

Background: FilGAP is a Rac GTPase-activating protein, but how it is regulated remains unclear.
Results: GTPase Arf6 binds to FilGAP and stimulates its RacGAP activity to induce plasma membrane blebbing.
Conclusion: GTPase Arf6 is a novel physiological regulator of FilGAP.
Significance: This study establishes a novel molecular link between Arf6 and FilGAP that may have a role in Arf6-dependent inactivation of Rac.
Keywords: Actin, Cell Migration, Cytoskeleton, PI 3-kinase, Rac, Rho, Signal Transduction, Small GTPases, Arf6, Cell Polarization
Abstract
The small GTP-binding protein Arf6 reorganizes the actin cytoskeleton through the regulation of Rac activity. We identified FilGAP, a Rac-specific Rho GTPase-activating protein that is recruited to plasma membranes by binding to activated Arf6. FilGAP binds to Arf6 through its pleckstrin homology domain. Activated Arf6 stimulated RacGAP activity of FilGAP, and knockdown of endogenous Arf6 by siRNA suppresses FilGAP-mediated bleb formation. Mutant FilGAP lacking phosphatidylinositol 3,4,5-trisphosphate (PIP3) binding (FilGAP R39C) binds to activated Arf6 and induces bleb formation. Moreover, bleb formation induced by wild-type FilGAP occurs in the presence of phosphatidylinositol 3-kinase inhibitors, suggesting a PIP3-independent interaction between FilGAP and Arf6. We propose that FilGAP may function as a mediator of the regulation of Rac by Arf6.
Introduction
Actin remodeling is essential for cell shape change and migration. Rho family small GTPases control actin cytoskeletal dynamics to induce particular actin-rich surface structures in cells. For example, RhoA promotes the formation of actin stress fibers and focal adhesions, Rac induces lamellae, and Cdc42 stimulates the formation of filopodia (1).
The ADP ribosylation factor (Arf1–6) proteins are small GTPases of the Ras superfamily and regulators of vesicle transport (2). Arf6 regulates the endocytosis and recycling of plasma membrane proteins and also regulates the cortical actin cytoskeleton (3–5). Previous studies have demonstrated that Arf6-dependent actin remodeling is mediated by cross-talk between Arf6 and Rac (6–10).
FilGAP is a Rho GTPase-activating protein (GAP)2 and binds to the actin filament cross-linking protein filamin A (FLNa) (11–17). FilGAP is a Rac-specific GAP that suppresses Rac-dependent lamellipodia formation and cell spreading (11). FilGAP is phosphorylated by Rho/ROCK, and this phosphorylation stimulates its RacGAP activity. Depletion of endogenous FilGAP induced a Rac-driven elongated mesenchymal morphology. Conversely, overexpression of FilGAP induced membrane blebbing and a rounded amoeboid morphology that required Rho/ROCK-dependent phosphorylation of FilGAP. Thus, FilGAP mediates the antagonism of Rac by Rho that suppresses a mesenchymal morphology and promotes ameboid migration (16).
In this study, we present a new molecular link between Arf6 and Rac. We identified FilGAP working downstream of activated Arf6 to specifically inactivate Rac. FilGAP may inactivate Rac as a downstream effector of Arf6.
EXPERIMENTAL PROCEDURES
Proteins and Plasmids
cDNAs encoding FilGAP (GAP, PH, and ΔPH) were amplified by polymerase chain reactions and ligated into the pCMV5-HA vector using the EcoRI site. GST-PAK-CRIB domain protein was produced in Escherichia coli using a pGEX2T-PAK-CRIB cDNA (11). cDNAs encoding FilGAP (full-length, R175A, and ΔGAP) were inserted into a pCMV5-HA vector (11). The HA-tagged Arf6 (wild-type, T27N, and Q67L) constructs in the pcDNA vector and the pGEX-GGA1 construct were provided by Dr. Nakayama (Kyoto University, Kyoto, Japan) (18, 19). Full-length FilGAP cDNA was inserted into the pCMV5-FLAG vector using the PstI and SalI sites. cDNA corresponding to the PH domain of FilGAP (amino acids 1–154) was isolated and ligated into the pEFBos-FLAG vector using the BamHI and NotI sites. Mutation of R39C of the FilGAP construct was achieved using the QuikChange mutagenesis protocol (Stratagene, La Jolla, CA), and the mutant cDNA was ligated into pCMV6C-FLAG vector using the EcoRI and SalI sites. FilGAP cDNAs (wild-type and R39C) were inserted into the pEGFP-c1 vector (Clontech, Palo Alto, CA) using the SalI site. cDNA corresponding to the PH domain of Akt1 (amino acids 1–140) was inserted into the pAcGFP-c2 vector (Clontech) using the EcoRI and BamHI sites.
RhoGAP Assays
To determine GTP loading of Rac1 in vivo, HEK293 cells were transiently transfected with relevant plasmids for 24 h. For cotransfection of plasmid DNA and siRNA, cells were first transfected with siRNA for 24 h and then cotransfected with plasmid DNA for an additional 24 h. Then, the cells were washed twice in ice-cold Tris-buffered saline (20 mm Tris/HCl (pH 7.5), 120 mm NaCl) and lysed in radioimmune precipitation assay buffer (20 mm Tris/HCl (pH 7.5), 120 mm NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 10 mm MgCl2, 0.2 mm PMSF, 10 μg/ml aprotinin, and 10 μg/ml leupeptin) at 4 °C. The cell lysates were precleared, samples of supernatant fluids were withdrawn for determination of total GTPases, and the remaining supernatant was incubated with 40 μg of GST-PAK-CRIB protein in the presence of glutathione-Sepharose beads. The beads were washed, and the extent of GTP-bound Rac1 was determined by Western blot analysis using anti-Rac1 antibody.
Cell Culture
HEK293 cells and the human melanoma cell line A7 were maintained as described previously (11, 20). HeLa cells were grown at 37 °C in DMEM supplemented with 10% fetal calf serum. For transfection, cells were transfected using Lipofectamine 2000 as described by the manufacturer (Invitrogen). Transfected cells were incubated in the presence or absence of wortmannin (100 nm) or LY294002 (50 μm) for 1 h at 37 °C. Immunofluorescent staining was performed as described previously (11, 20). Briefly, cells plated on coverslips were fixed in 3.7% paraformaldehyde, permeabilized in 0.5% Triton X-100, and stained with anti-HA, anti-FLAG, anti-FilGAP, or other antibodies. Cells were examined under a fluorescence or phase-contrast optics (Olympus, Tokyo, Japan). Images were recorded and imported into the Adobe Photoshop program.
Association of FilGAP with Arf6
HEK293 cells were not treated or transfected with the pcDNA-HA-Arf6 and pCMV5-FLAG-FilGAP constructs (full-length, PH, or ΔPH). Twenty-four hours later, the cells were washed twice with 10 ml of PBS, incubated with 1 mm dithiobis(succinimidyl propionate) at 25 °C for 30 min, washed twice with ice-cold PBS, suspended in 1 ml of lysis buffer containing 50 mm Tris/HCl (pH 7.4), 100 mm NaCl, 1.0% Nonidet P-40, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 10 μg/ml pepstatin A, and 1 mm PMSF, and homogenized. The cell lysates were precleared, and supernatant fluid was subjected to immunoprecipitation with an M2 anti-FLAG antibody coupled to Sepharose beads to precipitate transfected FilGAP. Immunoprecipitates were washed five times with lysis buffer, and bound protein was detected by Western blot analysis using anti-FLAG antibody for FilGAP or anti-HA antibody for Arf6. HEK293 cells were transfected with pcDNA-HA-Arf6 constructs. After 24 h, the cells were washed and suspended in 0.65 ml of buffer (10 mm HEPES (pH 7.9), 150 mm NaCl, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 10 μg/ml pepstatin A, and 1 mm PMSF) and incubated on ice for 20 min to lyse the cells. The cell lysates were precleared, and supernatant fluid was incubated with the GST-PH domain of FilGAP protein (20 μg) coupled to glutathione-Sepharose beads and incubated at 25 °C for 30 min in the presence of 0.25 mm dithiobis(succinimidyl propionate). The beads were precipitated and incubated with a solution containing 50 mm Tris/HCl (pH 7.5) at 4 °C for 15 min to terminate the cross-linking reaction. The reacted beads were washed three times with ice-cold PBS, and bound HA-Arf6 was detected by Western blot analysis using anti-HA antibody.
RNA Interference
siRNA oligonucleotides were purchased from Invitrogen. The targeting sequence of Arf6 was 5′-GGCAAGACAACAAUCCUGUACAAGU-3′. A7 cells were transfected with Arf6 siRNA or control siRNA oligonucleotides in the presence of the pCMV5-FLAG-FilGAP vector using Lipofectamine 2000. Twenty-four hours after transfection, the level of Arf6 protein was measured by Western blot analysis using anti-Arf6 antibody.
In Vitro Lipid Binding Assay
HEK293 cells were transiently transfected with GFP-FilGAP constructs for 24 h. The transfected cells were washed twice in TBS and lysed in lysis buffer (50 mm Tris (pH 8.0), 10 mm EDTA, 100 mm NaCl, and 0.5% Triton X-100). The cell lysates were precleared, and samples of supernatant fluids were diluted 10-fold into 3% fatty acid-free BSA in TBS containing 0.1% Tween 20 (TBS-T). PIP strips (Echelon Biosciences, Salt Lake City, UT) were blocked in 3% BSA in TBS-T for 1 h and then incubated with cell lysates in 3% BSA in TBS-T for 1 h. After washing three times with 1% BSA in TBS-T, the strips were incubated with anti-GFP antibody in 1% BSA in TBS-T for 1 h. After washing three times with 1% BSA in TBS-T, the strips were incubated with HRP-conjugated secondary antibodies (Bio-Rad) in 1% BSA in TBS-T for 1 h. After washing three times with TBS-T, signals were visualized using an ECL detection kit according to the instructions of the manufacturer (Thermo Scientific, Rockford, IL).
Antibodies
Polyclonal antibodies against FilGAP were raised in rabbits and purified as described previously (11). Monoclonal antibodies were purchased from Sigma-Aldrich (anti-FLAG and anti-α-tubulin), Roche (anti-HA and anti-GFP), Upstate (anti-Rac1), and Santa-Cruz Biotechnology (anti-Arf6).
RESULTS
FilGAP Binds to Activated Arf6 and Colocalizes at the Plasma Membrane
FilGAP contains pleckstrin homology (PH), RhoGAP, and coiled coil domains (Fig. 1A). Forced expression of FilGAP in human melanoma A7 cells induced membrane blebbing around the cell periphery (Fig. 1B). FilGAP mutants lacking the PH domain failed to induce blebbing (Fig. 1B). Overexpression of the PH domain or the GAP domain alone did not induce blebbing (Fig. 1B). Bleb formation required Rac GAP activity because FilGAP mutants lacking RacGAP activity failed to induce blebbing (Fig. 1B). RhoGAP-deficient mutant FilGAP R175A was engineered on the basis of the observation that RhoGAPs have key arginine residues that mediate their catalytic activity and that arginine at position 175 is the relevant residue in FilGAP (11, 21). These observations demonstrate that, in addition to the RhoGAP domain, the PH domain is necessary for bleb formation induced by FilGAP.
FIGURE 1.

The PH domain of FilGAP is required for bleb formation. A, schematic of mutated FilGAP constructs. CC, coiled coil. B, formation of membrane blebbing induced by FilGAP and its mutants. A7 cells were transfected with HA-tagged FilGAP or its mutants for 24 h in growing medium. The cells were fixed and stained with anti-HA antibody. Scale bar = 10 μm. C, sequence alignment of the PH domains. Identical amino acids are shown as boldface letters. FilGAP has a di-Gly, high-affinity, selective binding site to PIP3, as indicated by red letters. Sequence alignment was performed with the assistance of Higgins multiple alignment programs.
A BLAST search revealed that the PH domain of FilGAP is highly homologous to the PH domains of the ARF-guanine nucleotide exchange factor ARNO and Grp1 (Fig. 1C). Previous studies demonstrated that the PH domain of ARNO directly binds to Arf6 or the Arf6-related small GTPase Arl4 in a GTP-dependent manner (22–24). We therefore examined whether FilGAP associates with Arf6. Full-length FilGAP binds to wild-type and constitutively activated Arf6 Q67L protein after immunoprecipitation from HEK cells transfected with cDNAs encoding both proteins (Fig. 2). FilGAP did not bind to dominant-negative Arf6 T27N protein (Fig. 2). Wild-type Arf6 as well as constitutively activated Arf6 Q67L, but not Arf6 T27N, were precipitated using GST-GGA protein, confirming their GTP-bound state in the cells (data not shown). The PH domain of FilGAP mediates a stable complex with Arf6 in intact cells. Constitutively activated Arf6 Q67L bound the PH domain that was immunoprecipitated from HEK cells transfected with DNA encoding the FLAG-tagged PH domain of FilGAP but not with a truncated FilGAP lacking the PH domain of FilGAP (Fig. 3A). The PH domain of FilGAP also binds to activated Arf6 in vitro. A recombinant GST-PH FilGAP construct bound activated Arf6 but not dominant-negative Arf6 protein (Fig. 3B). The coimmunoprecipitation shown was performed in the presence of the cross-linker dithiobis(succinimidyl propionate) but could be observed in the absence of cross-linking (data not shown). The interaction between ARNO and Arf6 was also enhanced significantly using the cross-linker dithiobis(succinimidyl propionate), suggesting relatively weak interactions between activated Arf6 and the PH domain of ARNO (22).
FIGURE 2.
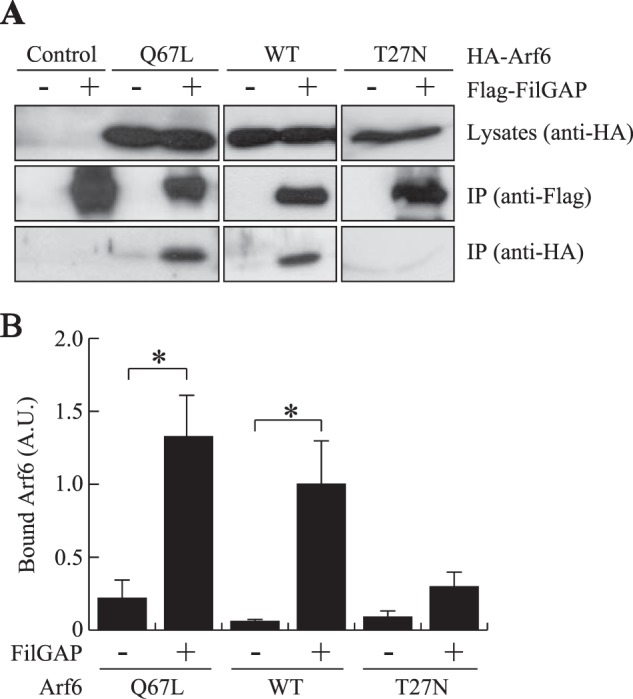
Binding of FilGAP to Arf6. A, association of FilGAP and Arf6 in intact cells. HEK cells were transiently transfected with FLAG-tagged FilGAP and HA-tagged Arf6. Cell extracts were prepared, and FLAG-FilGAP was immunoprecipitated (IP) using an anti-FLAG antibody. The washed immunoprecipitates were immunoblotted for the presence of FilGAP and Arf6. B, the relative amount of bound Arf6 proteins, treated as in A, was calculated, and the data are expressed as the mean ± S.E. (n = 4). *, p < 0.05. Statistical significance was determined by Student's t test. A. U., arbitrary units.
FIGURE 3.
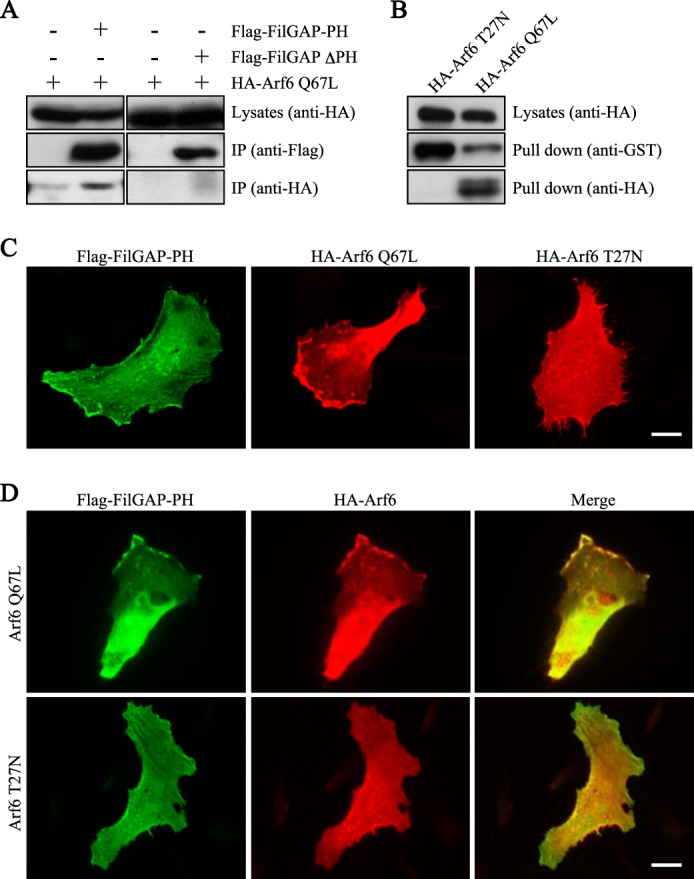
The PH domain of FilGAP interacts with Arf6. A, association of the PH domain of FilGAP and Arf6 in intact cells. HEK cells were transiently transfected with HA-Arf6 Q67L and the FLAG-tagged PH domain of FilGAP (FilGAP-PH) or the FLAG-tagged FilGAP mutant lacking the PH domain (FLAG-FilGAP-ΔPH). Cell extracts were prepared, and FilGAP-PH or FLAG-FilGAP-ΔPH protein was immunoprecipitated (IP) using an anti-FLAG antibody. The washed immunoprecipitates were immunoblotted for the presence of the PH domain or FilGAP-ΔPH (anti-FLAG) and Arf6 (anti-HA). B, association of FilGAP and Arf6 in vitro. HEK cells were transiently transfected with HA-Arf6 Q67L or HA-Arf6 T27N. Cell extracts were prepared and incubated with GST-FilGAP-PH fusion protein coupled to glutathione-Sepharose beads. The beads were washed, and the precipitates were immunoblotted for the presence of GST-FilGAP-PH (anti-GST) and Arf6 (anti-HA). C, localization of the PH domain of FilGAP and Arf6 mutants in migrating HeLa cells. HeLa cells were fixed 12 h after transfection with the PH domain of FilGAP or Arf6 mutants. The PH domains of FilGAP and Arf6 mutants were localized by staining the cells with polyclonal anti-FLAG antibody for FilGAP-PH (green) and monoclonal anti-HA antibody for Arf6 Q67L and Arf6 T27N (red). Scale bar =10 μm. D, HeLa cells were fixed 12 h after cotransfection with FLAG-FilGAP-PH and HA-Arf6 Q67L or HA-Arf6 T27N. The PH domains of FilGAP and Arf6 mutants were localized by staining the cells with polyclonal anti-FLAG antibody for FilGAP-PH (green) or monoclonal anti-HA antibody for Arf6 Q67L (red). Merged fluorescent images are also shown. Scale bar = 10 μm.
We next examined whether the PH domain of FilGAP colocalizes with activated Arf6 in intact cells. HeLa cells transfected with cDNAs encoding the FilGAP-PH, activated Arf6 Q67L, or dominant-negative Arf6 T27N were cultured to confluency, the cell monolayer was scratched, and then the migrating cells at the wound edge were fixed and observed. The PH domain of FilGAP is localized at the periphery of migrating cells (Fig. 3C). Constitutively activated Arf6 Q67L is predominantly localized at the leading lamella, whereas dominant-negative Arf6 T27N is localized diffusely (Fig. 3C). When the PH domain of FilGAP is cotransfected with activated Arf6, they are colocalized and concentrated at the leading edge of migrating HeLa cells, but this colocalization is not observed when the PH domain is cotransfected with the dominant-negative Arf6 T27N mutant (Fig. 3D). These results suggest that activated Arf6 binds to FilGAP and colocalizes at the plasma membrane.
Regulation of FilGAP Activity by Arf6
We showed that forced expression of FilGAP in human melanoma A7 cells induced blebs around the cell periphery (11, 16) and that the bleb formation requires RacGAP activity (GAP domain) of FilGAP (Fig. 1B). After transfection of the mutant Arf6 constructs in human melanoma A7 cells, constitutively activated Arf6 Q67L stimulated the surface protrusion and concentrated at the membrane, whereas the dominant-negative Arf6 T27N mutant failed to induce the protrusion and localized diffusely in the cells (Fig. 4, A and B). Because localization at the plasma membrane (Fig. 3, C and D) and regulation of surface structure (Fig. 4, A and B) induced by Arf6 is GTP-dependent, we examined whether Arf6 has any roles in the formation of blebs induced by FilGAP. siRNA targeting Arf6 reduced the expression of endogenous Arf6 in A7 cells (Fig. 4C). Depletion of Arf6 by siRNA suppressed bleb formation induced by FilGAP (Fig. 4, D and E).
FIGURE 4.

Involvement of Arf6 in bleb formation induced by FilGAP. A, localization of Arf6 in A7 cells. Human melanoma A7 cells were transfected with HA-tagged, constitutively activated Arf6 Q67L or dominant-negative Arf6 T27N. After 24 h, the cells were fixed, and Arf6 proteins were localized by staining the cells with anti-HA antibody (green). F-actin was localized by TRITC-phalloidin (red). Merged fluorescent images are also shown. Scale bar = 10 μm. B, the percentage of cells showing membrane-associated Arf6 proteins, as shown in A, was calculated, and the data are expressed as the mean ± S.E. (n = 4). *, p < 0.001. Statistical significance was determined by Student's t test. C, immunoblot analysis showing that Arf6 is depleted after 24 h of siRNA treatment of A7 cells. Anti-tubulin antibody was used as a loading control. D, reduction of bleb formation in cells treated with Arf6 siRNA. A7 cells were treated with or without Arf6 siRNA in the presence of an HA-tagged FilGAP construct. After 24 h, the cells were fixed, and FilGAP was stained with anti-HA antibody (green) and TRITC-labeled phalloidin for F-actin (red). Representative merged images are shown. Scale bar = 10 μm. E, the percentage of bleb-positive cells, as shown in D, was calculated, and the data are expressed as the mean ± S.E. (n = 4). *, p < 0.001. Statistical significance was determined by Student's t test. F, requirement of the PH and GAP domains of FilGAP for Arf6-induced bleb formation. A7 cells were transfected with HA-Arf6 Q67L and FLAG-tagged FilGAP or its mutants. After 24 h, the cells were fixed, and FilGAP was stained with rabbit polyclonal anti-FilGAP antibody (green), and HA-Arf6 Q67L was stained with mouse monoclonal anti-HA antibody (red). Representative merged images are shown. Scale bar = 5 μm.
The above result suggests that targeting of FilGAP to activated Arf6 at the plasma membrane facilitates the RacGAP activity of FilGAP and stimulates bleb formation at the plasma membrane by inactivating Rac. In agreement with this hypothesis, a FilGAP mutant incapable of interacting with activated Arf6 (FilGAP ΔPH) or a FilGAP mutant deficient in its Rac GAP activity (FilGAP R175A) failed to induce blebbing when cotransfected with Arf6 Q67L (Fig. 4F).
We next examined whether Arf6 could regulate the RacGAP activity of FilGAP. As observed in A7 cells, the forced expression of Arf6 stimulated the formation of surface protrusions induced by FilGAP in HEK cells (data not shown). The forced expression of constitutively activated Arf6 Q67L significantly decreases the amounts of active Rac when coexpressed with FilGAP in HEK cells, as determined by a GST-PBD pulldown assay (Fig. 5, A and B). On the other hand, depletion of endogenous Arf6 by siRNA reduced the RacGAP activity of FilGAP (Fig. 5, C and D). The bleb formation induced by FilGAP requires the inactivation of Rac. Constitutively activated Rac G12V, but not dominant-negative Rac T17N, stopped bleb formation induced by FilGAP (Fig. 5E). Thus, activated Arf6 stimulates FilGAP to down-regulate Rac and induce blebs in vivo.
FIGURE 5.
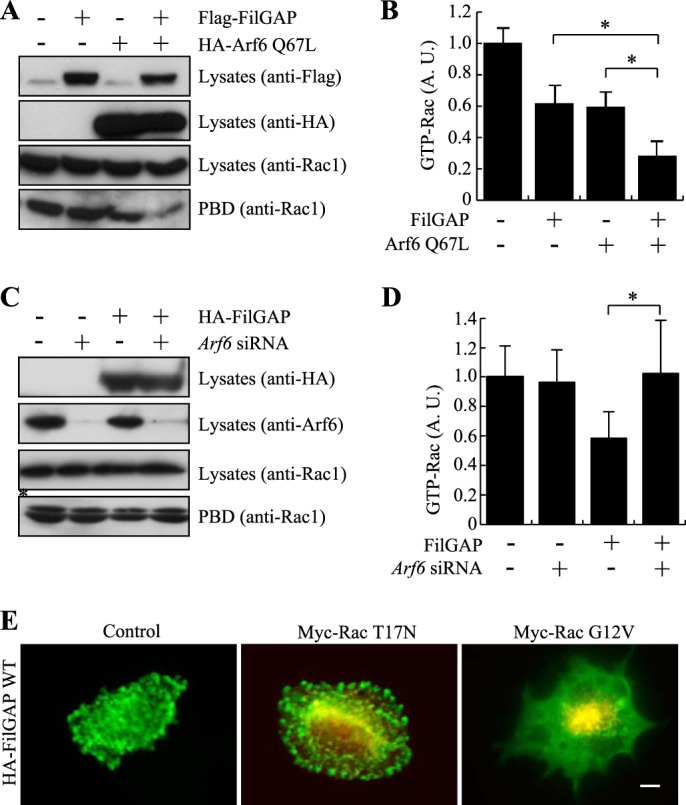
GTPase-stimulating activity of FilGAP and its regulation by Arf6. A, HEK cells were transfected with HA-Arf6 Q67L or FLAG-FilGAP. Cell extracts were incubated with GST-PBD that was immobilized on glutathione-Sepharose beads. The amount of Rac1 in cell lysates before pulldown and GTP (GST-PBD-bound) Rac1 was detected by immunoblotting. Expression levels of transfected Arf6 Q67L (anti-HA) and FilGAP (anti-FLAG) are also shown. B, the relative amount of GTP-Rac1 treated in A was calculated and expressed as the mean ± S.E. (n = 6). *, p < 0.05. Statistical significance was determined by Student's t test. A. U., arbitrary units. C, HEK cells were treated with or without Arf6 siRNA in the presence or absence of the HA-FilGAP construct. Cell extracts were incubated with GST-PBD that was immobilized on glutathione-Sepharose beads. The amount of Rac1 in cell lysates before pulldown and GTP (GST-PBD bound) Rac1 was detected by immunoblotting. Expression levels of transfected HA-FilGAP (anti-HA) and endogenous Arf6 (anti-Arf6) are also shown. The asterisk indicates a nonspecific band. D, the relative amount of GTP-Rac1 treated in C was calculated and expressed as the mean ± S.E. (n = 6). *, p < 0.05. Statistical significance was determined by Student's t test. E, FilGAP induces blebbing through inactivation of Rac1. A7 cells were transfected with HA-FilGAP in the presence or absence of dominant-negative Rac1 (myc-Rac T17N) or constitutively activated Rac1 (myc-Rac G12V) mutants. After 24 h, the cells were fixed, and FilGAP was stained with rabbit polyclonal anti-FilGAP antibody (green) and mouse monoclonal anti-myc antibody for myc-Rac1 mutants (red). Representative merged images of cells are shown. Scale bar = 5 μm.
FilGAP Binds to Arf6 in a PIP3-independent Manner
The PH domain of FilGAP contains two glycines (di-Gly) in a loop in the phosphoinositide-binding pocket (Fig. 1C). The di-Gly form binds to PIP3 with high affinity (25, 26). The PH domain of FilGAP binds to PIP3 in vitro (Fig. 6B). We therefore examined whether PIP3 is required for interaction between FilGAP and active Arf6. The PH domains binding to PIP3 with high affinity have key arginine residues that mediate their binding to PIP3 (26). On the basis of the sequence of FilGAP, an arginine of position 39 is the relevant residue, and we therefore engineered mutant FilGAP R39C. As expected, the mutant FilGAP R39C did not bind to PIP3 in vitro (Fig. 6, A and B). FilGAP R39C was coprecipitated with constitutively activated Arf6 Q67L when expressed in HEK cells (Fig. 6C). When mutant FilGAP R39C and activated Arf6 Q67L were overexpressed in A7 cells, mutant FilGAP R39C was able to induce blebs around the cell peripheries and colocalized with activated Arf6 (Fig. 6, D and E). Moreover, treatment of A7 cells with PI 3-kinase inhibitors such as wortmannin or LY294002 did not suppress bleb formation induced by overexpression of wild-type FilGAP (Fig. 6, F and G). Both PI3 kinase inhibitors stopped targeting of the Akt1-PH domain to the cell membrane, demonstrating that the inhibitors suppressed PIP3 production under our experimental conditions (data not shown). These results show that FilGAP is capable of interacting with activated Arf6 to induce blebs in the absence of PIP3.
FIGURE 6.
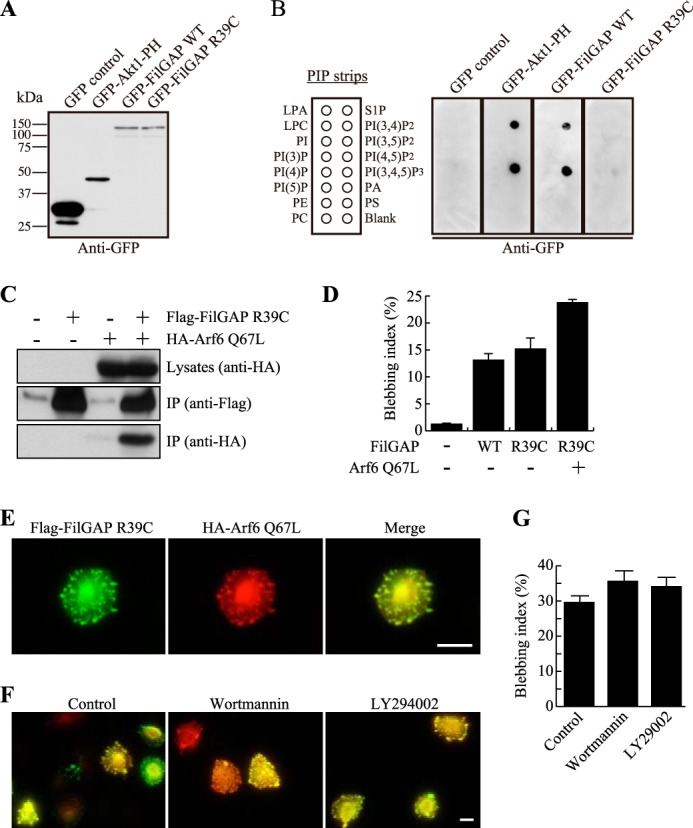
Arf6 interacts with FilGAP in a PIP3-independent manner. A, HEK cells were transiently transfected with a control plasmid (pEGFP), a pAcGFP plasmid encoding the PH domain of Akt1, or a pEGFP plasmids encoding FilGAP WT or FilGAP R39C. Cell lysates were prepared and immunoblotted with anti-GFP antibody. B, the FilGAP R39C mutant does not bind to PIP3. PIP strips were incubated with the cell lysates in A, and strip-bound proteins were detected using anti-GFP antibody. LPA, lysophosphatidic acid; LPC, lysophosphatidylcholine; PE, phosphatidylethanolamine; PC, phosphatidylcholine; S1P, sphingosine-1-phosphate; PA, phosphatidic acid; PS, phosphatidylserine. C, association of mutant FilGAP R39C and Arf6 in intact cells. HEK cells were transiently transfected with FLAG-FilGAP R39C and HA-Arf6 Q67L. Cell extracts were prepared, and FLAG-FilGAP R39C was immunoprecipitated (IP) using an anti-FLAG antibody. The washed immunoprecipitates were immunoblotted for the presence of FilGAP R39C (anti-FLAG) and Arf6 Q67L (anti-HA). D, bleb formation in cells transfected with FilGAP R39C. A7 cells were transfected with FLAG-FilGAP constructs and an HA-Arf6 Q67L construct. After 24 h, the cells were fixed, and FilGAP and Arf6 Q67L were stained with anti-FLAG (FilGAP) anti-HA (Arf6) antibodies. The percentage of bleb-positive cells was calculated, and the data are expressed as the mean ± S.E. (n = 4). E, localization of Arf6 and FilGAP R39C in A7 cells. A7 cells were transfected with FLAG-tagged mutant FilGAP R39C and constitutively activated Arf6 Q67L. After 24 h, the cells were fixed, and FilGAP R39C (green) and Arf6 Q67L proteins (red) were localized by staining the cells with anti-FLAG and anti-HA antibodies. A merged image is also shown. Scale bar = 10 μm. F, Arf6 stimulates FilGAP activity in a PIP3-independent manner. A7 cells were transfected with wild-type FLAG-FilGAP in the presence of HA-Arf6 Q67L. After 24 h, the cells were incubated with or without of wortmannin (100 nm) or LY294002 (50 μm) for 1 h. The cells were then fixed and stained for FilGAP (green) and Arf6 (red) using anti-FLAG and anti-HA antibodies. Representative merged images are shown. Scale bar = 10 μm. G, the percentage of bleb-positive cells was calculated, and the data are expressed as the mean ± S.E. (n = 4).
DISCUSSION
Arf6 controls actin remodeling, cell shape change, and cell migration, and many of the Arf6-dependent regulation of the actin cytoskeleton is mediated by Rac (2–4). Previous studies have demonstrated that Arf6 can activate Rac by activating RacGEFs such as DOCK180 and Karilin (9, 10). This study demonstrated that Arf6 could not only stimulate Rac activity but also down-regulate Rac through recruitment of RacGAP.
Inactivation of Rac by FilGAP downstream of Arf6 may have some physiological significance. First, the maintenance of proper Rac activity seems to be important for efficient cell migration. Microinjection of not only dominant-negative Rac but also constitutively activated Rac mutant inhibits the migration of macrophages (27). Both constitutively activated and dominant-negative Rac mutants inhibit PDGF-stimulated Rat1 fibroblast invasion into collagen matrices (28). Moreover, a small change in the total activity of Rac has a dramatic effect on the pattern of cell migration (29). Arf6 may, therefore, regulate cell migration by maintaining proper Rac activity through direct activation of RacGEF and RacGAP. Second, FilGAP could be involved in the Arf6-mediated down-regulation of Rac. Arf6 promotes adherens junction disassembly and epithelial cell scattering during the epithelial-to-mesenchymal transition of MDCK cells induced by HGF/SF (8). The adherens junction disassembly is caused by down-regulation of Rac induced by Arf6. It has been reported that the down-regulation of Rac is mediated, in part, by Arf6-dependent inactivation of RacGEF (Tiam1) (30). Because FilGAP is expressed in many epithelial cell lines and is localized at the membrane, Arf6 could inactivate Rac through direct activation of FilGAP during the epithelial-to-mesenchymal transition of epithelial cells.
This study provides strong evidence that FilGAP functions as a downstream effector of Arf6. First, FilGAP binds to Arf6 in a GTP-dependent manner in vitro and in vivo. Second, endogenous FilGAP colocalizes with activated Arf6 Q67L, but not dominant-negative Arf6 T27L, in HeLa cells. Third, activated Arf6 stimulates the RacGAP activity of FilGAP, as shown by a GTP-Rac1 pulldown assay. Finally, the PH domain of FilGAP appears to be the binding site for activated Arf6, and its primary structure is highly homologous to those of ArfGEFs, ARNO, and Grp1, which were also shown to bind to Arf6 in a GTP-dependent manner (22).
The affinity of the PH domain to phospholipids is dependent on the number of glycine residues in the PH domain. The triglycine form recognizes both phosphatidylinositol 4,5-bisphosphate and PIP3 with similar affinities, whereas the diglycine form has reduced affinity for phosphatidylinositol 4,5-bisphosphate and binds to PIP3 with high affinity (26). The PH domain of FilGAP contains diglycine and binds to PIP3 in vitro. Therefore, phospholipid signaling mediated by PI3-kinase could modulate FilGAP function through its PH domain. However, our study demonstrated that the FilGAP-PH domain does not require PIP3 to interact with Arf6 in vivo. Although Arf6 facilitates plasma membrane localization and activates the RacGAP activity of FilGAP, FilGAP is localized at the plasma membrane in the absence of Arf6 or PIP3. This is in contrast with the findings that translocation of ARNO to the plasma membrane requires PI3K signaling (23) and ARNO requires phospholipids to interact with Arf6 (22). These findings suggest that the PH domain of FilGAP has a unique structural property compared with that of ARNO, despite the fact that both bind to Arf6 in a GTP-dependent manner. The PH domain of FilGAP may have additional binding partners to localize at the plasma membrane.
Forced expression of FilGAP produced plasma membrane blebbing. The blebbing requires the PH and RacGAP domains of FilGAP, suggesting the down-regulation of Rac at the plasma membrane (31–33). Arf6 appears to play a role in FilGAP-dependent bleb formation as follows. Activated Arf6 colocalized with FilGAP at the plasma membrane, knockdown of endogenous Arf6 attenuates FilGAP-dependent bleb formation, activated Arf6 activates the RacGAP activity of FilGAP and stimulates down-regulation of Rac, and depletion of endogenous Arf6 reduced the RacGAP activity of FilGAP.
Although the FilGAP-dependent down-regulation of Rac and induction of membrane blebbing were facilitated by Arf6, Arf6 does not seem to be a requirement for FilGAP to inactivate Rac. Depletion of Arf6 failed to completely eliminate the ability of FilGAP to inactivate Rac and induce membrane blebbing (Figs. 4E and 5D). Therefore, it is likely that Arf6 and FilGAP can inactivate Rac independently. It has been shown that Arf6 down-regulates Rac through inactivation of RacGEF (Tiam1) (30). Arf6 could stimulate RacGAPs other than FilGAP.
We have shown previously that Rho is also involved in the formation of membrane blebbing through the activation of FilGAP (11, 16). FilGAP is phosphorylated by ROCK, and the phosphorylation activates RacGAP activity of FilGAP. Moreover, ROCK-dependent phosphorylation of FilGAP is required for bleb formation because forced expression of non-phosphorylatable mutant FilGAP failed to induce blebs (11). Therefore, both Rho and Arf6 appear to be involved in FilGAP-mediated bleb formation. The mechanism of activation of FilGAP by Rho and Arf6 is unclear, but both Rho and Arf6 stimulate the RacGAP activity of FilGAP in vivo. Specifically, ROCK-dependent phosphorylation of a cluster of serine and threonine residues at positions 573–577 activates FilGAP (11). As shown in this study, activated Arf6 directly binds to the PH domain of FilGAP and stimulates the RacGAP activity of FilGAP. Both phosphorylation by ROCK and direct binding of Arf6 could induce a change of FilGAP to activate its catalytic activity, as demonstrated by binding of Arf6 to the PH domain of Grp1 (34). It is also possible that Arf6 and Rho could change the localization of FilGAP so that FilGAP could efficiently inactivate Rac at the membrane to induce blebs. Further study is necessary to understand the mechanism of regulation of FilGAP by Rho and Arf6.
It is well established that activation of Rho/ROCK signaling induces plasma membrane blebbing (33, 35). Plasma membrane blebbing requires myosin II activity, and myosin II is phosphorylated and activated by ROCK. Therefore, both the ROCK inhibitor Y27632 and myosin II inhibitors, such as blebbistatin, suppress membrane blebbing (11, 36). On the other hand, forced expression of Arf6 or constitutive activation of its downstream effectors has not been reported to induce membrane blebbing. Therefore, Arf6 could cooperate Rho/ROCK-dependent membrane blebbing through the activation of FilGAP.
Arf6-dependent control of cell adhesion and migration seems to be important in the development of malignant transformation. For example, Arf6 regulates the disruption of E-cadherin-dependent cell adhesion during epithelial-to-mesenchymal transition, and Arf6 controls invadopodia formation (37). Moreover, Arf6-dependent regulation of Rac activity plays an important role in cancer invasion (38). Previous studies have demonstrated that some malignant tumor cells use blebs to move in three-dimensional environments (39, 40). Moreover, FilGAP and its close homolog ARHGAP22 are involved in the regulation of ameboid cancer cell migration in a three-dimensional environment (16, 17, 41, 42). It is unclear whether Arf6 is involved in the control of ameboid-type cancer cell migration. It will be interesting to study whether FilGAP is involved in the Arf6-dependent control of cell migration and cancer invasion.
Acknowledgments
We thank K. Nakayama (Kyoto University, Kyoto, Japan) for the HA-tagged Arf6 constructs and the pGEX-GGA1 construct.
This work was supported by grants-in-aid for scientific research from the Japan Society for the Promotion of Science and the Ministry of Education, Culture, Sports, Science, and Technology of Japan; by a grant for the All Kitasato Project Study; and by a Kitasato University research grant for young researchers.
- GAP
- GTPase-activating protein
- PH
- pleckstrin homology
- PIP3
- phosphatidylinositol 3,4,5-trisphosphate
- PI
- phosphatidylinositol
- TRITC
- tetramethylrhodamine isothiocyanate
- GEF
- guanine nucleotide exchange factor.
REFERENCES
- 1. Jaffe A. B., Hall A. (2005) Rho GTPases. Biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21, 247–269 [DOI] [PubMed] [Google Scholar]
- 2. Donaldson J. G., Honda A. (2005) Localization and function of Arf family GTPases. Biochem. Soc. Trans. 33, 639–642 [DOI] [PubMed] [Google Scholar]
- 3. D'Souza-Schorey C., Chavrier P. (2006) ARF proteins. Roles in membrane traffic and beyond. Nat. Rev. Mol. Cell Biol. 7, 347–358 [DOI] [PubMed] [Google Scholar]
- 4. Myers K. R., Casanova J. E. (2008) Regulation of actin cytoskeleton dynamics by Arf-family GTPases. Trends Cell Biol. 18, 184–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schweitzer J. K., Sedgwick A. E., D'Souza-Schorey C. (2011) ARF6-mediated endocytic recycling impacts cell movement, cell division and lipid homeostasis. Semin. Cell Dev. Biol. 22, 39–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Radhakrishna H., Al-Awar O., Khachikian Z., Donaldson J. G. (1999) ARF6 requirement for Rac ruffling suggests a role for membrane trafficking in cortical actin rearrangements. J. Cell Sci. 112, 855–866 [DOI] [PubMed] [Google Scholar]
- 7. Zhang Q., Calafat J., Janssen H., Greenberg S. (1999) ARF6 is required for growth factor- and Rac-mediated membrane ruffling in macrophages at a stage distal to Rac membrane targeting. Mol. Cell Biol. 19, 8158–8168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Palacios F., D'Souza-Schorey C. (2003) Modulation of Rac1 and Arf6 activation during epithelial cell scattering. J. Biol. Chem. 278, 17395–17400 [DOI] [PubMed] [Google Scholar]
- 9. Santy L. C., Ravichandran K. S., Casanova J. E. (2005) The DOCK180/Elmo complex couples ARNO-mediated arf6 activation to the downstream activation of Rac1. Curr. Biol. 15, 1749–1754 [DOI] [PubMed] [Google Scholar]
- 10. Koo T. H., Eipper B. A., Donaldson J. G. (2007) Arf6 recruits the Rac GEF Kalirin to the plasma membrane facilitating Rac activation. BMC Cell Biol. 8, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ohta Y., Hartwig J. H., Stossel T. P. (2006) FilGAP, a Rho- and ROCK-regulated GAP for Rac binds filamin A to control actin remodelling. Nat. Cell Biol. 8, 803–814 [DOI] [PubMed] [Google Scholar]
- 12. Nakamura F., Osborn T. M., Hartemink C. A., Hartwig J. H., Stossel T. P. (2007) Structural basis of filamin A functions. J. Cell Biol. 179, 1011–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shifrin Y., Arora P. D., Ohta Y., Calderwood D. A., McCulloch C. A. (2009) The role of FilGAP-filamin A interactions in mechanoprotection. Mol. Biol. Cell 20, 1269–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nakamura F., Heikkinen O., Pentikäinen O. T., Osborn T. M., Kasza K. E., Weitz D. A., Kupiainen O., Permi P., Kilpeläinen I., Ylänne J., Hartwig J. H., Stossel T. P. (2009) Molecular basis of filamin A-FilGAP interaction and its impairment in congenital disorders associated with filamin A mutations. PLoS ONE 4, e4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nieves B., Jones C. W., Ward R., Ohta Y., Reverte C. G., LaFlamme S. E. (2010) The NPIY motif in the integrin β 1 tail dictates the requirement for talin-1 in outside-in signaling. J. Cell Sci. 123, 1216–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Saito K., Ozawa Y., Hibino K., Ohta Y. (2012) FilGAP, a Rho/Rho-associated protein kinase-regulated GTPase-activating protein for Rac, controls tumor cell migration. Mol. Biol. Cell 23, 4739–4750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nakamura F. (2013) FilGAP and its close relatives: a mediator of Rho-Rac antagonism that regulates cell morphology and migration. Biochem. J. 453, 17–25 [DOI] [PubMed] [Google Scholar]
- 18. Shinotsuka C., Yoshida Y., Kawamoto K., Takatsu H., Nakayama K. (2002) Overexpression of an ADP-ribosylation factor-guanine nucleotide exchange factor, BIG2, uncouples brefeldin A-induced adaptor protein-1 coat dissociation and membrane tubulation. J. Biol. Chem. 277, 9468–9473 [DOI] [PubMed] [Google Scholar]
- 19. Hosaka M., Toda K., Takatsu H., Torii S., Murakami K., Nakayama K. (1996) Structure and intracellular localization of mouse ADP-ribosylation factor type 1 to type 6 (ARF1-ARF6). J. Biochem. 120, 813–819 [DOI] [PubMed] [Google Scholar]
- 20. Ohta Y., Suzuki N., Nakamura S., Hartwig J. H., Stossel T. P. (1999) The small GTPase RalA targets filamin to induce filopodia. Proc. Natl. Acad. Sci. U.S.A. 96, 2122–2128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Scheffzek K., Ahmadian M. R., Wittinghofer A. (1998) GTPase-activating proteins. Helping hands to complement an active site. Trends Biochem. Sci. 23, 257–262 [DOI] [PubMed] [Google Scholar]
- 22. Cohen L. A., Honda A., Varnai P., Brown F. D., Balla T., Donaldson J. G. (2007) Active Arf6 recruits ARNO/Cytohesin GEFs to the PM by binding their PH domains. Mol. Biol. Cell 18, 2244–2253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li C. C., Chiang T. C., Wu T. S., Pacheco-Rodriguez G., Moss J., Lee F. J. (2007) ARL4D recruits cytohesin-2/ARNO to modulate actin remodeling. Mol. Biol. Cell 18, 4420–4437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hofmann I., Thompson A., Sanderson C. M., Munro S. (2007) The Arl4 family of small G proteins can recruit the cytohesin Arf6 exchange factors to the plasma membrane. Curr. Biol. 17, 711–716 [DOI] [PubMed] [Google Scholar]
- 25. Klarlund J. K., Tsiaras W., Holik J. J., Chawla A., Czech M. P. (2000) Distinct polyphosphoinositide binding selectivities for pleckstrin homology domains of GRP1-like proteins based on diglycine versus triglycine motifs. J. Biol. Chem. 275, 32816–32821 [DOI] [PubMed] [Google Scholar]
- 26. Cronin T. C., DiNitto J. P., Czech M. P., Lambright D. G. (2004) Structural determinants of phosphoinositide selectivity in splice variants of Grp1 family PH domains. EMBO J. 23, 3711–3720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Allen W. E., Zicha D., Ridley A. J., Jones G. E. (1998) A role for Cdc42 in macrophage chemotaxis. J. Cell Biol. 141, 1147–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Banyard J., Anand-Apte B., Symons M., Zetter B. (2000) Motility and invasion are differentially modulated by Rho family GTPases. Oncogene 19, 580–591 [DOI] [PubMed] [Google Scholar]
- 29. Pankov R., Endo Y., Even-Ram S., Araki M., Clark K., Cukierman E., Matsumoto K., Yamada K. (2005) A Rac switch regulates random versus directionally persistent cell migration. J. Cell Biol. 170, 793–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Palacios F., Schweitzer J. K., Boshans R. L., D'Souza-Schorey C. (2002) ARF6-GTP recruits Nm23-H1 to facilitate dynamin-mediated endocytosis during adherence junctions disassembly. Nat. Cell Biol. 4, 929–936 [DOI] [PubMed] [Google Scholar]
- 31. Schwartz M. A., Meredith J. E., Kiosses W. B. (1998) An activated Rac mutant functions as a dominant negative for membrane ruffling. Oncogene 17, 625–629 [DOI] [PubMed] [Google Scholar]
- 32. Vidali L., Chen F., Cicchetti G., Ohta Y., Kwiatkowski D. J. (2006) Rac1-null mouse embryonic fibroblasts are motile and respond to platelet-derived growth factor. Mol. Biol. Cell 17, 2377–2390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fackler O. T., Grosse R. (2008) Cell motility through plasma membrane blebbing. J. Cell Biol. 181, 879–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Malaby A. W., van den Berg B., Lambright D. G. (2013) Structural basis for membrane recruitment and allosteric activation of cytohesin family Arf GTPase exchange factors. Proc. Natl. Acad. Sci. U.S.A. 110, 14213–14218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Charras G., Paluch E. (2008) Blebs lead the way. How to migrate without lamellipodia. Nat. Rev. Mol. Cell Biol. 9, 730–736 [DOI] [PubMed] [Google Scholar]
- 36. Charras G. T., Yarrow J. C., Horton M. A., Mahadevan L., Mitchison T. J. (2005) Non-equilibration of hydrostatic pressure in blebbing cells. Nature 435, 365–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sabe H., Hashimoto S., Morishige M., Ogawa E., Hashimoto A., Nam J. M., Miura K., Yano H., Onodera Y. (2009) The EGFR-GEP100-Arf6-AMAP1 signaling pathway specific to breast cancer invasion and metastasis. Traffic 10, 982–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hu B., Shi B., Jarzynka M. J., Yiin J. J., D'Souza-Schorey C., Cheng S. Y. (2009) ADP-ribosylation factor 6 regulates glioma cell invasion through the IQ-domain GTPase-activating protein 1-Rac1-mediated pathway. Cancer Res. 69, 794–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Friedl P., Wolf K. (2003) Tumor-cell invasion and migration. Diversity and escape mechanisms. Nat. Rev. Cancer 3, 362–374 [DOI] [PubMed] [Google Scholar]
- 40. Madsen C. D., Sahai E. (2010) Cancer dissemination. Lessons from leukocytes. Dev. Cell 19, 13–26 [DOI] [PubMed] [Google Scholar]
- 41. Sanz-Moreno V., Gadea G., Ahn J., Paterson H., Marra P., Pinner S., Sahai E., Marshall C. J. (2008) Rac activation and inactivation control plasticity of tumor cell movement. Cell 135, 510–523 [DOI] [PubMed] [Google Scholar]
- 42. Sanz-Moreno V., Marshall C. (2010) The plasticity of cytoskeletal dynamics underlying neoplastic cell migration. Curr. Opin. Cell Biol. 22, 690–696 [DOI] [PubMed] [Google Scholar]