

Background: HEXIM1 inhibits P-TEFb in the 7SK snRNP.
Results: The critical promoter driving HEXIM1 expression was identified and was found to respond to levels of free P-TEFb.
Conclusion: HEXIM1 expression levels rise to compensate for excessive P-TEFb activity.
Significance: Increased levels of free P-TEFb lead to induction of HEXIM1 to maintain transcriptional homeostasis.
Keywords: Cyclin-dependent Kinase (CDK), Cyclins, Promoters, RNA Polymerase II, Transcription
Abstract
By phosphorylating negative elongation factors and the C-terminal domain of RNA polymerase II (RNAPII), positive transcription elongation factor b (P-TEFb), which is composed of CycT1 or CycT2 and CDK9, activates eukaryotic transcription elongation. In growing cells, it is found in active and inactive forms. In the former, free P-TEFb is a potent transcriptional coactivator. In the latter, it is inhibited by HEXIM1 or HEXIM2 in the 7SK small nuclear ribonucleoprotein (snRNP), which contains, additionally, 7SK snRNA, methyl phosphate-capping enzyme (MePCE), and La-related protein 7 (LARP7). This P-TEFb equilibrium determines the state of growth and proliferation of the cell. In this study, the release of P-TEFb from the 7SK snRNP led to increased synthesis of HEXIM1 but not HEXIM2 in HeLa cells, and this occurred only from an unannotated, proximal promoter. ChIP with sequencing revealed P-TEFb-sensitive poised RNA polymerase II at this proximal but not the previously annotated distal HEXIM1 promoter. Its immediate upstream sequences were fused to luciferase reporters and were found to be responsive to many P-TEFb-releasing compounds. The superelongation complex subunits AF4/FMR2 family member 4 (AFF4) and elongation factor RNA polymerase II 2 (ELL2) were recruited to this proximal promoter after P-TEFb release and were required for its transcriptional effects. Thus, P-TEFb regulates its own equilibrium in cells, most likely to maintain optimal cellular homeostasis.
Introduction
Eukaryotic transcription by RNAPII4 is regulated at multiple steps, including initiation, promoter clearance, elongation, and termination of nascent transcripts (1). The assembly of the preinitiation complex is necessary before any of these subsequent steps can take place. The minimal preinitiation complex contains RNAPII, the mediator, and general transcription factors such as TFIIA, TFIIB, TFIID, TFIIE, TFIIF, and TFIIH (2).
RNAPII transcribes protein-coding genes in eukaryotes. Its C-terminal domain (CTD) contains multiple tandem repeats (52 in humans) of the consensus heptad sequence YSPTSPS (3). Many transcriptional regulatory components bind to the CTD. Modifications of the CTD, most notably by phosphorylation, affect the function of RNAPII, especially after promoter clearance and during cotranscriptional processing of RNA (4, 5). This phosphorylation occurs primarily on serines at positions 2 and 5 (Ser(P)-2 and Ser(P)-5) in the CTD (6, 7). Although Ser(P)-5 contributes to 5′ capping, Ser(P)-2 facilitates cotranscriptional processing of nascent transcripts, which include splicing, 3′ end cleavage, and polyadenylation (6, 7). CTD kinases that are responsible for this phosphorylation include TFIIH, P-TEFb, and CycK-CDK12 (8–12).
P-TEFb is a cyclin-dependent kinase composed of CycT1 or CycT2 and CDK9 (13, 14). It plays an essential role in regulating the elongation of transcription by RNAPII (15, 16). First, P-TEFb phosphorylates negative elongation factors, such as 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB) sensitivity-inducing factor (DSIF) and negative elongation factor (NELF), for the release of RNAPII from an arrested or stalled state at promoters (15, 16). Second, P-TEFb phosphorylates Ser(P)-2 of the elongating RNAPII, enabling it to recruit cotranscriptional RNA processing machineries (9). In growing cells, P-TEFb partitions between an active complex, alone or associated in a mutually exclusive fashion with the bromodomain-containing protein 4 (BRD4) or the superelongation complex (SEC) (free P-TEFb), and an inactive complex with 7SK snRNA, HEXIM1/2 proteins, LARP7, and MePCE (methyl phosphate-capping enzyme), also known as the 7SK small nuclear ribonucleoprotein (7SK snRNP) (16–20). Increased levels of HEXIM1 help to sequester P-TEFb in the 7SK snRNP (21). When bound to 7SK snRNA, HEXIM1 inhibits the kinase activity of CDK9 (22).
In this study, we examined the P-TEFb equilibrium (between the active and inactive states) and how 7SK snRNP is reassembled rapidly after the release of free P-TEFb. We found that free P-TEFb increases the synthesis of HEXIM1, which restores the 7SK snRNP. Of interest, only a previously unannotated HEXIM1 gene promoter responded to this release of free P-TEFb. This induction was robust and contributed to complex regulation of P-TEFb in growing cells.
EXPERIMENTAL PROCEDURES
Cell Lines, Antibodies, Chemicals, and Plasmids
HeLa cells and HEK293 cells were grown in DMEM with 4.5 g/liter glucose, l-glutamate, sodium pyruvate, and 10% FBS at 37 °C with 5% CO2. Antibodies for ChIP and western blotting were as follows. Anti-AFF4 (catalog no. ab57077), anti-BRD4 (catalog no. ab75898), anti-HEXIM1 (catalog no. ab25388), and anti-tubulin (catalog no. ab6046) antibodies were from Abcam. Anti-DSIF (catalog no. sc-28678), anti-NELF (catalog no. sc-23599), anti-CDK9 (catalog no. sc-484), and anti-RNAPII (catalog no. sc-899) antibodies were from Santa Cruz Biotechnology. Anti-ELL2 antibodies were from Bethyl Laboratories, Inc. Suberoylanilide hydroxamic acid (SAHA), tubastatin A (catalog no. S2627), and MS-275 (catalog no. S1053) were purchased from Selleck Chemicals. Hexamethylene bisacetamide (HMBA) and trichostatin A (TSA) were purchased from Sigma, and stock solutions were prepared in dimethyl sulfoxide (DMSO) or water. JQ1 was a gift from Dr. James Bradner (Harvard University), and stock solutions were prepared in DMSO. ST-80 was a gift from Prof. Manfred Jung (University of Freiburg, Germany). IBET was purchased from Tocris Bioscience. HEXIM1 gene upstream luciferase reporter constructs were made using pGL3-BASIC luciferase reporter vectors (Promega).
ChIP with Sequencing (ChIP-Seq)
HeLa cells were maintained in suspension at 5% CO2 and 37 °C in minimum essential medium Eagle Spinner modification (Invitrogen, catalog no. 11380-037) supplemented with 2 mm l-glutamine and 10% FBS. One liter of cells (5 × 105 cells/ml) was cross-linked with 1% formaldehyde in medium for 10 min before glycine addition to 125 mm. Steps after cross-linking and before elution used solutions with EDTA-free protease inhibitor mixture (Roche, catalog no. 11873580001), 0.5 mm PMSF, and 1 mm DTT. Cells were washed twice with PBS, and aliquots of 1 × 108 cells were frozen as pellets at −80 °C. Each pellet was thawed in 6 ml of PBS, repelleted, and then incubated for 10 min on ice in 6 ml of hypotonic buffer (20 mm HEPES (pH 7.9), 10 mm KCl, 1 mm EDTA, and 10% glycerol). After 30 strokes in a Dounce homogenizer, cells were pelleted, washed once in hypotonic buffer, and incubated on ice for 30 min with occasional vortexing in 2 ml of radioimmune precipitation assay buffer (50 mm Tris (pH 7.6), 150 mm NaCl, 1 mm EDTA, 0.25% sodium deoxycholate, and 1% IGEPAL® CA-630). Cells were sonicated in 1-ml aliquots on ice using a Fisher model 550 sonic dismembrator for 21 cycles of 30 s at intensity 4, followed by 90 s of off cycles. After sonication, samples were spun at 20,000 × g for 15 min at 4 °C.
For each immunoprecipitation, sonicated supernatant from 5 × 107 cells was incubated at 4 °C overnight with 10 μg of anti-RNAPII, anti-NELF, or anti-DSIF antibodies. 50-μl slurries of protein G-Sepharose 4B Fast Flow beads (Sigma, catalog no. P3296) were rinsed with 2 ml of radioimmune precipitation assay buffer in Bio-Spin disposable columns (Bio-Rad, catalog no. 732-6008) and then incubated with each sample at 4 °C for 1 h. Samples were washed with 15 ml of ice-cold radioimmune precipitation assay buffer, rinsed with 10 ml of ice-cold PBS, and eluted twice at 65 °C with 100 μl of elution buffer (10 mm Tris (pH 7.6), 1 mm EDTA, and 1% SDS). Cross-links were reversed overnight at 65 °C, and samples were treated with 40 μg of RNase A and 80 μg of proteinase K. DNA was isolated using a MinElute PCR purification kit (Qiagen, catalog no. 28004) and submitted to the University of Iowa DNA Facility for library preparation using the Ovation SP Ultralow DR Multiplex System (Nugen, catalog no. 8033-32) and sequencing on an Illumina HiSeq 2000 using 100-bp paired-end reads.
Paired sequences were aligned to the human genome 19 (hg19) from the UCSC assembly using the software Bowtie 2.0.5 (setting: minins=50, maxins=500, no-mixed, non-discordant). Mapped reads were analyzed using the software MACS 2.0.10 (setting: format: BAMPE, bdg) and visualized on the UCSC Genome Browser (setting: autoScale=on, windowingFunction=maximum, alwaysZero=on, yLineOnOff=on, smoothing-Window=off).
ChIP with Quantitative PCR (ChIP-qPCR)
ChIP-qPCR was carried out as described previously, with some modifications (23). Briefly, HEK293 cells were treated with 5 μm SAHA or DMSO for 1 h. Then, cells were cross-linked with 1% formaldehyde in PBS for 15 min at room temperature, followed by the addition of 125 mm glycine for 5 min at room temperature. Sonication of chromatin was carried out using a Fisher model 100 sonic dismembrator for 20 cycles of 15 s at intensity 4, followed by 30 s on ice. Sheared chromatin was precleared with 50 μl of protein G-Sepharose beads for 1 h at 4 °C. 2 μg of specific antibodies was added to the precleared lysate corresponding to 2 × 106 cells and incubated at 4 °C overnight. Lysates were then centrifuged at 10,000 × g for 10 min. 90% of the supernatant was used for further processing. 30 μl of protein G beads precoated with BSA and salmon sperm DNA were added to each tube and incubated at 4 °C for 1 h. The chromatin-protein-bead complexes were washed six times with the ChIP buffer. The DNA was purified with 10% Chelex beads (Bio-Rad) and used as a template for qPCR. The cDNA was quantified using the Stratagene Mx3004P real-time PCR system and Sensi-FAST SYBR Green reagents (Bioline) with specific primers. The primer sequences used in this study were as follows. For GAPDH normalization, primers were as described previously (12). For HEXIM1 mRNA quantification, the forward and reverse primers were GGCCCGAAAGATAACAACTACG and ACCT GCCAACTTCCAACTG, respectively.
Transient Transfection and Luciferase Assays
HEK293 or HEK293T cells (2 × 106) growing in log phase were transfected with 10 μg of plasmid DNA by X-tremeGENE transfection reagents. After transfection, cells were kept in 5% CO2 at 37 °C for 48 h. Luciferase activity in the cell lysate was determined using a luciferase assay system (Promega) according to the instructions of the manufacturer.
siRNA Knockdown
siRNA knockdown was performed in HEK293 cells using LipofectamineTM RNAiMAX (Invitrogen) as described in the accompanying protocol. siRNAs against CDK9, AFF4, and ELL2 were designed and purchased from Integrated DNA Technologies. The detailed siRNA sequences for CDK9, AFF4, and ELL2 are available upon request. The Silencer® negative control scrambled siRNA was purchased from Ambion®. A final concentration of 10 nm of siRNA was used in the experiment, and knockdown efficiency was checked 48 h after the transfection.
5′ Rapid Amplification of cDNA Ends (RACE)
5′ RACE was performed using a designed HEXIM1 gene-specific reverse primer (5′-CGGACGGCCACCCAACTGGGGGA-3′) and 5′ RACE System for Rapid Amplification of cDNA Ends, Version 2.0 (Invitrogen) as described in the accompanying protocol. HeLa cells were treated with either 5 μm SAHA, 10 mm HMBA, or DMSO and incubated at 5% CO2 at 37 °C for 1 h, followed by first-strand cDNA synthesis using the designed HEXIM1 gene-specific reverse primer.
Glycerol Gradients
Glycerol gradients were carried out to determine fractions of free P-TEFb and 7SK snRNP, as described previously, with some modifications (23). Glycerol gradients (10–30%) were established by pipetting 2 ml of each glycerol fraction (10, 15, 20, 25, and 30%, v/v) in medium-salt buffer A (20 mm HEPES-KOH, 0.1 m KCl, 0.2 mm EDTA, pH=7.8) into centrifugation tubes (Beckman, catalog no. 331372). Gradients were formed by standing for 6 h at 4 °C. HeLa cells (2 × 106) were untreated or treated with 5 μm JQ1 for 1 h and lysed in 0.6 ml of medium-salt buffer A for 30 min at 4 °C. Lysates were centrifuged at 20,000 × g for 5 min, and supernatants were loaded into tubes with preformed glycerol gradients. Protein complexes were then fractionated by centrifugation in an SW 40 Ti rotor (minimum radius, 6.67 cm; maximum radius, 15.88 cm; Beckman) at 186,000 × g for 21 h. 10 fractions (1 ml) were collected, precipitated with trichloroacetic acid, and analyzed with the indicated antibodies by western blotting.
RESULTS
Free P-TEFb Activates the Transcription of the HEXIM1 Gene
Human chromosome 17 contains two HEXIM genes, HEXIM1 and HEXIM2 (Fig. 1A). Although gene annotation in human genome browsers points to a single HEXIM1 promoter, previous data suggested the existence of two HEXIM1 transcripts (21). HEXIM1 is also one of the 50 most up-regulated genes after 7SK snRNA knockdown, which releases free P-TEFb (24). By our mapping and expressed sequence tag (EST) databases, the larger transcript originates from the RefSeq-annotated promoter and terminates in the distal poly(A) site (from positions −1158 to +3717, which is in agreement with genome browser data), and the smaller originates from an unannotated proximal promoter and terminates in the proximal poly(A) site (from positions +1 to +2453, new observations) (Fig. 1A). The two transcription start and poly(A) sites are 1156 and 1263 bp apart, respectively. The HEXIM1 coding sequence is contained within one exon (from positions 717–1796). Sequences from positions +29 to +674 are spliced out in both HEXIM1 transcripts (Fig. 1A). The HEXIM2 gene contains three exons, a very large intron 2, and one poly(A) site for a total length of 9340 bp.
FIGURE 1.

The HEXIM1 and HEXIM2 genes, HEXIM1 transcripts, and their responses to SAHA and HMBA. A, schematic of the HEXIM1 (Hex1) and HEXIM 2 genes and HEXIM1 transcripts. The HEXIM1 gene measures 4875 bp and contains only one exon (Hex1), but two transcripts are observed (the transcription start sites are labeled using ●). 645 bp are spliced out of both transcripts before this exon. Numbers start with the major transcription start sites of the HEXIM1 (this study) and HEXIM2 genes. The HEXIM1 gene also contains two polyadenylation (pA) sites, which are indicated by zigzag lines. The HEXIM2 gene measures 9340 bp and contains three exons. The annealing position of the promoter-proximal (pp) primer for 5′ RACE was at position 873 from the transcription start site (equivalent to position 157 of exon 1 in the HEXIM1 gene). B, SAHA releases free P-TEFb from the 7SK snRNP. Cell lysates of HeLa cells treated with DMSO or SAHA (5 μm for 90 min) were subjected to a 10–30% glycerol gradient sedimentation (the black bar indicates increasing glycerol concentrations). Anti-CycT1 or anti-CDK9 antibodies detected subunits of P-TEFb in each fraction by western blotting. C, only HEXIM1, but not HEXIM2, is inducible by SAHA in HeLa cells. The + and −signs indicate the presence and absence of SAHA (top and bottom panels) and flavopiridol (bottom panel), respectively. HeLa cells were treated with SAHA for 48 h. Western blot analyses were quantified, and data are presented as fold increase by SAHA. Only HEXIM1 was induced 3-fold by SAHA (top western blot and black bar), and its induction was abrogated by flavopiridol (bottom western blot and black bar). α-Tubulin was used as the loading control. These western blot analyses are representative of three independent experiments that yielded the same results. D, only the previously unannotated proximal HEXIM1 promoter is inducible by SAHA and HBMA in HeLa cells (lanes 1-3). The pp primer is presented in A. Lane 1 represents resting HeLa cells. Lane 2 is from HeLa cells treated with SAHA. Lane 3 is from HeLa cells treated with HMBA. Lane 4 is a blank control. Lane 5 is the DNA marker, defined as m in panel D.
Although it has been observed previously that levels of HEXIM1 transcripts and protein increase following the administration of the differentiation agent HMBA, which leads to a rapid release of free P-TEFb from the 7SK snRNP, it was unknown whether HEXIM2 behaves in the same way (21). The histone deacetylase inhibitor (HDACi) SAHA has the same effect as HMBA (23). Indeed, as presented in Fig. 1B, levels of free P-TEFb increased rapidly after the addition of SAHA to HeLa cells. By 90 min after the addition of 5 μm SAHA, CycT1 and CDK9 moved from higher (7SK snRNP) to lower (free P-TEFb) glycerol fractions (Fig. 1B, bottom SAHA rows). Next, we examined effects of SAHA on levels of HEXIM1 and HEXIM2 in these cells. Only those of HEXIM1 increased 3-fold (Fig. 1C, Hex1, first western blot and black bar). This increase was normalized to amounts of α-tubulin (Fig. 1C, third western blot). Of note, SAHA did not increase levels of HEXIM2 in HeLa cells (Fig. 1C, second western blot and white bar). Flavopiridol is a specific inhibitor of CDK9 (25). When SAHA and flavopiridol were administered together, the 3-fold increase of HEXIM1 recorded previously was no longer observed (Fig. 1C, fifth western blot and black bar). This finding confirms a previous study where anti-CDK9 and anti-CycT1 siRNAs abrogated increased levels of HEXIM1 transcripts after the administration of HMBA to HeLa cells (21). Thus, HEXIM1 gene expression could be induced by the release of free P-TEFb from the 7SK snRNP.
To determine which HEXIM1 promoter is inducible by free P-TEFb, we performed 5′ RACE following the administration of SAHA or HMBA to HeLa cells (Fig. 1D). The position of the 3′ reverse primer used in our 5′ RACE started at position 873 in the HEXIM1 gene. The size of amplified product measured 228 bp, and its levels increased following the administration of SAHA or HMBA to HeLa cells (Fig. 1C, lanes 1, 2, and 3). This fragment corresponds to the initiation of HEXIM1 transcription from the previously unannotated proximal promoter (Fig. 1A). This result indicates that it is this proximal HEXIM1 promoter that is activated by the release of free P-TEFb from the 7SK snRNP (20).
ChIP-Seq Data Confirm the Existence of the P-TEFb-inducible Proximal HEXIM1 Promoter
Most metazoan promoters have RNAPII paused slightly downstream of the transcription start site by NELF and DSIF (26–29). This pausing keeps these promoters available for rapid induction by P-TEFb, whose activity causes NELF to dissociate and RNAPII and DSIF to elongate on the gene (26, 27, 30, 31). To confirm which promoter was responsible for the observed P-TEFb-sensitive HEXIM1 transcription, ChIP-Seq was performed with anti-RNAPII, anti-NELF-A (NELF), and anti-SPT5 (DSIF) antibodies in HeLa cells.
Most of the dozen genes around the HEXIM1 gene displayed typical distributions of RNAPII, NELF, and DSIF (Fig. 2A). Peaks of all three proteins were seen within 100 bp of most of these promoters, which, when actively transcribed, also had downstream accumulations of RNAPII and DSIF but not NELF. Although the HEXIM1 gene showed high levels of transcription, pausing of RNAPII was negligible at the distal HEXIM1 gene promoter (Fig. 2B). Instead, the vast majority of RNAPII, NELF, and DSIF peaked at the previously unannotated proximal promoter. Not only do these data explain the P-TEFb-sensitive generation of the 228 bp 5′ RACE product in Fig. 1C, but the observed distribution of RNAPII and pausing factors together suggest that this newly identified promoter is the primary source of HEXIM1 gene expression in HeLa cells.
FIGURE 2.

Occupancy of RNAPII, NELF, and DSIF by ChIP-Seq reveals an unannotated proximal HEXIM1 promoter. RNAPII, NELF, and DSIF occupancy in HeLa cells was determined by ChIP-Seq analyses as described under “Experimental Procedures.” A and B are UCSC Genome Browser tracks displaying the density of RNAPII, NELF, and DSIF around the HEXIM1 gene. The vertical axes indicate the number of reads contributing to the signal density. The RefSeq annotation track displays the position of genes using thin blocks (untranslated regions), thick blocks (exons), lines (introns), and arrowheads (direction).
Mapping the Minimal Inducible HEXIM1 Promoter
To determine the minimal inducible HEXIM1 promoter, we investigated 3 kb upstream of the HEXIM1 coding region. When sequences of the annotated upstream HEXIM1 promoter were fused to the luciferase reporter gene (from positions −3000 to −1000), very low levels of luciferase expression were observed upon the addition of HMBA or SAHA (data not shown). In sharp contrast, the 1.2-kb upstream sequence that contained the proximal promoter led to 5-fold increased luciferase activity following the addition of SAHA to HEK293 cells (Fig. 3A, first black bar). Further deletions to positions −354, −104, and −54 were examined. As presented in Fig. 3A, second, third, and fourth black bars, only sequences that contained 104 bp of the proximal HEXIM1 promoter responded to SAHA (up to 20-fold). This 104-bp sequence, presented in Fig. 3B, contains putative DNA-binding motifs, including consensus specificity protein 1 (Sp1)-binding sites and a TATA box. A predicted transcription start site (+1) is indicated with an arrow. These GC and TATA boxes are very reminiscent of the HIV LTR, which is also activated by HMBA and SAHA (20).
FIGURE 3.

The minimal proximal HEXIM1 promoter required for transcription activation. A, 104 bp of the proximal HEXIM1 gene promoter are sufficient for its activation by SAHA in HEK293 cells. The schematic presents four HEXIM1 promoter-luciferase plasmid targets containing the indicated upstream promoter sequences. Right panel, basal and SAHA-induced luciferase activities for 48 h in HEK293 cells. Luc, luciferase; pA, polyadenylation. B, 104 bp of the proximal HEXIM1 gene promoter contains consensus Sp1-binding sites and a TATA box.
Compounds That Release Free P-TEFb Also Increase HEXIM1 Transcription
Compounds from diverse structural classes can reactivate HIV transcription from latency in cell lines and primary cells (23, 32). They include HMBA; the HDAC inhibitors SAHA, TSA, MS-275, and ST80; the bromodomain extraterminal (BET) bromodomain inhibitors JQ1 and iBET, and the PKC agonists phorbol 12-myristate 13-acetate, prostratin, and bryostatin. All compounds that released free P-TEFb also reactivated HIV transcription (23, 32). To examine the effects of these different compounds on HEXIM1 transcription, the same plasmid target (Hex1(-104)Luc) that responded best to SAHA was expressed transiently in HeLa and HEK293 cells (Fig. 4). We used concentrations of these compounds that were determined in previous studies (23, 32). Our results are presented as fold activation over DMSO using a logarithmic scale. Among the compounds examined, HMBA, MS-275, ST-80, SAHA, and TSA potently activated the Hex1(-104)Luc plasmid target. For example, TSA, a pan-HDACi, induced the luciferase activity 30-fold (Fig. 4A). ST-80 (an HDAC6 inhibitor) and MS-275 (an HDAC1, 2, and 3 inhibitor) each increased luciferase activity 10-fold (Fig. 4A). SAHA and HMBA each increased luciferase activity 5-fold (Fig. 4, A and B). Not all HDAC inhibitors released P-TEFb, e.g. tubastatin A (an HDAC6 inhibitor) (Fig. 4A). The BET bromodomain inhibitors JQ-1 and iBET each activated luciferase activity 3- to 4-fold (Fig. 4, A and B). The PKC agonist phorbol 12-myristate 13-acetate increased the luciferase activity 2-fold (Fig. 4A). These data follow closely those obtained with the HIV LTR using the same compounds (23, 32). Thus, the data suggest that HEXIM1 and HIV transcription respond similarly to the release of free P-TEFb from the 7SK snRNP.
FIGURE 4.
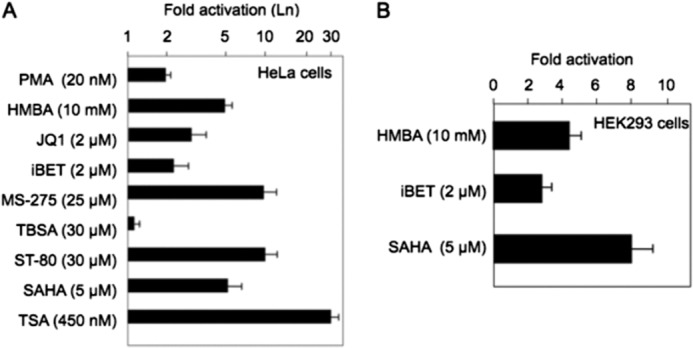
The proximal HEXIM1 promoter responds to different compounds that release free P-TEFb from the 7SK snRNP. The luciferase activity was measured for the Hex1(-104)Luc plasmid target after HeLa cells (A) or HEK293 cells (B) were incubated with the incubated compounds for 48 h. The following compounds activated the proximal HEXIM1 promoter: TSA, 30-fold; ST-80 and MS-275, 10-fold; SAHA and HMBA, 5-fold; BET gene bromodomain inhibitors JQ-1 and iBET, 2- to 3-fold; PKC agonist phorbol 12-myristate 13-acetate, 2-fold. Results for HeLa cells are presented in a logarithmic scale.
SAHA Increases Levels of P-TEFb at the HEXIM1 Promoter
Because SAHA activated HEXIM1 transcription in HeLa and HEK293 cells, we performed ChIPs on the inducible proximal HEXIM1 promoter. The design of primers is presented in Fig. 5A, top panel. Following the administration of SAHA, levels of CDK9 and RNAPII increased 3- to 5-fold, respectively (Fig. 5A, bottom panel, black columns 1 and 2). At the same time, although levels of AFF4 and ELL2 increased 2-fold, those of BRD4 decreased slightly (Fig. 5A, bottom panel, black columns 3, 4, and 5). Analogous to the situation with the HIV LTR, the administration of SAHA increased levels of components of the SEC at the HEXIM1 promoter.
FIGURE 5.

CDK9, RNAPII, AFF4, and ELL2 are increased at the proximal HEXIM 1 promoter after the addition of SAHA to HEK293 cells. A, schematic of the HEXIM1 (Hex1) gene, qPCR primers, and ChIP data. ChIPs were performed with anti-CDK9 (1), anti-RNAPII (2), anti-BRD4 (3), anti-AFF4 (4), and anti-ELL2 (5) antibodies. The fold enrichment was calculated by dividing qPCR signals obtained with each specific antibody after and before the addition of SAHA. They were also normalized to the IgG control. pA, polyadenylation. B, knockdown of CDK9, AFF4, or ELL2 with corresponding siRNAs abrogated SAHA-induced HEXIM1 transcription in HEK293 cells. Columns represent basal and SAHA-induced luciferase activities with the presence of specific siRNAs in HEK293 cells. C, scrambled siRNA (siSCR). The efficiency of our knockdowns was determined by western blotting for CDK9, ELL2, and AFF4. They are presented in the bottom panel.
To examine whether increased levels of SEC at the HEXIM1 promoter mediated effects of SAHA, we knocked down CDK9, ELL2, and AFF4 with specific siRNAs. First, HEK293 cells were transfected with Hex1(-104)Luc alone or cotransfected with siCDK9, siELL2, or siAFF4 RNAs for 48 h. These siRNAs have been validated previously (23, 33). Subsequently, cells were treated with SAHA or DMSO for 24 h, and luciferase activity was determined (Fig. 5B). The knockdown efficiencies were verified by western blot analyses (Fig. 5B, bottom panel). Of note, SAHA alone increased the luciferase activity from the Hex1(-104)luc plasmid target 6-fold (Fig. 5B). In contrast, when CDK9, ELL2, or AFF4 were knocked down, luciferase activity increased less than 2-fold (Fig. 5B). We conclude that free P-TEFb in the SEC is responsible for the effects of SAHA on the HEXIM1 promoter. These data parallel closely those obtained with the HIV LTR and reveal that the release of free P-TEFb from the 7SK snRNP activates transcription from the proximal HEXIM1 promoter in cells.
DISCUSSION
In this study, we confirmed that the shorter HEXIM1 gene transcripts are inducible by free P-TEFb (21). Neither longer HEXIM1 transcripts nor levels of HEXIM2 protein increased following the administration of SAHA in HeLa cells. Genome-wide ChIP-Seq data confirmed the location of the inducible promoter and the transcription start site of the HEXIM1 gene. Next, we mapped the minimal inducible HEXIM1 promoter to a 104-bp sequence that contained GC and TATA boxes that could respond to P-TEFb. This 104-bp fragment was inducible by HMBA, PKC agonists, HDAC inhibitors, and BET bromodomain inhibitors. Finally, ChIP also demonstrated increased levels of SEC components at the HEXIM1 promoter after the administration of SAHA. Knocking down these subunits abrogated the activation of HEXIM1 transcription by SAHA. We conclude that the release of free P-TEFb from the 7SK snRNP increases the synthesis of its inhibitor, HEXIM1, which facilitates the reassembly of the 7SK snRNP.
These data support and extend previous observations on HEXIM1 transcription and HIV reactivation by HMBA, SAHA, and related compounds. Importantly, they suggest that HIV and HEXIM1 transcription are regulated similarly, which has implications for cell growth, proliferation, and apoptosis as well as HIV latency and reactivation. In both cases, it is the release of P-TEFb and reassembly of the 7SK snRNP that play important roles in the mode of action of these compounds. Indeed, we have previously examined these effects directly with all available methods for measuring P-TEFb release. They include glycerol gradient centrifugation, low- and high-salt fractionation of nuclear extracts, and RNA immunoprecipitation (23). In all of these tests, only compounds that released free P-TEFb from the 7SK snRNP also activated HIV transcription. Of interest, the proximal HEXIM1 gene promoter and the HIV LTR not only respond similarly to these agonists but also contain very similar sequence motifs. Thus, sequences that contain Sp1 sites and a TATA box are very responsive to levels of free P-TEFb, which is recruited there via SEC rather than BRD4. This recruitment could be via Med 26 in the mediator or Paf1C on RNAPII itself (34, 35).
It is of some interest that increased synthesis of HEXIM1 helps to reassemble the 7SK snRNP. Upon the release of free P-TEFb from the 7SK snRNP, there is an abundance of HEXIM1/2 proteins in cells. Some of these HEXIM proteins appear in complexes with other RNA species, which include microRNAs (36). Additionally, the mode of release modifies HEXIM1 posttranslationally, e.g. by phosphorylation. For example, HMBA activates Akt, which phosphorylates HEXIM1 on a threonine and a serine at positions 270 and 278, respectively (20). PKC phosphorylates HEXIM1 on the serine at position 158 (37). Unless dephosphorylated, these modified HEXIM1 proteins cannot reassemble into the 7SK snRNP (38). Because exogenously expressed HEXIM1 protein, e.g. from transient transfection, also has a difficult time entering into the 7SK snRNP, it is possible that there are additional modifications of the HEXIM1 protein that are required for the assembly of the 7SK snRNP. Alternatively, it is likely that larger amounts of HEXIM1 are required for the optimal reassembly of the 7SK snRNP (39). Thus, cells precisely regulate their P-TEFb equilibrium for their growth and proliferation, and the newly synthesized HEXIM1 protein helps the reassembly of the 7SK snRNP (39, 40). Analogous to the situation with NF-κB, which activates the synthesis of its inhibitor, IκB, free P-TEFb also activates the synthesis of its inhibitor, HEXIM1. In this scenario, when levels of the 7SK snRNP are high, potent disruptors, such as HMBA, SAHA, etc., increase the synthesis of HEXIM1 protein to a degree that less free P-TEFb becomes available for cellular homeostasis, cells stop growing, and many undergo apoptosis. This latter phenotype represents the therapeutic potential of these compounds, which eliminate rapidly dividing cells.
Acknowledgements
We thank Drs. Manfred Jung and James Bradner for reagents.
This work was supported, in whole or in part, by National Institutes of Health Collaboratory of AIDS Researchers for Eradication (CARE) Center Grant U19 AI096113, HIV Accessory and Regulatory Complexes (HARC) Center Grant P50 GM082250, Grant RO1 AI049104 (to B. M. P.), and Grant GM35500 (to D. H. P.). This work was also supported by a University of Iowa Presidential Graduate Fellowship (to K. A. N.).
Paired sequences and processed bedGraph files are available in the Gene Expression Onnibus Repository under accession number GSE53008.
- RNAPII
- RNA polymerase II
- CTD
- C-terminal domain
- P-TEFb
- positive transcription elongation factor b
- DSIF
- DRB sensitivity-inducing factor
- NELF
- negative elongation factor
- SEC
- superelongation complex
- snRNP
- small nuclear ribonucleoprotein
- CDK
- cyclin-dependent kinase
- SAHA
- suberoylanilide hydroxamic acid
- HMBA
- hexamethylene bisacetamide
- DMSO
- dimethyl sulfoxide
- ChIP-Seq
- ChIP with sequencing
- qPCR
- quantitative PCR
- RACE
- rapid amplification of cDNA ends
- HDACi
- histone deacetylase inhibitor
- BET
- bromodomain extraterminal.
REFERENCES
- 1. Kapp L. D., Lorsch J. R. (2004) The molecular mechanics of eukaryotic translation. Annu. Rev. Biochem. 73, 657–704 [DOI] [PubMed] [Google Scholar]
- 2. Roeder R. G. (1991) The complexities of eukaryotic transcription initiation. Regulation of preinitiation complex assembly. Trends Biochem. Sci. 16, 402–408 [DOI] [PubMed] [Google Scholar]
- 3. McCracken S., Fong N., Yankulov K., Ballantyne S., Pan G., Greenblatt J., Patterson S. D., Wickens M., Bentley D. L. (1997) The C-terminal domain of RNA polymerase II couples mRNA processing to transcription. Nature 385, 357–361 [DOI] [PubMed] [Google Scholar]
- 4. O'Brien T., Hardin S., Greenleaf A., Lis J. T. (1994) Phosphorylation of RNA polymerase II C-terminal domain and transcriptional elongation. Nature 370, 75–77 [DOI] [PubMed] [Google Scholar]
- 5. Dahmus M. E. (1996) Reversible phosphorylation of the C-terminal domain of RNA polymerase II. J. Biol. Chem. 271, 19009–19012 [DOI] [PubMed] [Google Scholar]
- 6. Ahn S. H., Kim M., Buratowski S. (2004) Phosphorylation of serine 2 within the RNA polymerase II C-terminal domain couples transcription and 3′ end processing. Mol. Cell 13, 67–76 [DOI] [PubMed] [Google Scholar]
- 7. Komarnitsky P., Cho E. J., Buratowski S. (2000) Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev. 14, 2452–2460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Palancade B., Bensaude O. (2003) Investigating RNA polymerase II carboxyl-terminal domain (CTD) phosphorylation. Eur. J. Biochem. 270, 3859–3870 [DOI] [PubMed] [Google Scholar]
- 9. Peterlin B. M., Price D. H. (2006) Controlling the elongation phase of transcription with P-TEFb. Mol. Cell 23, 297–305 [DOI] [PubMed] [Google Scholar]
- 10. Feaver W. J., Svejstrup J. Q., Henry N. L., Kornberg R. D. (1994) Relationship of CDK-activating kinase and RNA polymerase II CTD kinase TFIIH/TFIIK. Cell 79, 1103–1109 [DOI] [PubMed] [Google Scholar]
- 11. Bartkowiak B., Liu P., Phatnani H. P., Fuda N. J., Cooper J. J., Price D. H., Adelman K., Lis J. T., Greenleaf A. L. (2010) CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 24, 2303–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Blazek D., Kohoutek J., Bartholomeeusen K., Johansen E., Hulinkova P., Luo Z., Cimermancic P., Ule J., Peterlin B. M. (2011) The cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 25, 2158–2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Price D. H. (2000) P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol. Cell. Biol. 20, 2629–2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin X., Taube R., Fujinaga K., Peterlin B. M. (2002) P-TEFb containing cyclin K and Cdk9 can activate transcription via RNA. J. Biol. Chem. 277, 16873–16878 [DOI] [PubMed] [Google Scholar]
- 15. Taube R., Peterlin M. (2013) Lost in transcription. Molecular mechanisms that control HIV latency. Viruses 5, 902–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marshall N. F., Price D. H. (1995) Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J. Biol. Chem. 270, 12335–12338 [DOI] [PubMed] [Google Scholar]
- 17. Barboric M., Nissen R. M., Kanazawa S., Jabrane-Ferrat N., Peterlin B. M. (2001) NF-κB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol. Cell 8, 327–337 [DOI] [PubMed] [Google Scholar]
- 18. Barboric M., Lenasi T., Chen H., Johansen E. B., Guo S., Peterlin B. M. (2009) 7SK snRNP/P-TEFb couples transcription elongation with alternative splicing and is essential for vertebrate development. Proc. Natl. Acad. Sci. U.S.A. 106, 7798–7803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Peterlin B. M., Brogie J. E., Price D. H. (2012) 7SK snRNA. A noncoding RNA that plays a major role in regulating eukaryotic transcription. RNA 3, 92–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Contreras X., Barboric M., Lenasi T., Peterlin B. M. (2007) HMBA releases P-TEFb from HEXIM1 and 7SK snRNA via PI3K/Akt and activates HIV transcription. PLoS Pathog. 3, 1459–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. He N., Pezda A. C., Zhou Q. (2006) Modulation of a P-TEFb functional equilibrium for the global control of cell growth and differentiation. Mol. Cell. Biol. 26, 7068–7076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Michels A. A., Fraldi A., Li Q., Adamson T. E., Bonnet F., Nguyen V. T., Sedore S. C., Price J. P., Price D. H., Lania L., Bensaude O. (2004) Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. EMBO J. 23, 2608–2619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bartholomeeusen K., Xiang Y., Fujinaga K., Peterlin B. M. (2012) Bromodomain and extra-terminal (BET) bromodomain inhibition activate transcription via transient release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein. J. Biol. Chem. 287, 36609–36616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Castelo-Branco G., Amaral P. P., Engström P. G., Robson S. C., Marques S. C., Bertone P., Kouzarides T. (2013) The non-coding snRNA 7SK controls transcriptional termination, poising, and bidirectionality in embryonic stem cells. Genome Biol. 14, R98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chao S. H., Price D. H. (2001) Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J. Biol. Chem. 276, 31793–31799 [DOI] [PubMed] [Google Scholar]
- 26. Core L. J., Waterfall J. J., Lis J. T. (2008) Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322, 1845–1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guo J., Price D. H. (2013) RNA polymerase II transcription elongation control. Chem. Rev. 113, 8583–8603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Deaton A. M., Bird A. (2011) CpG islands and the regulation of transcription. Genes Dev. 25, 1010–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Andrulis E. D., Guzmán E., Döring P., Werner J., Lis J. T. (2000) High-resolution localization of Drosophila Spt5 and Spt6 at heat shock genes in vivo. Roles in promoter proximal pausing and transcription elongation. Genes Dev. 14, 2635–2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Strobl L. J., Eick D. (1992) Hold back of RNA polymerase II at the transcription start site mediates down-regulation of c-myc in vivo. EMBO J. 11, 3307–3314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rasmussen E. B., Lis J. T. (1993) In vivo transcriptional pausing and cap formation on three Drosophila heat shock genes. Proc. Natl. Acad. Sci. U.S.A. 90, 7923–7927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bartholomeeusen K., Fujinaga K., Xiang Y., Peterlin B. M. (2013) Histone deacetylase inhibitors (HDACis) that release the positive transcription elongation factor b (P-TEFb) from its inhibitory complex also activate HIV transcription. J. Biol. Chem. 288, 14400–14407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. He N., Chan C. K., Sobhian B., Chou S., Xue Y., Liu M., Alber T., Benkirane M., Zhou Q. (2011) Human polymerase-associated factor complex (PAFc) connects the super elongation complex (SEC) to RNA polymerase II on chromatin. Proc. Natl. Acad. Sci. U.S.A. 108, E636–E645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Takahashi H., Parmely T. J., Sato S., Tomomori-Sato C., Banks C. A., Kong S. E., Szutorisz H., Swanson S. K., Martin-Brown S., Washburn M. P., Florens L., Seidel C. W., Lin C., Smith E. R., Shilatifard A., Conaway R. C., Conaway J. W. (2011) Human mediator subunit MED26 functions as a docking site for transcription elongation factors. Cell 146, 92–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim J., Guermah M., Roeder R. G. (2010) The human PAF1 complex acts in chromatin transcription elongation both independently and cooperatively with SII/TFIIS. Cell 140, 491–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li Q., Cooper J. J., Altwerger G. H., Feldkamp M. D., Shea M. A., Price D. H. (2007) HEXIM1 is a promiscuous double-stranded RNA-binding protein and interacts with RNAs in addition to 7SK in cultured cells. Nucleic Acids Res. 35, 2503–2512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fujinaga K., Barboric M., Li Q., Luo Z., Price D. H., Peterlin B. M. (2012) PKC phosphorylates HEXIM1 and regulates P-TEFb activity. Nucleic Acids Res. 40, 9160–9170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen R., Liu M., Li H., Xue Y., Ramey W. N., He N., Ai N., Luo H., Zhu Y., Zhou N., Zhou Q. (2008) PP2B and PP1α cooperatively disrupt 7SK snRNP to release P-TEFb for transcription in response to Ca2+ signaling. Genes Dev. 22, 1356–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Krueger B. J., Varzavand K., Cooper J. J., Price D. H. (2010) The mechanism of release of P-TEFb and HEXIM1 from the 7SK snRNP by viral and cellular activators includes a conformational change in 7SK. PLoS ONE 5, e12335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Blazek D., Barboric M., Kohoutek J., Oven I., Peterlin B. M. (2005) Oligomerization of HEXIM1 via 7SK snRNA and coiled-coil region directs the inhibition of P-TEFb. Nucleic Acids Research 33, 7000–7010 [DOI] [PMC free article] [PubMed] [Google Scholar]