

Background: NDRG1 is an important iron-regulated metastasis suppressor that plays an undefined role in the stress response.
Results: We demonstrate that NDRG1 suppresses the stress-induced, pro-survival autophagic pathway.
Conclusion: Suppression of the autophagic pathway by NDRG1 makes cells more susceptible to apoptosis.
Significance: These results indicate an important new mechanism through which NDRG1 exerts its metastasis suppressor activity.
Keywords: Iron, Iron Metabolism, Metastasis, Oxidative Stress, Stress, Dp44mT, Desferrioxamine, Iron Chelators, Iron-Protein Interactions, Ligand-Metal Interactions
Abstract
N-myc downstream regulated gene 1 (NDRG1) is a potent metastasis suppressor with an undefined role in the stress response. Autophagy is a pro-survival pathway and can be regulated via the protein kinase-like endoplasmic reticulum kinase (PERK)/eIF2α-mediated endoplasmic reticulum (ER) stress pathway. Hence, we investigated the role of NDRG1 in stress-induced autophagy as a mechanism of inhibiting metastasis via the induction of apoptosis. As thiosemicarbazone chelators induce stress and up-regulate NDRG1 to inhibit metastasis, we studied their effects on the ER stress response and autophagy. This was important to assess, as little is understood regarding the role of the stress induced by iron depletion and its role in autophagy. We observed that the chelator, di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mT), which forms redox-active iron and copper complexes, effectively induced ER stress as shown by activation of the PERK/eIF2α pathway. Dp44mT also increased the expression of the autophagic marker, LC3-II, and this was dependent on activation of the PERK/eIF2α axis, as silencing PERK prevented LC3-II accumulation. The effect of Dp44mT on LC3-II expression was at least partially due to iron-depletion, as this effect was also demonstrated with the classical iron chelator, desferrioxamine (DFO), and was not observed for the DFO-iron complex. NDRG1 overexpression also inhibited basal autophagic initiation and the ER stress-mediated autophagic pathway via suppression of the PERK/eIF2α axis. Moreover, NDRG1-mediated suppression of the pro-survival autophagic pathway probably plays a role in its anti-metastatic effects by inducing apoptosis. In fact, multiple pro-apoptotic markers were increased, whereas anti-apoptotic Bcl-2 was decreased upon NDRG1 overexpression. This study demonstrates the role of NDRG1 as an autophagic inhibitor that is important for understanding its mechanism of action.
Introduction
N-myc downstream regulated gene 1 (NDRG1)7 is a well established metastasis suppressor in a number of cancers including colon, prostate, and breast cancer (1–5). NDRG1 has been comprehensively characterized with the gene cloned and the protein sequenced (6–8). In addition, extensive information has been reported regarding its biochemistry and molecular genetics (8, 9). There are also data describing NDRG1 as predominantly localized in the cytosol and to a lesser extent the nucleus and plasma membrane (10, 11).
NDRG1 is also recognized as a significant stress response gene and is regulated by a number of stress stimuli (12–15). These include iron chelation (14), nickel compounds (15), cobalt (16), hypoxia (17), oxidative stress (11), and tunicamycin (13). NDRG1 has been shown to inhibit metastasis and tumor progression via its effects on a variety of critical cell signaling pathways involved in proliferation, locomotion, and oncogenesis e.g. AKT, TGF-β, ROCK1/pMLC2, Ras, WNT, etc. (18–21). The ability of NDRG1 to affect such a diverse range of signaling pathways indicates that it is well upstream of these pathways and, thus, can affect many different biological processes. Considering its effects on these molecular networks, further studies examining NDRG1 are essential to dissect its role in downstream cell survival pathways, such as autophagy, which can modulate metastasis (22).
Autophagy is an evolutionarily conserved catabolic process that occurs in response to stress that involves lysosomal degradation of unnecessary or damaged organelles, cellular constituents, and misfolded proteins (23). This recycling process is cyto-protective and helps to maintain cellular functioning under the influence of stress stimuli (24). Autophagy is crucial for maintenance of cellular homeostasis, “quality control,” defense against intra- and extracellular insults, and maintaining cellular energy status (25). This function is achieved through its ability to provide essential macromolecules derived from the degradation and recycling of intracellular components (25). The autophagic pathway has been previously shown to be an important regulator of tumor progression and metastasis (22, 23, 26).
The mechanism of autophagic degradation begins with sequestration of cytosolic proteins and/or organelles into a double membrane-lined structure called the autophagosome (27). The mature autophagosome fuses with a lysosome to form the autolysosome (27). The hydrolases in the lysosome catabolize the contents of the autophagosome and release nutrients back into the cytoplasm for reuse (27). This recycling of damaged proteins and organelles protects the cell from stress conditions. Under conditions where autophagy is inhibited, stress can result in irreparable cellular damage, leading to apoptosis (28).
The unfolded protein response is another pathway that is involved in maintenance of cellular homeostasis (29). It is up-regulated by the accumulation of excessive unfolded/misfolded proteins in the endoplasmic reticulum (ER) lumen (29). There are three well characterized pathways that can detect and respond to ER stress caused by unfolded/misfolded proteins: 1) protein kinase-like endoplasmic reticulum kinase (PERK), 2) inositol-requiring transmembrane kinase and endonuclease 1 (IRE1), and 3) activating transcription factor 6 (ATF6) (30). The PERK axis of the ER stress pathway has been described to up-regulate the autophagic “machinery” leading to cyto-protective effects (31). Moreover, ER stress-mediated through PERK directly induces increased phosphorylated eIF2α (p-eIF2α) levels, which leads to initiation of autophagosome formation (29, 31).
Iron is an essential element involved in a variety of metabolic pathways including DNA synthesis and energy generation (32). As such, iron is required in greater amounts by cancer cells than their normal counterparts (32). In contrast to traditional iron chelators such as desferrioxamine (DFO; Fig. 1A), recent studies have indicated that rationally designed iron chelators of the di-2-pyridylketone thiosemicarbazone class (e.g. di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mT); Fig. 1B) possess potent and selective anti-tumor activity (33) as well as marked anti-metastatic activity in vitro and in vivo (19, 21, 34). Unlike DFO, the novel thiosemicarbazones form intracellular redox-active iron and copper complexes that play a critical role in their anti-tumor activity (35). One of the important targets of these agents is the metastasis suppressor, NDRG1, which is up-regulated by hypoxia-inducible factor-1α (HIF-1α)-dependent and -independent mechanisms via cellular iron depletion (14, 36).
FIGURE 1.

Line drawings of the chemical structures of DFO (A), Dp44mT (B), and Dp2mT (C).
Because NDRG1 functions as a stress response protein and since autophagy is also up-regulated during stress, we examined the effect of NDRG1 on the autophagic pathway. Currently, nothing is known regarding the role of autophagy in the molecular mechanism of how NDRG1 is involved in inhibiting metastasis. In this study, we demonstrate that a novel redox-active iron chelator of the di-2-pyridylketone thiosemicarbazone class, namely, Dp44mT, effectively induces ER stress, leading to initiation of the autophagic pathway. Hence, we have used Dp44mT as a stress signal for the induction of this process. We show that NDRG1 overexpression suppresses the stress-induced autophagic response in cancer cells via suppression of the PERK/eIF2α stress pathway. Our studies also indicate that NDRG1-mediated suppression of the pro-survival autophagic pathway could play a role in its anti-metastatic effects by inducing apoptosis.
MATERIALS AND METHODS
Chelators
The chelator, Dp44mT, as well as the related di-2-pyridylketone thiosemicarbazone control compound, di-2-pyridylketone 2-methyl-3-thiosemicarbazone (Dp2mT; Fig. 1C), which cannot bind iron or other metals, were prepared and characterized as described previously (37, 38). DFO was purchased from Sigma.
Cell Culture
The human pancreatic cancer cell line PANC1, the human colon cancer cell line HCT116, and human prostate cancer cell line DU145 were purchased from the American Type Culture Collection (Manassas, VA) and grown under established conditions (37 °C, 95% air/5% CO2) in the presence of medium containing 10% fetal calf serum (FCS; Invitrogen). The cells were incubated with control medium or medium containing Dp44mT (0.1–5 μm), Dp2mT (5 μm), or DFO (250 μm) for 6–24 h at 37 °C.
Transfections
The NDRG1 overexpression clone of the PANC1 cell line and its vector-transfected counterpart were generated by standard procedures (20). The antibiotic, G418 (300 μg/ml; Sapphire Biosciences, Waterloo, Australia) was used as a selective agent to maintain the transfected population (20). The NDRG1 overexpression and knockdown clones for HCT116 were established as described (19). In this latter cell line, puromycin (2.5 μg/ml; Sigma) was used as a selection agent to obtain stable single clones (19). The NDRG1 overexpression clone of the DU145 cell line was established as described previously (21).
Gene Silencing by Small Interfering RNA
PERK knockdown using PERK small interfering RNA (siRNA) was performed using the manufacturer's protocol. Briefly, at 50–60% confluence, cells were transfected with either PERK siRNA (catalog #9024, Cell Signaling Technology, Beverly, MA) or a scrambled control (catalog #6568, Cell Signaling Technology) for 24 h at 37 °C using Lipofectamine 2000 (Invitrogen). Then the cells were further incubated in normal growth medium supplemented with FCS for an additional 48 h at 37 °C.
Immunofluorescence
Immunofluorescence was performed according to standard procedures (21). Briefly, cells seeded on coverslips were fixed with ice-cold methanol (Sigma) for 10 min followed by permeabilization with 0.1% Triton X-100 for 10 min at room temperature. The cells were then blocked with 10% bovine serum albumin in phosphate-buffered saline, 0.1% Tween followed by an overnight incubation (4 °C) with the primary antibody, anti-LC3 (Abacus, Brisbane, Australia; catalog #MBPM036; 1:1,000). This procedure was followed by an incubation with the fluorescent Alexa Fluor 488® (“green”) conjugated secondary anti-rabbit IgG (H+L; Invitrogen, catalog #11008; 1:1,000) for 1 h at room temperature.
After final washes with PBS, the coverslips were mounted using an anti-fade mounting solution containing 4′,6-diamidino-2-phenylindole (DAPI; catalog #P36935, Invitrogen), and images were examined and captured using an Olympus AxioObserver Z1 fluorescence microscope (Olympus; Mt. Waverley, VIC, Australia) with a 63× oil objective.
Western Blot Analysis
Preparation of cell lysates and Western blot analysis were performed via established protocols (39). Primary antibodies used include NDRG1 (catalog #ab37897; 1:1,000) from Abcam (Cambridge, UK), LC3 (catalog #MBPM036; 1:2,000) from Abacus (Brisbane, Australia); eIF2α (catalog #9722; 1:2,000), p-eIF2α (catalog #9721; 1:1,000), PERK (catalog #3192; 1:1,000), caspase-4 (catalog #4450; 1:1,000), caspase-3 (catalog #9665; 1:1,000), Bcl-2 (catalog #2876; 1:1,000), and cleaved-PARP (catalog #9541, 1:1,000) from Cell Signaling Technology; p-PERK (catalog #649401; 1:1000) from Australian Biosearch (Balcatta, Australia), and β-actin (catalog #A1978, 1:10,000) from Sigma. The secondary antibodies implemented include horseradish peroxidase-conjugated anti-goat (catalog # A5420; 1:10,000), anti-rabbit (catalog #A6154; 1:10,000), and anti-mouse (catalog #A4416; 1:10,000) antibodies from Sigma. To ensure equal loading of proteins, membranes were probed for β-actin.
Caspase-3/7 Activity
The activity of caspase-3/7 was assessed using the Caspase-Glo 3/7 assay kit (Promega, Madison, WI) using the manufacturer's protocol.
Statistical Analysis
Results are expressed as the means ± S.E. All experiments were performed at least three times and were compared using Student's t test. Data were considered statistically significant when p < 0.05.
RESULTS
NDRG1 Suppresses Iron Chelator-mediated Accumulation of the Autophagic Marker, LC3-II
NDRG1 is a well known stress response gene (12–15), and it has been documented that the survival pathway mediated by autophagy is up-regulated during stress (24). Considering the roles of iron chelation in inducing autophagy (40) and up-regulating the metastasis suppressor, NDRG1 (8, 14, 41), it was important to examine the interrelationships involved, as these have not been investigated. This aim was particularly important, considering that autophagy is a prosurvival response (25), and its regulation by NDRG1 could be relevant for understanding the effect of this latter molecule in suppressing metastasis (1–5).
We have identified a new group of potent thiosemicarbazones, known as the di-2-pyridylketone thiosemicarbazone series (e.g. Dp44mT), that up-regulate NDRG1 (14, 36) to inhibit metastasis (19, 21, 34). Dp44mT has a dual mechanism of action whereby it chelates intracellular metal ions to form cytotoxic redox-active complexes, which leads to cytotoxic reactive oxygen species (ROS) (37, 38, 42). The effects of Dp44mT on cell survival pathways, such as autophagy, remain unknown and are critical to investigate considering that iron depletion up-regulates NDRG1 and induces autophagy (14, 40). Previously it was shown that the iron chelator DFO induces autophagy, whereas other ligands, such as deferiprone and desferasirox, do not (40). However, the molecular mechanisms involved in this response have not been elucidated, and further studies are required to examine the effect of iron deprivation on autophagy and its role in metastasis suppression via NDRG1.
Initial studies were performed to examine the effect of NDRG1 overexpression on LC3-II expression as a function of Dp44mT concentration (Fig. 2, A and B) and incubation time (Fig. 2, C and D). In these experiments we utilized the well characterized PANC1 pancreatic cancer cell line stably overexpressing exogenous human NDRG1 (PANC1 N1) in comparison to their empty vector-transfected control cells (20) (PANC1 VC; Fig. 2). Previous studies have demonstrated that in this model NDRG1 plays an important anti-metastatic role by markedly affecting a variety of signaling pathways (20).
FIGURE 2.

NDRG1 overexpression decreases the classical marker of autophagy, LC3-II, and reduces the number of LC3-II-containing autophagosomes. PANC1 cells were incubated with the iron chelator Dp44mT to examine the effects of NDRG1 overexpression in these cells transfected with the empty vector (PANC1 VC) or NDRG1-containing vector (PANC1 N1). Western blotting was performed to investigate the alteration in expression of LC3-I/II and NDRG1. A, effect of a 24-h, 37 °C incubation of cells with Dp44mT (0.1–5 μm) on NDRG1 and LC3-I/II expression. B, densitometric analysis (arbitrary units) of the results in A. C, effect of incubation time (6–24 h at 37 °C) with 5 μm Dp44mT on NDRG1 and LC3-I/II expression in PANC1 VC and PANC1 N1 cells. D, densitometric analysis (arbitrary units) of the results in C. E, immunofluorescence studies with anti-LC3 antibody in PANC1 VC and N1 cells after a 24-h, 37 °C incubation with control media or this media containing Dp44mT (1 or 5 μm). Scale, 20 μm. The Western analysis and immunofluorescence images shown are typical of three experiments. Densitometric analysis in B and D is the mean ± S.E. (three experiments) normalized to β-actin. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus their respective controls; #, p < 0.05; ##, p < 0.01; ###, p < 0.001 versus their corresponding treatments in PANC1 VC cells.
In agreement with our earlier investigation (20), exogenous expression of FLAG-tagged NDRG1 in PANC1 cells was detected in Western blots as a band of ∼45 kDa (Fig. 2A). In addition, endogenously expressed NDRG1 (that is, NDRG1 expressed from the cell genome) was generally observed at ∼43 and ∼44 kDa (Fig. 2, A and C), indicating possible phosphorylation or cleavage of the protein (43–45). The endogenous 43-kDa band was clearly separated from the exogenous expressed NDRG1 at 45 kDa, enabling the assessment of their levels. In some blots an additional endogenous band appeared between the endogenous 43- and 44-kDa bands and, again, could be due to a post-translational modification. The densitometric analysis shown in this study represents the total of all NDRG1 bands. To examine the effects of iron chelation on NDRG1 and also autophagy, PANC1 VC and N1 cells were incubated with Dp44mT (0.1–5 μm) for 24 h at 37 °C. Dp44mT significantly (p < 0.05) increased the endogenous 44-kDa NDRG1 band relative to the control while having no significant (p > 0.05) effect on the lower 43-kDa endogenous NDRG1 band in the PANC1 VC cells (Fig. 2A). Similar results have been reported in our previous studies (20). Examining PANC1 N1 cells, the FLAG-tagged NDRG1 protein at 45 kDa was markedly expressed under all incubation conditions, and increasing concentrations of Dp44mT had no effect on its expression (Fig. 2A).
As an initial assessment of autophagy after iron chelation, LC3-II levels in cells were assessed by Western blotting. LC3-II is a classical marker of autophagy as it directly correlates with the autophagosome number (46, 47). In PANC1 VC and N1 cells, two bands were found at 18 and 16 kDa, which correspond to LC3-I and LC3-II, respectively (48). The conversion of LC3-I to LC3-II via lipidation is a critical step in the formation of the autophagosome (46). Upon incubation of PANC1 VC and N1 cells with Dp44mT, there was no significant (p > 0.05) increase in LC3-I expression as a function of concentration in each cell type (Fig. 2A). However, there was a slight, general decrease in the relative level of LC3-I in PANC1 N1 cells relative to PANC1 VC cells under most incubation conditions (Fig. 2A). Importantly, there was a marked and significant (p < 0.001–0.01) concentration-dependent increase in LC3-II after incubation with Dp44mT in both cell types, although this was significantly (p < 0.01) greater in PANC1 VC relative to PANC1 N1 cells at Dp44mT concentrations of 1–5 μm (Fig. 2, A and B). This observation indicates NDRG1 expression suppresses LC3-II levels. Similar results in terms of LC3-II expression were also found after 6 h of incubation with this concentration range of Dp44mT (data not shown).
Considering that LC3-II was highly expressed after an incubation of 24 h with Dp44mT at a concentration of 5 μm (Fig. 2A), studies then examined the effect of increasing incubation time (6–24 h) with this agent (5 μm) on NDRG1 and LC3-I/II levels (Fig. 2, C and D). Examining PANC1 VC cells, an increase in both NDRG1 and LC3-II levels was observed as a function of time, with this becoming significant (p < 0.01–0.05) at 24 and 6 h, respectively. Again, in NDRG1 overexpressing PANC1 N1 cells, the levels of LC3-II observed after incubation with Dp44mT were significantly (p < 0.01–0.05) reduced relative to PANC1 VC cells (Fig. 2, C and D).
Next, we directly examined LC3 expression using immunofluorescence to visualize autophagosomes (46) in PANC1 N1 and VC cells incubated for 24 h with control medium or medium containing Dp44mT at 1 or 5 μm (Fig. 2E). Formation of autophagosomes was visualized as green punctate staining in the cytoplasm (46) with the nucleus fluorescing blue using DAPI (21) (Fig. 2E). The level of LC3 staining in autophagosomes (Fig. 2E) revealed similar results to those observed from the Western blot experiments (Fig. 2A) when probed with the same antibody. Indeed, relative to control PANC1 VC cells, incubation with Dp44mT (1 or 5 μm) led to prominent punctate staining of LC3 consistent with its presence in autophagosomes (Fig. 2E). However, the intensity of the punctate LC3 staining seen in PANC1 N1 cells under all conditions was substantially less than that observed after incubation with Dp44mT in PANC1 VC cells (Fig. 2E). The investigations above demonstrate that NDRG1 expression suppresses the levels of LC3-II, suggesting the important role of this metastasis suppressor in inhibiting the pro-survival autophagic pathway in response to cell stressors.
NDRG1 Expression Also Decreases LC3-II Levels in Other Tumor Cell Types
To determine the generality of the above response, studies were also performed using the colonic tumor cell line, HCT116, stably overexpressing exogenous human NDRG1 (HCT116 N1) in comparison to their empty vector-transfected control cells (HCT116 VC; Fig. 3A). Very similar results were observed in HCT116 VC or N1 cells compared with PANC1 cells in terms of NDRG1 and LC3-II levels after incubation with Dp44mT (5 μm) for 24 h. LC3-II expression was significantly (p < 0.01) increased after incubation with Dp44mT in HCT116 VC cells, whereas NDRG1 overexpression in HCT116 N1 cells significantly (p < 0.05) suppressed LC3-II up-regulation (Fig. 3A). Similar observations demonstrating the suppression of LC3-II levels by NDRG1 overexpression were also observed using DU145 prostate cancer cells (data not shown).
FIGURE 3.
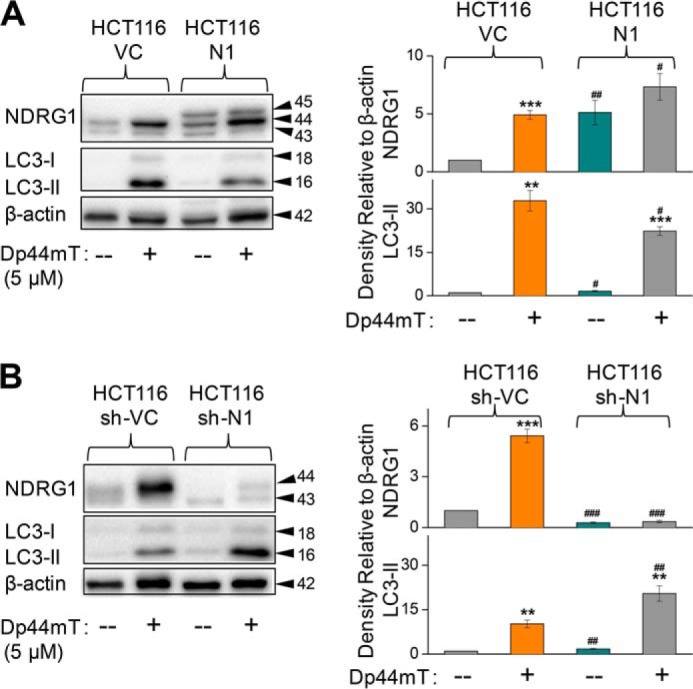
NDRG1 expression also decreases LC3-II levels in other cell types. A, HCT116 cells transfected with the empty vector (HCT116 VC) or NDRG1-containing vector (HCT116 N1) were incubated with control medium or this medium containing Dp44mT (5 μm for 24 h at 37 °C) to examine their effects on NDRG1 and LC3-II expression. B, HCT116 cells transfected with either sh-scrambled (HCT116 sh-VC) or sh-NDRG1 (HCT116 sh-N1) constructs were incubated with control media or this media containing Dp44mT (5 μm for 24 h at 37 °C) to investigate its effect on NDRG1 and LC3-II expression. The Western analysis shown is typical of three experiments. Densitometric analysis in A and B is in arbitrary units and is reported as the mean ± S.E. (three experiments) normalized to β-actin. **, p < 0.01; ***, p < 0.001 versus their respective controls; #, p < 0.05; ##, p < 0.01; ###, p < 0.001 versus their corresponding treatments in HCT116 VC for A or HCT116 sh-VC for B.
To further examine the hypothesis that NDRG1 negatively regulates LC3-II expression, we used an established model of HCT116 cells with stably silenced NDRG1 using a short hairpin (sh) construct against NDRG1 (HCT116 sh-N1; Fig. 3B) (19). These cells were compared with cells transfected with scrambled shRNA (HCT116 sh-VC). In concordance with the results for the NDRG1 overexpression model, the silencing of endogenous NDRG1 (i.e. HCT116 sh-N1 cells) led to a marked and significant (p < 0.001) decrease in NDRG1 expression for both the control and Dp44mT treatments relative to HCT116 sh-VC cells (Fig. 3B). Upon incubation with Dp44mT, silencing of NDRG1 significantly (p < 0.01) increased LC3-II expression in HCT116 sh-N1 cells relative to sh-VC cells treated with this agent (Fig. 3B). These studies indicate that NDRG1 overexpression decreases LC3-II levels in both PANC1 pancreatic and HCT116 colon cancer cells, suggesting that it is not a cell type-specific response.
Dp44mT Initiates and NDRG1 Suppresses Autophagic Flux in Cancer Cells
Because autophagy is a dynamic process (27), the observed increase in LC3-II expression and autophagosome formation upon treatment with Dp44mT (Fig. 2, A–E) could be due to either increased autophagosome formation (autophagic initiation) or decreased lysosomal-dependent breakdown of autophagosomes (autophagic degradation) (46). To determine if these processes are involved in the observed increase in LC3-II and autophagosome levels (Fig. 2E), we utilized a well characterized late-stage autophagic inhibitor, bafilomycin A1 (100 nm) (46, 48). This agent principally affects intra-lysosomal degradation by preventing autophagosome acidification and also autophagosome-lysosome fusion (46). The autophagic inhibitor and Dp44mT (5 μm) were implemented either alone or in combination, and their effects on the levels of LC3-II were investigated in PANC1 N1 cells relative to their PANC1 VC counterparts.
Incubation of PANC1 VC or N1 cells with bafilomycin A1 alone had no significant (p > 0.05) effect on NDRG1 expression relative to the control without this inhibitor (Fig. 4A). In contrast, incubation with Dp44mT alone (5 μm) significantly (p < 0.05) increased NDRG1 expression in VC cells but had not significant effect on its expression in N1 cells (Fig. 4A). Examining LC3-II levels in PANC1 VC cells, Dp44mT alone significantly (p < 0.01) increased its expression relative to the control, whereas bafilomycin A1 alone was significantly (p < 0.001) more effective an increasing the level of LC3-II than Dp44mT alone (Fig. 4A). The increase in LC3-II observed after incubation with bafilomycin A1 is indicative of the basal autophagic flux in cells, as it has been demonstrated that this agent is a late-stage autophagic inhibitor (46, 49). After incubation with the combination of Dp44mT and bafilomycin A1, the increase in LC3-II levels was significantly (p < 0.01) greater than that observed relative to bafilomycin A1 or Dp44mT alone (Fig. 4, A and B). These results indicate the significant (p < 0.01) increase in LC3-II levels observed after combination of both these agents was due to Dp44mT enhancing autophagic initiation.
FIGURE 4.

NDRG1 overexpression suppresses autophagic initiation as shown using the autophagic inhibitors bafilomycin A1 and chloroquine. A, PANC1 VC and PANC1 N1 cells were preincubated with control medium or this medium containing the late stage autophagy inhibitor bafilomycin A1 (100 nm) for 30 min at 37 °C followed by a 24-h, 37 °C incubation with either control medium or this medium containing Dp44mT (5 μm) alone, bafilomycin A1 (100 nm) alone, or the combination of Dp44mT (5 μm) and bafilomycin A1 (100 nm). Western blotting was then performed to assess NDRG1 and LC3-I/II expression. B, densitometric analysis (arbitrary units) of the results in A. C, Western blotting was performed to investigate the alteration in LC3-II and NDRG1 expression upon incubation with the autophagy inhibitor chloroquine (10 μm) and Dp44mT (5 μm) using the same incubation protocol as in A. D, densitometric analysis (arbitrary units) of the results in C. E, immunofluorescence studies with anti-LC3 antibody under the same incubation conditions used in A. Scale, 20 μm. The Western analysis and immunofluorescence images shown are typical of three experiments. Densitometric analysis is the mean ± S.E. (three experiments) normalized to β-actin: *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus their respective controls. #, p < 0.05; ##, p < 0.01; ###, p < 0.001 versus their corresponding treatments in PANC1 VC cells. †, p < 0.05; ††, p < 0.01 versus bafilomycin A1 or chloroquine alone in the respective cell lines.
Examining PANC1 N1 cells under all treatment conditions assessed, the levels of LC3-II were significantly (p < 0.001–0.05) suppressed by NDRG1 overexpression relative to PANC1 VC cells (Fig. 4, A and B). Nonetheless, a similar trend in the increase in LC3-II levels was observed in PANC1 N1 cells relative to the PANC1 VC cells, with LC3-II expression being significantly (p < 0.01–0.05) higher than the control after incubation with either Dp44mT or bafilomycin A1 or particularly the combination (Fig. 4, A and B). The LC3-II expression in PANC1 N1 cells as a result of bafilomycin A1 treatment was significantly (p < 0.01) less than that observed after the same treatment of PANC1 VC cells (Fig. 4, A and B). This observation suggested that NDRG1 suppresses the basal initiation of autophagy. Like the corresponding treatments in VC cells, after incubation of PANC1 N1 cells with the combination of Dp44mT and bafilomycin A1, there was slight but significant (p < 0.05) increase in LC3-II levels relative to bafilomycin A1 or Dp44mT alone (Fig. 4, A and B). These latter data suggest NDRG1 suppresses the Dp44mT-mediated initiation of autophagy.
In additional experiments, another well characterized, late-stage autophagic inhibitor, chloroquine, which affects intra-lysosomal degradation by preventing autophagosome acidification (46, 48), was used to compare the effects observed with bafilomycin A1 (Fig. 4, C and D). Assessing these results, chloroquine or bafilomycin A1 led to similar effects, suggesting Dp44mT stimulates initiation of autophagy and that NDRG1 suppresses this effect (Fig. 4, A–D). However, chloroquine did not inhibit late stage autophagy as much as bafilomycin A1, as suggested by the levels of LC3-II compared with the control after incubation with bafilomycin A1 (Fig. 4, A and B) compared with chloroquine (Fig. 4, C and D). This observation could be explained by the ability of chloroquine to affect only intra-lysosomal degradation via preventing acidification of autolysosomes (46), compared with bafilomycin A1, which also inhibits autophagosome-lysosomal fusion (46, 49).
Considering the findings with bafilomycin A1 and Dp44mT above, immunofluorescence studies (Fig. 4E) were performed under the same experimental conditions as described in Fig. 4A. As found for the Western analysis examining LC3 expression (Fig. 4A), similar results were also observed using immunofluorescence (Fig. 4E). Incubation with bafilomycin A1 (100 nm) alone led to green punctate staining that was markedly more intense than that found for either untreated control cells or cells incubated with Dp44mT alone (5 μm; Fig. 4E). The number of LC3-II-containing green puncta observed after incubation with bafilomycin A1 represents the basal level of autophagic flux (46, 48). In good agreement with the Western results (Fig. 4, A and B), incubation with the combination of bafilomycin A1 (100 nm) and Dp44mT led to markedly greater green LC3 punctate staining than that observed with bafilomycin A1 or Dp44mT alone and was consistent with marked autophagosome formation (Fig. 4E). In addition, these immunofluorescence studies demonstrate the intensity of punctate LC3 staining in N1 cells was substantially less than that observed in the corresponding treatments for VC cells (Fig. 4E). Again, these results suggest that the basal autophagic flux is suppressed by NDRG1 overexpression.
DFO, but Not Its Iron Complex, Increases LC3-II, Whereas Dp44mT and Its Iron and Copper Complexes Elevate LC3-II Expression
Studies were then initiated to investigate the mechanism through which Dp44mT mediates its effects on autophagic pathway. Dp44mT is a potent iron chelator that displays marked membrane permeability and iron chelation efficacy in terms of inducing cellular iron efflux and inhibiting iron uptake from the serum iron-binding protein, transferrin (38). However, once the ligand binds intracellular iron it becomes redox-active, and its effects can also be due to the generation of ROS (37, 38). In contrast, DFO shows lower membrane permeability and iron chelation efficacy than Dp44mT due to its lower lipophilicity (38, 50, 51), and once it has bound iron, the complex is redox-inactive and does not generate ROS (37).
To dissect the mechanisms involved in the up-regulation of LC3-II expression observed with Dp44mT and the potential roles of iron chelation in this process, studies were initiated to compare the effects of DFO to Dp44mT. In all studies, DFO was used at 250 μm due to its lower permeability and iron chelation efficacy (50, 51), whereas Dp44mT was examined at 5 μm. In addition, we also compared the effect of Dp44mT to its structurally related analog, Dp2mT (Fig. 1C), which has been specifically designed so that it does not chelate iron or other metals and acts as an appropriate negative control compound (21, 38).
Similar to the results found using Dp44mT and as demonstrated in previous studies (20), DFO significantly (p < 0.01) increased NDRG1 expression (upper, ∼44-kDa band only) in PANC1 VC cells relative to the control (Fig. 5A). As evident after incubation of PANC1 N1 cells with Dp44mT (Fig. 2, A and C), DFO had no significant (p > 0.05) effect on NDRG1 expression due to the marked exogenous NDRG1 levels at 45 kDa (Fig. 5A). In both PANC1 VC and N1 cells, Dp2mT (5 μm) had no significant (p > 0.05) effect on NDRG1 expression relative to their respective controls (Fig. 5A). Examining LC3-II expression in PANC1 VC cells, both Dp44mT and DFO significantly (p < 0.01) increased LC3-II levels, although Dp44mT was significantly (p < 0.01) more effective than DFO. In contrast, Dp2mT had no significant (p > 0.05) effect on LC3-II expression in PANC1 VC cells (Fig. 5A). NDRG1 overexpression significantly (p < 0.05) suppressed the increase in LC3-II levels mediated by Dp44mT or DFO, although again, Dp2mT had no significant effect on LC3-II expression (Fig. 5, A and B). Collectively, considering that Dp2mT cannot bind cellular iron (21, 38) and that DFO binds cellular iron without ROS generation (37), these studies demonstrated that iron deprivation was involved in increasing LC3-II expression after incubation with chelators. These results were confirmed using immunofluorescence studies where 24 h of incubation with either Dp44mT or DFO increased LC3-II punctate staining in cells relative to the control in both PANC1 VC and PANC1 N1 cells, although again, NDRG1 suppressed LC3-II expression with both chelators (Fig. 5C).
FIGURE 5.

Iron chelation by DFO increases LC3-II levels and LC3-II-containing autophagosome formation. A, PANC1 VC and N1 cells were incubated for 24 h at 37 °C with Dp2mT (5 μm; a non-metal-binding analog of Dp44mT (37, 38, 42)), Dp44mT (5 μm), or DFO (250 μm), and Western blotting was performed to examine their effect on NDRG1, LC3-I, or LC3-II levels. B, densitometric analysis (arbitrary units) of blots in A. C, immunofluorescence studies with anti-LC3 antibody using PANC1 VC and N1 cells after a 24-h, 37 °C incubation with Dp44mT (5 μm) or DFO (250 μm). Scale, 20 μm. The Western analysis and immunofluorescence images shown are typical of three experiments. Densitometric analysis is the mean ± S.E. (three experiments) normalized to β-actin: *, p < 0.05; **, p < 0.01 versus their respective controls. #, p < 0.05; ##, p < 0.01 versus their corresponding treatments in PANC1 VC cells.
Studies were then initiated to dissect the potential roles of iron deprivation and ROS generation in the increased LC3-II levels observed after incubation with Dp44mT (Figs. 2–5). Initial experiments compared the effects of Dp44mT and DFO to their respective iron complexes, which are redox-active and redox-inactive, respectively (37) (Fig. 6, A–F). Saturation of Dp44mT or DFO with iron leads to complexes that do not deplete cellular iron pools, and hence, these are relative controls to the ligands alone. To satisfy the coordination modes of these ligands to iron (27), ligand:metal ratios of 2:1 and 1:1 were used for Dp44mT and DFO, respectively. In the case of Dp44mT, we also examined the effects of its copper(II) complex (Cu2+-Dp44mT) at a metal:ligand molar ratio of 1:1 to satisfy the coordination geometry of this interaction (52). This was also important to assess, as Cu2+-Dp44mT plays a major role in the anti-proliferative efficacy of Dp44mT (53).
FIGURE 6.

Both iron chelation and redox activity leads to the greater efficacy of Dp44mT at increasing LC3-II levels relative to DFO which only chelates cellular iron. A, PANC1 VC and N1 cells were incubated for 24 h at 37 °C with control medium or this medium containing Dp44mT (5 μm), Fe3+-(Dp44mT)2 (5 μm), Cu2+-(Dp44mT) (5 μm), Fe3+ (as FeCl3; 2.5 μm), or Cu2+ (as CuCl2; 5 μm), and Western blotting performed to examine their effect on NDRG1, LC3-I or LC3-II levels. B, densitometric analysis (arbitrary units) of blots in A. C, PANC1 VC and N1 cells were incubated for 24 h at 37 °C with Dp44mT (1 μm), Cu2+-Dp44mT (1 μm), or Cu2+ (1 μm), and Western blotting was performed to examine their effect on NDRG1, LC3-I, or LC3-II levels. D, densitometric analysis (arbitrary units) of blots in C. E, PANC1 VC and N1 cells were incubated for 24 h at 37 °C with control medium or this medium containing DFO (250 μm), Fe3+-DFO (250 μm), or Fe3+ (as FeCl3; 250 μm), and Western blotting was performed to examine their effect on NDRG1, LC3-I, or LC3-II levels. F, densitometric analysis (arbitrary units) of blots in E. G, PANC1 VC and N1 cells were incubated for 24 h at 37 °C with control medium or this medium containing NAC (10 mm), Dp44mT (5 μm), or the combination of these agents, and Western blotting was performed to examine their effect on NDRG1, LC3-I, or LC3-II levels. H, densitometric analysis (arbitrary units) of blots in G. The Western analysis shown is typical of three experiments. Densitometric analysis is the mean ± S.E. (three experiments) normalized to β-actin: *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus their respective controls. #, p < 0.05; ##, p < 0.01; ###, p < 0.001 versus their corresponding treatments in PANC1 VC cells. †, p < 0.05; ††, p < 0.01 compared with cells incubated with Dp44mT alone in their respective cell lines.
Assessing PANC1 VC cells, both Dp44mT and its iron complex (Fe3+-(Dp44mT)2) significantly (p < 0.01–0.05) increased NDRG1 and LC3-II expression relative to the control (Fig. 6, A and B). The Cu2+-Dp44mT complex (5 μm) had no marked effect on NDRG1 but significantly (p < 0.05) increased LC3-II expression relative to the control, although at this concentration the agent was potently cytotoxic. Hence, further experiments were performed using lower levels (1 μm; described in Fig. 6, C and D, below). We observed a significant (p < 0.001–0.05) increase in the levels of LC3-II in PANC1 VC cells incubated with either Dp44mT (1 μm) or its copper complex (1 μm; Fig. 6, C and D). As relevant controls to the metal-containing complexes, both Fe3+ and Cu2+ were added at stoichiometric equivalent concentrations and had no effect on the expression of all proteins compared with the relative controls using PANC1 VC and PANC1 N1 cells (Fig. 6, A–D). In PANC1 N1 cells, both Dp44mT and Fe3+-(Dp44mT)2 had no effect on NDRG1 expression relative to the control but significantly (p < 0.001–0.01) increased LC3-II (Fig. 6, A and B). At the higher concentrations (5 μm), Cu2+-Dp44mT markedly reduced NDRG1 overexpression in PANC1-N1 cells (Fig. 6, A and B), which is consistent with its cytotoxic activity. In contrast, no significant (p > 0.05) effect of Cu2+-Dp44mT on NDRG1 overexpression was observed at a concentration of 1 μm (Fig. 6, C and D). In PANC N1 cells, NDRG1 overexpression significantly (p < 0.001–0.05) suppressed the increase in LC3-II levels observed with Dp44mT, Fe3+-(Dp44mT)2, or Cu2+-Dp44mT compared with corresponding treatments in PANC1 VC cells (Fig. 6, A–D).
Experiments then examined the effect of DFO (250 μm), which significantly (p < 0.05) increased NDRG1 but also LC3-II levels in PANC1 VC cells (Fig. 6, E and F). In contrast, Fe3+-DFO had no significant effect at increasing NDRG1 or LC3-II relative to the control in PANC1 N1 cells. Additionally, compared with the relative controls, the Fe3+-DFO complex did not significantly (p > 0.05) affect NDRG1 or LC3-II expression in PANC1-VC or PANC1 N1 cells (Fig. 6, E and F). These studies indicate that iron chelation is required to induce LC3-II expression, whereas for Dp44mT, the generation of its redox-active iron complex can also increase NDRG1 and LC3-II, and this ability may explain its greater effect relative to DFO.
NAC Decreases the Effect of Dp44mT on Inducing LC3-II Levels
To further elucidate the role of redox activity of Dp44mT in the up-regulation of LC3-II, studies were conducted to examine the effect of the anti-oxidant, N-acetylcysteine (NAC; 10 mm), which effectively inhibits the cytotoxic redox activity of Dp44mT (53). As shown previously (Fig. 2A), Dp44mT (5 μm) markedly and significantly (p < 0.01) increased LC3-II levels relative to the control, whereas NAC alone had no significant (p > 0.05) effect (Fig. 6, G and H). Incubation of PANC1 VC cells with the combination of Dp44mT and NAC significantly (p < 0.01) decreased LC3-II expression relative to Dp44mT alone. The same trend of NAC, significantly (p < 0.05) decreasing LC3-II expression in the presence of Dp44mT, was also observed in PANC1 N1 cells (Fig. 6, G and H). However, as shown previously, NDRG1 overexpression significantly (p < 0.05) reduced the Dp44mT-induced up-regulation of LC3-II in PANC1 N1 cells relative to PANC1 VC cells (Fig. 6, G and H). These results are consistent with the hypothesis that Dp44mT-mediated redox activity plays an important role in increasing LC3-II expression. Furthermore, these observations explain the increased efficacy of Dp44mT at inducing autophagy relative to DFO, which does not form redox-active complexes.
NDRG1 Suppresses the PERK/eIF2α ER Stress Pathway
The ER stress pathway has been previously shown to play a vital role in the regulation of autophagy via the PERK/eIF2α axis (29, 31). To determine the role of this pathway in Dp44mT-mediated autophagic initiation, we examined the levels of key proteins (i.e. PERK and eIF2α) involved in this process (Fig. 7). Both these proteins are activated by phosphorylation (54), and hence, their phosphorylated and total levels were assayed to study the effect of NDRG1 and Dp44mT on the ER stress pathway. Incubation of PANC1 VC cells with Dp44mT (5 μm) for 24 h significantly (p < 0.001) increased p-PERK levels compared with control cells, whereas there was no significant (p > 0.05) effect on total PERK expression (Fig. 7, A and B). As found for studies examining LC3-II levels (Fig. 2, A–D), NDRG1 overexpression in PANC1 N1 cells significantly (p < 0.01) suppressed Dp44mT-mediated phosphorylation of PERK relative to the corresponding treatment of PANC1 VC cells (Fig. 7, A and B). There was a slight but significant (p < 0.05) suppression of total PERK levels in PANC1 N1 cells incubated with or without Dp44mT (Fig. 7, A and B).
FIGURE 7.

PERK/eIF2α is involved in the NDRG1-mediated suppression of the autophagic pathway. A, PANC1 VC and N1 cells were incubated for 24 h at 37 °C in the presence or absence of Dp44mT (5 μm) to examine its effects on the levels of NDRG1, p-PERK, total PERK, p-eIF2α, total eIF2α, LC3-I, and LC3-II. B, densitometric analysis (arbitrary units) of the blots in A. C, HCT116 sh-VC and sh-N1 cells were incubated for 24 h at 37 °C in the presence or absence of Dp44mT (5 μm) to examine its effects on the levels of NDRG1, p-PERK, total PERK, p-eIF2α, total eIF2α, LC3-I, and LC3-II. D, densitometric analysis (arbitrary units) of the blots in C. E, PANC1 VC cells were preincubated for 48 h at 37 °C with either scrambled (Scr) or PERK siRNA. Cells were then subsequently incubated with Dp44mT (5 μm) for 24 h at 37 °C, and Western analysis performed to investigate levels of NDRG1, total PERK, p-eIF2α, total eIF2α, LC3-I, and LC3-II. F, densitometric analysis (arbitrary units) of the blots in E. The Western analysis shown is typical of three experiments. Densitometric analysis is the mean ± S.E. (three experiments) normalized to β-actin: *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus their respective controls. #, p < 0.05; ##, p < 0.01; ###, p < 0.001 versus their corresponding treatments in PANC1 VC for A or E or HCT116 sh-VC for C cells; ††, p < 0.01; †††, p < 0.001 versus their corresponding treatments in cells treated with the scrambled control.
Phosphorylation of eIF2α, which is a downstream target of PERK (55), was also significantly (p < 0.05) increased by Dp44mT compared with the control in PANC1 VC cells (Fig. 7, A and B). Importantly, NDRG1 overexpression in PANC1 N1 cells significantly (p < 0.05) reduced the Dp44mT-mediated increase of p-eIF2α levels (Fig. 7, A and B). However, in contrast, NDRG1 overexpression led to no significant (p > 0.05) alteration in total eIF2α levels (Fig. 7, A and B). These results demonstrate that Dp44mT can initiate the ER stress pathway via the PERK/eIF2α axis, and NDRG1 suppresses this pathway. Hence, these data can explain the observed effects of Dp44mT and NDRG1 on the initiation and suppression of autophagy, respectively.
To further elucidate the role of NDRG1 in suppressing PERK/eIF2α pathway, we used an established model of HCT116 cells with stably silenced NDRG1 (HCT116 sh-N1; Fig. 7, C and D) as shown previously in Fig. 3B. These cells were compared with cells transfected with scrambled shRNA (HCT116 sh-VC). Silencing of NDRG1 significantly (p < 0.01) increased the levels of phosphorylated PERK in HCT116 sh-N1 cells relative to the corresponding treatment (control and Dp44mT) in sh-VC cells (Fig. 7, C and D). Incubation of cells with Dp44mT also led to a significant (p < 0.01–0.05) increase in total PERK levels in both HCT116 sh-VC and sh-N1 cells compared with their respective controls. Furthermore, we observed significantly (p < 0.001) increased levels of p-eIF2α and the classical autophagy marker, LC3-II, in HCT116 sh-N1 cells treated with Dp44mT compared with corresponding treatment in HCT116 sh-VC. No significant change (p > 0.05) in levels of total eIF2α expression was observed under all incubation conditions. Together, these studies indicate that NDRG1 suppresses the PERK/eIF2α ER stress pathway. As this pathway is known to regulate autophagy, these results demonstrate a possible mechanism via which NDRG1 suppresses Dp44mT-mediated autophagic initiation.
Dp44mT Increases LC3-II Initiation via Its Effect on the PERK/eIF2α Axis
To determine the role of PERK/eIF2α pathway in Dp44mT-mediated autophagic initiation, we studied the effect of silencing PERK via transient transfection of siRNA in PANC1 VC cells relative to transfection of a scrambled control (Fig. 7, E and F). Silencing of PERK led to a marked and significant (p < 0.001–0.01) reduction in the expression of total PERK relative to the scrambled PANC1 VC control cells. As shown previously (Fig. 7, A and B), NDRG1 overexpression in PANC1 N1 cells led to a significant (p < 0.01–0.05) decrease in total PERK levels (Fig. 7, E and F) as compared with corresponding treatments in PANC1 VC cells.
We then examined the effect of PERK silencing on the downstream target of PERK, namely eIF2α (Fig. 7, E and F). In good agreement with studies in Fig. 7, A and B, Dp44mT significantly (p < 0.05) increased the levels of p-eIF2α relative to the control, and silencing PERK largely inhibited this effect (Fig. 7, E and F). Moreover, PERK silencing also slightly decreased endogenous p-eIF2α levels relative to the scrambled control. As shown previously, NDRG1 overexpression in PANC1 N1 cells also significantly (p < 0.01) prevented the Dp44mT-mediated increase in p-eIF2α levels relative to the corresponding treatment in PANC1 VC cells (Fig. 7, E and F). In contrast, there was no significant (p > 0.05) effect of either PERK silencing or NDRG1 overexpression on total eIF2α levels.
Next, we assessed the effect of PERK silencing on Dp44mT-mediated increase in LC3-II levels in PANC1 VC cells (Fig. 7, E and F). In these studies silencing of PERK significantly (p < 0.01) reduced the Dp44mT-mediated increase in LC3-II expression relative to the scrambled control. Similarly, NDRG1 overexpression in PANC1 N1 cells also showed a marked and significant (p < 0.001) reduction in LC3-II levels in the presence and absence of Dp44mT relative to PANC1 VC cells (Fig. 7, E and F). Collectively, the PERK/eIF2α ER stress pathway plays an important role in the Dp44mT-mediated induction of autophagy.
NDRG1 Suppresses Tunicamycin- and Serum Starvation-mediated Accumulation of the ER-Stress Marker, eIF2α, and the Autophagic Marker, LC3-II
Considering the effects of NDRG1 on Dp44mT-mediated ER stress and autophagy, we also examined whether NDRG1 affects the general cellular response to other classical stress stimuli. Tunicamycin (Tm) is well characterized to induce ER stress and autophagy via the PERK/eIF2α axis through its ability to inhibit N-linked glycosylation (29, 55). As shown previously (Fig. 2, A–D), incubation of PANC1 VC cells with Dp44mT significantly (p < 0.05) increased NDRG1 expression compared with the control (Fig. 8, A and B). However, Tm did not have any significant (p > 0.05) effect on NDRG1 expression. In PANC1 N1 cells, incubation with Dp44mT had no significant effect on NDRG1 levels compared with the PANC1 N1 control, whereas Tm (2.5 and 5.0 μg/ml) slightly, but significantly (p < 0.01), decreased NDRG1 expression relative to the control (Fig. 8, A and B).
FIGURE 8.

NDRG1 suppresses Tm- or serum starvation-mediated accumulation of the ER stress marker p-eIF2α and the autophagic marker LC3-II. PANC1 VC and N1 cells were incubated with either Dp44mT, Tm, or serum-free medium to examine the effects of NDRG1 overexpression on the levels of p-eIF2α, total eIF2α, LC3-I, or LC3-II by Western analysis or immunofluorescence assessing LC3-II containing autophagosomes. A, effect of a 24-h, 37 °C incubation with either control medium or this medium containing Dp44mT (5 μm) or Tm (2.5 or 5 μg/ml). B, densitometric analysis (arbitrary units) of blots in A. C, immunofluorescence studies with anti-LC3 antibody using PANC1 VC and N1 cells after a 24-h, 37 °C incubation with control medium or this medium containing Tm (5 μg/ml). Scale, 20 μm. D, effect of a 0.5–2-h, 37 °C incubation with serum-free medium (0.5–2 h). E, densitometric analysis (arbitrary units) of blots in D. F, immunofluorescence studies with anti-LC3 antibody using cells incubated with serum-free medium for 1 h at 37 °C relative to the control (0-h time point; no starvation). The Western analysis and immunofluorescence images shown are typical of three experiments. Densitometric analysis is the mean ± S.E. (three experiments) normalized to β-actin: *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus their respective controls. #, p < 0.05; ##, p < 0.01; ###, p < 0.001 versus their corresponding treatments in PANC1 VC cells.
As found for Dp44mT, incubation of PANC1 VC cells with Tm significantly (p < 0.01) increased p-eIF2α and LC3-II levels compared with the control (Fig. 8, A and B). However, although Tm acted similarly to Dp44mT in terms of its effect on p-eIF2α levels, it was significantly (p < 0.05) less active than the chelator at increasing LC3-II expression. Neither Dp44mT nor Tm had any significant effect on total eIF2α in either PANC1 VC or PANC1 N1 cells. Examining PANC1 N1 cells, NDRG1 overexpression significantly (p < 0.01–0.05) suppressed the increase in the levels of p-eIF2α and LC3-II mediated by Dp44mT or Tm (Fig. 8, A and B). Considering the Western blot results assessing Tm in Fig. 8, A and B, immunofluorescence studies examining LC3 staining were then performed (Fig. 8C). Incubation of PANC1 VC cells with Tm (5.0 μg/ml) led to green punctate staining consistent with LC3-containing autophagosomes (Fig. 8C). In contrast, upon incubation of PANC1 N1 cells with Tm, markedly less green punctate LC3 staining was observed relative to PANC1 VC cells. These results indicated that NDRG1 acts to suppress the stress stimulus from Tm in a similar way as that observed with Dp44mT, which led to suppressed expression of classical markers of ER stress and autophagy.
The data above indicate that NDRG1 had similar effects at suppressing the cellular response to very different stress stimuli, namely Dp44mT and Tm. To examine the hypothesis that the observed suppressive effects of NDRG1 on the ER stress response and autophagy was a general response, we assessed the effect of serum starvation. Starvation of serum induces the ER stress pathway by activating eIF2α phosphorylation and decreasing ATP levels and, thus, also stimulates autophagy (56). Hence, PANC1 VC and N1 cells were serum-starved over incubations of 0.5, 1, and 2 h, which have been previously well characterized to induce autophagy (46). Serum starvation of PANC1 VC cells over 1–2 h slightly but significantly (p < 0.05) increased expression of the 43-kDa NDRG1 band compared with the respective control (0 h; Fig. 8, D and E). In contrast, this treatment had no significant (p > 0.05) effect on the expression of the endogenous or exogenous NDRG1 bands at 43 and 45 kDa in PANC1 N1 cells.
After serum starvation of PANC1 VC cells for 0.5 and 1 h, p-eIF2α levels were significantly (p < 0.05) enhanced relative to the control (0 h; Fig. 8, D and E). Moreover, the level of p-eIF2α was highest after serum starvation of 1 h, there being a relative decrease at 2 h of starvation where the p-eIF2α levels were only slightly greater (p > 0.05) than the control (Fig. 8, D and E). When comparing PANC1 VC to N1 cells, p-eIF2α levels were significantly (p < 0.01–0.05) suppressed by NDRG1 overexpression at all time points (Fig. 8, D and E). Serum starvation had no effect on total eIF2α under all conditions.
Examining the autophagy marker, LC3, serum starvation of PANC1 VC cells led to significantly (p < 0.01) increased LC3-II levels after 1 h compared with the control (Fig. 8, D and E). No significant difference in LC3-II (p > 0.05) expression was observed at either 0.5 or 2 h compared with the control (0 h). Overexpression of NDRG1 in PANC1 N1 cells led to significant (p < 0.05) suppression of LC3-II expression at the 1-h time point relative to that found in PANC1 VC cells (Fig. 8, D and E). These findings after serum starvation for 1 h were also evident using immunofluorescence, where PANC1 VC cells showed increased LC3 punctate staining relative to the control cells (Fig. 8F). Moreover, NDRG1 overexpression in PANC1 N1 cells markedly suppressed LC3 punctate staining after starvation relative to PANC1 VC under these conditions (Fig. 8F). In summary, NDRG1 suppresses the response to stress induced by serum starvation, leading to decreased expression of LC3-II. Thus, the effect of NDRG1 at reducing autophagy and ER stress markers appears consistent among a variety of stress stimuli.
NDRG1 Expression Leads to Increased Susceptibility of PANC1 Cells to Dp44mT-induced Apoptosis
The studies above indicate that NDRG1 inhibits autophagy after a range of stress stimuli in a similar way to that observed with Dp44mT. As inhibition of autophagy would result in a decrease in the recycling of nutrients required for cellular metabolism and growth, it may be expected that there could be an increase in apoptosis under these conditions (28). This could explain the anti-metastatic activity of NDRG1. To assess this, PANC1 VC and PANC1 N1 cells were incubated for 24 h at 37 °C with a range of Dp44mT concentrations (5–50 μm), and the effect on the expression of a series of classical apoptotic markers (57) was assessed.
To examine the effect of Dp44mT and NDRG1 on apoptosis, we examined the protein levels of active caspases 3 and 4. The levels of caspase-3 cleavage product (at 19 kDa) and caspase-4 were significantly (p < 0.001–0.05) increased under all conditions (control and all Dp44mT concentrations) in PANC1 N1 compared with control PANC1 VC cells (Fig. 9, A and B). We then examined the levels of cleavage product of poly(ADP-ribose) polymerase (PARP), which is a known target for caspase-3 in vivo and is a classical marker of caspase-3 activity (58). The increased levels of cleaved PARP were only observed in PANC1 VC cells at a Dp44mT concentration of 25–50 μm (p < 0.05), whereas PARP cleavage was significantly (p < 0.01–0.001) increased in PANC1 N1 cells at a Dp44mT concentration of 15–50 μm (Fig. 9, A and B). There were marked and significantly (p < 0.001) higher levels of PARP in PANC1 N1 cells incubated with 50 μm Dp44mT compared with PANC1 VC. Moreover, we demonstrated that caspase-3/7 activity was significantly (p < 0.05) higher in PANC1 N1 cells compared with PANC1 VC cells incubated with 50 μm Dp44mT (Fig. 9C). These results indicate higher susceptibility of PANC1 N1 cells toward apoptosis.
FIGURE 9.

NDRG1 expression in PANC1 N1 cells enhances apoptotic marker expression in the presence of Dp44mT relative to PANC1 VC cells. A, PANC1 VC and N1 cells were incubated with either control medium (control) or this medium containing Dp44mT (5–50 μm) for 24 h at 37 °C to examine the effects of NDRG1 overexpression on the levels of caspase-3 (and the caspase-3 cleavage product), caspase-4, cleaved PARP, and anti-apoptotic Bcl-2. B, densitometric analysis (arbitrary units) of blots in A. C, caspase-3/7 activity after incubating PANC1 VC or PANC1 N1 cells with medium containing Dp44mT (25 or 50 μm) relative to cells incubated with control medium after a 24-h, 37 °C incubation. The Western analysis shown is typical of three experiments. Densitometric analysis is the mean ± S.E. (three experiments) normalized to β-actin. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus control PANC1 VC; #, p < 0.05 versus its corresponding treatments in PANC1 VC cells.
We further examined the effect of these incubation conditions on the expression of Bcl-2, which plays a role as an apoptosis antagonist (59). We observed using PANC1 VC cells that Bcl-2 levels were initially increased relative to the control upon incubation with lower concentrations of Dp44mT (5–10 μm; Fig. 9, A and B). The levels of Bcl-2 then subsequently decreased but remained significantly (p < 0.01–0.05) greater than the control at Dp44mT concentrations of 15 and 50 μm (Fig. 9, A and B). Bcl-2 was significantly (p < 0.001–0.05) decreased under all incubation conditions in PANC1 N1 cells relative to PANC1 VC cells (Fig. 9, A and B). In conclusion, these studies indicate that NDRG1 increases the induction of apoptosis after incubation with Dp44mT.
DISCUSSION
Autophagy plays a critical role in cellular metabolism and has also been shown to be an important process in cancer pathobiology depending upon the stage of the tumor (23, 26). Moreover, autophagy has been shown to be induced as a protective mechanism after stress stimuli (24). Interestingly, autophagy has also been linked to the process of metastasis due to its pro-survival role that is mediated through its ability to recycle crucial nutrients (22, 60). The metastasis suppressor, NDRG1, has been shown to play a critical role in regulating cellular signaling (18–21), but its role in autophagy has not been defined. For the first time in this investigation NDRG1 has been demonstrated to inhibit autophagy-induced by a range of ER stress stimuli mediated through a mechanism involving suppression of the PERK/eIF2α axis of the unfolded protein response.
In this study, we have primarily examined the effect of the recently developed anti-tumor agent, Dp44mT, that chelates intracellular metal ions to form cytotoxic, redox-active iron and copper complexes (37, 38, 53) on ER stress-mediated autophagy. Intriguingly, this ligand acts to markedly up-regulate NDRG1 and has been shown to potently inhibit tumor cell growth and metastasis in vitro and in vivo (19, 21, 34). Upon incubation with Dp44mT in a variety of tumor cell types, a marked and significant increase in the expression of both NDRG1 and the classical marker of autophagy, LC3-II, was observed.
Considering NDRG1 functions as a stress response gene (12–15) and because autophagy is also known to be up-regulated during stress (24), we examined the nature of the relationship between Dp44mT-induced NDRG1 and LC3-II expression. In these studies we utilized NDRG1 overexpression and silencing models. Dp44mT initially induces NDRG1 and LC3-II levels and thus autophagic initiation after a 24-h incubation. In contrast, NDRG1 overexpression reduces Dp44mT-induced LC3-II levels over this time frame (Fig. 2). Although these observations may initially appear contradictory, it is important to note that the ability of Dp44mT to induce LC3-II/autophagy is suppressed by endogenous NDRG1 expression. This was demonstrated by showing that the silencing of endogenous NDRG1 (sh-N1) in cells incubated with Dp44mT leads to increased LC3-II levels relative to the sh-scrambled control (sh-VC; Fig. 3B). Therefore, whereas NDRG1 and LC3-II are concurrently up-regulated by Dp44mT, NDRG1 nonetheless acts a suppressor of LC3-II expression/autophagy under conditions of stress. This can be rationalized by the emerging notion that NDRG1 is a stress-response protein, and consequently, its up-regulation works to suppress stress-induced LC3-II expression and autophagy.
To investigate the stage of autophagic pathway at which these chelators have their effect, we employed late stage autophagy inhibitors bafilomycin A1 or chloroquine (46, 48). By blocking the degradation stage of autophagy with bafilomycin A1 and chloroquine (46, 48) it was found that the increase in LC3-II levels observed after incubation with Dp44mT was due to increased synthesis rather than a decrease in degradation of LC3-II containing autophagosomes. Using these inhibitors we also demonstrated that NDRG1 suppresses both basal as well as Dp44mT-induced autophagic initiation (Fig. 4).
Previous studies have shown that Dp44mT exerts its anti-metastatic effects via a “double punch” mechanism, whereby this agent forms redox active iron or copper complexes resulting in the generation of free radicals (37, 38, 53). Additional experiments were initiated to understand the role of both iron depletion and redox activity in Dp44mT-mediated autophagic up-regulation. The formation of redox active Dp44mT metal complexes (37, 53) leads to hydroxyl radical generation from hydrogen peroxide (61). Our current studies show that the anti-oxidant NAC decreases the efficacy of Dp44mT at inducing the downstream indicator of ER stress, LC3-II, in PANC1 VC and PANC1 N1 cells (Fig. 6G). A previous investigation from our laboratory also demonstrated the efficacy of NAC at inhibiting the redox activity of Dp44mT-metal complexes (53). Thus, peroxide-generated hydroxyl radicals in part induce ER stress and autophagy observed after incubation with Dp44mT. However, as NAC does not totally prevent the up-regulation of LC3-II (Fig. 6G), this suggests that iron depletion by Dp44mT also leads to ER stress and up-regulated LC3-II. Hence, the combined effect of Dp44mT in depleting cellular iron and generating redox-active complexes is important in inducing ER stress. The role of iron depletion alone in increasing ER stress-mediated LC3-II expression was demonstrated using DFO. In these experiments, DFO, which binds cellular iron to form redox-inactive complexes, induced ER stress-mediated LC3-II expression, whereas its iron complex did not (Fig. 6E). Previous studies have shown that hydrogen peroxide initiates ER stress (as demonstrated by increased p-eIF2α), whereas incubation with NAC reduced elevated p-eIF2α levels (62). In summary, redox activity resulted in ER stress, which can be prevented by anti-oxidants such as NAC, but iron depletion alone is also sufficient to induce ER stress-mediated LC3-II expression.
A variety of stress stimuli including tunicamycin, serum starvation, and iron depletion mediated by DFO also demonstrated a similar response in terms of the decrease of LC3-II levels by NDRG1 expression. These later observations indicated the effect of NDRG1 on potentially suppressing the autophagic pathway was not only limited to Dp44mT-mediated stress. Of relevance, previous studies have demonstrated that c-Myc and N-myc promote ER stress-mediated autophagy (29), and both of these proto-oncogenes are known to inhibit NDRG1 expression (63). However, this previous work by others did not elucidate the downstream mechanism described herein, indicating that NDRG1 suppresses autophagy via inhibition of an ER stress pathway.
To understand the mechanism involved in NDRG1-mediated suppression of autophagy, our studies focused initially on the PERK-eIF2α axis of the unfolded protein response that plays a role in regulating autophagy (29, 31). We demonstrated that Dp44mT increases the phosphorylation of PERK/eIF2α, whereas NDRG1 overexpression decreased phosphorylation of these proteins. In contrast, silencing of NDRG1 led to increased PERK phosphorylation (Fig. 7, C and D). This observation may be explained by previous studies demonstrating that c-myc and N-myc promote ER stress-mediated autophagy (29), potentially by down-regulation of NDRG1. Furthermore, inhibition of the PERK/eIF2α axis by silencing PERK showed similar effects as NDRG1 overexpression. That is, there was a decrease in LC3-II levels in response to incubation with Dp44mT. These observations illustrate the importance of PERK and eIF2α in NDRG1-mediated suppression of the autophagic response.
There are >20 well characterized metastasis suppressor proteins (64, 65), but their interactions with important cell survival pathways such as autophagy have not been comprehensively investigated. However, studies assessing the metastasis suppressor, KAI1, have demonstrated that its overexpression induces autophagy, which is in direct contrast to the current work examining NDRG1 (66). Hence, these investigations indicate the differential effect of metastasis suppressors on autophagy that could be dependent on tumor stage, tumor type, and tumor microenvironment.
There has been increasing evidence suggesting an important role for autophagy in promoting metastatic progression, particularly in established tumors (60). Clinical studies have shown a positive correlation between increased autophagy and patients suffering from highly metastatic pancreatic and colon cancer with a poor prognosis (67, 68). A significant recent investigation has demonstrated a critical role of ROCK1 kinase in the regulation of beclin1-mediated autophagy (69). Our previous studies have shown that NDRG1 mediates its metastatic suppressor functions via suppression of ROCK1 kinase (19). Furthermore, the NDRG1 target, neural-precursor-cell-expressed developmentally down-regulated 4 (Nedd4; Ref. 20), polyubiquitinates the autophagy initiator, Beclin-1 (70), leading to its proteasomal degradation and, thus, inhibition of autophagy. These studies suggest a potential role of ROCK1 kinase and Nedd4 in the mechanism of autophagic suppression via NDRG1 documented herein.
A number of other investigations have demonstrated the importance of autophagy in metastatic progression via modulation of key steps involved in the metastasis-invasion cascade. Studies have shown that autophagy plays a protective role in cells undergoing anoikis due to extracellular matrix detachment, as observed in metastatic cells (71). Other reports suggest involvement of the autophagic inducer, β-1 integrin, in the survival of dormant cells (72). These dormancy characteristics are usually observed in cancer cells that undergo metastasis to form micro-metastases in distal host microenvironments (73). Thus, autophagy under these conditions may promote the survival of dormant cancer cells in distant tissues and can lead to tumor progression at a later stage.
An important aspect in understanding the marked anti-metastatic effect of NDRG1 after treatment with Dp44mT in vitro (19, 21) and in vivo (34) may be related to its ability to induce apoptosis markers and decrease the apoptosis antagonist, Bcl-2 (Fig. 9). This can be suggested to be due in part to the ability of NDRG1 to suppress autophagy after incubation with iron chelators (Figs. 2–5), which would decrease recycling of nutrients to facilitate apoptotic cell death. Previous studies examining NDRG1 have demonstrated that this protein is necessary, but not sufficient, for p53-mediated caspase activation and apoptosis (74). Furthermore, using pancreatic cancer cells, it has been demonstrated that NDRG1 inhibits pancreatic cancer growth in vitro and in vivo by induction of apoptosis, although the mechanism involved was not deciphered (75). Clearly, understanding the mechanism of action of Dp44mT is crucial, as such ligands are actively being developed to enter clinical trials (44), and understanding their efficacy could lead to the creation of improved agents.
In conclusion, this is the first investigation to establish the mechanistic relationship between NDRG1 and ER stress-mediated autophagy (Fig. 10). This process occurs through an interaction between NDRG1 and the PERK/eIF2α axis and is evident for a variety of stress stimuli, including iron-deprivation, ROS generation, tunicamycin treatment, and serum starvation. The suppression of autophagy could be a significant factor in the ability of NDRG1 to induce apoptosis. Hence, this investigation is important for further understanding the mechanisms by which NDRG1 can exert suppression of metastasis.
FIGURE 10.
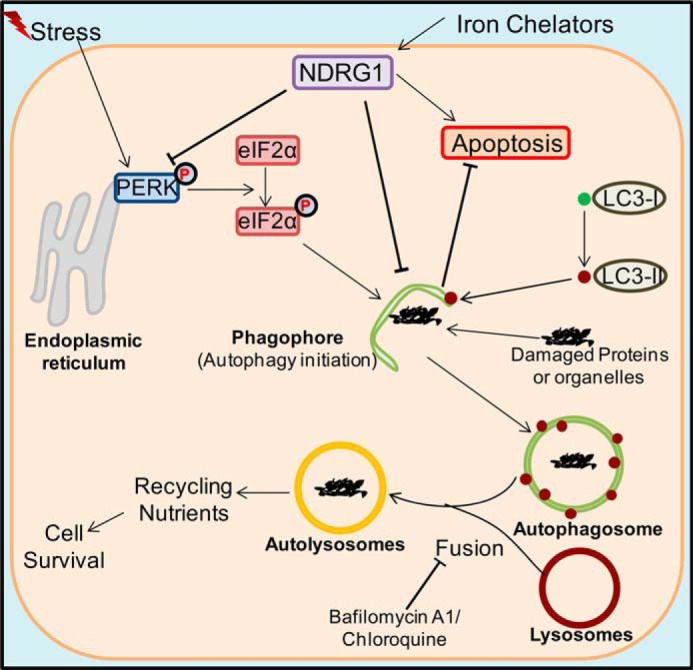
Schematic overview of relationships between the ER stress pathway, autophagy, and NDRG1. Stress stimuli are known to lead to the phosphorylation of PERK and its downstream effector eIF2α, which can lead to autophagic initiation through phagophore formation. This latter process involves the conversion of LC3-I into LC3-II and its incorporation into the phagophore membrane (27). Iron chelators can increase expression of the metastasis suppressor NDRG1 through HIF1α-dependent and -independent pathways (14, 36). The current study demonstrates that NDRG1 suppresses LC3-II expression and the formation of autophagosomes containing this latter protein. It also demonstrated that NDRG1 increases the susceptibility of the cell toward apoptosis. Thus, it is hypothesized that the NDRG1-mediated decrease in the autophagic flux results in decreased nutrient recycling that could play a role in the ability of NDRG1 to enhance apoptosis and inhibit metastasis.
Footnotes
- NDRG1
- N-myc downstream-regulated gene 1
- DFO
- desferrioxamine
- Dp2mT
- di-2-pyridylketone 2-methyl-3-thiosemicarbazone
- Dp44mT
- di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone
- ER
- endoplasmic reticulum
- NAC
- N-acetylcysteine
- Nedd4
- neural-precursor-cell-expressed developmentally down-regulated 4
- PARP
- poly(ADP-ribose) polymerase
- PERK
- protein kinase-like endoplasmic reticulum kinase
- ROS
- reactive oxygen species
- sh-
- short hairpin
- Tm
- tunicamycin
- p-eIF2α
- phosphorylated eIF2α.
REFERENCES
- 1. Bandyopadhyay S., Pai S. K., Gross S. C., Hirota S., Hosobe S., Miura K., Saito K., Commes T., Hayashi S., Watabe M. (2003) The Drg-1 gene suppresses tumor metastasis in prostate cancer. Cancer Res. 63, 1731–1736 [PubMed] [Google Scholar]
- 2. Bandyopadhyay S., Pai S. K., Hirota S., Hosobe S., Takano Y., Saito K., Piquemal D., Commes T., Watabe M., Gross S. C. (2004) Role of the putative tumor metastasis suppressor gene Drg-1 in breast cancer progression. Oncogene 23, 5675–5681 [DOI] [PubMed] [Google Scholar]
- 3. Guan R. J., Ford H. L., Fu Y., Li Y., Shaw L. M., Pardee A. B. (2000) Drg-1 as a differentiation-related, putative metastatic suppressor gene in human colon cancer. Cancer Res. 60, 749–755 [PubMed] [Google Scholar]
- 4. Hickok J. R., Sahni S., Mikhed Y., Bonini M. G., Thomas D. D. (2011) Nitric oxide suppresses tumor cell migration through N-Myc downstream-regulated gene-1 (NDRG1) expression. Role of chelatable iron. J. Biol. Chem. 286, 41413–41424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shah M. A., Kemeny N., Hummer A., Drobnjak M., Motwani M., Cordon-Cardo C., Gonen M., Schwartz G. K. (2005) Drg1 expression in 131 colorectal liver metastases. Correlation with clinical variables and patient outcomes. Clin. Cancer Res. 11, 3296–3302 [DOI] [PubMed] [Google Scholar]
- 6. van Belzen N., Dinjens W. N., Diesveld M. P., Groen N. A., van der Made A. C., Nozawa Y., Vlietstra R., Trapman J., Bosman F. T. (1997) A novel gene which is up-regulated during colon epithelial cell differentiation and down-regulated in colorectal neoplasms. Lab. Invest. 77, 85–92 [PubMed] [Google Scholar]
- 7. van Belzen N., Dinjens W. N., Eussen B. H., Bosman F. T. (1998) Expression of differentiation-related genes in colorectal cancer. Possible implications for prognosis. Histol. Histopathol. 13, 1233–1242 [DOI] [PubMed] [Google Scholar]
- 8. Fang B. A., Kovačević Z., Park K. C., Kalinowski D. S., Jansson P. J., Lane D. J., Sahni S., Richardson D. R. (2014) Molecular functions of the iron-regulated metastasis suppressor, NDRG1, and its potential as a molecular target for cancer therapy. Biochim. Biophys. Acta 1845, 1–19 [DOI] [PubMed] [Google Scholar]
- 9. Ellen T. P., Ke Q., Zhang P., Costa M. (2008) NDRG1, a growth and cancer related gene. Regulation of gene expression and function in normal and disease states. Carcinogenesis 29, 2–8 [DOI] [PubMed] [Google Scholar]
- 10. Kovacevic Z., Sivagurunathan S., Mangs H., Chikhani S., Zhang D., Richardson D. R. (2011) The metastasis suppressor, N-myc downstream regulated gene 1 (NDRG1), upregulates p21 via p53-independent mechanisms. Carcinogenesis 32, 732–740 [DOI] [PubMed] [Google Scholar]
- 11. Lachat P., Shaw P., Gebhard S., van Belzen N., Chaubert P., Bosman F. T. (2002) Expression of NDRG1, a differentiation-related gene, in human tissues. Histochem. Cell Biol. 118, 399–408 [DOI] [PubMed] [Google Scholar]
- 12. Chen B., Nelson D. M., Sadovsky Y. (2006) N-myc down-regulated gene 1 modulates the response of term human trophoblasts to hypoxic injury. J. Biol. Chem. 281, 2764–2772 [DOI] [PubMed] [Google Scholar]
- 13. Kokame K., Kato H., Miyata T. (1996) Homocysteine-respondent genes in vascular endothelial cells identified by differential display analysis GRP78/BiP and novel genes. J. Biol. Chem. 271, 29659–29665 [DOI] [PubMed] [Google Scholar]
- 14. Le N. T., Richardson D. R. (2004) Iron chelators with high antiproliferative activity up-regulate the expression of a growth inhibitory and metastasis suppressor gene. A link between iron metabolism and proliferation. Blood 104, 2967–2975 [DOI] [PubMed] [Google Scholar]
- 15. Salnikow K., Blagosklonny M. V., Ryan H., Johnson R., Costa M. (2000) Carcinogenic nickel induces genes involved with hypoxic stress. Cancer Res. 60, 38–41 [PubMed] [Google Scholar]
- 16. Salnikow K., Donald S. P., Bruick R. K., Zhitkovich A., Phang J. M., Kasprzak K. S. (2004) Depletion of intracellular ascorbate by the carcinogenic metals nickel and cobalt results in the induction of hypoxic stress. J. Biol. Chem. 279, 40337–40344 [DOI] [PubMed] [Google Scholar]
- 17. Cangul H. (2004) Hypoxia upregulates the expression of the NDRG1 gene leading to its overexpression in various human cancers. BMC Genet. 5, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dixon K. M., Lui G. Y., Kovacevic Z., Zhang D., Yao M., Chen Z., Dong Q., Assinder S. J., Richardson D. R. (2013) Dp44mT targets the AKT, TGF-β and ERK pathways via the metastasis suppressor NDRG1 in normal prostate epithelial cells and prostate cancer cells. Br. J. Cancer 108, 409–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sun J., Zhang D., Zheng Y., Zhao Q., Zheng M., Kovacevic Z., Richardson D. R. (2013) Targeting the metastasis suppressor, NDRG1, using novel iron chelators. Regulation of stress fiber-mediated tumor cell migration via modulation of the ROCK1/pMLC2 signaling pathway. Mol. Pharmacol. 83, 454–469 [DOI] [PubMed] [Google Scholar]
- 20. Kovacevic Z., Chikhani S., Lui G. Y., Sivagurunathan S., Richardson D. R. (2013) The iron-regulated metastasis suppressor NDRG1 targets NEDD4L, PTEN, and SMAD4 and inhibits the PI3K and Ras signaling pathways. Antioxid. Redox Signal. 18, 874–887 [DOI] [PubMed] [Google Scholar]
- 21. Chen Z., Zhang D., Yue F., Zheng M., Kovacevic Z., Richardson D. R. (2012) The iron chelators Dp44mT and DFO inhibit TGF-β-induced epithelial-mesenchymal transition via up-regulation of N-Myc downstream-regulated gene 1 (NDRG1). J. Biol. Chem. 287, 17016–17028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kenific C. M., Thorburn A., Debnath J. (2010) Autophagy and metastasis. Another double-edged sword. Curr. Opin. Cell Biol. 22, 241–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsuchihara K., Fujii S., Esumi H. (2009) Autophagy and cancer. Dynamism of the metabolism of tumor cells and tissues. Cancer Lett. 278, 130–138 [DOI] [PubMed] [Google Scholar]
- 24. Korolchuk V. I., Rubinsztein D. C. (2012) On signals controlling autophagy: it's time to eat yourself healthy. Biochemist 34, 8–13 [Google Scholar]
- 25. Ravikumar B., Sarkar S., Davies J. E., Futter M., Garcia-Arencibia M., Green-Thompson Z. W., Jimenez-Sanchez M., Korolchuk V. I., Lichtenberg M., Luo S. (2010) Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 90, 1383–1435 [DOI] [PubMed] [Google Scholar]
- 26. Morselli E., Galluzzi L., Kepp O., Mariño G., Michaud M., Vitale I., Maiuri M. C., Kroemer G. (2011) Oncosuppressive functions of autophagy. Antioxid. Redox Signal. 14, 2251–2269 [DOI] [PubMed] [Google Scholar]
- 27. Hamacher-Brady A. (2012) Autophagy regulation and integration with cell signaling. Antioxid. Redox Signal. 17, 756–765 [DOI] [PubMed] [Google Scholar]
- 28. Maiuri M. C., Zalckvar E., Kimchi A., Kroemer G. (2007) Self-eating and self-killing. Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 8, 741–752 [DOI] [PubMed] [Google Scholar]
- 29. Hart L. S., Cunningham J. T., Datta T., Dey S., Tameire F., Lehman S. L., Qiu B., Zhang H., Cerniglia G., Bi M. (2012) ER stress mediated autophagy promotes Myc-dependent transformation and tumor growth. J. Clin. Invest. 122, 4621–4634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Malhotra J. D., Kaufman R. J. (2007) The endoplasmic reticulum and the unfolded protein response. Semin. Cell. Dev. Biol. 18, 716–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. He C., Klionsky D. J. (2009) Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kalinowski D. S., Richardson D. R. (2005) The evolution of iron chelators for the treatment of iron overload disease and cancer. Pharmacol. Rev. 57, 547–583 [DOI] [PubMed] [Google Scholar]
- 33. Whitnall M., Howard J., Ponka P., Richardson D. R. (2006) A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc. Natl. Acad. Sci. U.S.A. 103, 14901–14906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu W., Xing F., Iiizumi-Gairani M., Okuda H., Watabe M., Pai S. K., Pandey P. R., Hirota S., Kobayashi A., Mo Y.-Y., Fukuda K., Li Y., Watabe K. (2012) N-myc downstream regulated gene 1 modulates Wnt-β-catenin signalling and pleiotropically suppresses metastasis. EMBO Mol. Med. 4, 93–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lovejoy D. B., Sharp D. M., Seebacher N., Obeidy P., Prichard T., Stefani C., Basha M. T., Sharpe P. C., Jansson P. J., Kalinowski D. S. (2012) Novel second-generation di-2-pyridylketone thiosemicarbazones show synergism with standard chemotherapeutics and demonstrate potent activity against lung cancer xenografts after oral and intravenous administration in vivo. J. Med. Chem. 55, 7230–7244 [DOI] [PubMed] [Google Scholar]
- 36. Lane D. J., Saletta F., Suryo Rahmanto Y., Kovacevic Z., Richardson D. R. (2013) N-myc downstream regulated 1 (NDRG1) is regulated by eukaryotic initiation factor 3a (eIF3a) during cellular stress caused by iron depletion. PloS ONE 8, e57273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Richardson D. R., Sharpe P. C., Lovejoy D. B., Senaratne D., Kalinowski D. S., Islam M., Bernhardt P. V. (2006) Dipyridyl thiosemicarbazone chelators with potent and selective antitumor activity form iron complexes with redox activity. J. Med. Chem. 49, 6510–6521 [DOI] [PubMed] [Google Scholar]
- 38. Yuan J., Lovejoy D. B., Richardson D. R. (2004) Novel di-2-pyridyl-derived iron chelators with marked and selective antitumor activity. In vitro and in vivo assessment. Blood 104, 1450–1458 [DOI] [PubMed] [Google Scholar]
- 39. Gao J., Richardson D. R. (2001) The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents, IV. The mechanisms involved in inhibiting cell-cycle progression. Blood 98, 842–850 [DOI] [PubMed] [Google Scholar]
- 40. De Domenico I., Ward D. M., Kaplan J. (2009) Specific iron chelators determine the route of ferritin degradation. Blood 114, 4546–4551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kovacevic Z., Richardson D. R. (2006) The metastasis suppressor, Ndrg-1. A new ally in the fight against cancer. Carcinogenesis 27, 2355–2366 [DOI] [PubMed] [Google Scholar]
- 42. Kalinowski D. S., Richardson D. R. (2007) Future of toxicology iron chelators and differing modes of action and toxicity. The changing face of iron chelation therapy. Chem. Res. Toxicol. 20, 715–720 [DOI] [PubMed] [Google Scholar]
- 43. Ghalayini M. K., Dong Q., Richardson D. R., Assinder S. J. (2013) Proteolytic cleavage and truncation of NDRG1 in human prostate cancer cells, but not normal prostate epithelial cells. Biosci. Rep. 33, 451–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kovacevic Z., Chikhani S., Lovejoy D. B., Richardson D. R. (2011) Novel thiosemicarbazone iron chelators induce up-regulation and phosphorylation of the metastasis suppressor N-myc down-stream regulated gene 1. a new strategy for the treatment of pancreatic cancer. Mol. Pharmacol. 80, 598–609 [DOI] [PubMed] [Google Scholar]
- 45. Murray J. T., Campbell D. G., Morrice N., Auld G. C., Shpiro N., Marquez R., Peggie M., Bain J., Bloomberg G. B., Grahammer F., Lang F., Wulff P., Kuhl D., Cohen P. (2004) Exploitation of KESTREL to identify NDRG family members as physiological substrates for SGK1 and GSK3. Biochem. J. 384, 477–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mizushima N., Yoshimori T., Levine B. (2010) Methods in mammalian autophagy research. Cell 140, 313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Klionsky D. J., Cuervo A. M., Seglen P. O. (2007) Methods for monitoring autophagy from yeast to human. Autophagy 3, 181–206 [DOI] [PubMed] [Google Scholar]
- 48. Klionsky D. J., Abdalla F. C., Abeliovich H., Abraham R. T., Acevedo-Arozena A., Adeli K., Agholme L., Agnello M., Agostinis P., Aguirre-Ghiso J. A. (2012) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8, 445–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yamamoto A., Tagawa Y., Yoshimori T., Moriyama Y., Masaki R., Tashiro Y. (1998) Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct. Funct. 23, 33–42 [DOI] [PubMed] [Google Scholar]
- 50. Richardson D. R., Milnes K. (1997) The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents II. The mechanism of action of ligands derived from salicylaldehyde benzoyl hydrazone and 2-hydroxy-1-naphthylaldehyde benzoyl hydrazone. Blood 89, 3025–3038 [PubMed] [Google Scholar]
- 51. Richardson D., Ponka P., Baker E. (1994) The effect of the iron (III) chelator, desferrioxamine, on iron and transferrin uptake by the human malignant melanoma cell. Cancer Res. 54, 685–689 [PubMed] [Google Scholar]
- 52. Jansson P. J., Sharpe P. C., Bernhardt P. V., Richardson D. R. (2010) Novel thiosemicarbazones of the ApT and DpT series and their copper complexes. Identification of pronounced redox activity and characterization of their antitumor activity. J. Med. Chem. 53, 5759–5769 [DOI] [PubMed] [Google Scholar]
- 53. Lovejoy D. B., Jansson P. J., Brunk U. T., Wong J., Ponka P., Richardson D. R. (2011) Antitumor activity of metal-chelating compound Dp44mT is mediated by formation of a redox-active copper complex that accumulates in lysosomes. Cancer Res. 71, 5871–5880 [DOI] [PubMed] [Google Scholar]
- 54. Tsai Y. C., Weissman A. M. (2010) The unfolded protein response, degradation from the endoplasmic reticulum, and cancer. Genes Cancer 1, 764–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kouroku Y., Fujita E., Tanida I., Ueno T., Isoai A., Kumagai H., Ogawa S., Kaufman R. J., Kominami E., Momoi T. (2007) ER stress (PERK/eIF2α phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 14, 230–239 [DOI] [PubMed] [Google Scholar]
- 56. Kroemer G., Mariño G., Levine B. (2010) Autophagy and the integrated stress response. Mol. Cell 40, 280–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vaux D. L., Strasser A. (1996) The molecular biology of apoptosis. Proc. Natl. Acad. Sci. U.S.A. 93, 2239–2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jiang C., Wang Z., Ganther H., Lu J. (2001) Caspases as key executors of methyl selenium-induced apoptosis (anoikis) of DU-145 prostate cancer cells. Cancer Res. 61, 3062–3070 [PubMed] [Google Scholar]
- 59. Chao D. T., Korsmeyer S. J. (1998) BCL-2 family. Regulators of cell death. Annu. Rev. Immunol. 16, 395–419 [DOI] [PubMed] [Google Scholar]
- 60. Xu Y., Xia X., Pan H. (2013) Active autophagy in the tumor microenvironment. A novel mechanism for cancer metastasis (review). Oncol. Lett. 5, 411–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jansson P. J., Hawkins C. L., Lovejoy D. B., Richardson D. R. (2010) The iron complex of Dp44mT is redox-active and induces hydroxyl radical formation. An EPR study. J. Inorg. Biochem. 104, 1224–1228 [DOI] [PubMed] [Google Scholar]
- 62. Yan M., Shen J., Person M. D., Kuang X., Lynn W. S., Atlas D., Wong P. K. (2008) Endoplasmic reticulum stress and unfolded protein response in Atm-deficient thymocytes and thymic lymphoma cells are attributable to oxidative stress. Neoplasia 10, 160–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li J., Kretzner L. (2003) The growth-inhibitory Ndrg1 gene is a Myc negative target in human neuroblastomas and other cell types with overexpressed N-or c-myc. Mol. Cell Biochem. 250, 91–105 [DOI] [PubMed] [Google Scholar]
- 64. Stafford L. J., Vaidya K. S., Welch D. R. (2008) Metastasis suppressors genes in cancer. Int. J. Biochem. Cell Biol. 40, 874–891 [DOI] [PubMed] [Google Scholar]
- 65. Shoushtari A. N., Szmulewitz R. Z., Rinker-Schaeffer C. W. (2011) Metastasis-suppressor genes in clinical practice. Lost in translation? Nat. Rev. Clin. Oncol. 8, 333–342 [DOI] [PubMed] [Google Scholar]
- 66. Wu C.-Y., Yan J., Yang Y.-F., Xiao F.-J., Li Q.-F., Zhang Q.-W., Wang L.-S., Guo X.-Z., Wang H. (2011) Overexpression of KAI1 induces autophagy and increases MiaPaCa-2 cell survival through the phosphorylation of extracellular signal-regulated kinases. Biochem. Biophys. Res. Commun. 404, 802–808 [DOI] [PubMed] [Google Scholar]
- 67. Fujii S., Mitsunaga S., Yamazaki M., Hasebe T., Ishii G., Kojima M., Kinoshita T., Ueno T., Esumi H., Ochiai A. (2008) Autophagy is activated in pancreatic cancer cells and correlates with poor patient outcome. Cancer Sci. 99, 1813–1819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Giatromanolaki A., Koukourakis M. I., Harris A. L., Polychronidis A., Gatter K. C., Sivridis E. (2010) Prognostic relevance of light chain 3 (LC3A) autophagy patterns in colorectal adenocarcinomas. J. Clin. Pathol. 63, 867–872 [DOI] [PubMed] [Google Scholar]
- 69. Gurkar A. U., Chu K., Raj L., Bouley R., Lee S.-H., Kim Y.-B., Dunn S. E., Mandinova A., Lee S. W. (2013) Identification of ROCK1 kinase as a critical regulator of Beclin1-mediated autophagy during metabolic stress. Nat. Commun. 4, 2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Platta H. W., Abrahamsen H., Thoresen S. B., Stenmark H. (2012) Nedd4-dependent lysine-11-linked polyubiquitination of the tumour suppressor Beclin1. Biochem. J. 441, 399–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fung C., Lock R., Gao S., Salas E., Debnath J. (2008) Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell 19, 797–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. White D. E., Kurpios N. A., Zuo D., Hassell J. A., Blaess S., Mueller U., Muller W. J. (2004) Targeted disruption of β1-integrin in a transgenic mouse model of human breast cancer reveals an essential role in mammary tumor induction. Cancer Cell 6, 159–170 [DOI] [PubMed] [Google Scholar]
- 73. Nguyen D. X., Bos P. D., Massagué J. (2009) Metastasis. From dissemination to organ-specific colonization. Nat. Rev. Cancer 9, 274–284 [DOI] [PubMed] [Google Scholar]
- 74. Stein S., Thomas E. K., Herzog B., Westfall M. D., Rocheleau J. V., Jackson R. S., 2nd, Wang M., Liang P. (2004) NDRG1 is necessary for p53-dependent apoptosis. J. Biol. Chem. 279, 48930–48940 [DOI] [PubMed] [Google Scholar]
- 75. Angst E., Dawson D. W., Stroka D., Gloor B., Park J., Candinas D., Reber H. A., Hines O. J., Eibl G. (2011) N-myc downstream regulated gene-1 expression correlates with reduced pancreatic cancer growth and increased apoptosis in vitro and in vivo. Surgery 149, 614–624 [DOI] [PubMed] [Google Scholar]