

Background: Dimerization of cystatin C initiates formation of amyloid fibrils in vascular walls.
Results: Reactive oxygen species regulate cystatin C dimerization in cells of the hematopoietic system, which are major contributors to the total pool of circulating cystatin C.
Conclusion: Cystatin C dimerization is regulated in hematopoietic cells.
Significance: Cystatin C synthesis and dimerization might be manipulated for therapeutic gain.
Keywords: Dendritic Cells, Macrophages, Protease Inhibitor, Protein Folding, Reactive Oxygen Species (ROS), Amyloidosis, Vascular Angiopathy
Abstract
The cysteine protease inhibitor cystatin C is thought to be secreted by most cells and eliminated in the kidneys, so its concentration in plasma is diagnostic of kidney function. Low extracellular cystatin C is linked to pathologic protease activity in cancer, arthritis, atherosclerosis, aortic aneurism, and emphysema. Cystatin C forms non-inhibitory dimers and aggregates by a mechanism known as domain swapping, a property that reportedly protects against Alzheimer disease but can also cause amyloid angiopathy. Despite these clinical associations, little is known about the regulation of cystatin C production, dimerization, and secretion. We show that hematopoietic cells are major contributors to extracellular cystatin C levels in healthy mice. Among these cells, macrophages and dendritic cells (DC) are the predominant producers of cystatin C. Both cell types synthesize monomeric and dimeric cystatin C in vivo, but only secrete monomer. Dimerization occurs co-translationally in the endoplasmic reticulum and is regulated by the levels of reactive oxygen species (ROS) derived from mitochondria. Drugs or stimuli that reduce the intracellular concentration of ROS inhibit cystatin C dimerization. The extracellular concentration of inhibitory cystatin C is thus partly dependent on the abundance of macrophages and DC, and the ROS levels. These results have implications for the diagnostic use of serum cystatin C as a marker of kidney function during inflammatory processes that induce changes in DC or macrophage abundance. They also suggest an important role for macrophages, DC, and ROS in diseases associated with the protease inhibitory activity or amyloidogenic properties of cystatin C.
Introduction
Cystatin C is the most studied of the type II cystatins, a family of secreted small proteins (∼120 amino acids) that inhibit cysteine proteases of the papain family, and legumain (1). Cystatin C inhibits proteases by forming tight, non-covalent associations with its target enzymes. Cystatin C has long been considered ubiquitously expressed and is abundant in body fluids. Due to these characteristics cystatin C is regarded as a major regulator of protease activity in the extracellular milieu.
Cystatin C influences the progression of diseases where extracellular proteolysis plays a role, for instance, cancer metastasis, atherosclerosis, aortic aneurisms, emphysema, or arthritis (2–6). It is important to identify which cells contribute to local cystatin C levels and to characterize mechanisms involved in regulation of cystatin C expression and secretion. Another property that underpins the clinical interest of cystatin C is its value as a diagnostic marker of kidney function (7, 8). This value is predicated on the assumption that cystatin C is produced by most cells in the body at a relatively constant rate so that its serum concentration is primarily regulated by removal in the glomerulus (9). However, previous reports by Tamura and collaborators (10) and ourselves (11, 48) demonstrated that the promoter of the gene encoding mouse cystatin C, cst3, contains binding elements for transcription factors IRF8 and PU.1 (Sfpi-1). Co-expression of IRF8 and PU.1 drives high cystatin C expression in macrophages and a population of dendritic cells, which in mice are characterized by CD8 expression (CD8+ DC)5 (48). Furthermore, we found that regulation of IRF-8 expression by inflammation-associated signals controls cystatin C production and serum concentration (11).
The third clinically relevant feature of cystatin C is its association with neurodegenerative disease and cerebral angiopathy (12). Cystatin C can form homodimers by a process known as “domain swapping”(13). Cystatin C dimers cannot inhibit cysteine proteases because their inhibitory region is hidden within the dimer interface (13). These dimers can “grow” by additional rounds of swapping to form toxic amyloid deposits similar to those composed of prion proteins or the β-amyloid peptide (Aβ) (14). Indeed, cystatin C is present in Aβ amyloid deposits in the brain and associated vascular tissue of Alzheimer disease patients and may contribute to pathology (15). A mutant form of human cystatin C has a much higher propensity to dimerize and form fibrils in the brain vasculature, and these fibrils cause hemorrhage, dementia, and eventually death in people carrying this mutation, a condition known as hereditary cystatin C amyloid angiopathy (HCCAA) (16). On the other hand, wild-type cystatin C reportedly plays a protective role in Alzheimer disease through its association with Aβ (17). For these pathologies, it is important to characterize not only the mechanisms that control cystatin C expression but also those that regulate its dimerization. However, most of the studies that have examined cystatin C dimerization were based on in vitro assays employing recombinant protein; little is known about the generation of dimers in vivo (18).
Here, we first extend our studies on the pattern of cystatin C expression and show that bone marrow-derived cells are major contributors to the steady-state level of serum cystatin C. We also show that CD8+ DC and macrophages synthesize both cystatin C monomers and dimers in vivo under non-pathological conditions, but regulate differently the secretion of the monomeric and dimeric forms. Finally, we demonstrate that the ratio of cystatin C monomer/dimer synthesized by macrophages and DC is regulated by the amount of intracellular reactive oxygen species (ROS) produced by mitochondria. We discuss the implications of these results for the application of serum cystatin C measurements as a diagnostic marker of kidney function, the contribution of different cell types to pathologies associated with cystatin C, and the potential therapeutic application of regulators of cystatin C synthesis, dimerization, and secretion, or ROS formation, for the treatment of these pathologies.
EXPERIMENTAL PROCEDURES
Mice and Reagents
C57BL/6 and cystatin C-deficient (19) mice were bred in the animal facilities of the Walter and Eliza Hall Institute (WEHI) and the Bio21 Institute at the University of Melbourne. All animal breeding and experimentation was conducted according to institutional guidelines and approved by WEHI Animal Care and Users Committee and the University of Melbourne Animal Ethics Committee. Anti-human cystatin C rabbit sera were from DAKO (Glostrup, Denmark) or Upstate Biotechnology (Lake Placid, NY). Goat anti-human cystatin C serum was from R&D Systems (Minneapolis, MN). Carboxymethylated papain bound to agarose beads was purchased from Calbiochem (San Diego, CA) or MP Biomedical (Solon, OH). Hydrogen peroxide (H2O2) was purchased from Cell Biolabs, Inc. (San Diego, CA). Ethacrynic acid (EA) and antimycin A (AA) was from Sigma.
Purification of Splenic DC and Lymphocytes
Splenic DC were purified as described before (20). Briefly, spleen fragments were digested with DNase I (Boehringer Mannheim) and collagenase (Worthington Biochemicals, Freehold, NJ) and enriched for light-density cells by centrifugation in 1.077 g/cm3 Nycodenz (Nycomed Pharma, Oslo, Norway). Non-DC were depleted using antibodies against CD3 (KT3–1.1), Thy-1 (T24/31.7), Ter 119, Ly6G (RB68C5), and CD45R (RA36B2), followed by incubation with anti-rat IgG-coupled magnetic beads (Dynal, Oslo, Norway) according to the manufacturer's instructions. CD8+ DC were further isolated by positive selection using immuno-magnetic beads (MACS, Miltenyi Biotec, Bergisch Gladbach, Germany) after staining with anti-CD8 (YTS 169.4) antibody. Other purified lymphocytes were sorted from DNase/collagenase-digested spleen single-cell suspensions by flow cytometry.
Dendritic Cell and Macrophage Cultures from Bone Marrow Precursors
Bone marrow (BM)-derived DC or macrophages were generated as described (21, 22). Briefly, BM was harvested, and red cells were removed by a 30-s exposure to red cell removal buffer and washed three times. Cells were then cultured at 1.5 × 106 cells/ml for 8 days in complete medium containing 300 ng/ml of mouse Flt-3L (WEHI) for DC or for 7 days in complete medium containing 25 ng/ml of recombinant human M-CSF (R&D Systems) for macrophages.
Bone Marrow Transplantations
Bone marrow chimeras were generated as described (23). Briefly, Ly5.1 recipient mice were γ-irradiated (2 × 0.55 gray) and reconstituted with 1 × 106 T and B cell-depleted bone marrow cells from wild-type or cystatin C−/− mice. Chimeras were then treated with neomycin (1.1 g/ml) in the drinking water for the next 6 weeks and used for subsequent experiments after at least 8 weeks from reconstitution. Confirmation of transplantation was performed by FACS analysis of splenocytes or blood cells measuring the percentage of donor cells.
ELISA
The concentration of cystatin C in serum was determined using the mouse DuoSet ELISA kit (R&D Systems) according to the manufacturer's instructions.
Radioactive Pulse-Chase Analysis
Pulse-chase experiments were carried out as previously described (24). Briefly, purified DC populations were incubated at a maximum of 107 cells/ml in methionine/cysteine-deficient DMEM for 30 min at 37 °C. DC were pulsed for 30–60 min with 1 mCi/ml of [35S]-labeled Protein Labeling Mix (73% l-[35S]methionine and 22% l-[35S]cysteine) (PerkinElmer Life Sciences) in the presence or absence of BFA, washed, and chased for varying amounts of time in RPMI-10. At each time point, cells were pelleted, re-suspended in 50 μl of PBS, snap frozen on dry ice, and stored at −80 °C for later use.
Cell Lysis and Immunoprecipitation
All steps for immunoprecipitation were performed at 4 °C as described before (25). Metabolically radiolabeled or non-radiolabeled cells were lysed in up to 1 ml of lysis buffer (50 mm Tris-HCl, pH 7.4, 5 mm MgCl2, 0.5% Nonidet P-40 and protease inhibitors (Roche Applied Science)), incubated for 15 min at 4 °C on a roller, and then centrifuged for 10 min at 13,000 × g in a Heraeus BiofugeTM (DJB Labcare) to remove nuclei and cell debris. For radioactively labeled samples the efficiency of [35S]Met/Cys incorporation was determined by trichloroacetic acid (TCA) (Sigma) precipitation whereby 5 μl of cell lysate was spotted on filter paper and washed three times in 1% TCA. The radioactivity was measured in duplicate spots using a TopCount scintillation counter (Canberra Packard) and the amount of cell lysate used for the immunoprecipitation was normalized accordingly. Cell lysates, serum-free cell culture supernatants, or gel filtration fractions were pre-cleared once for 60 min with 2 μl of normal rabbit or goat serum (Sigma) and 30 μl of 60% Protein G-Sepharose slurry (GE Healthcare), followed by a second 45-min pre-clearing step with Protein G-Sepharose beads. Immunoprecipitations were carried out for 2 h with 2 μl of anti-cystatin C serum and 30 μl of Protein G beads. Immunoprecipitates were washed three times with NET buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 5 mm EDTA, 0.5% Nonidet P-40 and protease inhibitors (Roche Applied Science)), resuspended in Laemmli SDS sample buffer, boiled, and analyzed by SDS-PAGE followed by Western blot or autoradiography.
Gel Filtration Chromatography
Samples (400 μl) of cell lysates or serum-free cell culture supernatants were fractionated by gel filtration chromatography using a Waters HPLC system on a pre-packed Superdex 200 column (300 × 10-mm internal diameter, GE Healthcare) equilibrated in 50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 5 mm MgCl2, 0.5% Nonidet P-40 and protease inhibitors (Roche Applied Science). Initial experiments included 0.5% Nonidet P-40 in the column buffer, which had no effect on the retention times of proteins and was subsequently omitted. The column was calibrated with Bio-Rad gel filtration standard containing vitamin B12 (1.35 kDa), equine myoglobin (17.5 kDa), chicken ovalbumin (44 kDa), bovine γ-globulin (158 kDa), and thyroglobulin dimer (670 kDa). The flow rate was 0.4 ml/min and 0.4-ml fractions were collected. Two consecutive fractions were combined and assayed for the presence of cystatin C by immunoprecipitation and Western blot analysis.
Papain-Agarose Precipitation
Cell lysates or cell culture supernatants were incubated with a saturating amount of (proteolytically inactive) carboxymethylated papain bound to agarose beads at 4 °C for 2 h with gentle mixing. The papain-agarose beads were sedimented by centrifugation and washed three times with NET buffer. Papain-bound cystatin C was recovered by boiling in Laemmli sample buffer and analyzed by Western blot or autoradiography as applicable. Where indicated, the samples were subjected to a new round of papain-agarose precipitation followed by immunoprecipitation of the remaining cystatin C as described above.
SDS-PAGE, Western Blot Analysis, and Autoradiography
Frozen cell pellets, immunoprecipitates, or papain-agarose precipitates were re-suspended in Laemmli sample buffer and resolved by 11.5% SDS-PAGE. Reducing agents were omitted from the sample buffer where indicated. Western blot analysis was performed as described (24). After SDS-PAGE, proteins were electroblotted using a HoeferTM semi-dry transfer unit (Amersham Biosciences) onto an Hybond-PTM PVDF membrane (Amersham Biosciences) and blocked for 2 h with 5% (w/v) skim milk in PBS containing 0.2% (v/v) Tween 20 (PBS-T). The membranes were rinsed in PBS-T and incubated with the appropriate primary antibody for 1–1.5 h. Bound proteins were detected using appropriate secondary antibodies conjugated to horseradish peroxidase (GE Healthcare). Detection was performed using SuperSignal® chemiluminescence substrate (Pierce, Rockford, IL). All Western blot quantitation was performed with a Molecular Dynamics densitometer using ImageQuantTM 5.0 software. For autoradiography, gels were then treated with dimethyl sulfoxide containing 4–5 g/liter of 2,5-diphenyloxazole (Sigma) scintillant. Radioactive polypeptides were visualized using a phosphorimager (Molecular Dynamics) or Scientific Imaging Film (Hyperfilm, Amersham Biosciences or Biomax, Kodak). Quantitation was performed with a Molecular Dynamics densitometer using ImageQuantTM 5.0 software.
Intracellular ROS Detection
Cells were incubated or not with the indicated reagents and then washed with PBS. Detection of intracellular ROS was conducted by incubating the cells with 2 mm of the redox sensitive probe 5- (and 6-)chloromethyl 2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (Molecular Probes) for 15 min at 37 °C. Oxidation of the probe was detected by the increase in FITC fluorescence using flow cytometry.
Statistics
Mean ± S.D. values, and two-tailed Student's t tests (unpaired) were calculated using Microsoft Excel software. A p level ≤ 0.05 was considered significant. Error bars represent mean ± S.E.
RESULTS
Bone Marrow-derived Cells Are Major Contributors to Extracellular Cystatin C Concentration
Expression of the gene encoding cystatin C, cst3, is stimulated by transcription factor IRF-8 and further enhanced by another transcription factor, PU.1 (Sfpi-1) (10, 11). As IRF-8 and PU.1 are predominantly expressed in hematopoietic cells, we first examined to what extent the overall levels of extracellular cystatin C depend on expression by bone marrow-derived cells. We transferred bone marrow from wild-type or cystatin C−/− mice into sublethally irradiated cystatin C+/+ mice. More than 90% of the hematopoietic compartment in the resulting chimeric mice was derived from the donor bone marrow (data not shown). The amount of cystatin C in spleen cell lysates of chimeras reconstituted with cystatin C−/− bone marrow was lower than in mice reconstituted with wild-type bone marrow (Fig. 1A). This was not surprising because most of the splenic cells are bone marrow-derived. More strikingly, the concentration of cystatin C in the serum of mice reconstituted with cystatin C−/− bone marrow was 30% lower than in mice reconstituted with wild-type bone marrow (Fig. 1B). We also generated chimeras where cystatin C−/− hosts received wild-type bone marrow. In these chimeric mice the serum concentration of cystatin C was 18% of the amount present in mice where all cells expressed cystatin C (Fig. 1C). These results demonstrate an important contribution of bone marrow-derived cells to the overall extracellular level of cystatin C.
FIGURE 1.

Contribution of bone marrow-derived cells to the serum levels of Cst C. A–C, wild-type (CstC+/+) or cystatin C knock-out (CstC−/−) mice were reconstituted with wild-type or cystatin C-deficient BM as indicated by the arrows. The total splenocytes (A) or blood (B and C) of the reconstituted mice were examined for cystatin C contents by Western blot (A) or ELISA (B and C). Blood samples from cystatin C knock-out mice were included as negative control. ELISA results are presented as average values of duplicates from eight (B) or three (C) mice per group. Error bars represent S.E., and n.d. indicates not detected. **, p < 0.01. D, total cell lysates from purified splenic cell populations as indicated, or DC or macrophages generated in culture from BM precursors (BM-MΦ, BM-DC) were run in SDS-PAGE and their cystatin C contents were examined by Western blot. The same membrane was probed with anti-Actin antibody as loading control. E, purified cell populations as indicated were metabolically labeled and newly synthesized cystatin C was immunoprecipitated from cell lysates (lanes C) or the culture supernatant (lanes S), visualized by autoradiography and quantitated in a phosphorimager. All results are representative of two (MΦ) or multiple (DC) independent experiments performed.
Macrophages and CD8+ DC Are the Major Bone Marrow-derived Producers of Cystatin C
To further characterize the pattern of expression of cystatin C among bone marrow-derived cells, we used Western blot to compare its abundance in purified B lymphocytes, CD4+ and CD8+ T lymphocytes, and DC. The DC were subdivided into the two major subgroups present in lymphoid organs, CD8+ DC and CD11b+ DC. Expression was highest in CD8+ DC (11) and negligible in all lymphocyte populations (Fig. 1D) (48). Macrophages have also been reported to express high levels of cystatin C (10, 26), but have not been compared side by side to other cell types. We generated bone marrow-derived macrophages or DC in vitro and included them in this comparison, which revealed an intermediate level of cystatin C expression in macrophages (Fig. 1D). To verify that the contrast in intracellular content of cystatin C was due to differences in synthesis, we metabolically radiolabeled purified CD11b+ and CD8+ DC and peritoneal macrophages and immunoprecipitated cystatin C from normalized amounts of radiolabeled cell lysates. The CD8+ DC synthesized the largest amount, followed by macrophages (Fig. 1E). The amount of cystatin C secreted by the three cell types to the culture supernatant during radiolabeling followed the same pattern (Fig. 1E). These results suggest that CD8+ DC and macrophages are major contributors to extracellular cystatin C concentration. In the rest of this article we will refer to CD8+ DC as DC.
Constitutive Formation of Inactive Cystatin C Dimers in Primary DC
The main function of cystatin C, and the basis for its reported role in cancer metastasis, atherosclerosis, aortic aneurisms, emphysema, or arthritis (27), is the inhibition of cysteine proteases. All of the cystatin C contained in DC lysates could be immunoprecipitated using a rabbit antiserum (data not shown). To assess how much of the protein could bind to papain, the canonical cysteine protease inhibited by cystatin C, we used immobilized carboxymethylated papain to precipitate this fraction from freshly purified DC lysates. Carboxymethylation inactivates the protease activity of papain but does not prevent high-affinity binding of cystatin C. We were surprised to find that we could only recover 60% of the protein. The remaining 40% could be retrieved sequentially by immunoprecipitation using anti-cystatin C antibodies (Fig. 2A). We wondered if the fraction that could not be recognized by papain corresponded to domain-swapped cystatin C dimers, which do not bind to cysteine proteases (13). To investigate this possibility we fractionated whole DC lysates by gel filtration chromatography, immunoprecipitated cystatin C from the column fractions, and detected cystatin C by Western blot. Approximately one-half of the cystatin C contained in the DC lysate eluted at a position corresponding to a monomer (13 kDa), whereas the other half eluted at a position consistent with a 26-kDa dimer (Fig. 2B). The cystatin C dimers denatured in SDS-PAGE and migrated to the same position in the gel as the monomers. Chromatographic analysis of a DC lysate pre-depleted of papain-reactive cystatin C confirmed that only the papain-unreactive cystatin C species eluted as a dimer (Fig. 2B). The dimer was not disulfide bound as revealed by Western blot analysis of a DC lysate run in SDS-PAGE in reducing versus non-reducing conditions (Fig. 2C). To address the possibility that the species that eluted in size-exclusion chromatography as a dimer might in fact be cystatin C bound to some other protein, we resorted to mass spectrometry. We depleted the papain-reactive fraction from cell lysates. The remaining cystatin C dimer was then immunoprecipitated and loaded in SDS-PAGE. The regions of the gel containing proteins with molecular mass <25 kDa were excised, trypsinized, and analyzed by mass spectrometry to identify the proteins therein. We clearly detected cystatin C (5 peptides, 39% coverage) but no other mouse proteins were identified (data not shown). As these results were obtained with primary DC directly purified from lymphoid tissue, without any incubation in vitro, they show that DC contain both active (monomeric) and inactive (dimeric) cystatin C under non-pathological conditions.
FIGURE 2.
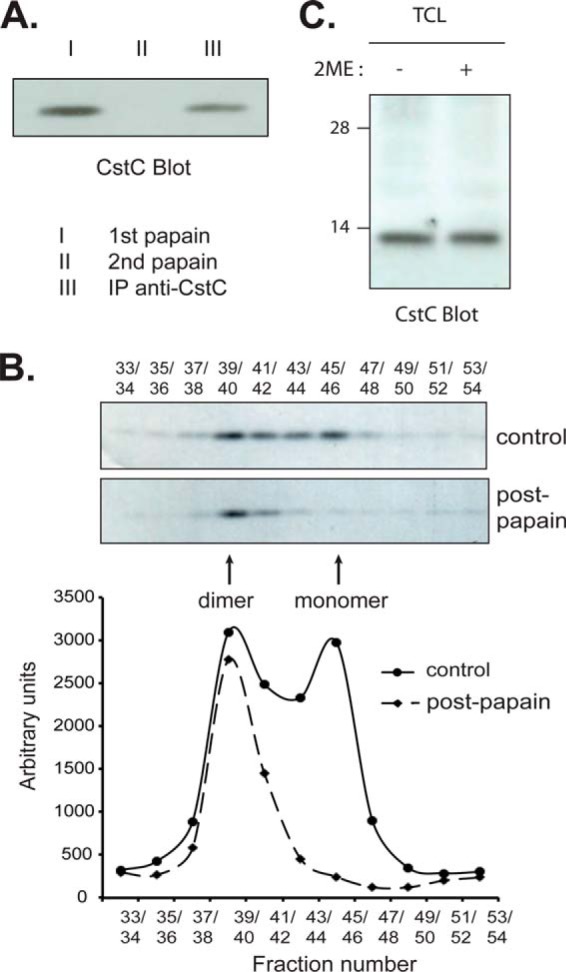
Primary DC contain inactive cystatin C dimers. A, lysates of freshly isolated splenic CD8+ DC were incubated with immobilized, carboxymethylated (proteolytically inactive) papain to precipitate active cystatin C (I). A saturating amount of papain was used as confirmed by the lack of additional cystatin C recovered during a second round of precipitation with the same reagent (II). Remaining (inactive) cystatin C was retrieved by immunoprecipitation using an anti-cystatin C rabbit serum (III). Both the papain-reactive and inactive fractions of cystatin C were run in SDS-PAGE and visualized by Western blot. B, lysate of DC as above was either left untreated (control, upper gel) or incubated with carboxymethylated papain to deplete the active cystatin C (Post-papain, lower gel) before gel filtration chromatography. Cystatin C was retrieved from the fractions by immunoprecipitation using an anti-cystatin C rabbit serum. The immunoprecipitates were separated by 11.5% SDS-PAGE, and cystatin C was visualized by Western blot. The results were quantitated by densitometry (bottom graph). C, total cell lysates (TCL) of CD8+ DC were mixed with sample buffer lacking (−) or containing (+) 2-mercaptoethanol (2ME) as a reducing agent, fractionated by SDS-PAGE and analyzed by Western blot to detect cystatin C. All results are representative of two to three independent experiments.
Cystatin C Dimerizes in the ER during Synthesis, but Is Secreted Only in Monomeric Form
The cystatin C dimer detected inside primary DC by Western blot might be of endogenous origin or captured from the extracellular environment. To distinguish between these two possibilities we investigated cystatin C synthesis and dimerization in splenic DC. Both papain-reactive monomer and non-reactive dimer could be recovered from DC metabolically radiolabeled (pulsed) for 30 min with [35S]Met/Cys (Fig. 3, A and B). Brefeldin A (BFA) is an inhibitor of protein export out of the endoplasmic reticulum (ER) and its addition during the pulse resulted in accumulation of both monomer and dimer, indicating that cystatin C dimerization occurred within the ER during or shortly after translation (Fig. 3C). To determine the fate of the two molecular species, we incubated the radiolabeled DC in non-radioactive medium (chase) for up to 120 min and recovered the papain-reactive and non-reactive cystatin C fractions from the cells and the culture supernatant (this pulse-chase experiment was performed without BFA, Fig. 3, A and B). We found that the intracellular cystatin C monomer and dimer were both greatly reduced during the chase period, but almost all of the cystatin C recovered from the culture medium was monomeric (Fig. 3, A and B). Furthermore, the amount of monomeric cystatin C secreted to the medium was higher than that detected intracellularly after the pulse (Fig. 3A). This suggested that at least some of the newly synthesized dimer had been converted to monomer along the secretory pathway or soon after secretion. However, not all of the cystatin C synthesized during the pulse could be accounted for at the end of the 120-min chase, suggesting that some of the dimer was degraded intracellularly. Indeed, in cells pulse-chased in the presence of BFA to prevent egress of cystatin C out of the ER, the amount of monomer remained stable but the amount of dimer decreased gradually (Fig. 3D). Together, these data indicate that DC synthesized both monomeric and dimeric cystatin C, but only secreted the monomeric form. Some of the dimeric cystatin C was secreted and converted to monomer and the rest was degraded intracellularly. We could not prevent intracellular cystatin C dimer degradation with lysosomal or proteasomal protease inhibitors (Fig. 3, D and E), so the identity of the proteases involved in degradation of dimeric cystatin C remains unknown.
FIGURE 3.
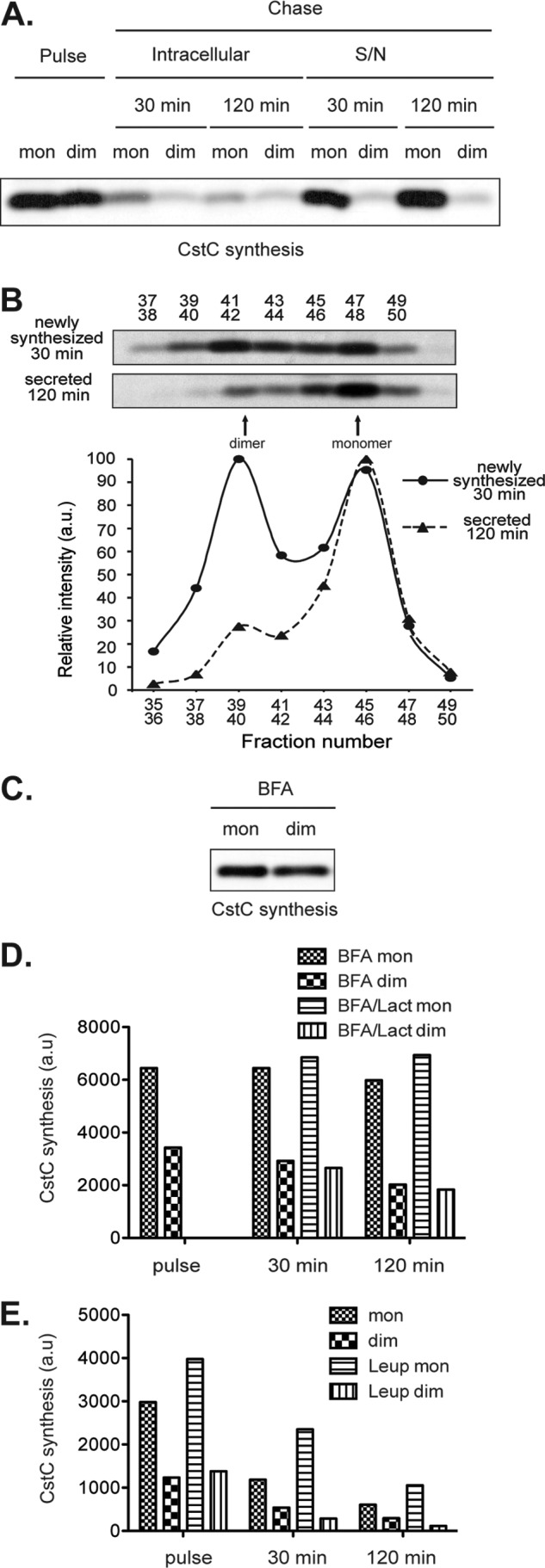
Cystatin C dimerizes in the ER, but is mostly secreted as monomers. A, newly synthesized proteins from splenic CD8+ DC were metabolically labeled with [35S]Met/Cys. The cells were equally divided into pulsed and chased samples. The pulsed samples were lysed immediately, whereas the chased samples were incubated for the indicated times before lysis. Cystatin C monomer (mon) and dimer (dim) from either cell lysates (intracellular) or cell culture supernatants (S/N) were retrieved by precipitation with immobilized papain and then by immunoprecipitation with anti-cystatin C serum, respectively. The immunoprecipitates were fractionated by SDS-PAGE, and revealed by autoradiography. B, freshly isolated CD8+ DC were treated as in A. Cell lysates from pulsed samples (top gel) or culture supernatants after a 120-min chase (bottom gel) were fractionated by gel-filtration chromatography. Cystatin C was retrieved from the fractions by immunoprecipitation using an anti-cystatin C rabbit serum. The immunoprecipitates were separated by 11.5% SDS-PAGE and cystatin C was visualized by autoradiography. The band intensities were quantitated in a phosphorimager (bottom graph). C, newly synthesized proteins from splenic CD8+ DC were metabolically labeled with 35S in the presence of the ER-Golgi protein transport inhibitor BFA, and cystatin C was retrieved as in A (pulse). D, Splenic CD8+ DC were metabolically labeled in the presence of BFA and chased in the presence of BFA plus, where indicated, the proteasome inhibitor Lactacystin (Lact). Papain-reactive monomers and non-reactive dimers were immunoprecipitated and run in SDS-PAGE as in A. The amount of radioactive cystatin C in each lane was quantitated in a phosphorimager. E, as in D, but cells were pulsed without drugs and chased in the presence or absence of leupeptin. All results are representative of three or more independent experiments performed.
Intracellular ROS Levels Determine the Extent of Cystatin C Dimerization
We extended the analysis of cystatin C dimerization to macrophages and found that in this cell type most of the cystatin C detected intracellularly by Western blot was dimeric (Fig. 4A). Pulse-chase analysis confirmed that in macrophages cystatin C synthesis is biased toward production of the dimeric form, but like DC, macrophages only secreted monomeric cystatin C (Fig. 4B). This result suggested that the amount of cystatin C dimer produced during or shortly after synthesis in the ER might vary depending on the physico-chemical conditions of this compartment in each cell type, and that these conditions were more conductive to dimerization in macrophages.
FIGURE 4.

Intracellular ROS levels correlate with cystatin C dimerization. A, macrophages (MΦ) or DC generated in vitro from BM precursors were lysed and cystatin C was precipitated with carboxymethylated papain followed by immunoprecipitation with anti-cystatin C serum as described in the legend to Fig. 2A. The samples were fractionated in SDS-PAGE and cystatin C detected by Western blot. B, cystatin C monomer and dimer synthesis and secretion by MΦ was analyzed as described for DC in the legend to Fig. 3A. C, macrophages and DC were loaded with the cell-permeable, redox-sensitive dye CM-H2DCFDA (5- (and 6-)chloromethyl 2′,7′-dichlorodihydrofluorescein diacetate), and their intracellular ROS levels were examined by flow cytometry. The graph shows the mean fluorescence intensity (MFI) in the FITC channel. The data are presented as the mean ± S.E. of three independent experiments performed. D, the intracellular ROS levels in CD8+ DC freshly isolated from spleens (immature), or in their counterparts cultured overnight (mature), were measured by FACS. E, lysates of purified immature (left) and mature (right) CD8+ DC were fractionated by gel filtration chromatography. Cystatin C was immunoprecipitated from the fractions, run in SDS-PAGE, visualized by Western blot, and quantitated by densitometry (bottom graphs). F, lysates of mature CD8+ DC were analyzed as described in the legend to Fig. 2A to retrieve cystatin C monomer (papain-binding) and dimer (sequentially immunoprecipitated with anti-cystatin C serum). G, mature CD8+ DC were metabolically labeled/chased, and processed as described in the legend to Fig. 3B to measure cystatin C monomer and dimer synthesis and secretion. All results are representative of two to three independent experiments performed.
One factor that might influence cystatin C dimerization is the intracellular level of ROS, which promotes protein dimerization (28). Macrophages have higher levels of ROS than DC (Fig. 4C), so a correlation between the ROS level and intracellular cystatin C dimerization did exist. We predicted that decreasing the level of intracellular ROS would reduce cystatin C dimerization in DC. This could be achieved by culturing splenic DC in vitro, which leads to spontaneous activation of the DC (29). DC activation is accompanied with a reduction in ROS levels (Fig. 4D). Resting (freshly purified) and mature (cultured overnight) DC synthesized similar amounts of cystatin C (data not shown), but the latter down-regulated the formation of papain non-reactive cystatin C dimers (Fig. 4, E–G). This result supported the notion of a direct correlation between the production of ROS and cystatin C dimerization.
Next we investigated the effect of raising the intracellular ROS level. For this test we could not use primary splenic DC as in the previous experiment because upon incubation in vitro these cells undergo spontaneous activation and down-regulate ROS production. Instead we used DC generated by culture of bone marrow precursors in medium supplemented with the DC growth factor, Flt3L. These DC are considered the functional equivalent of splenic DC (30). Incubation of these DC with hydrogen peroxide (H2O2) increased their intracellular ROS levels (Fig. 5A, upper panel) and promoted accumulation of the papain non-reactive cystatin C dimers (Fig. 5A, lower panel). These dimers were not disulfide linked as they were not stable in non-reducing SDS-PAGE (Fig. 5B). As an alternative treatment to increase the intracellular ROS levels in the DC, we incubated the cells with EA, an agent that depletes intracellular glutathione (31). A burst of intracellular ROS levels was observed following treatment with EA, which was accompanied with a gradual increase in the proportion of dimeric cystatin C within the cells (Fig. 5C). This increase was due to enhanced formation of cystatin C dimers co-translationally or shortly after translation, although the dimers disassembled upon secretion so that all the cystatin C released to the culture supernatant of EA-treated DC was monomeric (Fig. 5D). DC treated with EA thus resembled untreated macrophages (Fig. 4, A and B).
FIGURE 5.

Intracellular ROS from mitochondria promote cystatin C dimerization. A, DC generated in vitro from BM precursors (BM-DC) were incubated with or without the oxidant H2O2 (0.8 mm) for 30 min. ROS levels in the cells were measured by flow cytometry (upper panel) and cystatin C monomers (papain-binding) and dimers (non-binding) were retrieved from cell lysates as described in the legend to Fig. 2A. The data in the histogram shows the mean ± S.E. (n = 3). B, total cell lysates of the H2O2-treated samples were analyzed by non-reducing SDS-PAGE and Western blot. C, BM-DC were incubated at 37 °C with or without the antioxidant-depleting agent EA at 60 μm. Intracellular ROS levels were measured by flow cytometry (upper panel) and cystatin C monomers and dimers were retrieved and visualized by Western blot (lower panel) as in A. D, BM-DC were pulsed chased as described in the legend to Fig. 3A but in the presence or absence of 60 mm EA. Cystatin C monomers and dimers were retrieved from cell lysates after the pulse, and from the culture supernatant after the chase (S/N) as indicated. Newly synthesized cystatin C was visualized by autoradiography. The result is representative of two independent experiments. The intensity of the bands containing cystatin C in the two independent experiments were quantitated by densitometry and shown as mean ± S.E. (bottom graphs). E, BM-DC were either left untreated or treated with 60 μm EA, or 60 μm EA plus 50 nm mitochondria respiration chain complex inhibitor AA in triplicates before they were harvested and loaded with redox sensitive dye to check for intracellular ROS levels by flow cytometry. The data shows the mean ± S.E., which is representative of two independent experiments. F, BM-DC were lysed and their intracellular cystatin C species examined by Western blot as in C. BM-DC from cystatin C-deficient mice (CstC−/−) were included as negative control. All results are representative of two (A, B, D, and E), three (F), or five (C) independent experiments performed.
Blocking the Mitochondrial Respiration Chain Antagonizes EA-induced Cystatin C Dimerization
Mitochondria are major producers of ROS as by-products of the respiratory chain (32). To investigate the contribution of mitochondria to the EA-mediated induction of cystatin C dimerization we treated the cells with AA, a specific inhibitor of the mitochondrial respiratory complex III. This drug antagonized the effect of EA on ROS formation (Fig. 5E) and induction of cystatin C dimerization (Fig. 5F). Neither EA alone nor EA plus AA affected cell viability during the assay (data not shown). These results indicate that the production of ROS by mitochondria plays an important role in the magnitude of intracellular cystatin C dimerization within cells.
DISCUSSION
The first important conclusion of the results presented here is that cystatin C is not as ubiquitously expressed as generally assumed. We showed that bone marrow-derived cells contribute up to 30% of the steady-state concentration of cystatin C in mouse serum. Among these cells, the major producers were CD8+ DC and macrophages. Immunofluorescence microscopy analysis of Langerhans cells also revealed high levels of cystatin C in this DC type of the skin (data not shown). This pattern of expression correlates with the results of the Immunological Genome Project and appears conserved in humans, with DC and macrophages showing the highest expression of the CST3 gene (49).
One important implication of these observations is that the local concentration of cystatin C in a particular tissue may vary depending on the abundance of DC and macrophages. These two cell types accumulate at sites of inflammation, including atherosclerotic lesions, aortic aneurysms, and the brain (4, 33–35). Furthermore, CD8+ DC and monocyte-derived DC are present in healthy aortas and both expand during atherosclerosis (36). It is plausible that the control of pathological extracellular proteolysis at sites of inflammation depends in part on secretion of cystatin C by DC and macrophages. Indeed, DC reportedly play protective roles in atherosclerosis (36). We previously showed that IL-10 released during inflammation down-regulates Irf-8 and hence cst3 expression in DC and macrophages (11). As discussed below, the control of cystatin C dimerization and secretion by ROS introduces an additional layer of regulation. This complex interplay of regulatory mechanisms of cystatin C activity may partially explain the contrasting results of studies that examined the correlation between serum cystatin C and vascular diseases (4). Recruitment of macrophages and DC at incipient vascular lesions may increase cystatin C secretion both locally and systemically, protecting against pathological degradation of the extracellular matrix by cysteine proteases. However, as the disease progresses and inflammation sets in, production of anti-inflammatory IL-10 might cause reduced production of cystatin C (11). Increased ROS levels at lesion sites (37) would also promote cystatin C dimerization and intracellular retention (see below). These effects may contribute to reduce cystatin C secretion and tip the local proteolytic balance toward extracellular matrix destruction (38). It is in this situation where the low cystatin C concentration in plasma would correlate with susceptibility to vascular disease (3, 38).
The level of cystatin C in plasma is used as a diagnostic marker of kidney disease (7–9, 39). Our results are also relevant to the interpretation of these measurements. Clearly, the assumption that cystatin C is constitutively produced by most nucleated cells at stable levels is not correct because some cell types express much more than others and expression is regulated by external stimuli (IL-10, ROS). Our results predict that infectious/inflammatory processes that lead to expansion of macrophages and DC, even if the increments are modest in the context of the whole body, might cause a significant change in serum cystatin C levels. Indeed, it has been shown that serum cystatin C concentration correlates with increased numbers of monocytes, the precursor of macrophages and DC (40, 41). This correlation has been interpreted as evidence of kidney dysfunction by the same inflammatory processes that triggered monocyte recruitment. We propose that the increase in serum cystatin C might be a direct consequence of monocytosis, independently of the damage that the inflammatory process may have induced in the kidneys.
The third area where our results have clinical implications is cerebral amyloid angiopathy and neurodegeneration. Cystatin C plays beneficial and pathologic roles in Alzheimer disease (17, 42–44). The beneficial roles have been attributed to the protease inhibitory function of cystatin C (42), to its largely uncharacterized neuroprotective or neurogenic activities (43, 44), or to its capacity to inhibit formation of Aβ amyloid (17). The pathologic roles have been attributed to the propensity of cystatin C to form dimers and amyloid deposits in the brain vasculature, by itself or with other proteins (12). This is particularly true in the case of HCCAA, caused by a mutant form of cystatin C much more prone to domain swapping and amyloid formation (12). As discussed above for other pathologies, bone marrow-derived cells, in particular DC and macrophages, may contribute to these protective or deleterious effects. We propose that transplantation of HCCAA patients with non-mutant bone marrow may provide a viable therapy to avert the fatal consequences of the disease. Another implication is that inflammatory processes that induce DC and/or macrophage recruitment to the brain may increase the local concentration of cystatin C with potential pathologic or beneficial effects.
We have also described what we believe is the first study of cystatin C dimerization in primary cells expressing physiological levels of the protein. The biochemistry of cystatin C domain swapping has been studied in vitro employing recombinant protein (45). Dimerization has previously been observed intracellularly, but in transfected cells overexpressing the protein, so the physiological relevance of this observation was unclear (46). Here we showed that >40% of the total amount of cystatin C produced by primary DC and macrophages in healthy mice dimerizes in the ER lumen during or soon after translation. Dimerization did not correlate with the level of expression. The intracellular ROS levels, in contrast, exerted a major influence, with higher levels favoring dimer formation. We could not detect secretion of dimers; they were either converted into monomers in the extracellular medium and/or degraded intracellularly by unknown proteases. Control of dimerization may be a mechanism to regulate the secretion of protease inhibitory (monomeric) cystatin C, but it cannot be excluded that dimeric cystatin C may have specific intracellular functions. The effect of ROS on cystatin C dimerization is relevant in the context of atherosclerosis and aortic aneurisms because ROS increase in these lesions (37), which might result in lesser secretion of monomeric cystatin C and increased extracellular cysteine protease activity and pathology.
Our observations prompt the hypothesis that intracellular processes may play a bigger role in the initial steps of cystatin C oligomerization/amyloidogenesis than generally recognized. Oxidative stress or cardiomyopathy induces cardiac tissue to release intracellular cystatin C, resulting in elevated serum cystatin C levels (47). Events that promote increases in intracellular ROS levels in the brain or its associated vasculature might induce formation of cystatin C dimers. Such events are usually linked with insults that promote cellular stress and death, which might cause release of intracellular cystatin C dimers that could seed the formation of extracellular oligomers and eventually amyloid. Monocyte-derived macrophages and DC recruited to the site of tissue damage might exacerbate the process by contributing to raise the local concentration of cystatin C, and by themselves undergoing cell death and releasing cystatin C dimers induced by the oxidative stress. This process would be exacerbated in individuals expressing the dimerization prone, mutant form of cystatin C associated with HCCAA (16). Strikingly, the lifespan of carriers of the mutant form of cystatin C that causes HCCAA decreased drastically in the 19th century, and this has been attributed to changes in lifestyle, specifically to a diet richer in carbohydrates and salt (33). We speculate that such changes may increase the levels of ROS and the incidence of inflammatory processes in the brain vasculature, both of which would promote increased local cystatin C expression and dimerization. Changes in lifestyle patterns to reduce ROS formation or administration of antioxidants might slow down the formation of cystatin C amyloid.
This work was supported in part by National Health and Medical Research Council (NHMRC) of Australia Grants 1006428 (to Y. X.) and 461219 (to J. G. Z.) and the Victorian Government's Operational Infrastructure Support Program.
- DC
- dendritic cells
- ER
- endoplasmic reticulum
- ROS
- reactive oxygen species
- Aβ
- amyloid β
- HCCAA
- hereditary cystatin C amyloid angiopathy
- CAA
- cerebral amyloid angiopathy
- Flt-3L
- fms-related tyrosine kinase 3 ligand
- EA
- ethacrynic acid
- AA
- antimycin A
- BM
- bone marrow
- BFA
- brefeldin A.
REFERENCES
- 1. Abrahamson M., Alvarez-Fernandez M., Nathanson C. M. (2003) Cystatins. Biochem. Soc. Symp. 70, 179–199 [DOI] [PubMed] [Google Scholar]
- 2. Keppler D. (2006) Towards novel anti-cancer strategies based on cystatin function. Cancer Lett 235, 159–176 [DOI] [PubMed] [Google Scholar]
- 3. Shi G. P., Sukhova G. K., Grubb A., Ducharme A., Rhode L. H., Lee R. T., Ridker P. M., Libby P., Chapman H. A. (1999) Cystatin C deficiency in human atherosclerosis and aortic aneurysms. J. Clin. Invest. 104, 1191–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu J., Sukhova G. K., Sun J. S., Xu W. H., Libby P., Shi G. P. (2004) Lysosomal cysteine proteases in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 24, 1359–1366 [DOI] [PubMed] [Google Scholar]
- 5. Rokadia H. K., Agarwal S. (2012) Serum cystatin C and emphysema: results from the National Health and Nutrition Examination Survey (NHANES). Lung 190, 283–290 [DOI] [PubMed] [Google Scholar]
- 6. Bird P. I., Trapani J. A., Villadangos J. A. (2009) Endolysosomal proteases and their inhibitors in immunity. Nat. Rev. Immunol. 9, 871–882 [DOI] [PubMed] [Google Scholar]
- 7. Grubb A. (2010) Non-invasive estimation of glomerular filtration rate (GFR). The Lund model: simultaneous use of cystatin C- and creatinine-based GFR-prediction equations, clinical data and an internal quality check. Scand. J. Clin. Lab. Invest. 70, 65–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shlipak M. G., Matsushita K., Ärnlöv J., Inker L. A., Katz R., Polkinghorne K. R., Rothenbacher D., Sarnak M. J., Astor B. C., Coresh J., Levey A. S., Gansevoort R. T., and CKD Prognosis Consortium (2013) Cystatin C versus creatinine in determining risk based on kidney function. N. Engl. J. Med. 369, 932–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smith E. R. (2013) Cystatin C. More than a filtration marker? Atherosclerosis 230, 73–75 [DOI] [PubMed] [Google Scholar]
- 10. Tamura T., Thotakura P., Tanaka T. S., Ko M. S., Ozato K. (2005) Identification of target genes and a unique cis element regulated by IRF-8 in developing macrophages. Blood 106, 1938–1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xu Y., Schnorrer P., Proietto A., Kowalski G., Febbraio M. A., Acha-Orbea H., Dickins R. A., Villadangos J. A. (2011) IL-10 controls cystatin C synthesis and blood concentration in response to inflammation through regulation of IFN regulatory factor 8 expression. J. Immunol. 186, 3666–3673 [DOI] [PubMed] [Google Scholar]
- 12. Kaur G., Levy E. (2012) Cystatin C in Alzheimer's disease. Front. Mol. Neurosci. 5, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Janowski R., Kozak M., Jankowska E., Grzonka Z., Grubb A., Abrahamson M., Jaskolski M. (2001) Human cystatin C, an amyloidogenic protein, dimerizes through three-dimensional domain swapping. Nat. Struct. Biol. 8, 316–320 [DOI] [PubMed] [Google Scholar]
- 14. Wahlbom M., Wang X., Lindström V., Carlemalm E., Jaskolski M., Grubb A. (2007) Fibrillogenic oligomers of human cystatin C are formed by propagated domain swapping. J. Biol. Chem. 282, 18318–18326 [DOI] [PubMed] [Google Scholar]
- 15. Mi W., Jung S. S., Yu H., Schmidt S. D., Nixon R. A., Mathews P. M., Tagliavini F., Levy E. (2009) Complexes of amyloid-β and cystatin C in the human central nervous system. J. Alzheimers Dis. 18, 273–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Palsdottir A., Abrahamson M., Thorsteinsson L., Arnason A., Olafsson I., Grubb A., Jensson O. (1988) Mutation in cystatin C gene causes hereditary brain haemorrhage. Lancet 2, 603–604 [DOI] [PubMed] [Google Scholar]
- 17. Mi W., Pawlik M., Sastre M., Jung S. S., Radvinsky D. S., Klein A. M., Sommer J., Schmidt S. D., Nixon R. A., Mathews P. M., Levy E. (2007) Cystatin C inhibits amyloid-beta deposition in Alzheimer's disease mouse models. Nat. Genet. 39, 1440–1442 [DOI] [PubMed] [Google Scholar]
- 18. Levy E., Jaskolski M., Grubb A. (2006) The role of cystatin C in cerebral amyloid angiopathy and stroke: cell biology and animal models. Brain Pathol. 16, 60–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huh C. G., Håkansson K., Nathanson C. M., Thorgeirsson U. P., Jonsson N., Grubb A., Abrahamson M., Karlsson S. (1999) Decreased metastatic spread in mice homozygous for a null allele of the cystatin C protease inhibitor gene. Mol. Pathol. 52, 332–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vremec D., Pooley J., Hochrein H., Wu L., Shortman K. (2000) CD4 and CD8 expression by dendritic cell subtypes in mouse thymus and spleen. J. Immunol. 164, 2978–2986 [DOI] [PubMed] [Google Scholar]
- 21. Xu Y., Zhan Y., Lew A. M., Naik S. H., Kershaw M. H. (2007) Differential development of murine dendritic cells by GM-CSF versus Flt3 ligand has implications for inflammation and trafficking. J. Immunol. 179, 7577–7584 [DOI] [PubMed] [Google Scholar]
- 22. Zhan Y., Vega-Ramos J., Carrington E. M., Villadangos J. A., Lew A. M., Xu Y. (2012) The inflammatory cytokine, GM-CSF, alters the developmental outcome of murine dendritic cells. Eur. J. Immunol. 42, 2889–2900 [DOI] [PubMed] [Google Scholar]
- 23. Belz G. T., Wilson N. S., Smith C. M., Mount A. M., Carbone F. R., Heath W. R. (2006) Bone marrow-derived cells expand memory CD8+ T cells in response to viral infections of the lung and skin. Eur. J. Immunol. 36, 327–335 [DOI] [PubMed] [Google Scholar]
- 24. Villadangos J. A., Riese R. J., Peters C., Chapman H. A., Ploegh H. L. (1997) Degradation of mouse invariant chain: roles of cathepsins S and D and the influence of major histocompatibility complex polymorphism. J. Exp. Med. 186, 549–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu Y., Huntington N. D., Harder K. W., Nandurkar H., Hibbs M. L., Tarlinton D. M. (2012) Phosphatidylinositol-3 kinase activity in B cells is negatively regulated by Lyn tyrosine kinase. Immunol. Cell Biol. 90, 903–911 [DOI] [PubMed] [Google Scholar]
- 26. Warfel A. H., Zucker-Franklin D., Frangione B., Ghiso J. (1987) Constitutive secretion of cystatin C (γ-trace) by monocytes and macrophages and its down-regulation after stimulation. J. Exp. Med. 166, 1912–1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Henskens Y. M., Veerman E. C., Nieuw Amerongen A. V. (1996) Cystatins in health and disease. Biol. Chem. Hoppe Seyler 377, 71–86 [DOI] [PubMed] [Google Scholar]
- 28. Gotoh Y., Cooper J. A. (1998) Reactive oxygen species- and dimerization-induced activation of apoptosis signal-regulating kinase 1 in tumor necrosis factor-α signal transduction. J. Biol. Chem. 273, 17477–17482 [DOI] [PubMed] [Google Scholar]
- 29. Wilson N. S., El-Sukkari D., Belz G. T., Smith C. M., Steptoe R. J., Heath W. R., Shortman K., Villadangos J. A. (2003) Most lymphoid organ dendritic cell types are phenotypically and functionally immature. Blood 102, 2187–2194 [DOI] [PubMed] [Google Scholar]
- 30. Naik S. H., Proietto A. I., Wilson N. S., Dakic A., Schnorrer P., Fuchsberger M., Lahoud M. H., O'Keeffe M., Shao Q. X., Chen W. F., Villadangos J. A., Shortman K., Wu L. (2005) Cutting edge: generation of splenic CD8+ and CD8− dendritic cell equivalents in Fms-like tyrosine kinase 3 ligand bone marrow cultures. J. Immunol. 174, 6592–6597 [DOI] [PubMed] [Google Scholar]
- 31. Keelan J., Allen N. J., Antcliffe D., Pal S., Duchen M. R. (2001) Quantitative imaging of glutathione in hippocampal neurons and glia in culture using monochlorobimane. J. Neurosci. Res. 66, 873–884 [DOI] [PubMed] [Google Scholar]
- 32. Murphy M. P. (2009) How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Palsdottir A., Helgason A., Palsson S., Bjornsson H. T., Bragason B. T., Gretarsdottir S., Thorsteinsdottir U., Olafsson E., Stefansson K. (2008) A drastic reduction in the life span of cystatin C L68Q carriers due to life-style changes during the last two centuries. PLoS Genet. 4, e1000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bobryshev Y. V. (2010) Dendritic cells and their role in atherogenesis. Lab. Invest. 90, 970–984 [DOI] [PubMed] [Google Scholar]
- 35. Terry R. L., Getts D. R., Deffrasnes C., van Vreden C., Campbell I. L., King N. J. (2012) Inflammatory monocytes and the pathogenesis of viral encephalitis. J. Neuroinflammation 9, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Choi J. H., Cheong C., Dandamudi D. B., Park C. G., Rodriguez A., Mehandru S., Velinzon K., Jung I. H., Yoo J. Y., Oh G. T., Steinman R. M. (2011) Flt3 signaling-dependent dendritic cells protect against atherosclerosis. Immunity 35, 819–831 [DOI] [PubMed] [Google Scholar]
- 37. Rosenfeld M. E. (1998) Inflammation, lipids, and free radicals: lessons learned from the atherogenic process. Semin. Reprod. Endocrinol. 16, 249–261 [DOI] [PubMed] [Google Scholar]
- 38. Sukhova G. K., Wang B., Libby P., Pan J. H., Zhang Y., Grubb A., Fang K., Chapman H. A., Shi G. P. (2005) Cystatin C deficiency increases elastic lamina degradation and aortic dilatation in apolipoprotein E-null mice. Circ. Res. 96, 368–375 [DOI] [PubMed] [Google Scholar]
- 39. Randers E., Erlandsen E. J. (1999) Serum cystatin C as an endogenous marker of the renal function. A review. Clin. Chem. Lab. Med. 37, 389–395 [DOI] [PubMed] [Google Scholar]
- 40. Evangelopoulos A. A., Vallianou N. G., Bountziouka V., Katsagoni C., Bathrellou E., Vogiatzakis E. D., Bonou M. S., Barbetseas J., Avgerinos P. C., Panagiotakos D. B. (2012) Association between serum cystatin C, monocytes and other inflammatory markers. Intern. Med. J. 42, 517–522 [DOI] [PubMed] [Google Scholar]
- 41. Ganda A., Magnusson M., Yvan-Charvet L., Hedblad B., Engström G., Ai D., Wang T. J., Gerszten R. E., Melander O., Tall A. R. (2013) Mild renal dysfunction and metabolites tied to low HDL cholesterol are associated with monocytosis and atherosclerosis. Circulation 127, 988–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sun B., Zhou Y., Halabisky B., Lo I., Cho S. H., Mueller-Steiner S., Devidze N., Wang X., Grubb A., Gan L. (2008) Cystatin C-cathepsin B axis regulates amyloid beta levels and associated neuronal deficits in an animal model of Alzheimer's disease. Neuron 60, 247–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nishiyama K., Konishi A., Nishio C., Araki-Yoshida K., Hatanaka H., Kojima M., Ohmiya Y., Yamada M., Koshimizu H. (2005) Expression of cystatin C prevents oxidative stress-induced death in PC12 cells. Brain Res. Bull. 67, 94–99 [DOI] [PubMed] [Google Scholar]
- 44. Tizon B., Sahoo S., Yu H., Gauthier S., Kumar A. R., Mohan P., Figliola M., Pawlik M., Grubb A., Uchiyama Y., Bandyopadhyay U., Cuervo A. M., Nixon R. A., Levy E. (2010) Induction of autophagy by cystatin C: a mechanism that protects murine primary cortical neurons and neuronal cell lines. PLoS One 5, e9819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nilsson M., Wang X., Rodziewicz-Motowidlo S., Janowski R., Lindström V., Onnerfjord P., Westermark G., Grzonka Z., Jaskolski M., Grubb A. (2004) Prevention of domain swapping inhibits dimerization and amyloid fibril formation of cystatin C: use of engineered disulfide bridges, antibodies, and carboxymethylpapain to stabilize the monomeric form of cystatin C. J. Biol. Chem. 279, 24236–24245 [DOI] [PubMed] [Google Scholar]
- 46. Merz G. S., Benedikz E., Schwenk V., Johansen T. E., Vogel L. K., Rushbrook J. I., Wisniewski H. M. (1997) Human cystatin C forms an inactive dimer during intracellular trafficking in transfected CHO cells. J. Cell Physiol. 173, 423–432 [DOI] [PubMed] [Google Scholar]
- 47. Xie L., Terrand J., Xu B., Tsaprailis G., Boyer J., Chen Q. M. (2010) Cystatin C increases in cardiac injury: a role in extracellular matrix protein modulation. Cardiovasc. Res. 87, 628–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. El-Sukkari D., Wilson N. S., Hakansson K., Steptoe R. J., Grubb A., Shortman K., Villadangos J. A. (2003) The protease inhibitor cystatin C is differentially expressed among dendritic cell populations, but does not control antigen presentation. J. Immunol. 171, 5003–5011 [DOI] [PubMed] [Google Scholar]
- 49. Kim C. C., Lanier L. L. (2013) Beyond the transcriptome: completion of act one of the immunological genome project. Curr. Opin. Immunol. 25, 593–597 [DOI] [PMC free article] [PubMed] [Google Scholar]