

Background: Selenoproteins play a critical role in neuroprotection.
Results: Deletion of selenocysteine lyase (Scly) in combination with selenoprotein P (Sepp1) further aggravates the phenotype of Sepp1−/− mice, as Scly−/−Sepp1−/− mice have impaired survival and surviving mice exhibit neurological dysfunction.
Conclusion: Sepp1 and Scly work cooperatively to maintain selenoprotein function in the brain.
Significance: Deficient brain selenoprotein levels may contribute to epilepsy and neurodegeneration.
Keywords: Epilepsy, Neurodegeneration, Oxidative Stress, Selenocysteine, Selenoprotein, Inferior Colliculus, Parvalbumin Interneurons, Selenocysteine Lyase, Selenoprotein P
Abstract
Selenoproteins are a unique family of proteins, characterized by the co-translational incorporation of selenium as selenocysteine, which play key roles in antioxidant defense. Among selenoproteins, selenoprotein P (Sepp1) is particularly distinctive due to the fact that it contains multiple selenocysteine residues and has been postulated to act in selenium transport. Within the brain, Sepp1 delivers selenium to neurons by binding to the ApoER2 receptor. Upon feeding a selenium-deficient diet, mice lacking ApoER2 or Sepp1 develop severe neurological dysfunction and exhibit widespread brainstem neurodegeneration, indicating an important role for ApoER2-mediated Sepp1 uptake in normal brain function. Selenocysteine lyase (Scly) is an enzyme that plays an important role in selenium homeostasis, in that it catalyzes the decomposition of selenocysteine and allows selenium to be recycled for additional selenoprotein synthesis. We previously reported that constitutive deletion of Scly results in neurological deficits only when mice are challenged with a low selenium diet. To gain insight into the relationship between Sepp1 and Scly in selenium metabolism, we created novel transgenic mice constitutively lacking both genes (Scly−/−Sepp1−/−) and characterized the neurobehavioral phenotype. We report that deletion of Scly in conjunction with Sepp1 further aggravates the phenotype of Sepp1−/− mice, as these mice needed supraphysiological selenium supplementation to survive, and surviving mice exhibited impaired motor coordination, audiogenic seizures, and brainstem neurodegeneration. These findings provide the first in vivo evidence that Scly and Sepp1 work cooperatively to maintain selenoprotein function in the mammalian brain.
Introduction
Elevated levels of oxidative stress have been implicated as an important contributing factor to the development of a variety of neurological and cognitive impairments, including Alzheimer disease, Parkinson disease, epilepsy, and schizophrenia. One of the main lines of defense against oxidative stress is the selenoprotein family, which includes the glutathione peroxidases, thioredoxin reductases, and iodothyronine deiodinases. Selenoproteins are characterized by the co-translational incorporation of selenium in the form of the 21st amino acid, selenocysteine, at UGA codons, which otherwise serve as stop codons. These proteins play critical roles in redox reactions neutralizing oxidative stress, where selenocysteine residues act as catalytic sites. Selenoproteins are also essential for neuronal function, as neuron-specific ablation of selenoprotein synthesis in mice results in extensive neurodegeneration, seizures, and death by 2 weeks of age (1).
Among selenoproteins, selenoprotein P (Sepp1)3 has the distinctive characteristic that it contains multiple selenocysteine residues and this unique protein is thought to play a key role in selenium transport and delivery. Sepp1 is an extracellular protein that contains the majority of selenium found in plasma and has been demonstrated to deliver selenium within the brain, primarily by binding to the lipoprotein receptor, ApoER2 (2). In mice, Sepp1 deletion results in a neurological phenotype that includes deficits in motor coordination and cognition, and increased susceptibility to seizures (3, 4). Moreover, deletion of either Sepp1 or ApoER2 results in diminished brain selenium levels, and upon feeding a selenium-deficient diet both Sepp1 and ApoER2 KO mice exhibit severe neurological dysfunction and brainstem neurodegeneration (5, 6).
Upon delivery of selenium to neurons via ApoER2-mediated endocytosis of Sepp1, the selenocysteine residues must be processed prior to utilization in selenoprotein synthesis. Selenocysteine lyase (Scly) is an enzyme that plays a critical role in selenium homeostasis by decomposing selenocysteine into l-alanine and selenide (7). This breakdown of selenocysteine by Scly promotes the production of selenophosphate, which is an essential precursor for the specific selenocysteyl-tRNA(Ser)Sec used in selenoprotein translation. To investigate the physiological role of Scly in vivo, our laboratory previously generated Scly KO (Scly−/−) mice. These mice exhibited mild obesity with no discernible neurological deficits when fed a standard laboratory diet (8, 9). However, upon challenge with a low selenium diet, we observed cognitive deficits, reduced glutathione peroxidase activity in the brain, and the development of metabolic syndrome in these mice.
To gain a clearer understanding of the relationship between Sepp1 and Scly in selenium metabolism, we generated a novel transgenic mouse in which both genes were constitutively absent (Scly−/−Sepp1−/−) and subsequently characterized the neurobehavioral phenotype. Our results show that deletion of Scly in combination with Sepp1 exacerbates the neurological phenotype of Sepp1−/− mice, as we observed reduced survival, impaired motor coordination, audiogenic seizures, and brainstem neurodegeneration in selenium-supplemented Scly−/−Sepp1−/− mice. These findings demonstrate that Sepp1 and Scly have complementary roles in maintaining selenium homeostasis in the brain.
EXPERIMENTAL PROCEDURES
Animals
Scly−/−Sepp1−/− mice were generated at the University of Hawaii vivarium through three generations of selective breeding. First, Scly−/−Sepp1+/+ and Scly+/+Sepp1+/− mice were cross-bred to obtain mice heterozygous for both genes. Heterozygotes were then crossed with Scly−/−Sepp1+/+ mice to generate Scly−/−Sepp1+/− mice. Mating of Scly−/−Sepp1+/− then yielded male Scly−/−Sepp1−/− subjects. This breeding strategy was necessary, as male Sepp1−/− mice have been previously demonstrated to be infertile (10). Age and sex-matched Sepp1−/− and C57BL/6 wild-type mice were used as controls. All animals were raised on standard lab chow diet (∼0.25 ppm selenium) and selenium-supplemented water (1.0 mg/ml sodium selenite). Littermates were group housed until 12–13 weeks of age and then single housed 5 days to acclimatize before behavioral experiments. All behavioral testing was conducted with single-housed adult male mice between the ages of 12 and 18 weeks during the light cycle. Procedures and experimental protocols were approved by the University of Hawaii's Institutional Animal Care and Use Committee. All efforts were made to minimize animal discomfort and the number of animals used.
Rotorod
Motor coordination was tested using a standard rotorod apparatus. Mice were placed on a horizontal rod that began rotating at 4 rpm and increased to 40 rpm over a 5-min period. Mice were tested four times daily for two consecutive days with an intertrial interval of 30 min. The latency to fall off the rod was recorded and used to calculate an average for each day.
Open Field Test
Mice were placed in the center of an open field apparatus (50 × 50 cm) with 40-cm high opaque walls and allowed to explore for 5 min. The field was divided into 16 equal squares (12.5 × 12.5 cm), partitioning the space into 12 outer squares and 4 inner squares. Animal movement was recorded by an overhead video camera connected to a PC and analyzed by video tracking software (VideoMot 2, TSE Systems). The amount of time spent in the inner and outer squares and the total distance traveled were measured.
Elevated Plus Maze
The elevated plus maze consists of two opposing open arms and two opposing closed arms, connected by a central platform and elevated above the ground. Mice were placed in the center platform of the maze and allowed to explore for 5 min. The amount of time spent in open arms, closed arms, and center region were measured, as well as the total distance traveled during the 5 min test.
Protein Extraction and Immunoblotting
A representative sample of mice (n = 3–4 per genotype) was selected for protein analysis. Mice were asphyxiated with CO2 and their brains were quickly removed. Brains were cut in half along the longitudinal fissure, with one hemisphere being snap-frozen in liquid nitrogen for later use as a whole brain sample. For the remaining hemisphere, the cerebellum, brain stem, and hippocampus were dissected out and snap-frozen in liquid nitrogen for later analysis. Frozen tissues were lysed by sonication in CelLytic MT buffer (Sigma) containing protease inhibitors (Calbiochem) and centrifuged at 14,000 × g for 10 min at 4 °C. Supernatants were collected and the protein concentrations were measured using Bradford assay (Bio-Rad). For Western blotting, samples consisting of 40 μg of total protein were separated by 4–20% SDS-PAGE on gradient gels (Bio-Rad), transferred to Immobilon-FL polyvinylidene difluoride membranes (Millipore), and probed for 2 h at room temperature with specific antibodies. Membranes were washed with phosphate-buffered saline (PBS) containing 0.01% Tween 20 and incubated in the dark with secondary antibodies coupled to infrared fluorophores (LI-COR Biosciences). After additional washes in PBS, blots were imaged and analyzed using an Odyssey infrared imager (LI-COR Biosciences).
Glutathione Peroxidase Activity Assay
Soluble proteins/peptides were extracted as described above. Total glutathione peroxidase activity in whole brain tissue was then measured by the reduction rate of tert-butylhydroperoxide catalyzed by the samples upon the oxidation of glutathione (GSH) and reduced nicotinamide adenine dinucleotide phosphate (NADPH). A unit of enzyme activity was defined as the consumption of 1 μmol of NADPH per min, calculated from the expression (Vmax × Vt/Vs)/(0.0062 × D), using 0.0062 μm−1 cm−1 as the extinction coefficient for NADPH at 340 nm.
Thioredoxin Reductase Activity Assay
Total thioredoxin reductase activity in whole brain tissue was measured using a commercially available kit (Cayman Chemical).
Oxidative Stress Assays
Oxidative stress in whole brain tissue was assessed using commercially available kits for protein carbonyl content (Cayman Chemical) and lipid peroxidation (Oxiselect HNE Adduct Elisa Kit, Cell Biolabs).
Histology and Immunohistochemistry
Following behavioral testing, a representative sample (n = 3- 4 per genotype) of WT, Sepp1−/−, and Scly−/−Sepp1−/− mice was selected for histological analysis. Mice were deeply anesthetized (1.2% Avertin; 0.7 ml/mouse) and perused with ice-cold 0.1 m PBS followed by 4% paraformaldehyde in PBS. Brains were removed, stored in 4% paraformaldehyde for 24 h then immersed in graded solutions of sucrose (10, 20, and 30%) until they sank. Brains were cut into 40-μm sections on a cryostat and stored as free-floating sections in a cryoprotective solution (0.05 m PBS, 25% glycerol, 25% polyethylene glycol) at 4 °C. For diaminobenzidine tetrahydrochloride immunohistochemistry, free floating sections were treated with 0.3% H2O2 to inactivate endogenous peroxidases, blocked, and incubated overnight at 4 °C with the appropriate primary antibody. The following day, sections were probed with the appropriate biotinylated secondary antibody followed by incubation in avidin-biotin-peroxidase complex (Vector ABC kit, Vector Laboratories), and immunoreactivity was visualized by peroxidase detection using diaminobenzidine tetrahydrochloride (DAB Substrate kit, Vector Laboratories) as a chromogen substrate. Sections were rinsed in PBS several times then mounted on slides, dehydrated with graded solutions of ethanol followed by xylene, and coverslipped with Permount (Fischer Scientific, Fair Lawn, NJ).
Antibodies
The primary antibodies used for immunohistochemistry were rabbit anti-parvalbumin (1:10,000; Swant, Bellizona, Switzerland, PV 25), mouse anti-GAD67 (1:5000; EMD Millipore, MAB5406), rabbit anti-calretinin (1:2000; Swant, CR 7699/3H), and rabbit anti-c-Fos (1:5000; EMD Millipore Calbiochem, Darmstadt, Germany, PC38). The primary antibodies used for Western blotting were mouse anti-β-actin (1:5000; Sigma, A2228), mouse anti-Gad67 (1:5000; EMD Millipore, MAB5406), goat anti-glutathione peroxidase 1 (1:1000; R&D Systems, Minneapolis, MN, AF3798), rabbit anti-thioredoxin reductase 1 (1:1000; Novus Biologicals, Littleton, CO, NBP1-96738), rabbit anti-SelM (1:500; Sigma, HPA019601), goat anti-parvalbumin (1:500; Swant, PVG 214), and rabbit anti-calretinin (1:1000; Swant).
Stereology
Quantitative analysis of cell numbers was performed on a Zeiss microscope (Axioskop2) equipped with Micro-Brightfield Stereo Investigator software (MBF Bioscience). An optical dissector (counting box) was used to analyze and count cells. The following regions were outlined using a 5× objective with the aid of a mouse brain atlas (11) at specified coronal levels relative to bregma: somatosensory cortex (1.18 mm); medial septum (0.50 mm); CA1, CA2/3, and dentate gyrus of the dorsal hippocampus (−1.82 mm); medial geniculate nucleus (−3.40 mm); and inferior colliculus (−5.02 mm). Following contour selection, optical fractionator analysis was conducted at high magnification (×20 objective) using a 300 × 300-μm counting frame to quantify the number of positively stained cells.
Assessment of Audiogenic Seizures
Mice were placed in the open field apparatus for 5 min in a sound attenuating room equipped with a glass window for observation. The first 150 s of the test was a silent habituation period, after which an 85 dB white noise was played through a loudspeaker for the remainder of the trial. Animals were scored for pre- and post-sound incidences of wild running, seizures, and death. Trials were stopped early if animals seized continuously for more than 15 s. Average speed of locomotion during the pre-sound period and in the initial 10-s period following the onset of white noise was measured using video tracking software. We chose to measure locomotion only in the 10-s period following sound onset because several Scly−/−Sepp1−/− mice developed seizures and the test had to be terminated early. Ninety min after testing, mice were deeply anesthetized with Avertin and perfused with 4% paraformaldehyde. Brains were extracted and later processed for histological analysis.
Silver Staining
Coronally cut sections were stored in 0.1 m phosphate buffer (pH 7.4) containing 4% paraformaldehyde at 4 °C for 5 days prior to silver staining. Silver staining was performed using the FD NeurosilverTM Kit II (FD Neurotechnologies, Inc., Baltimore, MD) according to the manufacturer's instructions. Following staining, sections were mounted, dehydrated in ethanol, cleared in xylene and coverslipped with Permount.
Quantification of Gad67 Immunoreactivity and Silver Staining
Brains were processed for Gad67 immunohistochemistry as previously described. The density for Gad67 immunoreactivity in the inferior colliculus (IC) was quantified at the same coronal level (−5.02) as used for stereology. Bright-field images (×5) were captured with a digital camera mounted on a Zeiss microscope (Axioskop2), imported into ImageJ analysis software and converted to black and white images. Using a mouse brain atlas (11), contours were drawn around the IC and the adjacent lateral periaqueductal gray. The contour around the IC was selected for positive Gad67 IR, whereas the adjacent lateral periaqueductal gray was used as a background control. Mean optical density was determined as the difference between the IC and lateral periaqueductal gray density. Relative levels of silver staining were determined using a similar methodology in 4 selected regions that exhibited consistent staining in Scly−/−Sepp1−/− mice. The following regions were evaluated with the aid of a mouse brain atlas at the following specified coronal levels relative to bregma: red nucleus parvicellular part (−3.28), decussation of the superior cerebellar peduncle (−4.48), ventral nucleus of the lateral lemniscus (−4.48), and inferior colliculus (−4.96). Gray scale images were acquired using a ×2.5 objective lens and imported into ImageJ 1.37 and inverted to simulate dark-field illumination. For each image, contours were drawn around the region of interest and the optical density was measured. Also, for each image analyzed, a contour was drawn around the dorsal raphe region immediately ventral to the aqueduct of Sylvius, which served as a background control region. This region was chosen because it exhibited no staining in any of the genotypes analyzed. Mean optical density was derived by subtracting the optical density of the background control region from the optical density of the region of interest.
Statistical Analysis
Data were analyzed and plotted using Prism version 4.0c (GraphPad Software, Inc., San Diego, CA). One-way ANOVAs were used to compare data between genotypes for a single brain region for the following analyses: protein levels in Western blots, immunohistochemical staining (PV, Gad67, and CR), and average optical density of silver staining. One-way ANOVAs were also used to examine the distance traveled in open field and elevated plus maze tests. Two-way ANOVAs were used for all other experiments. Results are represented as mean ± S.E.
RESULTS
Generation of Scly−/−Sepp1−/− Mice
We generated a novel transgenic double knock-out mouse lacking genes for both Scly and Sepp1 (Scly−/−Sepp1−/−) by crossing homozygous Scly−/− mice with heterozygous Sepp1+/− mice. When fed a standard laboratory chow diet, we found that Scly−/−Sepp1−/− exhibited severe neurological dysfunction and did not survive into adulthood. Upon supplementation with sodium selenite in the drinking water, we observed increased viability of Scly−/−Sepp1−/− mice. Even when supplemented with selenium, we found that ∼60% of male Scly−/−Sepp1−/− mice failed to survive until 14 weeks of age (Fig. 1A). In contrast, we found that all female Scly−/−Sepp1−/− mice survived until adulthood when they were selenium supplemented. Due to the fact that neurological dysfunction was more pronounced in male Scly−/−Sepp1−/− mice compared with females, we chose to focus our initial behavioral and biochemical studies on selenium-supplemented male Scly−/−Sepp1−/− mice that survived into adulthood.
FIGURE 1.
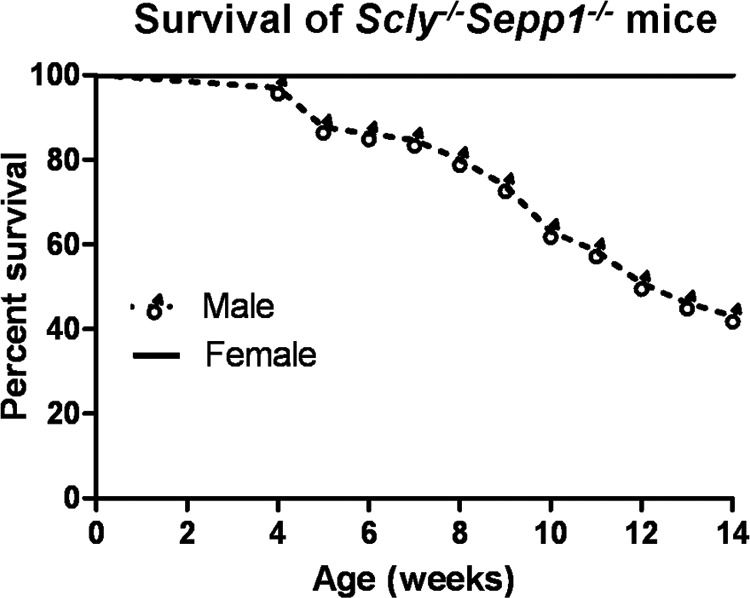
Diminished survival of male Scly−/−Sepp1−/− mice. Survival curve of male (n = 65) and female (n = 20) Scly−/−Sepp1−/− mice raised on standard lab chow and selenium-supplemented water (1.0 mg/ml of sodium selenite in drinking water).
Impaired Motor Coordination and Diminished Locomotion in Scly−/−Sepp1−/− Mice
We first conducted a series of standard neurobehavioral tests to evaluate motor coordination, locomotion, and anxiety-like behavior in Scly−/−Sepp1−/− mice (Fig. 2A). Motor coordination, as measured by the latency to fall off a rotating rod in the rotorod test, was significantly impaired in the double knock-out mice in comparison to both wild-type and Sepp1−/− mice (Fig. 2B). Two-way ANOVA revealed significant main effects for both genotype (F(2,29) = 6.6, p < 0.01) and trial day (F(1,29) = 11.6, p < 0.01). Post hoc analysis showed significant differences between WT and Scly−/−Sepp1−/− mice on both the first (p < 0.01) and second (p < 0.01) day of rotorod testing. No significant differences in anxiety-like behavior were observed between genotypes, as assessed by the amount of time spent in the center of the open field apparatus (Fig. 2C) or in the open arms of the elevated plus maze (Fig. 2E). For the open field test, no significant differences were observed in locomotion, although Scly−/−Sepp1−/− mice exhibited a non-significant trend toward less distance traveled (Fig. 2D). One-way ANOVA of locomotor activity during elevated plus maze testing revealed significant differences between genotypes (F(2,33) = 9.2, p < 0.001) and Tukey's post hoc comparisons showed that Scly−/−Sepp1−/− mice traveled significantly less distance than wild-type controls (p < 0.001, Fig. 2F). Collectively, these results demonstrate impaired motor coordination and diminished locomotion in Scly−/−Sepp1−/− mice.
FIGURE 2.

Impaired motor coordination and locomotion in Scly−/−Sepp1−/− mice. A, experimental timeline for behavioral testing. B, mean (± S.E.) latency to fall off rotorod. C, mean (±S.E.) time spent in inner and outer regions of the open field test. D, mean (± S.E.) distance traveled during the open field test. E, mean (± S.E.) time spent in the open and closed arms as well as the center during the elevated plus maze test. F, mean (± S.E.) distance traveled during the open field test. **, p < 0.01; ***, p < 0.001 compared with wild-type; #, p < 0.05 compared with Sepp1−/−. Error bars represent S.E.
Diminished Whole Brain Selenoenzyme Activity without Concomitant Increases in Markers of Oxidative Stress in Scly−/−Sepp1−/− Mice
We next evaluated the activity of selenium-dependent antioxidant enzymes by measuring glutathione peroxidase (GPx) and thioredoxin reductase (Txnrd) activity in whole brain samples. One-way ANOVA revealed significant differences in GPx activity between genotypes (F(2,10) = 31.20, p < 0.001), with Scly−/−Sepp1−/− mice having significantly lower activity than both wild-type (p < 0.001) and Sepp1−/− counterparts (p < 0.001) (Fig. 3A). For Txnrd activity, Scly−/−Sepp1−/− mice showed a trend toward reduced levels, which approached significance (F(2,14) = 3.470, p = 0.0647) (Fig. 3B). We also measured the levels of two markers of oxidative stress, protein carbonyls and 4-hydroxynonenal (4HNE), to determine whether diminished selenium-dependent antioxidant activity results in elevated oxidative stress. Surprisingly, we observed no differences in levels of protein carbonyls (Fig. 3C) or 4HNE (Fig. 3D) between genotypes for whole brain samples.
FIGURE 3.

Diminished whole brain selenoenzyme activity without concomitant increase in oxidative stress in Scly−/−Sepp1−/− mice. A, mean (± S.E.) GPx activity. B, mean (± S.E.) Txnrd activity. C, mean (± S.E.) levels of protein carbonyl content. D, mean (± S.E.) levels of 4HNE. ***, p < 0.001 compared with wild-type; ###, p < 0.001 compared with Sepp1−/−. Error bars represent S.E.
Diminished Selenoprotein Levels in Scly−/−Sepp1−/− Mice
To corroborate the findings of our GPx and Txnrd activity assays, we then used Western blotting to quantify the relative amount of three selenoproteins: Txnrd1, GPx1, and selenoprotein M (SelM). We also looked at three distinct brain regions: the hippocampus, brain stem, and cerebellum, to determine whether there were any regional differences in selenoprotein expression (Fig. 4, A-D). For both the brainstem and cerebellum, Scly−/−Sepp1−/− mice exhibited significantly reduced expression of Txnrd1, GPx1, and SelM relative to both wild-type and Sepp1−/− mice. In addition, Scly−/−Sepp1−/− mice had significantly attenuated expression of Txnrd1 and SelM in the hippocampus relative to the other genotypes. GPx1 levels in the hippocampus of Scly−/−Sepp1−/− mice were significantly lower than Sepp1−/− mice and showed a non-significant trend toward diminished expression when compared with wild-type mice. These results confirm the findings of our GPx and Txnrd activity assays and further demonstrate that combined deletion of Scly and Sepp1 results in a global reduction of selenoprotein levels within the brain.
FIGURE 4.

Decreased brain selenoprotein levels in Scly−/−Sepp1−/− mice. A, representative Western blot showing relative levels of thioredoxin reductase 1 (Txnrd1), glutathione peroxidase 1 (GPx1), and SelM in three brain regions of wild-type (WT), Sepp1−/− (KO), and Scly−/−Sepp1−/− (DKO) mice. B, mean (± S.E.) levels of Txnrd1. C, mean (± S.E.) levels of GPx1. D, mean (± S.E.) levels of SelM. Abbreviations: BS, brain stem; CB, cerebellum; HC, hippocampus. *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with wild-type; #, p < 0.05; ##, p < 0.01; ###, p < 0.001 compared with Sepp1−/−. Error bars represent S.E.
Decreased Expression of Parvalbumin and Gad67 in the Inferior Colliculus of Scly−/−Sepp1−/− Mice
Several previous studies have demonstrated an important role for selenoproteins in the development and maintenance of parvalbumin (PV) interneurons, a class of GABAergic interneurons with fast-spiking properties (1, 12, 13). PV interneurons have also been shown to be susceptible to oxidative stress (14, 15) and defective PV interneuron networks have been implicated in both epilepsy and schizophrenia (16, 17). We hypothesized that the frequent seizures we observed in Scly−/−Sepp1−/− mice resulted from dysfunctional networks of PV interneurons. To investigate this matter, we first performed Western blotting to measure levels of PV, glutamic acid decarboxylase 67 (Gad67), and calretinin (CR) in the same regions that we previously evaluated selenoprotein expression. Contrary to our expectations, the expression levels of PV, Gad67, and CR did not significantly differ between genotypes for any of the brain regions that we investigated (Fig. 5, A–D). We next performed immunohistochemistry to further probe for regional differences in PV interneuron density. For stereological analysis of PV interneuron density, we chose to focus our analyses upon four brain regions: the somatosensory cortex, medial septum, hippocampus, and inferior colliculus (Fig. 6, A and B). We selected to investigate the septohippocampal regions because of previous studies showing that Sepp1−/− mice exhibit impairments in spatial learning and hippocampal synaptic plasticity (4). The somatosensory cortex and inferior colliculus were also selected because they were found to display neurodegeneration in Sepp1−/− mice fed a selenium-deficient diet (6, 18). One-way ANOVA showed a significant difference between genotypes in the inferior colliculus (F(2,11) = 9.54, p < 0.01), with Scly−/−Sepp1−/− mice having a reduced density of PV positive cells in comparison to wild-type mice (p < 0.01). No significant differences were found in any of the other regions. All gross morphological features appeared normal in the brains of Scly−/−Sepp1−/− mice, indicating that there were no major neurodevelopmental abnormalities in these mice. We also observed reduced levels of Gad67 in the inferior colliculus of Scly−/−Sepp1−/− relative to both wild-type and Sepp1−/− mice (p < 0.001) (Fig. 6, C and D). Conversely, within the inferior colliculus, the density of CR positive interneurons did not differ between genotypes (Fig. 6, C and E). These findings demonstrate a region-specific loss of PV and Gad67 expression in the inferior colliculus of Scly−/−Sepp1−/− mice and suggest that inhibitory neurotransmission within the inferior colliculus may be diminished in these mice.
FIGURE 5.

Normal levels of Gad67, calretinin and parvalbumin in Scly−/−Sepp1−/− mice as detected by Western blotting. A, representative Western blot showing relative levels of Gad67, CR, and PV in three brain regions of wild-type (WT), Sepp1−/− (KO) and Scly−/−Sepp1−/− (DKO) mice. B, mean (± S.E.) levels of Gad67. C, mean (± S.E.) levels of CR. D, mean (± S.E.) levels of PV. Abbreviations: BS, brain stem; CB, cerebellum; HC, hippocampus. Error bars represent S.E.
FIGURE 6.

Reduced density of PV interneurons and Gad67 immunoreactivity in inferior colliculus of Scly−/−Sepp1−/− mice. A, representative images showing PV expression in the hippocampus (left column) and inferior colliculus (middle and right columns) of wild-type (top row), Sepp1−/− (middle row), and Scly−/−Sepp1−/− mice (bottom row) with higher magnification images of the inferior colliculus on the far right. B, mean (± S.E.) density of PV interneurons per mm3 (n = 3 per genotype) in brain regions investigated. C, representative images showing Gad67 (left column) and calretinin (right column) expression in the inferior colliculus of wild-type (top row), Sepp1−/− (middle row), and Scly−/−Sepp1−/− mice (bottom row). D, mean (± S.E.) level of Gad67 immunoreactivity in the inferior colliculus (n = 3 per genotype). E, mean (± S.E.) density of calretinin positive cells per mm3 in the inferior colliculus (n = 3 per genotype). Abbreviations: DG, dentate gyrus; MS, medial septum; SC, somatosensory cortex. *, p < 0.05; ***, p < 0.001 compared with wild-type; ###, p < 0.001 compared with Sepp1−/−. Error bars represent S.E.
Scly−/−Sepp1−/− Mice Are Prone to Audiogenic Seizures and Display Elevated c-Fos Immunoreactivity in the Inferior Colliculus
Several published reports have demonstrated an association between attenuated inhibitory neurotransmission in the inferior colliculus and audiogenic seizures (19, 20). To determine whether the frequent seizures observed in Scly−/−Sepp1−/− mice were audiogenic in nature, we subjected mice to a modified open field test in which mild white noise (85 dB) was played through an overhead loudspeaker during the second half of the test procedure. During the pre-sound period, the average speed of locomotion did not differ between genotypes, although Scly−/−Sepp1−/− mice exhibited a non-significant trend toward less movement. In response to the white noise, we observed significantly increased locomotion in Scly−/−Sepp1−/− mice relative to the other two genotypes (p < 0.001) (Fig. 7, A and B). Furthermore, a number of Scly−/−Sepp1−/− mice exhibited a brief bout of uncontrolled wild running behavior that was often immediately followed by a tonic-clonic seizure (Table 1). To further examine the brain regions involved in seizure activity, we looked at c-Fos expression in the brains of mice subjected to this behavioral procedure (Fig. 7, C and D). In comparison to the other two genotypes, Scly−/−Sepp1−/− mice had significantly elevated levels of c-Fos in the inferior colliculus (p < 0.05). No significant differences between genotypes were observed in any of the other brain regions that we examined. These results demonstrate that Scly−/−Sepp1−/− mice are especially prone to audiogenic seizures and suggest that the seizures result from excessive activation of the inferior colliculus in response to sound.
FIGURE 7.

Wild running behavior, audiogenic seizures, and elevated c-Fos expression in the inferior colliculus of Scly−/−Sepp1−/− mice. A, mean (± S.E.) speed prior to 85-dB white noise and in the 10-s period immediately after start of 85-dB white noise. B, images from video tracking software showing the path traveled prior to sound and during the 10-s following onset of sound. C, representative images showing c-Fos expression in the hippocampus (left column) and inferior colliculus (middle and right columns) of wild-type (top row), Sepp1−/− (middle row), and Scly−/−Sepp1−/− mice (bottom row) with higher magnification images of the inferior colliculus on the far right. D, mean (± S.E.) density of c-Fos positive cells per mm3 (n = 3 per genotype) in the brain regions investigated. Abbreviations: DG, dentate gyrus; MGN, medial geniculate nucleus; MS, medial septum; SC, somatosensory cortex; SN, substantia nigra. *, p < 0.05; ***, p < 0.001 compared with wild-type; ###, p < 0.001 compared with Sepp1−/−. Error bars represent S.E.
TABLE 1.
Summary of responses to 85 dB white noise in the open field
| Genotype | Wild running | Seizure | Death |
|---|---|---|---|
| WT | 0/10 | 0/10 | 0/10 |
| Sepp1−/− | 0/10 | 0/10 | 0/10 |
| Scly−/−Sepp1−/− | 5/7 | 4/7 | 0/7 |
Brainstem Neurodegeneration in Auditory and Motor Regions of Scly−/−Sepp1−/− Mice
We next performed silver staining to examine neurodegeneration in Scly−/−Sepp1−/− mice relative to wild-type and Sepp1−/− mice. Previous studies showed that when fed a selenium-deficient diet Sepp1−/− mice develop extensive neurodegeneration in hindbrain regions associated with auditory and motor function, and that the brain damage can largely be prevented by feeding a high selenium diet (6). We found that the brains of selenium-supplemented Scly−/−Sepp1−/− mice displayed a similar regional distribution of silver staining to that of Sepp1−/− mice fed a selenium-deficient diet (Fig. 8). Silver staining was not regularly observed in any forebrain regions of Scly−/−Sepp1−/− mice, including the hippocampus, a finding in unison with that previously reported for Sepp1−/− mice. For Scly−/−Sepp1−/− mice, consistent silver staining was observed in the red nucleus parvicellular part (Fig. 8B), decussation of the cerebellar peduncle (Fig. 8C), ventrolateral lemniscus (Fig. 8C), and the inferior colliculus (Fig. 8D). For selenium-supplemented wild-type and Sepp1−/− mice, silver staining was not consistently detectable in any of these regions. Densitometric analysis showed that the optical density of silver staining was significantly elevated in these four regions of Scly−/−Sepp1−/− mice relative to wild-type controls (Fig. 9A). We also observed some silver staining in other brain regions of Scly−/−Sepp1−/− mice, but the neurodegeneration was not consistently observed across all the mice in the group. These regions included the prerubral field (Fig. 8A), lateral portion of the medial geniculate nucleus (Fig. 8B), tectospinal tract (Fig. 8C), brachium of the inferior colliculus (Fig. 8C), superior cerebellar peduncle (Fig. 8D), inferior cerebellar peduncle (Fig. 8E), cerebellar white matter (Fig. 8E), and the gigantocellular reticular nucleus (Fig. 8E). Collectively, these findings indicate that Scly−/−Sepp1−/− mice exhibit neurodegeneration in several brainstem regions involved in auditory and motor function, and that selenium supplementation is not sufficient to prevent this damage.
FIGURE 8.

Brainstem neurodegeneration in Scly−/−Sepp1−/− mice. A–E, images of silver-stained brain sections from wild-type (left column), Sepp1−/− (middle column), and Scly−/−Sepp1−/− mice (right column). Images in the far right column are higher magnification images of silver-stained sections from Scly−/−Sepp1−/− mice. For Scly−/−Sepp1−/− mice, positive silver staining was observed in the prerubral field (A), red nucleus parvicellular part (B), medial geniculate nucleus (B), decussation of the cerebellar peduncle (C), brachium of the inferior colliculus (C), ventrolateral lemniscus (C), tectospinal tract (C), inferior colliculus (D), superior cerebellar peduncle (D), inferior cerebellar peduncle (E), cerebellar white matter (E), and gigantocellular reticular nucleus (E). Abbreviations: BIC, brachium of the inferior colliculus; cwm, cerebellar white matter; Gi, gigantocellular reticular nucleus; icp, inferior cerebellar peduncle; MGN, medial geniculate nucleus; PR, prerubral field; RPC, red nucleus parvicellular part; scp, superior cerebellar peduncle; ts, tectospinal tract; VLL, ventrolateral lemniscus; XSCP, decussation of the cerebellar peduncle. Scale bar = 200 μm.
FIGURE 9.

Quantification and schematic representation of silver staining in Scly−/−Sepp1−/− mice. A, mean (± S.E.) optical density of silver staining in the red nucleus parvicellular part, ventrolateral lemniscus, decussation of the cerebellar peduncle, and inferior colliculus. B–E, schematic representation of brain regions exhibiting silver staining in Scly−/−Sepp1−/− mice superimposed upon coronal Nissl-stained brain sections representing levels at 2.70 (B), 3.28 (C), 4.48 (D), 4.96 (E), and 5.88 mm (F) posterior to bregma. Regions shaded gray indicate areas where silver staining was quantified and observed to be significantly elevated in Scly−/−Sepp1−/− mice. Regions shaded white represent areas where silver staining was observed to a lesser degree in Scly−/−Sepp1−/− mice. Abbreviations: BIC, brachium of the inferior colliculus; cwm, cerebellar white matter; Gi, gigantocellular reticular nucleus; icp, inferior cerebellar peduncle; MGN, medial geniculate nucleus; PR, prerubral field; RPC, red nucleus parvicellular part; scp, superior cerebellar peduncle; ts, tectospinal tract; VLL, ventrolateral lemniscus; XSCP, decussation of the cerebellar peduncle. *, p < 0.05; **, p < 0.01 compared with wild-type mice; #, p < 0.05 compared with Sepp1−/− mice. Error bars represent S.E.
DISCUSSION
The aim of our study was to characterize the neurobehavioral phenotype of a novel mouse model in which the genes for selenoprotein P and selenocysteine lyase were both constitutively absent. We report that genetic ablation of both Scly and Sepp1, exacerbates the neurological phenotype of Sepp1−/− mice and that these deficits cannot be overcome by dietary selenium supplementation. Consistent with our previous study reporting that Sepp1 deletion affects males more than females (8), we note that neurological dysfunction is considerably more pronounced in male Scly−/−Sepp1−/− mice. We also found that mild white noise induced seizures in Scly−/−Sepp1−/− mice and this response coincided with increased c-fos activation and diminished expression of PV and Gad67 in the inferior colliculus. Finally, selenium-supplemented Scly−/−Sepp1−/− mice exhibited consistent silver staining in brainstem regions associated with auditory and motor function, displaying a pattern of neurodegeneration similar to that previously reported for selenium-deficient Sepp1−/− mice (6). These results clearly demonstrate that Scly and Sepp1 work cooperatively to maintain selenoprotein function in the brain.
Previous studies have documented the harmful effects of Sepp1 deletion on neurological function and also shown that this dysfunction can largely be mitigated by supraphysiological selenium supplementation in the diet or drinking water (3, 21). Furthermore, liver-specific ablation of selenoprotein synthesis does not reduce brain GPx activity or induce neurological dysfunction, indicating that the brain is not dependent on Sepp1 derived from the liver (22). These findings, along with studies showing that Sepp1 is expressed in the brain (23, 24), suggest a distinct pool of Sepp1 acts locally within the brain. Scly has been proposed to act in unison with Sepp1 to maintain brain selenoprotein levels by decomposing Sepp1-derived selenocysteine and allowing it to be recycled for selenoprotein synthesis (8, 22). This notion is supported by recent studies in mammalian cell culture demonstrating that Scly promotes selenoprotein biosynthesis when selenium is provided in the form of l-selenocysteine (25). Our data provides the first concrete evidence that Sepp1 and Scly work in tandem within the brain, as GPx activity and selenoprotein levels were significantly reduced in selenium-supplemented Scly−/−Sepp1−/− mice in comparison to wild-type and Sepp1−/− mice.
These findings also add to a growing body of evidence demonstrating the important supporting role of selenoproteins in the maintenance of fast-spiking GABAergic PV interneurons. PV interneurons produce sustained, high-frequency trains of action potentials that function to synchronize activity within populations of neurons (17). This type of interneuron has been reported to have higher levels of cytochrome oxidase c and greater numbers of mitochondria than other interneuron subpopulations (calretinin, cholecystokinin), which likely allows them to sustain high-frequency firing (26). PV interneurons also express high levels of the transcriptional co-activator peroxisome proliferator-activated receptor γ-coactivator-1α, which is critically involved in regulating energy metabolism and defense against oxidative stress (27). Moreover, mice lacking peroxisome proliferator-activated receptor γ-coactivator-1α exhibit reduced PV expression and GABAergic dysfunction (28). Several recent studies have also shown that PV interneurons have higher baseline levels of oxidative stress in comparison to other neuronal subtypes, and that excessive oxidative stress results in diminished PV expression (27, 29). Likewise, ablation of neuronal selenoprotein synthesis was observed to promote neurodegeneration and disrupt development of PV interneurons in the cortex and hippocampus (1), presumably because of diminished protection against oxidative stress. We recently reported that ApoER2 is highly expressed on PV interneurons, which suggests that Sepp1 may be preferentially targeted to this class of metabolically active interneurons to promote antioxidant defense (13).
Furthermore, we also observed reduced PV expression in the inferior colliculus of Sepp1−/− mice fed a standard laboratory diet, which corresponded with a regional increase in oxidative stress. Corroborating these results, expression of PV and Gad67 was significantly reduced in the inferior colliculus of selenium-supplemented Scly−/−Sepp1−/− mice. To our surprise, despite the fact that selenoprotein expression was greatly attenuated throughout the brain of Scly−/−Sepp1−/− mice, PV and Gad67 expression were maintained at normal levels in all other regions that we investigated. Moreover, the levels of two established markers of oxidative stress, protein carbonyl and 4HNE, also did not differ between genotypes in whole brain samples. This suggests that compensatory mechanisms may act to counterbalance the effects of diminished selenoprotein levels. These may include increased production of selenium-independent antioxidants and/or decreased cell metabolism to limit production of reactive oxygen species. However, whereas the vast majority of the Scly−/−Sepp1−/− brain appeared normal, certain discrete brain regions were selectively affected by the combined loss of Sepp1 and Scly.
The inferior colliculus plays a critical role in processing auditory information and has also been reported to be the most metabolically active region in the rodent brain (30, 31). The high metabolic rate of the inferior colliculus may make this region more vulnerable to redox imbalance and, thus, provide a plausible rationale as to why we observe a region-specific loss of PV and Gad67 expression in Scly−/−Sepp1−/− mice. ApoER2-mediated uptake of Sepp1 has an important neuroprotective role in the inferior colliculus, as both ApoER2−/− and Sepp1−/− mice exhibit neurodegeneration in the inferior colliculus when fed a selenium-deficient diet (6). Based upon these findings and our previous work showing diminished PV expression in the inferior colliculus of Sepp1−/− mice, we hypothesized that the frequent seizures we observed in Scly−/−Sepp1−/− mice were audiogenic in nature and due to reduced GABAergic inhibition in the inferior colliculus. Audiogenic seizures have been reported in several rodent strains and have been associated with alterations in GABA-mediated inhibition in the inferior colliculus (32, 33). We found that in response to mild sound (85 dB white noise), Scly−/−Sepp1−/− mice exhibited a brief bout of uncontrolled wild running that was often immediately followed by tonic-clonic seizure activity. This behavior was found to correspond with increased c-Fos activation and diminished expression of PV and Gad67 within the inferior colliculus. These findings are consistent with previous literature showing an association between decreased GABAergic inhibition in the inferior colliculus and audiogenic seizures.
Rare clinical cases of individuals with mutations in genes involved in selenoprotein function and metabolism exhibit some symptoms analogous to those observed in Scly−/−Sepp1−/− mice. Patients with mutations in the selenocysteine insertion sequence-binding protein 2 (SBP2), an RNA-binding protein required for the translation of selenoprotein mRNAs (34), were found to have deficient selenoprotein levels and to exhibit multiple deficits, including hearing loss at high frequencies, muscle weakness, gait abnormalities, and cognitive impairments (35). Similarly, mutations in the selenocysteine synthase (SepSecS) gene were reported to cause progressive cerebello-cerebral atrophy, a syndrome characterized by progressive microcephaly, mental retardation, severe spasticity, and generalized seizures (36, 37). In the clinical literature, there is also evidence for a relationship between low selenium status and epilepsy in children (38–40). The possibility exists that several of these cases may be influenced by genetic differences in genes involved in selenium metabolism. Indeed, in some cases selenium supplementation has been reported to attenuate pediatric epilepsy (39, 40). This uniquely parallels the finding that the neurological phenotype of Sepp1−/− mice can be mitigated by selenium supplementation.
In summary, our study provides the first evidence that Scly and Sepp1 work cooperatively to maintain selenoprotein function in vivo within the mammalian brain. We report that male Scly−/−Sepp1−/− mice do not survive until adulthood unless selenium supplemented, and that supplemented mice exhibit impaired motor function, audiogenic seizures, and brainstem neurodegeneration. Consistent with previous literature demonstrating that diminished GABAergic inhibition within the inferior colliculus increases susceptibility to audiogenic seizures, expression of Gad67 and PV within the inferior colliculus was greatly reduced in Scly−/−Sepp1−/− mice. These results provide further evidence of the important supporting role of selenoproteins in PV interneurons and demonstrate the collaborative roles of selenoprotein P and selenocysteine lyase in keeping this class of highly metabolic neurons healthy.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 DK47320, G12 MD007601, and G12 RR003061.
- Sepp1
- selenoprotein P
- Scly
- selenocysteine lyase
- GPx
- glutathione peroxidase
- Txnrd
- thioredoxin reductase
- 4HNE
- 4-hydroxynonenal
- SelM
- selenoprotein M
- Gad67
- glutamic acid decarboxylase 67
- PV
- parvalbumin
- CR
- calretinin
- IC
- inferior colliculus
- ANOVA
- analysis of variance
- dB
- decibel.
REFERENCES
- 1. Wirth E. K., Conrad M., Winterer J., Wozny C., Carlson B. A., Roth S., Schmitz D., Bornkamm G. W., Coppola V., Tessarollo L., Schomburg L., Köhrle J., Hatfield D. L., Schweizer U. (2010) Neuronal selenoprotein expression is required for interneuron development and prevents seizures and neurodegeneration. FASEB J. 24, 844–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Burk R. F., Hill K. E. (2009) Selenoprotein P. Expression, functions, and roles in mammals. Biochim. Biophys. Acta 1790, 1441–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hill K. E., Zhou J., McMahan W. J., Motley A. K., Burk R. F. (2004) Neurological dysfunction occurs in mice with targeted deletion of the selenoprotein P gene. J Nutr 134, 157–161 [DOI] [PubMed] [Google Scholar]
- 4. Peters M. M., Hill K. E., Burk R. F., Weeber E. J. (2006) Altered hippocampus synaptic function in selenoprotein P deficient mice. Mol. Neurodegener. 1, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Burk R. F., Hill K. E., Olson G. E., Weeber E. J., Motley A. K., Winfrey V. P., Austin L. M. (2007) Deletion of apolipoprotein E receptor-2 in mice lowers brain selenium and causes severe neurological dysfunction and death when a low-selenium diet is fed. J. Neurosci. 27, 6207–6211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Valentine W. M., Abel T. W., Hill K. E., Austin L. M., Burk R. F. (2008) Neurodegeneration in mice resulting from loss of functional selenoprotein P or its receptor apolipoprotein E receptor 2. J. Neuropathol. Exp. Neurol. 67, 68–77 [DOI] [PubMed] [Google Scholar]
- 7. Esaki N., Nakamura T., Tanaka H., Soda K. (1982) Selenocysteine lyase, a novel enzyme that specifically acts on selenocysteine. Mammalian purification and distribution and properties of pig liver enzyme. J. Biol. Chem. 257, 4386–4391 [PubMed] [Google Scholar]
- 8. Raman A. V., Pitts M. W., Seyedali A., Hashimoto A. C., Seale L. A., Bellinger F. P., Berry M. J. (2012) Absence of selenoprotein P but not selenocysteine lyase results in severe neurological dysfunction. Genes Brain Behav. 11, 601–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Seale L. A., Hashimoto A. C., Kurokawa S., Gilman C. L., Seyedali A., Bellinger F. P., Raman A. V., Berry M. J. (2012) Disruption of the selenocysteine lyase-mediated selenium recycling pathway leads to metabolic syndrome in mice. Mol. Cell. Biol. 32, 4141–4154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hill K. E., Zhou J., McMahan W. J., Motley A. K., Atkins J. F., Gesteland R. F., Burk R. F. (2003) Deletion of selenoprotein P alters distribution of selenium in the mouse. J. Biol. Chem. 278, 13640–13646 [DOI] [PubMed] [Google Scholar]
- 11. Paxinos G., Franklin K. (2004) The mouse brain in stereotaxic coordinates. 2nd Ed., Elsevier Academic Press, New York [Google Scholar]
- 12. Carlson B. A., Schweizer U., Perella C., Shrimali R. K., Feigenbaum L., Shen L., Speransky S., Floss T., Jeong S. J., Watts J., Hoffmann V., Combs G. F., Gladyshev V. N., Hatfield D. L. (2009) The selenocysteine tRNA STAF-binding region is essential for adequate selenocysteine tRNA status, selenoprotein expression, and early age survival of mice. Biochem. J. 418, 61–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pitts M. W., Raman A. V., Hashimoto A. C., Todorovic C., Nichols R. A., Berry M. J. (2012) Deletion of selenoprotein P results in impaired function of parvalbumin interneurons and alterations in fear learning and sensorimotor gating. Neuroscience 208, 58–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kinney J. W., Davis C. N., Tabarean I., Conti B., Bartfai T., Behrens M. M. (2006) A specific role for NR2A-containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons. J. Neurosci. 26, 1604–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Behrens M. M., Ali S. S., Dao D. N., Lucero J., Shekhtman G., Quick K. L., Dugan L. L. (2007) Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science 318, 1645–1647 [DOI] [PubMed] [Google Scholar]
- 16. Lewis D. A., Hashimoto T., Volk D. W. (2005) Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 6, 312–324 [DOI] [PubMed] [Google Scholar]
- 17. Freund T. F., Katona I. (2007) Perisomatic inhibition. Neuron 56, 33–42 [DOI] [PubMed] [Google Scholar]
- 18. Caito S. W., Milatovic D., Hill K. E., Aschner M., Burk R. F., Valentine W. M. (2011) Progression of neurodegeneration and morphologic changes in brains of juvenile mice with selenoprotein P deleted. Brain Res. 1398, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Faingold C. L., Gehlbach G., Caspary D. M. (1986) Decreased effectiveness of GABA-mediated inhibition of the inferior colliculus of the genetically epilepsy-prone rat. Exp. Neurol. 93, 145–159 [DOI] [PubMed] [Google Scholar]
- 20. Faingold C. L., Anderson C. A. (1991) Loss of intensity-induced inhibition in inferior colliculus neurons leads to audiogenic seizure susceptibility in behaving genetically epilepsy-prone rats. Exp. Neurol. 113, 354–363 [DOI] [PubMed] [Google Scholar]
- 21. Schweizer U., Michaelis M., Köhrle J., Schomburg L. (2004) Efficient selenium transfer from mother to offspring in selenoprotein-P-deficient mice enables dose-dependent rescue of phenotypes associated with selenium deficiency. Biochem. J. 378, 21–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schweizer U., Streckfuss F., Pelt P., Carlson B. A., Hatfield D. L., Köhrle J., Schomburg L. (2005) Hepatically derived selenoprotein P is a key factor for kidney but not brain selenium supply. Biochem. J. 386, 221–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Scharpf M., Schweizer U., Arzberger T., Roggendorf W., Schomburg L., Köhrle J. (2007) Neuronal and ependymal expression of selenoprotein P in the human brain. J. Neural Transm. 114, 877–884 [DOI] [PubMed] [Google Scholar]
- 24. Schomburg L., Schweizer U., Holtmann B., Flohé L., Sendtner M., Köhrle J. (2003) Gene disruption discloses the role of selenoprotein P in selenium delivery to target tissues. Biochem. J. 370, 397–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kurokawa S., Takehashi M., Tanaka H., Mihara H., Kurihara T., Tanaka S., Hill K., Burk R., Esaki N. (2011) Mammalian selenocysteine lyase is involved in selenoprotein biosynthesis. J. Nutr. Sci. Vitaminol. 57, 298–305 [DOI] [PubMed] [Google Scholar]
- 26. Gulyás A. I., Buzsáki G., Freund T. F., Hirase H. (2006) Populations of hippocampal inhibitory neurons express different levels of cytochrome c. Eur. J. Neurosci. 23, 2581–2594 [DOI] [PubMed] [Google Scholar]
- 27. Jiang Z., Rompala G. R., Zhang S., Cowell R. M., Nakazawa K. (2013) Social isolation exacerbates schizophrenia-like phenotypes via oxidative stress in cortical interneurons. Biol. Psychiatry 73, 1024–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lucas E. K., Markwardt S. J., Gupta S., Meador-Woodruff J. H., Lin J. D., Overstreet-Wadiche L., Cowell R. M. (2010) Parvalbumin deficiency and GABAergic dysfunction in mice lacking PGC-1α. J. Neurosci. 30, 7227–7235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cabungcal J. H., Steullet P., Kraftsik R., Cuenod M., Do K. Q. (2013) Early-life insults impair parvalbumin interneurons via oxidative stress: reversal by N-acetylcysteine. Biol. Psychiatry 73, 574–582 [DOI] [PubMed] [Google Scholar]
- 30. Jay T. M., Lucignani G., Crane A. M., Jehle J., Sokoloff L. (1988) Measurement of local cerebral blood flow with [14C]iodoantipyrine in the mouse. J. Cereb. Blood Flow Metab. 8, 121–129 [DOI] [PubMed] [Google Scholar]
- 31. Sokoloff L. (1981) Localization of functional activity in the central nervous system by measurement of glucose utilization with radioactive deoxyglucose. J. Cereb. Blood Flow Metab. 1, 7–36 [DOI] [PubMed] [Google Scholar]
- 32. Faingold C. L. (2002) Role of GABA abnormalities in the inferior colliculus pathophysiology: audiogenic seizures. Hear. Res. 168, 223–237 [DOI] [PubMed] [Google Scholar]
- 33. Ross K. C., Coleman J. R. (2000) Developmental and genetic audiogenic seizure models: behavior and biological substrates. Neurosci. Biobehav. Rev. 24, 639–653 [DOI] [PubMed] [Google Scholar]
- 34. Copeland P. R., Fletcher J. E., Carlson B. A., Hatfield D. L., Driscoll D. M. (2000) A novel RNA-binding protein, SBP2, is required for the translation of mammalian selenoprotein mRNAs. EMBO J. 19, 306–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schoenmakers E., Agostini M., Mitchell C., Schoenmakers N., Papp L., Rajanayagam O., Padidela R., Ceron-Gutierrez L., Doffinger R., Prevosto C., Luan J., Montano S., Lu J., Castanet M., Clemons N., Groeneveld M., Castets P., Karbaschi M., Aitken S., Dixon A., Williams J., Campi I., Blount M., Burton H., Muntoni F., O'Donovan D., Dean A., Warren A., Brierley C., Baguley D., Guicheney P., Fitzgerald R., Coles A., Gaston H., Todd P., Holmgren A., Khanna K. K., Cooke M., Semple R., Halsall D., Wareham N., Schwabe J., Grasso L., Beck-Peccoz P., Ogunko A., Dattani M., Gurnell M., Chatterjee K. (2010) Mutations in the selenocysteine insertion sequence-binding protein 2 gene lead to a multisystem selenoprotein deficiency disorder in humans. J. Clin. Invest. 120, 4220–4235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Agamy O., Ben Zeev B., Lev D., Marcus B., Fine D., Su D., Narkis G., Ofir R., Hoffmann C., Leshinsky-Silver E., Flusser H., Sivan S., Söll D., Lerman-Sagie T., Birk O. S. (2010) Mutations disrupting selenocysteine formation cause progressive cerebello-cerebral atrophy. Am. J. Hum. Genet. 87, 538–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schweizer U., Dehina N., Schomburg L. (2011) Disorders of selenium metabolism and selenoprotein function. Curr. Opin. Pediatr. 23, 429–435 [DOI] [PubMed] [Google Scholar]
- 38. Ashrafi M. R., Shams S., Nouri M., Mohseni M., Shabanian R., Yekaninejad M. S., Chegini N., Khodadad A., Safaralizadeh R. (2007) A probable causative factor for an old problem: selenium and glutathione peroxidase appear to play important role in epilepsy pathogenesis. Epilepsia 48, 1750–1755 [DOI] [PubMed] [Google Scholar]
- 39. Ramaekers V. T., Calomme M., Vanden Berghe D., Makropoulos W. (1994) Selenium deficiency triggering intractable seizures. Neuropediatrics 25, 217–223 [DOI] [PubMed] [Google Scholar]
- 40. Weber G. F., Maertens P., Meng X. Z., Pippenger C. E. (1991) Glutathione peroxidase deficiency and childhood seizures. Lancet 337, 1443–1444 [DOI] [PubMed] [Google Scholar]