

Background: The mechanisms by which H2S regulates inflammation remain unclear.
Results: H2S inhibits NF-κB p65 phosphorylation, nuclear translocation, DNA binding activity, and recruitment to MCP-1 promoter in ox-LDL-treated macrophages by targeting the free sulfhydryl group on cysteine 38 in p65.
Conclusion: H2S inhibits macrophage inflammation by suppressing NF-κB activation.
Significance: These findings reveal mechanisms for regulation of NF-κB pathway by H2S.
Keywords: Hydrogen Sulfide, Inflammation, Macrophages, NF-κB, Sulfhydryl
Abstract
This study was designed to examine the role of hydrogen sulfide (H2S) in the generation of oxidized low-density lipoprotein (ox-LDL)-stimulated monocyte chemoattractant protein 1 (MCP-1) from macrophages and possible mechanisms. THP-1 cells and RAW macrophages were pretreated with sodium hydrosulfide (NaHS) and hexyl acrylate and then treated with ox-LDL. The results showed that ox-LDL treatment down-regulated the H2S/cystathionine-β-synthase pathway, with increased MCP-1 protein and mRNA expression in both THP-1 cells and RAW macrophages. Hexyl acrylate promoted ox-LDL-induced inflammation, whereas the H2S donor NaHS inhibited it. NaHS markedly suppressed NF-κB p65 phosphorylation, nuclear translocation, DNA binding activity, and recruitment to the MCP-1 promoter in ox-LDL-treated macrophages. Furthermore, NaHS decreased the ratio of free thiol groups in p65, whereas the thiol reductant DTT reversed the inhibiting effect of H2S on the p65 DNA binding activity. Most importantly, site-specific mutation of cysteine 38 to serine in p65 abolished the effect of H2S on the sulfhydration of NF-κB and ox-LDL-induced NF-κB activation. These results suggested that endogenous H2S inhibited ox-LDL-induced macrophage inflammation by suppressing NF-κB p65 phosphorylation, nuclear translocation, DNA binding activity, and recruitment to the MCP-1 promoter. The sulfhydration of free thiol group on cysteine 38 in p65 served as a molecular mechanism by which H2S inhibited NF-κB pathway activation in ox-LDL-induced macrophage inflammation.
Introduction
Atherosclerosis is the most common cardiovascular disease and the leading cause of morbidity and mortality in many countries. However, the precise mechanism of atherosclerosis remains unclear. Inflammation is a pivotal factor throughout the whole process of atherosclerosis (1, 2). The accumulation of cholesterol-rich lipoproteins in the artery wall results in the recruitment of circulating monocytes, their adhesion to the endothelium, and their differentiation into tissue macrophages. This initial process is predominantly mediated by monocyte chemoattractant protein 1 (MCP-1), which is a potent chemotactic factor for monocytes (3) and is also secreted by monocytes/macrophages (4, 5).
Hydrogen sulfide (H2S), which has long been known as a toxic gas, is now regarded as a novel gasotransmitter (6). Endogenous H2S is generated from l-cysteine, and the key enzymes that catalyze this process include cystathionine-β-synthase (CBS),3 cystathionine-γ-lyase (CSE), and 3-mercaptopyruvate sulfurtransferase (MPST) (7–9). Endogenous H2S plays important regulatory roles in the development of a variety of cardiovascular diseases (10–12). Most interestingly, in recent years, evidence has accumulated to suggest important roles for H2S as a regulator of inflammation. H2S donors can inhibit lipopolysaccharide-induced formation of inflammatory mediators in macrophages (13) and decrease the levels of inflammatory cytokines such as IL-6 and TNF-α and other cytokines in rats with acute lung injury and endotoxic shock (14–16). Our research group demonstrated that H2S exerted an antiatherogenic effect in apoE−/− mice (16). However, the mechanisms by which H2S regulates inflammatory molecules remain unclear. This study, therefore, was undertaken to examine the inhibitory role of H2S in MCP-1 expression induced by ox-LDL in macrophages and to determine whether NF-κB signaling is involved as a pivotal regulatory pathway in this process.
MATERIALS AND METHODS
Cell Culture of THP-1 Monocytes and RAW Macrophages
THP-1 (a promonocytic cell line) and RAW macrophages were obtained from the ATCC (catalog no. TIB-202). Cells were grown in RPMI 1640 medium containing 10% FBS, 100 units/ml penicillin, and 100 units/ml streptomycin. THP-1 cells were maintained at 37 °C in a humidified atmosphere of 5% CO2 and induced to differentiate into macrophages by incubation with phorbol 12-myristate 13-acetate (50 nmol/liter) for 24 h (17). Macrophages were pretreated with NaHS for 30 min and stimulated with oxidized LDL (50 mg/liter) for the indicated times (18).
Plasmids and Transfection
PIRES2-EGFP expression vectors for human p65 (wild-type) and human p65 C38S (a cysteine-to-serine mutation at the 38th position of p65) were purchased from JingSai Co. (Wuhan, China). THP-1-derived macrophages were plated 12–16 h before transfection at a density of 1 × 106 cells/10-cm plate. The cells in each plate were transiently transfected with 8 μg of p65 or p65 C38S plasmid using 20 μl of Lipofectamine 2000 reagent (Invitrogen) per plate as described by the manufacturer. After 24 h, the cells were exposed to the indicated concentrations of ox-LDL, NaHS, and the sulfhydryl modifiers diamide or DTT. Total or nuclear proteins were then extracted after a designated time. Additionally, THP-1-derived macrophages were transfected with vehicle using the method described above. All transfection experiments were performed in duplicate and repeated separately at least three times.
Determination of CBS, CSE, MPST, and MCP-1 mRNA Expression by Quantitative Real-time PCR
Total RNA was extracted using TRIzol reagent and transcribed into cDNA using oligo(dT)15 primer and moloney murine leukemia virus reverse transcriptase. Quantitative real-time PCR was performed on an ABI Prism 7300 instrument (Applied Biosystems, Foster, CA). The amplification conditions for the cDNA for CBS, CSE, MPST, and MCP-1 were as follows: denaturing at 94 °C for 15 s, annealing at 60 °C for 1 min, and polymerizing at 72 °C for 30 s for 40 cycles. Samples and standards were determined in duplicate. TaqMan probes were modified by 5′-carboxyfluorescein and 3′-carboxytetramethylrhodamine. The primers used for human CBS were 5′-CCCCTATGGTCAGAATCAACAAG-3′ (sense), 5′-GGCTGATGCGGTCCTTCA-3′ (antisense), and 5′-AGTGTGAGTTCTTCAACGCGGGCG-3′ (TaqMan probe), and the expected product length was 111 base pairs. The primers used for human CSE were 5′-CCCAGTTCCTGGAATCTAATCCT-3′ (sense), 5′-CACTGACGCTTCACCAACTCA-3′ (antisense), and 5′-TAGAAAAGGTTATTTATCCTGGGCTGCCCTCT-3′ (TaqMan probe), and the expected product length was 91 base pairs. The primers used for human MPST were 5′-CCGAGACGGCATTGAACCT-3′ (sense), 5′-CCTGGCTCAGGAAGTCTGTGA-3′ (antisense), and 5′-CCACATCCCAGGTACCGTGAACATCC-3′ (TaqMan probe), and the expected product length was 71 base pairs. The primers for human MCP-1 were 5′-CAGCAAGTGTCCCAAAGAAGCT-3′ (sense), 5′-TGCTTGTCCAGGTGGTCCAT-3′ (antisense), and 5′-TGATCTTCAAGACCATTGTGGCCAAGG-3′ (TaqMan probe), and the expected product length was 111 base pairs. GAPDH was used for normalization. The 5′ and 3′ primers for GAPDH were 5′-CAGTCAGCCGCATCTTCTTTT-3′, 5′-GTGACCAGGCGCCCAATAC-3′, and 5′-CGTCGCCAGCCGAGCCACA-3′ (TaqMan probe), respectively, and the expected product length was 110 base pairs.
Measurement of CBS, CSE, and MPST Protein Expression by Western Blotting
Total protein from THP-1-derived macrophages was extracted in ice-cold protein lysis buffer containing 50 mmol/liter Tris-Cl (pH 7.4), 150 mmol/liter NaCl, 1 mmol/liter ethylenediamine tetraacetic acid, 1% Nonidet P-40, 0.25% sodium deoxycholate, 1 mmol/liter PMSF, and protease and phosphatase inhibitors. Protein samples (20 μg) were separated by 10% SDS-PAGE and transferred onto a PVDF membrane. The membrane was incubated overnight with primary antibodies against CBS (Santa Cruz Biotechnology, Santa Cruz, California), CSE (Abnova Corp., Abnova, Taiwan, China), MPST (Santa Cruz Biotechnology), or GAPDH (Lab Version Corp., Fremont, CA) at 4 °C and then incubated in HRP-conjugated secondary antibody. Immunoreactions were visualized by ECL and exposed to Kodak x-ray film (Eastman Kodak Co., Rochester, NY). GAPDH was used for normalization.
Measurement of H2S Content in Cell Supernatant
Cell supernatant H2S was measured using a sulfite electrode (PXS-270, Shanghai, China) (19, 20). Briefly, 0.5 ml of antioxidant buffer (2.35 m NaOH and 0.27 m EDTA) was mixed with 0.5 ml of cell supernatant or standard S2− solution (10, 20, 40, 60, or 80 μmol/liter, respectively) in the same volume. After being rinsed with distilled water and dried, the electrode was immersed into the sample. The electrode potential (voltage value, in millivolt) was recorded when the reading stabilized. The H2S concentration was calculated according to standard curve plotting by voltage value versus concentration on the basis of standard S2− solution.
Measurement of Endogenous H2S Content in Cells
Endogenous H2S in cells was measured using a fluorescent probe (provided by Prof. Xinjing Tang, Peking University Health Science Centre, Beijing, China) as described previously (21). Briefly, the culture supernatant of THP-1-derived macrophages was discarded and washed with PBS (0.01 μmol/liter) three times. Then the cells were stained in the working liquid of the fluorescent probe for 30 min at 37 °C, washed with PBS, and detected as blue fluorescence by confocal microscopy.
Measurement of MCP-1, TNF-α, MIF, and IL-10 Content in Cell Supernatant by ELISA
Cell supernatant concentrations of MCP-1, TNF-α, migration inhibition factor (MIF), and IL-10 were measured in duplicate by quantitative sandwich ELISA (R&D Systems, Minneapolis, MN) according to the instructions of the manufacturer. The kits for the assay utilized two monoclonal antibodies directed against separate antigens of human MCP-1, TNF-α, MIF, and IL-10. The concentrations in the supernatant were calculated from the linear range of a standard curve.
Determination of MCP-1 Expression and NF-κB p65 Nuclear Translocation by Immunofluorescence
For determination of MCP-1 expression and NF-κB nuclear translocation, THP-1-derived macrophages on slides were pretreated with 100 μmol/liter or 500 μmol/liter NaHS for 30 min and stimulated with ox-LDL for 6 h or 30 min. They were fixed in 4% paraformaldehyde for 30 min, washed with PBS, blocked with 5% BSA in PBS for 30 min at 37 °C, and then reacted with rabbit anti-human MCP-1 or NF-κB p65 antibody (1:15 dilution in PBS) overnight at 4 °C. After washing, slides were incubated for 1 h at 37 °C with FITC-conjugated goat anti-rabbit IgG and propidium iodide and then viewed on a confocal fluorescent microscope. Green fluorescence indicated MCP-1 and NF-κB p65 expressions, and red fluorescence represented nuclei. NF-κB translocation occurred in cells in which green and red overlapped.
Measurement of Phosphorylation of NF-κB p65 and IκBα Protein by Western Blotting
Total protein in THP-1-derived macrophages was extracted in ice-cold protein lysis buffer containing 50 mmol/liter Tris-Cl (pH 7.4), 150 mmol/liter NaCl, 1 mmol/liter ethylenediamine tetraacetic acid, 1% Nonidet P-40, 0.25% sodium deoxycholate, 1 mmol/liter PMSF, and protease and phosphatase inhibitors. Protein samples (20 μg) were separated by 10% sodium SDS-PAGE and transferred onto a PVDF membrane. The membrane was incubated overnight with primary antibodies against phosphorylated NF-κB or IκBα (Cell Signaling Technology, Boston, MA) at 4 °C and then incubated in HRP-conjugated secondary antibody. Immunoreactions were visualized by ECL and exposed on Kodak x-ray film (Eastman Kodak Co.). After exposure, the membrane was stripped with stripping solution for 30 min and then incubated overnight with primary antibodies against NF-κB or IκBα (Cell Signaling Technology) at 4 °C. It was then incubated in HRP-conjugated secondary antibody and exposed.
Measurement of Sulfhydryl Group NF-κB p65 Protein Expression by Western Blotting
Total protein in THP-1-derived macrophages was extracted in ice-cold protein lysis buffer, and cell lysates were incubated with EZ-linkTM PEO-iodoacetyl biotin (10 mg/ml, Pierce) for 12 h at 4 °C and then incubated with 30 μl of UltraLinkTM Immobilized NeutravidinTM (Pierce) for 4 h on a roller system at 4 °C. The beads were washed twice with 1 ml of lysis buffer and three times with 1 ml of PBS. The measurement of sulfhydryl groups in the NF-κB p65 protein was conducted by Western blotting and probed with rabbit anti-NF-κB antibody (Cell Signaling Technology).
Modified Biotin Switch Assay for Sulfhydration
Sulfhydration of p65 was monitored by the modified biotin switch assay as described previously (22). THP-1-derived macrophages were transfected with wild-type or mutant p65 (C38S) plasmid and then treated with or without NaHS for 30 min. The cells were homogenized in cell lysis buffer and centrifuged at 10,000 × g for 10 min at 4 °C. Cell lysates were added to blocking buffer (lysis buffer supplemented with 2.5% SDS and 20 mm S-methyl methanethiosulfonate) at 50 °C for 20 min with frequent vortexing. Then the MMTS was removed by acetone, and proteins were precipitated at −20 °C for 20 min. The proteins were resuspended in lysis buffer and incubated with EZ-linkTM PEO-iodoacetyl biotin (10 mg/ml). After incubation for 12 h at 4 °C, biotinylated proteins were precipitated by 30 μl of UltraLinkTM Immobilized NeutravidinTM for 4 h on a roller system at 4 °C. The beads were washed twice with 1 ml of lysis buffer and three times with 1 ml of PBS. The biotinylated proteins were eluted by SDS-PAGE sample buffer, subjected to Western blotting, and probed with rabbit anti-NF-κB antibody.
Measurement of NF-κB p65 Binding Activity by EMSA
Nuclear extracts were prepared using a commercial kit (catalog no. 78835) of NE-PER nuclear and cytoplasmic extraction reagents (Thermo Scientific). In brief, cells were washed twice with ice-cold phosphate-buffered saline after incubation and suspended in buffer cytoplasmic extraction reagent I (CER I). The cells were then incubated on ice for 10 min, after which ice-cold buffer cytoplasmic extraction reagent II (CER II) was added. The tube was mixed thoroughly using a Vortex mixer for 10 s before centrifugation (16,000 × g) at 4 °C for 5 min. The nuclear pellets obtained were resuspended in ice-cold buffer nuclear extraction reagent and kept on ice for 35 min with intermittent agitation. The samples were then subjected to centrifugation for 10 min at 4 °C, and the supernatant was stored at −70 °C. The protein concentration was determined using a Bio-Rad protein assay kit.
The EMSA was carried out using a digoxigenin (DIG) gel shift kit (Roche Applied Science) according to the instructions of the manufacturer. Briefly, an oligonucleotide with the NF-κB consensus binding sequence 5′-AGTTGAGGGACTTTCCCAGGC-3′ was used. The consensus oligonucleotide was labeled with DIG-11-dUTP according to the description of the manufacturer. The DNA probe was then incubated with 10 μg of the nuclear extract at room temperature for 15 min. The protein-DNA complexes were then separated on a 6% polyacrylamide gel and electrically transferred to a nylon membrane (Roche Applied Science Biochemica) for chemiluminescent band detection. To examine the specificity of the binding, we used a cold competition experiment in which a 125-fold excess of the unlabeled oligonucleotide with the same sequence was added.
Measurement of NF-κB p65 Binding Activity by ELISA
Nuclear extract preparation was performed as in the preceding section. Equal quantities of nuclear protein (10 μg) were assayed for p65 NF-κB activity by using the Trans-AM NF-κB p65 transcription factor assay kit (Active Motif, Carlsbad, CA) according to the instructions of the manufacturer. Briefly, nuclear extracts were incubated in 96-well plates coated with immobilized oligonucleotide containing a consensus binding site for p65 NF-κB. NF-κB binding to the target oligonucleotide was detected by a primary antibody specific for the activated form of p65, visualized by anti-IgG horseradish peroxidase conjugate, and quantified at the wavelength of 450 nm.
Measurement of NF-κB p65 Binding to the MCP-1 Promoter by ChIP
ChIP assays were performed according to the protocol of the EpiQuikTM chromatin immunoprecipitation kit (Epigentek, Farmingdale, NY). Cells were washed twice in PBS (10 mm, pH 7.4) and fixed with fresh fixation solution (1% formaldehyde) for 10 min at room temperature, followed by glycine stop-fix solution. Cells were washed twice with cold PBS, and then the PBS was poured off and discarded. Cells were scraped into ice-cold lysis buffer, followed by 10 min incubation on ice. Chromatin was sheared using 25% sonication power under optimized conditions (10 pulses of 20 s each with a 30-s rest on ice between pulses) to an average DNA size of 600 bp, and lysates were cleared by centrifugation at 14,000 × g for 10 min at 4 °C. Immunoprecipitations were performed overnight at 4 °C with 10 μl of the NF-κB p65 antibody, and chromatin was eluted as described in the instructions of the manufacturer. The cross-links were reversed, and DNA was purified with the addition of proteinase K. DNA was analyzed by PCR. Primer sequences for the amplification of MCP-1 were as follows: 5′-CCAGCCAAATGCATTCTCTTCTA-3′ (forward) and 5′-GAGGTCAGTGCTGGCGTGA-3′ (reverse).
Statistical Analysis
SPSS13.0 for Windows was used to analyze the data (SPSS Inc., Chicago, IL). Results were expressed as mean ± S.D. Comparisons among groups involved one-way analysis of variance followed by the least significant difference test. p < 0.05 was considered statistically significant.
RESULTS
Changes in Endogenous H2S Pathway in Ox-LDL-induced, THP-1-derived Macrophages
We detected the changes in the H2S level and H2S synthase expression depending on the duration of treatment with ox-LDL. Using a sulfite electrode, we found that H2S levels in the supernatant were much lower in THP-1-derived macrophages treated with ox-LDL for 6, 12, and 24 h than in untreated THP-1-derived macrophages (Fig. 1A). In agreement, the endogenous H2S in THP-1-derived macrophages was also decreased after ox-LDL treatment for 3–24 h (Fig. 1B). We further explored the expression of CBS, CSE, and MPST, the key enzymes catalyzing endogenous H2S, in ox-LDL-induced, THP-1-derived macrophages. Western blotting showed that CBS protein expression was much lower in THP-1-derived macrophages treated with ox-LDL for 6, 12, and 24 h than in untreated THP-1-derived macrophages (Fig. 1C). Real-time PCR and agarose gel electrophoresis indicated that CBS mRNA expression was much lower in THP-1-derived macrophages treated with ox-LDL for 3, 6, and 12 h than in untreated THP-1-derived macrophages (Fig. 1D), whereas CSE and MPST mRNA and protein expressions did not change (Fig. 1, E and F).
FIGURE 1.

The endogenous H2S pathway was down-regulated in ox-LDL-induced, THP-1-derived macrophages (mean ± S. D.). A, changes of endogenous H2S-generating enzymes in ox-LDL-induced, THP-1-derived macrophages (mean ± S.D.). The H2S level was analyzed by sensitive sulfite electrode. *, p < 0.05 compared with a control group. B, representative fluorescent staining (blue) of endogenous H2S in THP-1-derived macrophages treated with ox-LDL for various time periods. C and D, CBS protein (C, analysis by Western blotting) and mRNA (D, analysis by real-time PCR) expressions decreased in ox-LDL-induced, THP-1-derived macrophages. *, p < 0.05 compared with a control group. E, mRNA and protein expression of CSE in THP-1-derived macrophages after ox-LDL treatment for 12 and 24 h. F, mRNA and protein level of MPST in THP-1-derived macrophages after ox-LDL treatment for 12 and 24 h. Data are means ± S.D. of three independent experiments performed in triplicate.
H2S Inhibited MCP-1, TNF-α, MIF, and IL-10 Content and MCP-1 Expression in Ox-LDL-stimulated, THP-1-derived Macrophages and RAW Macrophages
To explore the effect of H2S on MCP-1, TNF-α, MIF, and IL-10 content, THP-1-derived macrophages were pretreated with NaHS (100 μmol/liter or 500 μmol/liter) and hexyl acrylate (HA, a CBS inhibitor, 50 μmol/liter), and then stimulated with ox-LDL (50 mg/liter) for 24 h. Firstly, we detected endogenous H2S expression in these groups and found that HA stimulation indeed inhibited H2S expression in THP-1-derived macrophages, whereas NaHS treatment up-regulated ox-LDL-suppressed endogenous H2S levels (Fig. 2A). Then, ELISA analysis showed that the MCP-1, TNF-α, and MIF contents in the supernatant were much higher, whereas IL-10 content was much lower, in THP-1-derived macrophages treated with ox-LDL for 24 h than in untreated THP-1-derived macrophages (all p < 0.05). HA treatment increased MCP-1, TNF-α, and MIF contents and decreased IL-10 content, whereas NaHS treatment at 100 μmol/liter and 500 μmol/liter decreased MCP-1, TNF-α, and MIF contents but increased the IL-10 content in cell supernatant in ox-LDL-stimulated, THP-1-derived macrophages (Fig. 2B). We also repeated the evaluation of the effect of H2S on MCP-1 content in mouse RAW macrophages and found similar results (Fig. 2C). Confocal images showed that MCP-1 protein expression increased greatly when treated with ox-LDL for 6 h, whereas NaHS treatment at 100 μmol/liter and 500 μmol/liter significantly suppressed MCP-1 protein expression in ox-LDL-stimulated THP-1-derived macrophages (Fig. 2D).
FIGURE 2.

H2S treatment decreased MCP-1 generation in ox-LDL-induced, THP-1-derived macrophages and RAW macrophages. A, representative fluorescent staining (blue) of endogenous H2S in THP-1-derived macrophages. B, MCP-1, TNF-α, and MIF contents increased, but IL-10 content decreased, in ox-LDL-induced, THP-1-derived macrophages by ELISA (mean ± S.D.). C, MCP-1 content in RAW macrophages. D and E, MCP-1 protein (green) and mRNA expression in THP-1-derived macrophages. Nuclei were stained with propidium iodide (red). Data are means ± S.D. of three independent experiments performed in triplicate. *, p < 0.05 compared with a control group; #, p < 0.05 compared with an ox-LDL-treated group.
We also examined MCP-1 mRNA expression by real-time PCR. The results showed that MCP-1 mRNA expression increased markedly when treated with ox-LDL for 2 h. HA treatment further increased MCP-1 mRNA expression, whereas NaHS treatment at 100 μmol/liter and 500 μmol/liter significantly suppressed MCP-1 mRNA expression in ox-LDL-stimulated, THP-1-derived macrophages (Fig. 2E).
H2S Inhibited the NF-κB p65 Pathway during Ox-LDL-stimulated Macrophage Inflammation
Considering that the NF-κB pathway is the key element mediating inflammation, we studied the effect of H2S on the NF-κB pathway during ox-LDL-stimulated macrophage inflammation by examining p65 phosphorylation, nuclear translocation, DNA binding activity, and recruitment to the promoter of MCP-1. Firstly, we examined the effect of H2S on NF-κB p65 phosphorylation in ox-LDL-stimulated THP-1-derived macrophages and RAW macrophages. THP-1-derived macrophages and RAW macrophages were stimulated with ox-LDL in the presence or absence of NaHS (at 100 μmol/liter or 500 μmol/liter) for 30 min. Western blot analysis showed that phosphorylation of NF-κB p65 was increased markedly during stimulation with ox-LDL, whereas NaHS treatment significantly suppressed NF-κB p65 phosphorylation in ox-LDL-stimulated, THP-1-derived macrophages and RAW macrophages (Fig. 3, A and B).
FIGURE 3.
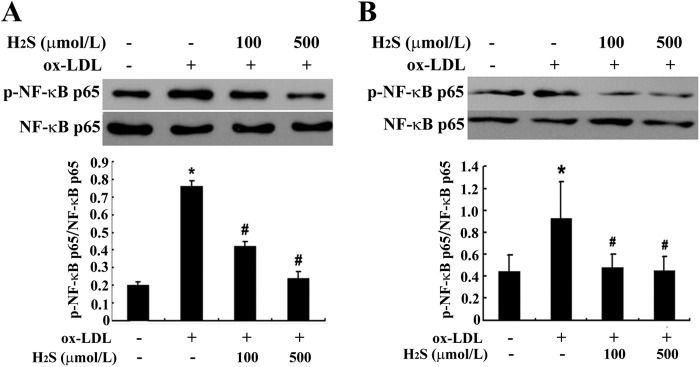
H2S treatment inhibited NF-κB phosphorylation in ox-LDL-induced, THP-1-derived macrophages and RAW macrophages (mean ± S. D.). A, NF-κB phosphorylation in THP-1-derived macrophages. B, NF-κB phosphorylation in RAW macrophages. Data are means ± S.D. of three independent experiments performed in triplicate. *, p < 0.05 compared with a control group; #, p < 0.05 compared with an ox-LDL-treated group.
Furthermore, we tested the effect of H2S on NF-κB p65 nuclear translocation in ox-LDL-stimulated THP-1-derived macrophages and RAW macrophages. THP-1-derived macrophages and RAW macrophages were stimulated with ox-LDL in the presence or absence of NaHS (at 100 μmol/liter or 500 μmol/liter) for 30 min. Immunofluorescence analysis showed that NF-κB p65 nuclear translocation was promoted by ox-LDL stimulation, whereas NaHS treatment significantly inhibited NF-κB p65 nuclear translocation in ox-LDL-stimulated, THP-1-derived macrophages and RAW macrophages (Fig. 4, A and B).
FIGURE 4.
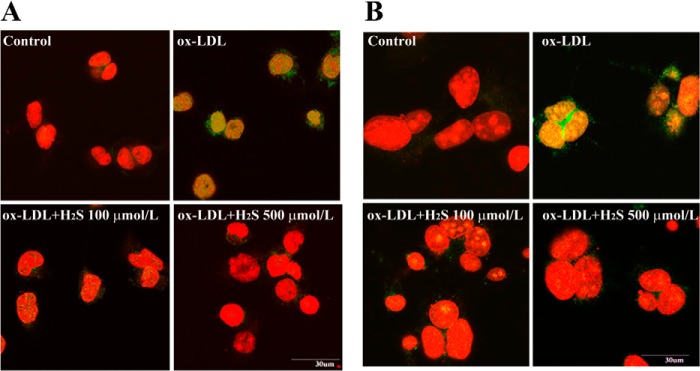
H2S treatment inhibited NF-κB translocation to the nucleus in ox-LDL-induced, THP-1-derived macrophages and RAW macrophages. A, H2S inhibited NF-κB (green) translocated to the nucleus in ox-LDL-induced, THP-1-derived macrophages. Nuclei were stained with propidium iodide (red). B, H2S inhibited NF-κB (green) translocated to the nucleus in ox-LDL-induced RAW macrophages. Nuclei were stained with propidium iodide (red). Data are from three independent experiments performed in triplicate.
We then examined the effect of H2S on NF-κB p65 DNA binding activity in ox-LDL-stimulated THP-1-derived macrophages. THP-1-derived macrophages were stimulated with ox-LDL in the presence or absence of NaHS (at 50 μmol/liter, 100 μmol/liter, or 200 μmol/liter) for 30 min. Analysis by EMSA showed that NF-κB p65 DNA binding activity increased significantly during ox-LDL treatment, whereas NaHS treatment significantly suppressed NF-κB p65 DNA binding activity in ox-LDL-stimulated, THP-1-derived macrophages (Fig. 5A). To further confirm the inhibitory effect of H2S on NF-κB p65 DNA binding activity, we performed ELISA, a more sensitive analysis, and found that NF-κB p65 DNA binding activity was promoted significantly by ox-LDL treatment. Meanwhile, NaHS treatment significantly inhibited NF-κB p65 DNA binding activity in ox-LDL-stimulated, THP-1-derived macrophages (Fig. 5B).
FIGURE 5.

H2S treatment inhibited NF-κB binding activity in ox-LDL-induced, THP-1-derived macrophages. A, NF-κB binding activity by EMSA. B, NF-κB binding activity by ELISA. C, NF-κB binding activity with the MCP-1 promoter by ChIP. Data are means ± S.D. of three independent experiments performed in triplicate. *, p < 0.05 compared with a control group; #, p < 0.05 compared with an ox-LDL-treated group.
Next, we used a ChIP assay to investigate whether the inhibitory effect of H2S resulted from disrupting NF-κB p65 recruitment to the promoter region of MCP-1 genes in ox-LDL-stimulated, THP-1-derived macrophages. THP-1-derived macrophages were stimulated with ox-LDL in the presence or absence of NaHS (at 100 μmol/liter) for 30 min. The result showed that the recruitment of NF-κB p65 to the promoter region of MCP-1 genes was increased significantly with ox-LDL treatment, whereas NaHS treatment significantly suppressed the recruitment of NF-κB p65 to the promoter region of MCP-1 genes in ox-LDL-stimulated, THP-1-derived macrophages (Fig. 5C).
H2S Functions on NF-κB p65 via Free Sulfhydryl Groups in Ox-LDL-stimulated, THP-1-derived Macrophages
To examine whether H2S acts on NF-κB p65 via its free sulfhydryl groups, we detected the free sulfhydryl groups of NF-κB p65 by Western blotting after stimulating THP-1-derived macrophages with ox-LDL in the presence or absence of NaHS (at 100 μmol/liter) for 30 min. The results showed that free sulfhydryl groups in a number of NF-κB p65 were increased markedly when treated with ox-LDL, whereas NaHS treatment significantly suppressed the level of free sulfhydryl groups in ox-LDL-stimulated, THP-1-derived macrophages (Fig. 6A).
FIGURE 6.

H2S inhibited the free sulfhydryl groups of NF-κB p65. A, free sulfhydryl groups of NF-κB p65 assayed by Western blotting. B, NF-κB binding activity assayed by EMSA. C, NF-κB binding activity assayed by ELISA. Data are means ± S.D. of three independent experiments performed in triplicate. *, p < 0.05 compared with a control group; #, p < 0.05 compared with an ox-LDL-treated group.
Next, we utilized the sulfhydryl modifiers DTT and diamide to determine the role of the sulfhydryl groups of NF-κB p65 in the function of H2S on the NF-κB p65 pathway. We overexpressed p65 by transfecting a p65-expressing plasmid into THP-1 derived macrophages and then treated them with an H2S donor in the presence or absence of DTT and diamide. The EMSA and ELISA results showed that H2S suppressed the DNA binding activity of p65 and that DTT could reverse this effect, whereas diamide mimicked it (Fig. 6, B and C). The results suggested that H2S functions on NF-κB p65 via free sulfhydryl groups in ox-LDL-stimulated, THP-1-derived macrophages.
The Free Sulfhydryl Group on Cysteine 38 in NF-κB p65 Is a Target of H2S
Next, to test how H2S sulfhydrated the NF-κB p65 subunit, we transfected THP-1-derived macrophages with a wild-type or mutant p65 (C38S) plasmid for 48 h and then treated them with NaHS for 30 min. Sulfhydration of p65 was monitored by a modified biotin switch assay. As expected, H2S dramatically increased sulfhydration of p65 in THP-1-derived macrophages transfected with the wild-type p65 plasmid, and the process was concentration-dependent. However, mutation of cysteine-38 at the p65 subunit totally abolished H2S-elicited sulfhydration of NF-κB, indicating that H2S sulfhydrated the p65 at cysteine 38 (Fig. 7A).
FIGURE 7.

Cysteine 38 of NF-κB p65 is a possible target of H2S. A, Western blot analysis and quantification of p65 sulfhydration (s-NF-κB) in THP-1-derived macrophages transfected with wild-type or mutant p65 C38S. B, NF-κB p65 phosphorylation (p-NF-κB) in cells transfected with the wild-type or mutant p65 C38S plasmid before ox-LDL and H2S treatment. C, nucleus translocation of NF-κB (green). Nuclei were stained with propidium iodide (red). D, NF-κB (wild-type or mutant p65 C38S) binding activity assayed by EMSA. E, NF-κB (wild-type or mutant p65 C38S) binding activity assayed by ELISA. Data are means ± S.D. of three independent experiments performed in triplicate.
To examine whether H2S inhibited NF-κB activation by sulfhydrating cysteine 38 of NF-κB p65, THP-1-derived macrophages were transfected with a wild-type or mutant p65 (C38S) plasmid for 48 h and treated with ox-LDL and NaHS for 30 min. We then detected the effect of H2S on p65 phosphorylation, nuclear translocation, DNA binding activity, and recruitment to the promoter region of MCP-1 genes in ox-LDL-stimulated, THP-1-derived macrophages.
Compared with cells transfected with vehicle, NF-κB p65 phosphorylation, nuclear translocation and DNA binding activity were increased markedly in cells transfected with the wild-type or mutant p65 (C38S) plasmid, whereas H2S significantly prevented ox-LDL-induced NF-κB activation in cells transfected with the wild-type p65 plasmid. Notably, the inhibitory effects of H2S on p65 phosphorylation, nuclear translocation, and DNA binding activity were abolished in cells expressing the mutant p65 (C38S) plasmid (Fig. 7, B–E). Thus, these results indicated that H2S inhibited NF-κB activation by targeting the free sulfhydryl group on cysteine 38 in the NF-κB p65 subunit.
H2S Inhibited IκBα Degradation and Phosphorylation
The translocation of NF-κB to the nucleus is preceded by the phosphorylation and degradation of IκBα. To determine whether the inhibition of ox-LDL-induced NF-κB activation by H2S was related to IκBα degradation, we pretreated THP-1-derived macrophages with NaHS and then exposed the cells to ox-LDL for 30 min. Western blot analysis revealed that ox-LDL induced IκBα degradation and increased the phosphorylation of IκBα, whereas H2S blocked the effects of ox-LDL on IκBα degradation and phosphorylation (Fig. 8). These results indicated that H2S inhibited ox-LDL-induced NF-κB activation not only through the sulfhydration of p65 subunit but also suppressed IκBα degradation and phosphorylation.
FIGURE 8.

H2S inhibited the phosphorylation and degradation of IκBα in ox-LDL-induced, THP-1-derived macrophages. Western blot analysis and quantification of phospho-IκBα (p-IκBα) and IκBα protein expression in THP-1-derived macrophages. Data are means ± S.D. of three independent experiments performed in triplicate. *, p < 0.05 compared with a control group; #, p < 0.05 compared with an ox-LDL-treated group.
DISCUSSION
H2S is an active gasotransmitter with features of low molecular weight, continuous production, quick diffusion, and wide biological effects (23, 24). Previous studies have demonstrated that H2S can inhibit macrophage-derived foam cell formation (16, 25–26). Our research group confirmed that H2S generation in vessels was decreased in apoE −/− mice and that it was an important factor in the formation and development of atherosclerosis (16). Therefore, we inferred that there might be some changes in H2S levels in the cell supernatant of macrophages stimulated with ox-LDL.
We detected the alteration of the endogenous H2S pathway in THP-1-derived macrophages, including H2S level, in the cell and supernatant and the expression of the three key enzymes CBS, CSE, and MPST. We found that H2S levels, CBS mRNA, and protein expression were much lower in ox-LDL treated, THP-1-derived macrophages than in untreated, THP-1-derived macrophages, whereas CSE and MPST expression did not change. Therefore, we hypothesized that CBS, CSE, and MPST were all expressed in THP-1-derived macrophages and that a down-regulated endogenous CBS/H2S pathway was probably involved in the mechanisms responsible for the regulation of macrophage inflammation.
MCP-1, TNF-α, MIF, and IL-10 are important cytokines in the regulation of inflammation (27–31). We studied the possible impact of H2S on MCP-1, TNF-α, MIF, and IL-10 secretion in macrophages. The results show that HA treatment increased MCP-1, TNF-α, and MIF contents but decreased IL-10 content, whereas NaHS treatment (at 100 μmol/liter and 500 μmol/liter) decreased MCP-1, TNF-α, and MIF contents and increased IL-10 content in cell supernatant from ox-LDL-stimulated, THP-1-derived macrophages. In another cell line, mouse RAW macrophages, the inhibitory effect of H2S on MCP-1 content was similar. At the same time, NaHS treatment (at 100 μmol/liter and 500 μmol/liter) significantly decreased MCP-1 protein and RNA expression in ox-LDL-stimulated, THP-1-derived macrophages. These results suggest that H2S might inhibit secretion and expression of MCP-1 in ox-LDL-stimulated macrophages, which might be significant in the protection against atherosclerosis.
We then studied how H2S inhibits MCP-1 generation. It is widely accepted that NF-κB is required for full induction of MCP-1 (32–36). NF-κB is a key transcription factor widely distributed in most cell types. It mainly consists of dimers of the two subunits p50 and p65 (Rel A), and the p65 protein is the key transcriptionally active component of NF-κB. In non-stimulated cells, it exists in the cytoplasm in an inactive form associated with an inhibitory protein called IκB. The release of the inhibitory IκB subunit from the complex, followed by phosphorylation and translocation of the dimer to the nucleus, leads to the activation of NF-κB (37). In the nucleus, it regulates the transcription of many genes (37, 38). We hypothesize that NF-κB is likely to be involved in the underlying mechanism by which H2S inhibits secretion and expression of MCP-1 in ox-LDL-stimulated macrophages.
To test the hypothesis, we first examined the effect of H2S on the phosphorylation of NF-κB p65 in ox-LDL-stimulated, THP-1-derived macrophages. Phosphorylation of NF-κB p65 was increased greatly in ox-LDL-stimulated, THP-1-derived macrophages. In contrast, NaHS treatment at 100 μmol/liter and 500 μmol/liter significantly decreased the phosphorylation of NF-κB p65 in ox-LDL-stimulated, THP-1-derived macrophages. Furthermore, NF-κB p65 translocation to the nucleus was up-regulated in ox-LDL-stimulated, THP-1-derived macrophages, whereas NaHS treatment (at 100 μmol/liter and 500 μmol/liter) significantly suppressed NF-κB p65 translocation to the nucleus in ox-LDL-stimulated, THP-1-derived macrophages.
To further confirm the inhibitory effect of H2S on NF-κB p65 phosphorylation and translocation to the nucleus in macrophages, we performed the experiment in the RAW macrophage cell line and obtained similar results. These results suggested that H2S inhibited MCP-1 expression via suppression of NF-κB p65 phosphorylation and translocation to the nucleus in macrophages.
Following translocation to the nucleus, the p65-p50 dimer binds to the promoters of NF-κB-dependent inflammatory genes to induce their expression. Therefore, we further detected the inhibitory effect of H2S on NF-κB p65 DNA binding activity. An EMSA study showed that NF-κB p65 DNA binding activity was increased significantly in ox-LDL-stimulated, THP-1-derived macrophages, whereas NaHS pretreatment (at 50 μmol/liter, 100 μmol/liter, and 200 μmol/liter) significantly suppressed NF-κB p65 DNA binding activity in ox-LDL-stimulated, THP-1-derived macrophages.
ELISA, a more sensitive method for testing NF-κB p65 DNA binding activity, confirmed the inhibitory effect of H2S on NF-κB DNA binding activity. The results showed that NF-κB p65 DNA binding activity was up-regulated by ox-LDL treatment, whereas NaHS pretreatment significantly inhibited NF-κB p65 DNA binding activity in ox-LDL-stimulated, THP-1-derived macrophages. These results suggested that H2S inhibited MCP-1 expression via suppression of NF-κB p65 DNA binding activity in ox-LDL-stimulated, THP-1-derived macrophages.
We then tested whether H2S inhibited NF-κB p65 binding to the promoter region of MCP-1 genes in ox-LDL-stimulated, THP-1-derived macrophages. The ChIP assay is a powerful tool for identifying proteins associated with specific regions of the genome, which uses antibodies that recognize the specific protein or specific modification of the protein. The ChIP results showed that the recruitment of NF-κB p65 to the promoter region of MCP-1 genes increased significantly during ox-LDL treatment, whereas NaHS treatment at 100 μmol/liter significantly suppressed the recruitment of NF-κB p65 to the promoter region of MCP-1 genes in ox-LDL-stimulated, THP-1-derived macrophages. Therefore, we deduced that H2S inhibited MCP-1 expression by inhibiting the recruitment of NF-κB p65 to the promoter region of MCP-1 genes.
It is possible that H2S inhibits the binding of NF-κB p65 to DNA by modification of the NF-κB protein. Sulfhydration of NF-κB appears to be a posttranslation modification of p65, required for its transcriptional influence on antiapoptotic genes (39). Sulfhydration is a physiological process whereby H2S attaches an additional sulfur to the thiol (-SH) groups of cysteines, yielding a hydropersulfide (-SSH) (22, 40). The transformation of disulfide bridges into sulfhydryl groups of the cysteine-containing proteins at the center of cytochrome c oxidase was regarded as the mechanism for intoxication of H2S (41). Thus, the free sulfhydryl groups of the cysteine-containing proteins may be effective targets of H2S (42). We wondered whether H2S inhibited NF-κB by sulfhydration. To see whether H2S targeted the sulfhydryl groups in the NF-κB p65 protein, we first measured the ratio of NF-κB p65 containing free sulfhydryl groups to total NF-κB p65 protein in THP-1-derived macrophage cells incubated with an H2S donor. After treatment with the H2S donor, the ratio was decreased significantly. However, DTT could reverse this effect of H2S. These results suggest that H2S could target the sulfhydryl group and decrease the level of reduced thiol in the NF-κB protein in THP-1-derived macrophage cells. This effect could be reversed by thiol reductants, suggesting a role for the thiol group in the action of NF-κB by H2S.
Cysteine 38 in NF-κB p65 is highly sensitive to various agents (43–49). The importance of cysteine 38 in p65 has been appreciated for many years. Cysteine 38 in p65 interacts with the phosphate backbone of NF-κB binding sites (48), whereas its oxidation or nitrosylation is known to inhibit DNA binding (49). Moreover, it is the target of many NF-κB inhibitors (45–47). For example, Yadav et al. (45) found that 3-formylchromone, an anticancer agent, inhibited the direct binding of p65 to DNA and that this binding was reversed by a reducing agent, thus suggesting the role of the cysteine residue. Furthermore, in that study, mutation of Cys-38 to Ser in p65 abolished this effect of the chromone (45). Taken together, cysteine 38 in p65 seems to be a crucial residue regulating the NF-κB pathway. The above facts provide a necessary clue for our exploration to see whether the sulfhydryl group on cysteine 38 in p65 could be a possible target of H2S. This study showed that H2S dramatically increased sulfhydration of p65 in THP-1-derived macrophages transfected with the wild-type p65 plasmid, and the process was concentration-dependent. However, mutation of cysteine 38 at the p65 subunit totally abolished H2S-elicited sulfhydration of NF-κB. Furthermore, we found that H2S inhibited phosphorylation of p65, nuclear translocation, and the DNA binding of wild-type p65 but failed to do so when cysteine 38 was replaced with serine in p65, which suggested that the free sulfhydryl group on cysteine 38 in the p65 acted as a target of H2S.
Sen et al. (39) found that CSE knockout blocked the TNF-α-induced DNA-binding activity of NF-κB and cell death, which represented the physiological antiapoptotic mechanism for H2S. However, in this study, it was found that, under pathophysiological conditions, H2S inhibited nuclear translocation of NF-κB in ox-LDL-induced, THP-1 derived macrophage inflammation, which represented the pathophysiological anti-inflammation mechanism of H2S. Therefore, we supposed that the different effect of H2S on the NF-κB pathway might depend upon different conditions (pathophysiologic or physiologic).
In this study, the data also showed that H2S suppressed the ox-LDL-induced phosphorylation and degradation of IκBα, suggesting that H2S inhibited NF-κB activation not only through sulfhydrating p65 but also by means of preventing IκBα activation. However, how H2S inhibited IκBα activation remains to be elucidated.
In our previous study, we demonstrated that H2S exerted an antiatherogenic effect in apoE−/− mice (16). In this study, we extended the mechanistic understanding that H2S inhibited ox-LDL-induced macrophage inflammation. Studies of CBS−/− mice by Namekata et al. (50) showed that CBS deficiency led to abnormal lipid metabolism. Considering that H2S is generated from homocysteine and cysteine by CBS, CBS deficiency might led to an increase in homocysteine but a decrease in H2S. Therefore, abnormal lipid metabolism in CBS−/− mice might be partly caused by insufficient H2S production, suggesting that H2S probably plays an important role in lipid metabolism. On the whole, it is suggested that H2S is a multitarget, antiatherogenic molecule at least exerting functions of inhibiting inflammatory response and likely improving abnormal lipid metabolism.
In conclusion, we discovered an inhibitory effect of H2S on macrophage inflammation via inhibiting phosphorylation of p65, nuclear translocation, and DNA binding activity and recruitment of p65 to the promoter region of the target gene. Importantly, the sulfhydryl group on cysteine 38 in p65 is the possible target of H2S in this process.
This work was supported by Major Basic Research Development Program of People's Republic of China Grants 2011CB503904, 2012CB517806, and 2013CB933801; by National Natural Science Foundation of China Grants 30901620, 31130030, and 30821001; and by Beijing Natural Science Foundation Grants 7122184 and 7121014.
- CBS
- cystathionine-β-synthase
- CSE
- cystathionine-γ-lyase
- MPST
- 3-mercaptopyruvate sulfurtransferase
- ox-LDL
- oxidized LDL
- MIF
- migration inhibition factor
- HA
- hexyl acrylate.
REFERENCES
- 1. Ross R. (1999) Atherosclerosis. An inflammatory disease. N. Engl. J. Med. 340, 115–126 [DOI] [PubMed] [Google Scholar]
- 2. Hansson G. K., Libby P., Schönbeck U., Yan Z. Q. (2002) Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ. Res. 91, 281–291 [DOI] [PubMed] [Google Scholar]
- 3. Gong J. H., Ratkay L. G., Waterfield J. D., Clark-Lewis I. (1997) An antagonist of monocyte chemoattractant protein 1 (MCP-1) inhibits arthritis in the MRL-lpr mouse model. J. Exp. Med. 186, 131–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yoshimura T., Robinson E. A., Tanaka S., Appella E., Leonard E. J. (1989) Purification and amino acid analysis of two human monocyte chemoattractants produced by phytohemagglutinin-stimulated human blood mononuclear leukocytes. J. Immunol. 142, 1956–1962 [PubMed] [Google Scholar]
- 5. Yoshimura T., Yuhki N., Moore S. K., Appella E., Lerman M. I., Leonard E. J. (1989) Human monocyte chemoattractant protein-1 (MCP-1). Full-length cDNA cloning, expression in mitogen-stimulated blood mononuclear leukocytes, and sequence similarity to mouse competence gene JE. FEBS Lett. 244, 487–493 [DOI] [PubMed] [Google Scholar]
- 6. Wang R. (2002) Two's company, three's a crowd. Can H2S be the third endogenous gaseous transmitter? FASEB J. 16, 1792–1798 [DOI] [PubMed] [Google Scholar]
- 7. Shibuya N., Koike S., Tanaka M., Ishigami-Yuasa M., Kimura Y., Ogasawara Y., Fukui K., Nagahara N., Kimura H. (2013) A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nat. Commun. 4, 1366. [DOI] [PubMed] [Google Scholar]
- 8. Shibuya N., Kimura H. (2013) Production of hydrogen sulfide from D-cysteine and its therapeutic potential. Front. Endocrinol. 4, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kimura H. (2014) Production and physiological effects of hydrogen sulfide. Antioxid. Redox Signal. 20, 783–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tang C., Li X., Du J. (2006) Hydrogen sulfide as a new endogenous gaseous transmitter in the cardiovascular system. Curr. Vasc. Pharmacol. 4, 17–22 [DOI] [PubMed] [Google Scholar]
- 11. Du J., Zhang C., Yan H., Tang C. (2006) A newly found gasotransmitter, hydrogen sulfide, in the pathogenesis of hypertension and other cardiovascular diseases. Curr. Hypertens. Rev. 2, 123–126 [Google Scholar]
- 12. Lefer D. J. (2007) A new gaseous signaling molecule emerges. Cardioprotective role of hydrogen sulfide. Proc. Natl. Acad. Sci. U.S.A. 104, 17907–17908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Whiteman M., Li L., Rose P., Tan C. H., Parkinson D. B., Moore P. K. (2010) The effect of hydrogen sulfide donors on lipopolysaccharide-induced formation of inflammatory mediators in macrophages. Antioxid. Redox. Signal. 12, 1147–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li T., Zhao B., Wang C., Wang H., Liu Z., Li W., Jin H., Tang C., Du J. (2008) Regulatory effects of hydrogen sulfide on IL-6, IL-8 and IL-10 levels in the plasma and pulmonary tissue of rats with acute lung injury. Exp. Biol. Med. 233, 1081–1087 [DOI] [PubMed] [Google Scholar]
- 15. Li L., Salto-Tellez M., Tan C. H., Whiteman M., Moore P. K. (2009) GYY4137, a novel hydrogen sulfide-releasing molecule, protects against endotoxic shock in the rat. Free. Radic. Biol. Med. 47, 103–113 [DOI] [PubMed] [Google Scholar]
- 16. Wang Y., Zhao X., Jin H., Wei H., Li W., Bu D., Tang X., Ren Y., Tang C., Du J. (2009) Role of hydrogen sulfide in the development of atherosclerotic lesions in apolipoprotein E knockout mice. Arterioscler. Thromb. Vasc. Biol. 29, 173–179 [DOI] [PubMed] [Google Scholar]
- 17. Auwerx J. (1991) The human leukemia cell line, THP-1. A multifaceted model for the study of monocyte-macrophage differentiation. Experientia 47, 22–31 [DOI] [PubMed] [Google Scholar]
- 18. Cushing S. D., Berliner J. A., Valente A. J., Territo M. C., Navab M., Parhami F., Gerrity R., Schwartz C. J., Fogelman A. M. (1990) Minimally modified low density lipoprotein induces monocyte chemotactic protein 1 in human endothelial cells and smooth muscle cells. Proc. Natl. Acad. Sci. U.S.A. 87, 5134–5138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li W., Tang C., Jin H., Du J. (2011) Regulatory effects of sulfur dioxide on the development of atherosclerotic lesions and vascular hydrogen sulfide in atherosclerotic rats. Atherosclerosis 215, 323–330 [DOI] [PubMed] [Google Scholar]
- 20. Olson K. R. (2009) Is hydrogen sulfide a circulating “gasotransmitter” in vertebrate blood? Biochim. Biophys. Acta 1787, 856–863 [DOI] [PubMed] [Google Scholar]
- 21. Chen B., Li W., Lv C., Zhao M., Jin H., Jin H., Du J., Zhang L., Tang X. (2013) Fluorescent probe for highly selective and sensitive detection of hydrogen sulfide in living cells and cardiac tissues. Analyst 138, 946–951 [DOI] [PubMed] [Google Scholar]
- 22. Mustafa A. K., Gadalla M. M., Sen N., Kim S., Mu W., Gazi S. K., Barrow R. K., Yang G., Wang R., Snyder S. H. (2009) H2S signals through protein S-sulfhydration. Sci. Signal. 2, ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Baskar R., Bian J. (2011) Hydrogen sulfide gas has cell growth regulatory role. Eur. J. Pharmacol. 656, 5–9 [DOI] [PubMed] [Google Scholar]
- 24. Liu Y. H., Lu M., Hu L. F., Wong P. T., Webb G. D., Bian J. S. (2012) Hydrogen sulfide in the mammalian cardiovascular system. Antioxid. Redox Signal. 17, 141–185 [DOI] [PubMed] [Google Scholar]
- 25. Zhao Z. Z., Wang Z., Li G. H., Wang R., Tan J. M., Cao X., Suo R., Jiang Z. S. (2011) Hydrogen sulfide inhibits macrophage-derived foam cell formation. Exp. Biol. Med. 236, 169–176 [DOI] [PubMed] [Google Scholar]
- 26. Liu Z., Han Y., Li L., Lu H., Meng G., Li X., Shirhan M., Peh M. T., Xie L., Zhou S., Wang X., Chen Q., Dai W., Tan C. H., Pan S., Moore P. K., Ji Y. (2013) The hydrogen sulfide donor, GYY4137, exhibits anti-atherosclerotic activity in high fat fed apolipoprotein E−/− mice. Br. J. Pharmacol. 169, 1795–1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Standiford T. J., Kunkel S. L., Phan S. H., Rollins B. J., Strieter R. M. (1991) Alveolar macrophage-derived cytokines induce monocyte chemoattractant protein-1 expression from human pulmonary type II-like epithelial cells. J. Biol. Chem. 266, 9912–9918 [PubMed] [Google Scholar]
- 28. Brown Z., Strieter R. M., Neild G. H., Thompson R. C., Kunkel S. L., Westwick J. (1992) IL-1 receptor antagonist inhibits monocyte chemotactic peptide 1 generation by human mesangial cells. Kidney Int. 42, 95–101 [DOI] [PubMed] [Google Scholar]
- 29. Barna B. P., Pettay J., Barnett G. H., Zhou P., Iwasaki K., Estes M. L. (1994) Regulation of monocyte chemoattractant protein-1 expression in adult human non-neoplastic astrocytes is sensitive to tumor necrosis factor (TNF) or antibody to the 55-kDa TNF receptor. J Neuroimmunol. 50, 101–107 [DOI] [PubMed] [Google Scholar]
- 30. Fruebis J., Gonzalez V., Silvestre M., Palinski W. (1997) Effect of probucol treatment on gene expression of VCAM-1, MCP-1, and M-CSF in the aortic wall of LDL receptor-deficient rabbits during early atherogenesis. Arterioscler. Thromb. Vasc. Biol. 17, 1289–1302 [DOI] [PubMed] [Google Scholar]
- 31. Shioi A., Katagi M., Okuno Y., Mori K., Jono S., Koyama H., Nishizawa Y. (2002) Induction of bone-type alkaline phosphatase in human vascular smooth muscle cells. Roles of tumor necrosis factor-α and oncostatin M derived from macrophages. Circ. Res. 91, 9–16 [DOI] [PubMed] [Google Scholar]
- 32. Ueda A., Okuda K., Ohno S., Shirai A., Igarashi T., Matsunaga K., Fukushima J., Kawamoto S., Ishigatsubo Y., Okubo T. (1994) NF-κB and Sp1 regulate transcription of the human monocyte chemoattractant protein-1 gene. J. Immunol. 153, 2052–2063 [PubMed] [Google Scholar]
- 33. Freter R. R., Alberta J. A., Hwang G. Y., Wrentmore A. L., Stiles C. D. (1996) Platelet-derived growth factor induction of the immediate-early gene MCP-1 is mediated by NF-κB and a 90-kDa phosphoprotein coactivator. J. Biol. Chem. 271, 17417–17424 [DOI] [PubMed] [Google Scholar]
- 34. Martin T., Cardarelli P. M., Parry G. C., Felts K. A., Cobb R. R. (1997) Cytokine induction of monocyte chemoattractant protein-1 gene expression in human endothelial cells depends on the cooperative action of NF-κB and AP-1. Eur. J. Immunol. 27, 1091–1097 [DOI] [PubMed] [Google Scholar]
- 35. Marumo T., Schini-Kerth V. B., Busse R. (1999) Vascular endothelial growth factor activates nuclear factor-κB and induces monocyte chemoattractant protein-1 in bovine retinal endothelial cells. Diabetes 48, 1131–1137 [DOI] [PubMed] [Google Scholar]
- 36. Ha H., Yu M. R., Choi Y. J., Kitamura M., Lee H. B. (2002) Role of high glucose-induced nuclear factor-κB activation in monocyte chemoattractant protein-1 expression by mesangial cells. J. Am. Soc. Nephrol. 13, 894–902 [DOI] [PubMed] [Google Scholar]
- 37. Baeuerle P. A., Henkel T. (1994) Function and activation of NF-κB in the immune system. Annu. Rev. Immunol. 12, 141–179 [DOI] [PubMed] [Google Scholar]
- 38. Huang T. T., Miyamoto S. (2001) Postrepression activation of NF-κB requires the amino-terminal nuclear export signal specific to IκBα. Mol. Cell Biol. 21, 4737–4747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sen N., Paul B. D., Gadalla M. M., Mustafa A. K., Sen T., Xu R., Kim S., Snyder S. H. (2012) Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Mol. Cell. 45, 13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li L., Rose P., Moore P. K. (2011) Hydrogen sulfide and cell signaling. Annu. Rev. Pharmacol. Toxicol. 51, 169–187 [DOI] [PubMed] [Google Scholar]
- 41. Smith R. P., Abbanat R. A. (1966) Protective effect of oxidized glutathione in acute sulfide poisoning. Toxicol. Appl. Pharmacol. 9, 209–217 [DOI] [PubMed] [Google Scholar]
- 42. Zhang R., Sun Y., Tsai H., Tang C., Jin H., Du J. (2012) Hydrogen sulfide inhibits L-type calcium currents depending upon the protein sulfhydryl state in rat cardiomyocytes. PloS ONE 7, e37073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sandur S. K., Ichikawa H., Sethi G., Ahn K. S., Aggarwal B. B. (2006) Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) suppresses NF-κB activation and NF-κB-regulated gene products through modulation of p65 and IκBα kinase activation, leading to potentiation of apoptosis induced by cytokine and chemotherapeutic agents. J. Biol. Chem. 281, 17023–17033 [DOI] [PubMed] [Google Scholar]
- 44. Han Y., Englert J. A., Yang R., Delude R. L., Fink M. P. (2005) Ethyl pyruvate inhibits nuclear factor-κB-dependent signaling by directly targeting p65. J. Pharmacol. Exp. Ther. 312, 1097–1105 [DOI] [PubMed] [Google Scholar]
- 45. Yadav V. R., Prasad S., Gupta S. C., Sung B., Phatak S. S., Zhang S., Aggarwal B. B. (2012) 3-Formylchromone interacts with cysteine 38 in p65 protein and with cysteine 179 in IκBα kinase, leading to down-regulation of nuclear factor-κB (NF-κB)-regulated gene products and sensitization of tumor cells. J. Biol. Chem. 287, 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 46. García-Piñeres A. J., Castro V., Mora G., Schmidt T. J., Strunck E., Pahl H. L., Merfort I. (2001) Cysteine 38 in p65/NF-κB plays a crucial role in DNA binding inhibition by sesquiterpene lactones. J. Biol. Chem. 276, 39713–39720 [DOI] [PubMed] [Google Scholar]
- 47. García-Piñeres A. J., Lindenmeyer M. T., Merfort I. (2004) Role of cysteine residues of p65/NF-κB on the inhibition by the sesquiterpene lactone parthenolide and N-ethyl maleimide, and on its transactivating potential. Life Sci. 75, 841–856 [DOI] [PubMed] [Google Scholar]
- 48. Chen F. E., Huang D. B., Chen Y. Q., Ghosh G. (1998) Crystal structure of p50/p65 heterodimer of transcription factor NF-κB bound to DNA. Nature 391, 410–413 [DOI] [PubMed] [Google Scholar]
- 49. Kelleher Z. T., Matsumoto A., Stamler J. S., Marshall H. E. (2007) NOS2 regulation of NF-κB by S-nitrosylation of p65. J. Biol. Chem. 282, 30667–30672 [DOI] [PubMed] [Google Scholar]
- 50. Namekata K., Enokido Y., Ishii I., Nagai Y., Harada T., Kimura H. (2004) Abnormal lipid metabolism in cystathionine β-synthase-deficient mice, an animal model for hyperhomocysteinemia. J. Biol. Chem. 279, 52961–52969 [DOI] [PubMed] [Google Scholar]