levels. In each panel we show the number of H-ras-GTP in the different nanocluster configurations (top, left), the total number of H-ras-GTP in nanoclusters (top, right), the lifetime of the different nanocluster configurations (bottom, left), and the resulting RBD recruitment (bottom, right). Total numbers of H-ras-GTP in nanoclusters is calculated by adding the number of H-ras-GTP in each nanocluster configuration. These values can be easily converted into the nanoclustered fraction as the total number of H-ras is 1000. RBD recruitment is calculated by adding the fractional contribution of each nanocluster configuration weighted by their individual lifetimes. Bars in blue correspond to data from mutant H-rasG12V-R169A/K170A, in gray to H-rasG12V, and green to H-rasG12V-R128A/R135A. Individual nanocluster configurations are shown below the bars, with description shown in A. Note that except for the lifetime-plot, the scales in B are only half that of those in C and D.
In conclusion, our computational model could capture essential quantitative features of our experimental data. It clarified that Gal-1 induced stabilization of nanoclusters and the H-ras mutant, specifically affect the fraction and lifetime of the various nanocluster configurations. This allows for specific recruitment rates of the RBD, even if the overall nanoclustered fraction is identical.
DISCUSSION
We have demonstrated H-ras membrane conformer/orientation-specific nanoclustering responses that allosterically regulate effector recruitment. We here use allosterism following the concept advanced by Kuriyan and Eisenberg (56), who proposed a broader definition that includes any structural and organizational changes that impact on the conformation or active site organization (57). Classical signaling defects of Ras activity can be readily detected by biochemical methods in solution. However, our findings would have gone unnoticed by classical biochemical approaches, due to the membrane-associated re-tuning of the Ras nanocluster system-response that leads to profound and systematic alterations of Ras/MAPK signaling strength (17, 18).
Based on our results, we suggest the following model. Different H-ras orientation mutants that shift the conformational equilibrium of H-ras on the membrane have different abilities to form specific complexes that contain the nanoclustering scaffold protein Gal-1 (18). The exact structural details of this interaction are unknown, but the fact that membrane anchorage is required for a clear discrimination of the mutants is in agreement with different conformers having different complexation abilities. However, we cannot exclude at this point that the mutations on helix α4 or in the hvr directly perturb contacts between the two proteins. Alternatively, any complex that brings H-ras and Gal-1 into close proximity, i.e. within a FRET distance of ∼5–7 nm, could be changed. The stability of these complexes determines the incorporation probability of H-ras into nanocluster and the lifetime of nanocluster, as evidenced by the combination of our experimental and simulation data. Longer lived nanoclusters can recruit more effectors to the membrane (Fig. 6), which determines the input into the downstream MAPK signaling cascade. This implies that Ras signaling scales with the number and stability of nanoclusters. Thus, our results significantly extend the previous model (10, 16, 58) by revealing a complex, allosteric coupling between H-ras membrane conformers, Gal-1, and nanoclustering that dictates effector recruitment and signaling. The distinct dependence of H-ras orientation mutant nanoclustering on cellular Gal-1 levels may explain why differences in nanoclustering were not found significant by electron microscopic analysis (17). Only our titration of cellular Gal-1 levels clearly revealed the different nanoclustering response profiles of the H-ras mutants.
It is generally conceivable that the fluorescent protein tags affect the affinity (here assessed by the total FRET) of interactions of their fusion partners. Indeed, the residual dimerization tendency of enhanced GFP-like fluorescent proteins can lead to higher FRET in the specific case when FRET due to clustering of membrane anchors in the membrane is investigated (59). Although we are using full-length proteins or protein domains, we nevertheless employed the monomeric variant of enhanced GFP, termed mGFP, to minimize the chances of tag-induced binding artifacts in the membrane context. The effect of enhanced fluorescent protein interaction on FRET in protein-protein interaction studies was recently systematically addressed (60). The objective was to increase the FRET by either enhancing the “stickiness” of fluorescent proteins or even to introduce helper interactions. Although background FRET was not increased, the maximum FRET in a given protein interaction biosensor could be significantly improved, albeit only to a small extent (e.g. 12–15% FRET efficiency in Ras/Raf1). However, in our case the fluorescent proteins are all fused in the same way to H-ras. This does not systematically affect the RBD binding, as evidenced by the congruence of in vitro binding data (Fig. 1B) and our experiments in cells with compactin treatment (Fig. 2B). Conversely, the systematic modulation of the effective RBD binding of H-ras orientation mutants that is observed when they are bound to the cellular membrane (Fig. 2B) can only be explained by the introduced mutations. Moreover, results obtained with the SNAP tag, which is not known to have a dimerization tendency, lead to comparable results as those with the mGFP tags (compare Figs. 3C and 5C). We therefore conclude that the fluorescent protein tags do not affect our conclusions on the H-ras orientation mutant-specific RBD recruitment.
Importantly, our data suggest that Ras mutations can alter signaling not only by classical alterations of its biochemical properties but also by affecting nanoclustering. This has two broad implications for Ras biology. First, we previously showed that the systematic change of the activity of the H-ras orientation mutants is representative of several if not all Ras paralogs (18). Therefore, our current results corroborate the exciting possibility that Ras paralog-specific signaling is critically affected by allosteric processes associated with membrane nanoclustering.
Second, we formerly also showed that orientation mutations on helix α4 and the hvr are linked to mutations in a novel switch III region, suggesting that the switch III is also involved in mediating membrane orientation of H-ras (17, 21, 22). Therefore, based on the results presented here, mutations in this switch III region may lead to differences in Ras nanoclustering. Intriguingly, a number of cancer and RASopathy associated mutations can be found in the β2-β3 loop and on helix α5 of Ras, which constitute the switch III (61). We postulate that some of these mutations lead to increased nanoclustering and therefore augmented signaling output of Ras in a tumor growth-promoting fashion.
In summary, our results are significant, as they suggest that the Ras nanoclustering system broadly impacts on Ras/MAPK signaling output in the physiological and pathophysiological context. Recent evidence suggests that certain approved drugs can have an impact on Ras nanoclustering (41, 62). Therefore, we propose that targeting Ras nanoclustering to block or modulate aberrant Ras functioning may have exciting therapeutic potential for cancer and other diseases.
Finally, we anticipate that our findings on H-ras will also have relevance for other membrane-anchored signaling proteins. We have provided evidence for nanoclustering of heterotrimeric G proteins, Rho and Rab small GTPases, as well as Src family kinases (50, 63, 64). Therefore, our results may represent another case, where insight into allosteric regulation of Ras by nanoclustering will be paradigmatic for other signaling proteins.
Acknowledgments
We thank Christina Oetken-Lindholm for technical help. We acknowledge Veronika Mueller and Alf Honigmann (Max Planck Institute, Göttingen, Germany) for help with the STED-FCS measurements; Vladimir Belov and Gyuzel Mitronova (Max Planck Institute, Göttingen, Germany) for synthesis of the fluorescent lipid used for STED-FCS calibration, and Stefan Hell (Max Planck Institute, Göttingen, Germany) for ongoing generous support. We thank Alemayehu Gorfe (University of Texas at Houston) for the images in Fig. 1A.
This work was supported by an Academy of Finland fellowship grant, the Sigrid Juselius Foundation, the Cancer Society of Finland, and a Marie-Curie Reintegration Grant (to D. A.).
This article was selected as a Paper of the Week.

This article contains supplemental Tables S1 and S2.
- GAL-1
- galectin-1
- BHK
- baby hamster kidney
- FCS
- fluorescence correlation spectroscopy
- FLIM
- fluorescence-lifetime imaging microscopy
- FRAP
- fluorescence recovery after photobleaching
- MEF
- mouse embryonic-fibroblast
- RBD
- ras binding domain of c-Raf
- STED
- stimulated emission depletion microscopy
- STED-FCS
- fluorescence correlation spectroscopy enhanced with stimulated emission depletion
- mant
- N-methylanthraniloyl
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- hvr
- hypervariable region
- SiR
- silicon-containing rhodamine
- mGFP
- monomeric green fluorescent protein
- mRFP
- monomeric red fluorescent protein.
REFERENCES
- 1. Mor A., Philips M. R. (2006) Compartmentalized Ras/MAPK signaling. Annu. Rev. Immunol. 24, 771–800 [DOI] [PubMed] [Google Scholar]
- 2. Rocks O., Peyker A., Bastiaens P. I. (2006) Spatio-temporal segregation of Ras signals: One ship, three anchors, many harbors. Curr. Opin. Cell Biol. 18, 351–357 [DOI] [PubMed] [Google Scholar]
- 3. Vetter I. R., Wittinghofer A. (2001) The guanine nucleotide-binding switch in three dimensions. Science 294, 1299–1304 [DOI] [PubMed] [Google Scholar]
- 4. Downward J. (2003) Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 3, 11–22 [DOI] [PubMed] [Google Scholar]
- 5. Ahearn I. M., Haigis K., Bar-Sagi D., Philips M. R. (2012) Regulating the regulator: Post-translational modification of RAS. Nat. Rev. Mol. Cell Biol. 13, 39–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Plowman S. J., Muncke C., Parton R. G., Hancock J. F. (2005) H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc. Natl. Acad. Sci. U.S.A. 102, 15500–15505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hancock J. F., Parton R. G. (2005) Ras plasma membrane signalling platforms. Biochem. J. 389, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Abankwa D., Gorfe A. A., Hancock J. F. (2007) Ras nanoclusters: Molecular structure and assembly. Semin. Cell Dev. Biol. 18, 599–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Janosi L., Li Z., Hancock J. F., Gorfe A. A. (2012) Organization, dynamics, and segregation of Ras nanoclusters in membrane domains. Proc. Natl. Acad. Sci. U.S.A. 109, 8097–8102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rotblat B., Belanis L., Liang H., Haklai R., Elad-Zefadia G., Hancock J. F., Kloog Y., Plowman S. J. (2010) H-Ras nanocluster stability regulates the magnitude of MAPK signal output. PLoS ONE 5, e11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Belanis L., Plowman S. J., Rotblat B., Hancock J. F., Kloog Y. (2008) Galectin-1 is a novel structural component and a major regulator of H-Ras nanoclusters. Mol. Biol. Cell 19, 1404–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Prior I. A., Muncke C., Parton R. G., Hancock J. F. (2003) Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J. Cell Biol. 160, 165–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shalom-Feuerstein R., Plowman S. J., Rotblat B., Ariotti N., Tian T., Hancock J. F., Kloog Y. (2008) K-ras nanoclustering is subverted by overexpression of the scaffold protein galectin-3. Cancer Res. 68, 6608–6616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Paz A., Haklai R., Elad-Sfadia G., Ballan E., Kloog Y. (2001) Galectin-1 binds oncogenic H-Ras to mediate Ras membrane anchorage and cell transformation. Oncogene 20, 7486–7493 [DOI] [PubMed] [Google Scholar]
- 15. Rotblat B., Niv H., André S., Kaltner H., Gabius H. J., Kloog Y. (2004) Galectin-1 (L11A) predicted from a computed galectin-1 farnesyl-binding pocket selectively inhibits Ras-GTP. Cancer Res. 64, 3112–3118 [DOI] [PubMed] [Google Scholar]
- 16. Tian T., Harding A., Inder K., Plowman S., Parton R. G., Hancock J. F. (2007) Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nat. Cell Biol. 9, 905–914 [DOI] [PubMed] [Google Scholar]
- 17. Abankwa D., Hanzal-Bayer M., Ariotti N., Plowman S. J., Gorfe A. A., Parton R. G., McCammon J. A., Hancock J. F. (2008) A novel switch region regulates H-ras membrane orientation and signal output. EMBO J. 27, 727–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Abankwa D., Gorfe A. A., Inder K., Hancock J. F. (2010) Ras membrane orientation and nanodomain localization generate isoform diversity. Proc. Natl. Acad. Sci. U.S.A. 107, 1130–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gorfe A. A., Hanzal-Bayer M., Abankwa D., Hancock J. F., McCammon J. A. (2007) Structure and dynamics of the full-length lipid-modified H-Ras protein in a 1,2-dimyristoylglycero-3-phosphocholine bilayer. J. Med. Chem. 50, 674–684 [DOI] [PubMed] [Google Scholar]
- 20. Amor J. C., Horton J. R., Zhu X., Wang Y., Sullards C., Ringe D., Cheng X., Kahn R. A. (2001) Structures of yeast ARF2 and ARL1: distinct roles for the N terminus in the structure and function of ARF family GTPases. J. Biol. Chem. 276, 42477–42484 [DOI] [PubMed] [Google Scholar]
- 21. Grant B. J., McCammon J. A., Gorfe A. A. (2010) Conformational selection in G-proteins: Lessons from Ras and Rho. Biophys. J. 99, L87–L89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grant B. J., Gorfe A. A., McCammon J. A. (2009) Ras conformational switching: Simulating nucleotide-dependent conformational transitions with accelerated molecular dynamics. PLoS Comput. Biol. 5, e1000325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Güldenhaupt J., Rudack T., Bachler P., Mann D., Triola G., Waldmann H., Kötting C., Gerwert K. (2012) N-Ras forms dimers at POPC membranes. Biophys. J. 103, 1585–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mazhab-Jafari M. T., Marshall C. B., Stathopulos P. B., Kobashigawa Y., Stambolic V., Kay L. E., Inagaki F., Ikura M. (2013) Membrane-dependent modulation of the mTOR activator Rheb: NMR observations of a GTPase tethered to a lipid-bilayer nanodisc. J. Am. Chem. Soc. 135, 3367–3370 [DOI] [PubMed] [Google Scholar]
- 25. Kapoor S., Triola G., Vetter I. R., Erlkamp M., Waldmann H., Winter R. (2012) Revealing conformational substrates of lipidated N-Ras protein by pressure modulation. Proc. Natl. Acad. Sci. U.S.A. 109, 460–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kapoor S., Weise K., Erlkamp M., Triola G., Waldmann H., Winter R. (2012) The role of G-domain orientation and nucleotide state on the Ras isoform-specific membrane interaction. Eur. Biophys. J. 41, 801–813 [DOI] [PubMed] [Google Scholar]
- 27. Zhou Z., Cironi P., Lin A. J., Xu Y., Hrvatin S., Golan D. E., Silver P. A., Walsh C. T., Yin J. (2007) Genetically encoded short peptide tags for orthogonal protein labeling by Sfp and AcpS phosphopantetheinyl transferases. ACS Chem. Biol. 2, 337–346 [DOI] [PubMed] [Google Scholar]
- 28. Berrow N. S., Alderton D., Sainsbury S., Nettleship J., Assenberg R., Rahman N., Stuart D. I., Owens R. J. (2007) A versatile ligation-independent cloning method suitable for high-throughput expression screening applications. Nucleic Acids Res. 35, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tucker J., Sczakiel G., Feuerstein J., John J., Goody R. S., Wittinghofer A. (1986) Expression of p21 proteins in Escherichia coli and stereochemistry of the nucleotide-binding site. EMBO J. 5, 1351–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Taylor S. J., Shalloway D. (1996) Cell cycle-dependent activation of Ras. Curr. Biol. 6, 1621–1627 [DOI] [PubMed] [Google Scholar]
- 31. Wong K. A., Russo A., Wang X., Chen Y.-J., Lavie A., O'Bryan J. P. (2012) A new dimension to Ras function: a novel role for nucleotide-free Ras in class II phosphatidylinositol 3-kinase β (PI3KC2β) regulation. PLoS ONE 7, e45360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lakowicz J. R. (2006) Principles of Fluorescence Spectroscopy, 3rd Ed, pp. 353–366, Springer, New York [Google Scholar]
- 33. Zhang X.-D., Dou S.-X., Xie P., Hu J.-S., Wang P.-Y., Xi X. G. (2006) Escherichia coli RecQ is a rapid, efficient, and monomeric helicase. J. Biol. Chem. 281, 12655–12663 [DOI] [PubMed] [Google Scholar]
- 34. LiCata V. J., Wowor A. J. (2008) Applications of fluorescence anisotropy to the study of protein-DNA interactions. Methods Cell Biol. 84, 243–262 [DOI] [PubMed] [Google Scholar]
- 35. Levy R., Biran A., Poirier F., Raz A., Kloog Y. (2011) Galectin-3 mediates cross-talk between K-Ras and Let-7c tumor suppressor microRNA. PLoS ONE 6, e27490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Majoul I., Jia Y., Duden R. (2006) Handbook of Biological Confocal Microscopy, pp. 788–808, Springer, New York [Google Scholar]
- 37. Mueller V., Ringemann C., Honigmann A., Schwarzmann G., Medda R., Leutenegger M., Polyakova S., Belov V. N., Hell S. W., Eggeling C. (2011) STED nanoscopy reveals molecular details of cholesterol- and cytoskeleton-modulated lipid interactions in living cells. Biophys. J. 101, 1651–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lukinavičius G., Umezawa K., Olivier N., Honigmann A., Yang G., Plass T., Mueller V., Reymond L., Corrêa I. R., Jr., Luo Z.-G., Schultz C., Lemke E. A., Heppenstall P., Eggeling C., Manley S., Johnsson K. (2013) A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins. Nature Chemistry 5, 132–139 [DOI] [PubMed] [Google Scholar]
- 39. Keppler A., Pick H., Arrivoli C., Vogel H., Johnsson K. (2004) Labeling of fusion proteins with synthetic fluorophores in live cells. Proc. Natl. Acad. Sci. U.S.A. 101, 9955–9959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Feder T. J., Brust-Mascher I., Slattery J. P., Baird B., Webb W. W. (1996) Constrained diffusion or immobile fraction on cell surfaces: a new interpretation. Biophys. J. 70, 2767–2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cho K.-J., Kasai R. S., Park J.-H., Chigurupati S., Heidorn S. J., van der Hoeven D., Plowman S. J., Kusumi A., Marais R., Hancock J. F. (2012) Raf inhibitors target ras spatiotemporal dynamics. Curr. Biol. 22, 945–955 [DOI] [PubMed] [Google Scholar]
- 42. Hibino K., Watanabe T. M., Kozuka J., Iwane A. H., Okada T., Kataoka T., Yanagida T., Sako Y. (2003) Single- and multiple-molecule dynamics of the signaling from H-Ras to cRaf-1 visualized on the plasma membrane of living cells. Chemphyschem 4, 748–753 [DOI] [PubMed] [Google Scholar]
- 43. Li H., Gorfe A. A. (2013) Aggregation of lipid-anchored full-length H-Ras in lipid bilayers: simulations with the MARTINI force field. PLoS ONE 8, e71018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gurry T., Kahramanoğullari O., Endres R. G. (2009) Biophysical mechanism for ras-nanocluster formation and signaling in plasma membrane. PLoS ONE 4, e6148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li Z., Janosi L., Gorfe A. A. (2012) Formation and domain partitioning of H-ras peptide nanoclusters: effects of peptide concentration and lipid composition. J. Am. Chem. Soc. 134, 17278–17285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Spoerner M., Herrmann C., Vetter I. R., Kalbitzer H. R., Wittinghofer A. (2001) Dynamic properties of the Ras switch I region and its importance for binding to effectors. Proc. Natl. Acad. Sci. U.S.A. 98, 4944–4949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kiel C., Aydin D., Serrano L. (2008) Association rate constants of ras-effector interactions are evolutionarily conserved. PLoS Comput. Biol. 4, e1000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee H.-W., Kyung T., Yoo J., Kim T., Chung C., Ryu J. Y., Lee H., Park K., Lee S., Jones W. D., Lim D.-S., Hyeon C., Heo W. D., Yoon T.-Y. (2013) Real-time single-molecule co-immunoprecipitation analyses reveal cancer-specific Ras signalling dynamics. Nat. Commun. 4, 1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nguyen U. T., Guo Z., Delon C., Wu Y., Deraeve C., Fränzel B., Bon R. S., Blankenfeldt W., Goody R. S., Waldmann H., Wolters D., Alexandrov K. (2009) Analysis of the eukaryotic prenylome by isoprenoid affinity tagging. Nat. Chem. Biol. 5, 227–235 [DOI] [PubMed] [Google Scholar]
- 50. Köhnke M., Schmitt S., Ariotti N., Piggott A. M., Parton R. G., Lacey E., Capon R. J., Alexandrov K., Abankwa D. (2012) Design and application of in vivo FRET biosensors to identify protein prenylation and nanoclustering inhibitors. Chem. Biol. 19, 866–874 [DOI] [PubMed] [Google Scholar]
- 51. He H.-T., Marguet D. (2011) Detecting nanodomains in living cell membrane by fluorescence correlation spectroscopy. Annu. Rev. Phys. Chem. 62, 417–436 [DOI] [PubMed] [Google Scholar]
- 52. Billaudeau C., Mailfert S., Trombik T., Bertaux N., Rouger V., Hamon Y., He H.-T., Marguet D. (2013) Probing the plasma membrane organization in living cells by spot variation fluorescence correlation spectroscopy. Methods Enzymol. 519, 277–302 [DOI] [PubMed] [Google Scholar]
- 53. Eggeling C., Ringemann C., Medda R., Schwarzmann G., Sandhoff K., Polyakova S., Belov V. N., Hein B., von Middendorff C., Schönle A., Hell S. W. (2009) Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature 457, 1159–1162 [DOI] [PubMed] [Google Scholar]
- 54. Murakoshi H., Iino R., Kobayashi T., Fujiwara T., Ohshima C., Yoshimura A., Kusumi A. (2004) Single-molecule imaging analysis of Ras activation in living cells. Proc. Natl. Acad. Sci. U.S.A. 101, 7317–7322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lommerse P. H., Snaar-Jagalska B. E., Spaink H. P., Schmidt T. (2005) Single-molecule diffusion measurements of H-Ras at the plasma membrane of live cells reveal microdomain localization upon activation. J. Cell Sci. 118, 1799–1809 [DOI] [PubMed] [Google Scholar]
- 56. Kuriyan J., Eisenberg D. (2007) The origin of protein interactions and allostery in colocalization. Nature 450, 983–990 [DOI] [PubMed] [Google Scholar]
- 57. Nussinov R. (2013) The spatial structure of cell signaling systems. Phys. Biol. 10, 045004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tian T., Plowman S. J., Parton R. G., Kloog Y., Hancock J. F. (2010) Mathematical modeling of K-Ras nanocluster formation on the plasma membrane. Biophys. J. 99, 534–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zacharias D. A., Violin J. D., Newton A. C., Tsien R. Y. (2002) Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296, 913–916 [DOI] [PubMed] [Google Scholar]
- 60. Grünberg R., Burnier J. V., Ferrar T., Beltran-Sastre V., Stricher F., van der Sloot A. M., Garcia-Olivas R., Mallabiabarrena A., Sanjuan X., Zimmermann T., Serrano L. (2013) Engineering of weak helper interactions for high-efficiency FRET probes. Nat. Methods 10, 1021–1027 [DOI] [PubMed] [Google Scholar]
- 61. Prior I. A., Lewis P. D., Mattos C. (2012) A comprehensive survey of Ras mutations in cancer. Cancer Res. 72, 2457–2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhou Y., Cho K.-J., Plowman S. J., Hancock J. F. (2012) Nonsteroidal anti-inflammatory drugs alter the spatiotemporal organization of Ras proteins on the plasma membrane. J. Biol. Chem. 287, 16586–16595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Abankwa D., Vogel H. (2007) A FRET map of membrane anchors suggests distinct microdomains of heterotrimeric G proteins. J. Cell Sci. 120, 2953–2962 [DOI] [PubMed] [Google Scholar]
- 64. Najumudeen A. K., Köhnke M., Solman M., Alexandrov K., Abankwa D. (2013) Cellular FRET-biosensors to detect membrane targeting inhibitors of N-myristoylated proteins. PLoS ONE 8, e66425. [DOI] [PMC free article] [PubMed] [Google Scholar]