

Abstract
B-cell leukemia/lymphoma 2 (BCL-2) prevents commitment to programmed cell death at the mitochondrion. It remains a challenge to identify those tumors that are best treated by inhibition of BCL-2. Here we demonstrate that acute myeloid leukemia (AML) cell lines, primary patient samples, and murine primary xenografts are very sensitive to treatment with the selective BCL-2 antagonist ABT-199. In primary patient cells, the median IC50 was approximately 10 nM, and cell death occurred within 2 h. Our ex vivo sensitivity results compare favorably with those observed for chronic lymphocytic leukemia (CLL), a disease for which ABT-199 has demonstrated consistent activity in clinical trials. Moreover, mitochondrial studies using BH3 profiling demonstrate activity at the mitochondrion that correlates well with cytotoxicity, supporting an on target mitochondrial mechanism of action. Our protein and BH3 profiling studies provide promising tools that can be tested as predictive biomarkers in any clinical trial of ABT-199 in AML.
Keywords: AML, apoptosis, BCL-2, targeted therapy, BH3 profiling
Introduction
Acute myelogenous leukemia (AML) is a hematopoietic neoplasia characterized by the rapid, clonal growth of the myeloid lineage of blood cells. The disease affects approximately 14,000 adults in the United States each year and unfortunately, despite recent advances in the treatment of AML, 10,400 people die from their disease (1). Most AML patients become resistant to chemotherapy at some point in their course and succumb to their disease. Therefore, it is necessary to prevent chemo-resistance or enhance chemosensitivity in a selective fashion to lead to a higher cure rate and a lower toxic burden.
A novel strategy to treat cancer cells is to directly stimulate the mitochondrial apoptotic pathway in them. The mitochondrial apoptotic pathway is regulated by the B-cell leukemia/ lymphoma 2 (BCL-2) family of proteins. These proteins respond to upstream apoptotic signals that control mitochondrial outer membrane permeabilization (MOMP) and the release of cytochrome c, a hallmark of mitochondrial apoptosis (2). The family consists of pro-apoptotic BH3-only proteins, pro-apoptotic multi-domain effector proteins and anti-apoptotic proteins which together act as a rheostat to control MOMP. The BH3-only proteins are further subdivided into two groups based on function - the “activators” (BID, BIM and PUMA) and “sensitizers” (such as BAD, BIK, HRK and NOXA) (3–5). The activators bind to and inhibit the anti-apoptotics (such as BCL-2, BCL-XL and MCL-1), and also directly interact with the effector proteins causing the proteins to oligomerize at the mitochondria and cause MOMP (6, 7). However, the BH3-only sensitizers can only bind to the anti-apoptotics which allows for the release of the activator and effector proteins to drive MOMP (3).
Navitoclax (ABT-263), which functions as a small molecule mimetic of the BH3 domain of the BH3-only sensitizer protein BAD, efficiently binds to BCL-2, BCL-XL and BCL-W, releasing bound pro-apoptotic proteins and causing MOMP in BCL-2 dependent cancer cells (8). In early clinical trials, navitoclax showed potency in the treatment of chronic lymphocytic leukemia (CLL) and small-cell lung cancer (9, 10). However, treatment with navitoclax causes on-target, dose-limiting thrombocytopenia because platelets are dependent on the anti-apoptotic protein BCL-XL for their survival (11). The dose-dependent thrombocytopenia limited navitoclax’s use in many malignancies, particularly leukemias where patients often present with pre-existing thrombocytopenia. This prompted the development of ABT-199, a modified BH3-mimetic derivative of ABT-263 which maintains specificity for BCL-2, but lacks affinity for BCL-XL (12). The remodeled drug has shown cancer killing efficacy in CLL in vivo, myc-driven lymphomas in mice and estrogen receptor-positive breast cancer while sparing platelets (12–14).
AML bulk and stem cells are dependent on BCL-2 for survival and BCL-2 inhibition by ABT-737 (an in vitro tool compound with activity very similar to navitoclax) causes cell death in AML cells (15). Importantly, BCL-2 inhibition relatively spares normal hematopoietic stem cells which are more dependent on MCL-1 for their survival (16, 17). Thus, the first goal of the present study is to evaluate the anti-cancer effects of ABT-199 on AML and compare its efficacy with ABT-737/navitoclax, drugs that have both shown activity in the ex vivo treatment of AML cell lines and AML primary patient samples and in human clinical trials (15). The second goal is to determine if BH3-profiling can be used as a tool to predict cellular response to ABT-199 treatment.
BH3-profiling is a method to determine the mitochondrial priming level of a cell by exposing cellular mitochondria with standardized amounts of peptides derived from the BH3 domains of BH3-only proteins and determining the rate of MOMP, as measured by either cytochrome c release or depolarization across the inner mitochondrial membrane (18). Previously, we have shown that the priming status of the cell is predictive of the cell’s chemo-responsiveness in that the more primed the cell is, the more sensitive the cell is to various chemotherapeutics (16, 19). Furthermore, BH3-profiling can also identify anti-apoptotic addictions (16, 19, 20). For instance, the BAD BH3-only peptide binds with high affinity with BCL-2, BCL-XL and BCL-W, while the HRK BH3 peptide binds with high affinity only to BCL-XL. Thus, MOMP following BAD peptide incubation suggests an anti-apoptotic dependency on BCL-2, BCL-XL or BCL-W, while MOMP following HRK peptide incubation indicated dependency on BCL-XL. Using this tool, we can identify AML cells which depend on BCL-2 for survival and that are more likely to die following BCL-2 inhibition. Thus, we hypothesize that cells that are addicted to BCL-2 for survival will be sensitive to ABT-199 and that we can predict this response by BH3 profiling.
Results
ABT-199 Kills AML Cell Lines Potently and Quickly in vitro and in vivo
As an initial test of the potential utility of ABT-199 in AML, we exposed AML cell lines to increasing concentrations of ABT-199 for 48 h and then determined the IC50 values. Comparisons were made with ABT-737. As shown in Fig. 1A, the IC50 of ABT-199 ranged from <10 nM to >1000 nM, and sensitivity to ABT-737 roughly tracked sensitivity to ABT-199. It is notable that in sensitive AML cell lines (IC50 < 0.1 μM), ABT-199 is more potent than ABT-737 (Supplemental Figure S1A and S1B), probably due to ABT-199’s 5-fold higher affinity to BCL-2 protein (12). Prior experience with CLL, a disease for which excellent clinical activity of ABT-199 has been observed, has revealed that CLL cells are killed in an on-target fashion, and that the killing was evident within 4 h (25). Therefore, we tested whether ABT-199 could rapidly induce apoptosis in a sensitive AML cell line - MOLM-13. In Figure 1B and 1C, it can be seen that cell growth is inhibited and cell apoptosis is observed within just a few hours of exposure to ABT-199.
Figure 1. Selective inhibition of BCL-2 by ABT-199 kills AML cell lines quickly and effectively.

A). AML cell lines were treated with ABT-199 or ABT-737 for 48 h. Calcusyn software was used to calculate the IC50 values based on the number of viable cells (i.e., Annexin V−/PI−) determined by FACS analysis. B). MOLM-13 AML cells were treated with indicated concentrations of ABT-199. Apoptosis induction was determined by Annexin V/PI flow cytometry. C). Viable (i.e., Annexin V−/PI−) cell counts were quantified by FACS analysis using CountBright counting beads. D). Serial bioluminescence images of mice bearing MOLM-13 tumors treated with the vehicle or ABT-199 (treatment started on day 4, administered by oral gavage at dose of 100 mg/kg). E). Kaplan-Meier survival curves for mice treated as described in E (n = 7 per arm). Statistical significance was calculated using Log-rank (Mantel-Cox) test (p < 0.0004 ). F). H&E staining of histological sections of liver, spleen, and bone marrow 15 d post leukemia cell injection. Age- and sex-matched mice without tumor were used as controls. Representative MOLM-13 cells are indicated by arrows. Representative engraftment areas are circled in green. All pictures were taken under the same magnification; scale bar equals 50 μm. G). Immunohistochemical staining of histological sections of liver, spleen, and bone marrow with human CD45 antibody 15 d post leukemia cell injection. Scale bar equals 50 μm.
To demonstrate that the efficacy seen was consistent with tolerable in vivo delivery of ABT-199, we tested the effect of ABT-199 on an aggressive mouse xenograft model of MOLM-13. NOD SCID gamma (NSG) mice were injected with luciferase-labeled MOLM-13 cells and monitored by bioluminescence imaging (BLI) for tumor development. After confirmation of AML engraftment in the bone marrow (Figure 1D, day 4), the mice were treated with ABT-199 (100 mg/kg) by daily oral gavage for 2 weeks. Serial BLI images showed that ABT-199 treatment markedly inhibited leukemia progression, which translated into prolonged overall survival when compared to vehicle-treated mice (p = 0.0004, Figure 1E). ABT-199 treated mice also carried significantly lower leukemia burden in bone marrow, spleen and liver as indicated by hematoxylin and eosin staining (H&E, Figure 1F) and immunohistochemical analysis of human CD45 (Figure 1G).
ABT-199 Sensitivity Correlates with BCL-2 Protein Level
Next we tested whether there were correlates of cell line sensitivity to ABT-199 that supported an on-target action of killing via competition for the BH3 binding site selectively of BCL-2. Relative levels of several BCL-2 family proteins were measured by Western blot and densitometry (Figure 2A). Spearman analysis was performed to evaluate the correlation between IC50 values and protein expression. Levels of BCL-2 correlated with sensitivity to ABT-199, while levels of BCL-XL inversely correlated with ABT-199 sensitivity (Figure 2B). Levels of MCL-1 demonstrated a trend to anti-correlation with sensitivity to ABT-199, but the trend was not statistically significant (Figure 2B). These observations supported the on-target effects of ABT-199.
Figure 2. Sensitivity to ABT-199 positively correlates with endogenous BCL-2 protein level and negatively correlates with BCL-XL protein level in AML cell lines.

A) Western blot analysis of BCL-2 family proteins in untreated AML cells. The band intensity was quantified using Odyssey v2.0 software, and displayed numerically as a ratio of the band intensity detected in the OCI-AML3 cells. B) Significant correlations were observed between ABT-199 IC50 values and BCL-2/BCL-XL protein levels. The non-parametric one-tailed Spearman test was used to determine the correlation coefficient. The p values provided are nominal p values not corrected for multiple comparisons. C). MCL-1 knockdown by 85% was achieved by lentiviral shRNA. D). MCL-1 knockdown significantly sensitized OCI-AML3 cells to ABT-199. E). Western blot analysis showing HL-60 AML cells transfected to stably overexpress BCL-XL or BCL-2. F). Overexpression of BCL-XL or BCL-2 in HL-60 cells confers complete resistance to ABT-199-induced apoptosis.
The OCI-AML3 cell line was relatively insensitive to ABT-199 and ABT-737 (Figure 1A). A quantitative immunoblot showed that OCI-AML3 cells had high expression of BCL-2 and MCL-1 and relatively low level of BCL-XL (Figure 2A). If ABT-199 is a BH3 mimetic specific for BCL-2, then MCL-1 knockdown should significantly sensitize OCI-AML3 cells to this compound. To test this, MCL-1 protein level was reduced by 85% in OCI AML3 cells by lentiviral transduction using a previously validated MCL1-specific shRNA, without affecting BCL-2 protein expression (Figure 2C). Indeed, MCL-1 knockdown greatly increased sensitivity to ABT-199 (Figure 2D) as well as to ABT-737 (Supplemental Figure S2). HL-60 cells with high levels of BCL-2 protein and relatively low BCL-XL and MCL-1 expression are very sensitive to ABT-199 (Figure 1A). BCL-XL overexpression conferred resistance to ABT-199 in HL-60 cells, while BCL-2 overexpression made HL-60 cells moderately resistant to ABT-199 (Figure 2E-F). All these results are consistent with a killing mechanism operating via selective targeting of BCL-2 in AML cells.
ABT-199-resistant MOLM-13 cells express lower BCL-2 levels compared to the parental cells
Although ABT-199 effectively induced apoptosis in MOLM-13 cells, a fraction of MOLM-13 cells remained alive after 24 h treatment with 50 nM ABT-199 (Figure 2B). To determine whether surviving cells represent a sub-clone with low target (Bcl-2) expression levels, we selected ABT-199-resistant MOLM-13 cells by culturing parental cells in medium containing 50nM ABT-199. Intracellular flow cytometry was performed to measure cellular BCL-2 protein in the parental and ABT199-resistant MOLM-13 cells. Although no distinct sub-populations were observed, the BCL-2 associated MFI of the parental MOLM-13 cells distributed between ~1 to ~20 (Supplemental Figure S3A), indicating a heterogeneity of BCL-2 expression in the parental cells. In sum, resistant cells expressed lower levels of BCL-2 protein compared to the parental cells (resistant cells: MFI = 3.48; parental cells: MFI = 5.51; P < 0.0001, Supplemental Figure S3B).
ABT-199 Operates Selectively on BCL-2 Dependent Mitochondria
If ABT-199 is killing cancer cells via displacement of pro-apoptotic proteins from BCL-2, it should be operating on mitochondria. As one would expect if this were the case, we observed a correlation between direct mitochondrial toxicity and cellular toxicity for ABT-199 and ABT-737 in the 12 cell lines studied in Figure 1A (Figure 3A, B). We also tested whether detection of BCL-2 dependence using mitochondrial exposure to the BAD BH3 peptide correlated with cellular sensitivity to these agents. We found that while there was a good correlation between mitochondrial sensitivity to the BAD peptide and cellular drug sensitivity for the most sensitive cell lines, there was a group of relatively drug -resistant cell lines that still demonstrated mitochondrial sensitivity to the BAD BH3 peptide (Figure 3 C, D). A clue to the reason for this was revealed by the tendency of these cell lines to have mitochondria that were also quite sensitive to the HRK peptide, an indicator of BCL-XL dependence. To ensure that we were studying BCL-2 dependence specifically, especially in these less drug-sensitive cells, we made a correction, by subtracting the HRK signal from the BAD signal. In Figure 3E and F, we used this modified metric to observe a good correlation between mitochondrial BCL-2 dependence and cellular sensitivity.
Figure 3. ABT-199 functions selectively on BCL-2 dependent mitochondria in AML cell lines.

A). The IC50 values of AML cell lines treated with ABT-737 from Figure 1A were correlated with the mitochondrial response of ABT-737 (1μM). Mitochondrial response was measured by JC1 based BH3 profiling. B). IC50 values of cell lines treated with ABT-199 from Figure 1A were correlated with the mitochondrial response of mitochondrial ABT-199 (0.1μM)C). IC50 values of AML cells treated with ABT-737 were correlated with the response to the BAD BH3 (80μM). The mitochondrial responses to the BAD BH3 peptide were measured by JC1 based BH3 profiling. D). IC50 values of AML cell treated with ABT-199 from Figure 1A were correlated with the mitochondrial response of the BAD BH3 (80uM) peptide. E). IC50 values of AML cells treated with ABT-737 were correlated with the response to the BAD BH3 (80μM) – HRK BH3 (80μM). The mitochondrial responses to the BAD and HRK BH3 peptides were measured by JC1 based BH3 profiling. F). IC50 values of AML cells treated with ABT-199 from Figure 1A were correlated with the mitochondrial response of the BAD BH3 (80uM) – HRK BH3 (80uM) peptide. Statistical correlation was performed using a one-tailed Spearman r using GraphPad Prism 6.
ABT-199 Efficiently Kills Primary AML Myeloblasts
We observed ABT-199 selectively kills BCL-2 dependent cell lines; therefore, we next wanted to test the sensitivity of primary patient AML samples to treatment with ABT-199. AML myeloblasts from patient bone marrow or peripheral blood were exposed to ABT-199 for 48 h in minimum essential medium alpha supplemented with cytokines. It is notable that the primary cells were quite sensitive, with median IC50 values less than 10 nM (Figure 4A). Note that this is significantly lower than the IC50 observed in the AML cell lines (Figure 1A). Note also that this is comparable to the sensitivity observed for ex vivo exposure of CLL cells, a disease in which ABT-199 has shown clinical activity in most patients treated (25).
Figure 4. ABT-199 efficiently kills primary AML myeloblasts as a single agent.

A). IC50 determination for ABT-199 and ABT-737 treatment of primary AML samples. Fresh mononuclear cells from AML patients were isolated from bone marrow or peripheral blood and treated with ABT-199 and ABT-737 for 48 h. The IC50 values were calculated based on viable (i.e., Annexin V−/PI−) cell numbers determined by FACS analysis. Samples with ABT-199 IC50 < 0.1 μM were defined as “sensitive”, while those with ABT-199 IC50 > 1 μM were defined as “resistant”. B). Frozen primary AML myeloblasts were thawed treated with ABT-199 and ABT-263 for 8 h in the absence of fetal bovine serum. Viability was assessed by Annexin-/PI- via FACS analysis and IC50 values were calculated using GraphPad Prism software. C). Thawed primary AML samples were treated for 2 h with 1–1000 nm of ABT-199 and viability was assessed by Annexin V-PI- by FACS analysis D). Nonparametric Spearman correlation analysis shows a significant (p = 0.017) negative correlation between ABT-199 IC50 values and BCL-2 protein levels. E) A non-significant (p = 0.069) positive correlation was observed between ABT-199 IC50 values and BCL-XL protein levels. F). Boxplots represent the quartiles and range of log2 values of mRNA expression for BCL-2 genes in different subgroups of AML and normal bone marrows. The median is indicated by the black line in each box. Numbers on top indicate number of patients in each specified subgroup. Differences in gene expression with P values ≤ 0.005 were considered statistically significant, as denoted by *. G). Patient AML samples treated with 100 nM ABT-199 for 24 hours were subjected to FACS analysis of specific apoptosis based on Annexin V staining in the bulk AML myleoblast and CD34+/CD38−/CD123+ LSC-containing population. P value determined via paired t-test.
Because prolonged ex vivo culture by itself can promote sensitivity to apoptosis of AML myeloblasts, we wanted to test whether we would see sensitivity at shorter time points as well. Another potential confounding element is that the ABT class of compounds tend to be highly bound by serum proteins (26). We found that culturing AML cell lines or primary cells in the absence of serum for 8 h did not alter the mitochondrial priming or BCL-2 dependence, compared to culture in the presence of serum (Supplemental Figure S4). Therefore, we tested sensitivity of AML myeloblasts to ABT-199 at 8 h in the absence of serum (Figure 4B). Clinical and genetic data for both sets of AML samples is available in Supplemental Table 1. Again, AML myeloblasts proved to be sensitive to ABT-199 with a median IC50 of 20 nm. Indeed, when we reduced exposure times further, to 2 hours, we could still see induction of apoptosis by ABT-199 in AML myeloblasts (Figure 4C). Similarly rapid induction of cell death has been observed for the clinically sensitive CLL, consistent with a direct action of ABT-199 on AML myeloblast mitochondria, promoting apoptosis in the absence of a requirement for additional cell signaling extrinsic to the mitochondria.
Upon testing of additional cryopreserved AML patient samples, including AML cells with diploid cytogenetics and mutations in FLT3, NRAS, and NPM1 genes, 20 out of 25 (80%) were sensitive to ABT-199 (100 nM), while 5 samples were resistant to both ABT-199 and ABT-737 (Table S2). However, samples from patients with complex cytogenetics and JAK2 mutation (n = 9) were largely insensitive to ABT-199 (1 of the 9, or 11.1% response rate, p = 0.0005 by two-tailed Fisher exact test). Further we found no correlation between ABT-199 sensitivity and FAB classification (Supplemental Figure S5A) or NPM1 (Supplemental Figure S5B) or FLT-3 mutational status (Supplemental Figure S5C). There was no difference in ABT-199 sensitivity between samples sensitive or resistant to conventional induction chemotherapy (Supplemental Figure S5D), consistent with prior findings with ABT-737 (16).
We next tested whether sensitivity to ABT-199 correlated with protein expression for primary AML myeloblasts. As we found with AML cell lines, sensitivity to ABT-199 correlated directly with BCL-2 expression and inversely with BCL-XL expression as measured by quantitative Western blot (Figure 4D-E). No significant relationship was observed between MCL-1 level and sensitivity to ABT-199 in primary AML myeloblasts (Supplemental Figure S6). AML myeloblasts also demonstrate higher BCL-2 mRNA expression than normal bone marrow (Figure 4F). Higher expression of mRNA for MCL-1, BCL-XL, and BIM in AML myeloblasts was not observed (Supplemental Figure S7).
ABT-199 Induces Apoptosis in AML Stem/Progenitor Cells (LSPCs)
We next tested whether ABT-199 is capable of inducing cell death not only in AML blasts, but also in the phenotypically defined AML stem/progenitor cells characterized by CD34+CD38− CD123+ immunophenotype (27). Samples from six ABT-199-sensitive AML patients with high blast counts were incubated with ABT-199 or ABT-737 for 24 h, and apoptosis induction was determined by Annexin V flow cytometry in electronically gated AML blasts (CD45dimSSClow) and AML stem/progenitor cells (CD45dimSSClow CD34+CD38− CD123+). ABT-199 (Figure 4G) and ABT-737 (Supplemental Figure S8) induced apoptotic cell death in both bulk AML blasts and AML stem/progenitor cells.
BH3 Profiling Predicts AML Myeloblast Killing by ABT-199
We next tested whether killing of primary AML myeloblasts by ABT-199 acted as a true BH3 mimetic in an on-target fashion on BCL-2 dependent mitochondria. If this is the case, we would expect that mitochondria sensitive to the BAD BH3 peptide should also be sensitive to the ABT-199 peptide. Indeed, we found an extremely tight correlation between mitochondrial sensitivity to BAD BH3 and ABT-199 across 30 independent patient samples (Figure 5A). Supporting the on-target effect of this class of drugs, a similar correlation was found for ABT-263 (Supplemental Figure S9). No such correlation was observed for the comparison of the BCL-XL selective peptide HRK BH3 and the IC50 of ABT-199, supporting BCL-2 selective action of ABT-199 (Figure 5B). We observed a weak anti-correlation between cellular sensitivity to ABT-199 and sensitivity to the MCL-1 selective peptide NOXA BH3 (Figure 5C). This suggests that there is a minor tendency for MCL-1 dependent mitochondria to be less sensitive to ABT-199.
Figure 5. BH3 profiling predicts AML myeloblast killing by ABT-199.

A). Intracellular BH3 (iBH3) profiling was performed on thawed primary AML cells using the BAD BH3 (80 μM) and ABT-199 (1μM). The mitochondrial sensitivity to BAD BH3 and ABT-199 were positively correlated. B). There is no correlation between the IC50 of primary AML samples from Figure 4B with the BCL-XL specific BH3 peptide HRK (80 μM). C). The IC50 of primary AML samples from Figure 4B were correlated with the NOXA (80μM), a MCL-1 specific NOXA BH3 peptide. D). The ABT-199 IC50 of primary AML samples from Figure 4B were correlated with the BAD BH3 peptide (80uM). E). The ABT-199 IC50 from Figure 4B was correlated with the ABT-199 mitochondrial response (1μM). All correlations were tested using a one-tailed Spearman r correlation using GraphPad Prism software.
In other diseases, BH3 profiling has proven a useful tool for predicting the cytotoxic effect of BH3 mimetic small molecules (28, 29). Here we tested whether BH3 profiling using the BAD BH3 peptide predicted cytotoxicity from ABT-199, and found that the correlation was very good (Figure 5D). In addition, the mitochondrial effect of ABT-199 correlated well with the cytotoxic effect (Figure 5E), again supporting a direct mitochondrial effect of ABT-199, consistent with a mechanism of action of direct competition for the BH3 binding site of BCL-2 on mitochondria.
BH3 Profiling Predicts Response to ABT-199 in an AML Xenograft Model
Tumor xenograft models established by inoculation of cancer cell lines into immunodeficient mice have been used widely for testing novel therapies. However, cultured tumor cells can undergo changes in their gene expression patterns after prolonged passage in in vitro culture. Therefore, the preclinical results obtained from patient-tumor derived xenograft (PDX) models may offer superior modeling of the human disease, especially for testing target-oriented therapies. We have shown that ABT-199 was very effective in a murine AML cell line xenograft model (Figure 1E). As a more clinically relevant test of ABT-199’s anti-leukemic efficacy in vivo, NSG mice were injected with primary AML cells from two different patients (R and S) and monitored for leukemia engraftment by measurements of human CD45+ cells in peripheral blood. After confirmation of AML engraftment, the mice were randomly divided into vehicle and treatment groups. Treated mice received ABT-199 for 2 weeks, after which all the mice were sacrificed, and bone marrows were examined for AML tumor burden by human CD45 flow cytometry. FACS analysis showed that 2-wks of ABT-199 treatment significantly reduced leukemia burden in murine bone marrows in mice injected with cells from patient S (mean, 70 ± 16% human CD45+ cells in bone marrow of control mice (n = 9) and 32.7 ± 12% in ABT-199 treated mice (n = 11, p = 0.0004, Figure 6A). We did not observe a decrease in tumor burden in mice injected with cells from patient R (mean 70.3 ± 8.1% human CD45+ cells in bone marrow of control mice (n = 8) and 74.3 ± 6.4% in ABT-199 treated mice (n = 8, p = 0.1930, Figure 6B).
Figure 6. BH3 profiling predicts AML progression in a primary AML xenograft model.
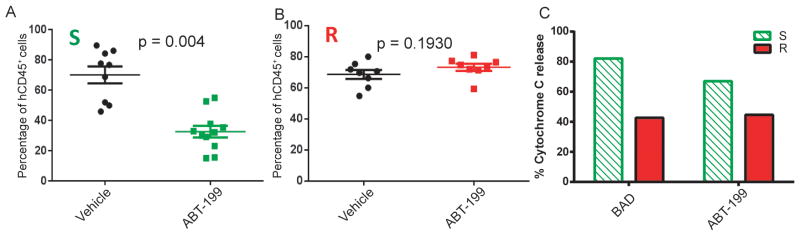
A–B). NSG mice were injected with primary AML cells as described under Methods. Mice were treated with ABT-199 100 mg/kg oral daily dose starting 3 weeks after AML cell injection, for two weeks. The graph represents % of human CD45+ leukemic cells in the murine bone marrow in mice sacrificed upon completion of the therapy. A non-parametric, unpaired, two-tailed t-test was used to evaluate the significance of mean difference. C). Intracellular BH3 profiling using the BAD BH3 (80 μM) and ABT-199 (10 μM) was performed on pre-treatment patient samples.
Since we observed a difference in response in the xenograft model following ABT-199 treatment, we asked whether the response to ABT-199 could be predicted by BH3 profiling. In blinded fashion, pre-treatment AML myeloblasts from each model were subjected to BH3 profiling in which mitochondria were exposed to the BAD BH3 peptide as well as ABT-199 itself. We found that mitochondria from AML myeloblasts from patient S released more cytochrome c following incubation with the BAD peptide or ABT-199 compared to patient R (Figure 6C). These results provide evidence that ABT-199 kills AML myeloblasts by the expected mechanism of inhibition of mitochondrial BCL-2. Furthermore, these results suggest that BH3 profiling might predict the response of AML primary cells to ABT-199 in vivo.
Discussion
Relapsed AML is a difficult cancer to treat effectively; therefore, there is need for improved treatment options for refractory AML. Here, we show that selective inhibition of BCL-2 by ABT-199 kills AML cell lines and primary patient cells both ex vivo and in in vivo mouse xenografts as a single agent in the low nano-molar range. The concentrations used in our studies here are in the 0.001–1 μg/ml range, a range readily achievable in clinical trials where serum concentrations of 3–4 μg/ml have been observed (25). Moreover, the drug acts very quickly in vitro, killing cells within 2 hours of drug exposure. We also show that as in CLL, ABT-199 functions on-target at the mitochondria. This is consistent with the observation that AML myeloblasts from chemorefractory patients showed no difference in their BCL-2 dependence, as measured by BH3 profiling, or sensitivity to ABT-199 compared to chemosensitive cells (16).
Our in vitro results suggest there will be heterogeneity in clinical response (IC50s ranged from 0.43 to >1000 nm), so that a predictive biomarker would be of great utility. Here we present four methods that may be predictive of clinical response to ABT-199. The first method is cytogenetics. The cellular death response to ABT-199 appears to be largely independent of cytogenetic and genetic mutation status, except perhaps for complex karyotype and JAK2 mutant patients, suggesting that treatment with ABT-199 could be useful for patients who have poor prognostic factors. The utility of cytogenetics as a more general predictive biomarker for response to ABT-199 needs to be examined across many more samples.
A second method is ex vivo short term culture of the primary patient samples with ABT-199. The disadvantage of this method is that it is difficult to reliably culture primary AML cells for the requisite time frame to observe cell death. We observed that even after a short 8 h culture there could be upwards of 60% spontaneous apoptotic death in the control un-treated primary AML cells. Therefore, it would not be ideal to rely on an in vitro cell death assay where many samples could be potentially lost due to spontaneous cell death during culture.
The third predictive biomarker method is to measure BCL-2 levels by Western blot. We show that increased expression of BCL-2 is associated with increased sensitivity to ABT-199. However, given the complex interactions of the BCL-2 family members, individual measurements of the various anti-apoptotics alone may not provide accurate data on the in vivo biology of the anti-apoptotic dependencies in AML. Many of the BCL-2 family members are regulated by post-translational modifications and interactions with other proteins. These types of interactions are difficult to capture in static Western blot measurements.
The fourth method, BH3 profiling, may prove useful as a predictive biomarker. BH3 profiling is a functional assay which accounts for the relative amounts and interactions of all of the BCL-2 family members. We show here that the mitochondrial response to the BAD peptide as well as mitochondrial ABT-199 correlates with the ex vivo drug treatment. Most significantly, BH3 profiling could discriminate in vivo sensitivity of human AML cells to ABT-199 (Figure 6). Thus, we may be able to use BH3 profiling of pre-treatment AML samples to direct ABT-199 treatment to AML cases that are most BCL-2 dependent. While the assay is less familiar to many, it is a straightforward protocol using reagents and equipment available in most clinical and research laboratories. Moreover, results are available the same day the sample is acquired. As for the other putative biomarkers, empiric testing in the clinical setting is the only way to truly validate BH3 profiling as a useful predictive biomarker.
Although we focused largely on the expression of BCL-2 and BCL-XL in AML, we also detected the expression of MCL-1. It has been recently reported that the anti-apoptotic MCL-1 is necessary for the development and sustained growth of AML (30). Since ABT-199 does not inhibit MCL-1, increased expression of MCL-1 could be a potential source of upfront resistance to BCL-2 inhibition by ABT-199. However, we show that the majority of AML patient samples tested did not show MCL-1 dependence (as indicated by the NOXA response). This suggests that although MCL-1 may be necessary for the development of AML, most cases of AML may not depend on MCL-1 for survival as much as on BCL-2. Indeed, in a pertinent direct comparison, we have found that most AML myeloblasts are more BCL-2 dependent and less MCL-1 dependent than HSC, though there are about 20% exceptions (16). It may well be that dependence on individual anti-apoptotic proteins varies with myeloid differentiation state.
BCL-2 was discovered in lymphoid cancer cells, and much of the research on this protein has been conducted in lymphoid cells, where it is highly expressed (31). It is therefore understandable that clinical testing of ABT-199 has so far focused on lymphomas and CLL. Here we demonstrate that selective, on-target BCL-2 inhibition using a clinically active drug is a promising avenue for clinical investigation in the myeloid malignancy AML. It is particularly important to recognize that even AML myeloblasts that are resistant to conventional therapies appear to be quite sensitive to BCL-2 inhibition. Thus, BCL-2 inhibition by ABT-199 offers hope to those AML cases that most need novel therapeutic intervention. Our results strongly support the testing of ABT-199 for treatment of AML patients as the majority of patient samples were sensitive to the drug in ex vivo culture. Furthermore, our results support the testing of BH3-profiling as a predictive biomarker for ABT-199 response in the clinic.
Methods
Cell lines
The AML cell lines were purchased from the American Type Culture Collection (Manassas, VA) or Deutsche Sammlung von Mikroorganismen und Zellkulturen (Braunschweig, Germany) or were kindly provided by Dr. James Griffin (Dana-Farber Cancer Institute, Boston, MA). HL-60, MOLM-13, OCI-AML2, OCI-AML3, KG-1, U937 and Kasumi-1 were validated in September 2010 by short tandem repeat DNA fingerprinting using the AmpFlSTR Identifiler kit according to manufacturer’s instructions (Applied Biosystems). HL-60, MOLM-13, OCI-AML2, OCI-AML3, KG-1, U937 and Kasumi-1 were validated in September 2010 by short tandem repeat DNA fingerprinting using the AmpFlSTR Identifiler kit according to manufacturer’s instructions (Applied Biosystems). HL-60 cell lines with stable overexpression of BCL-2 or BCL-XL, and the control cell line with empty vector, were kindly provided by Dr. Kapil N. Bhalla (The Methodist Hospital Research Institute, Houston, TX). AML cell lines were cultured in RPMI 1640 medium supplemented with 10% or 20% fetal bovine serum, 10mM L-glutamine, 100 U/ml penicillin and 10 mg/ml streptomycin. Cells were kept at 37°C in a humidified atmosphere of 5% CO2.
Treatment of AML Cell Lines with ABT-737 and ABT-199
AML cells were incubated for appropriate time in RPMI 1640- medium supplemented with 10% FBS and titrated concentrations of ABT-199 or ABT-737. Viable AML cells were enumerated by flow cytometry using counting beads with concurrent Annexin-V and propidium iodide (PI) staining. IC50 values were calculated using Calcusyn software (Biosoft, Cambridge, MA) based on the number of live cells (Annexin-V−/PI−). For specific details, see Supplemental Methods.
Quantitative Western Blot
Cell lysates were prepared and immunoblotted as previously described (22). Antibodies used for quantitative Western blot were: BCL-2 (#M0887, Dako, Carpinteria, CA), BCL-XL (# 2764, Cell Signaling Technology, Danvers, MA), MCL-1 (#559027, BD Biosciences, San Diego, CA), α-Tubulin and β-Actin (loading controls, #T6199 and #A5441, Sigma-Aldrich, St. Louis, MO). Blots were scanned with Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE). The band intensity was quantified using Odyssey software v2.0. The ratio of band intensity of BCL-2 proteins relative to that of loading control was normalized to the ratio in untreated OCI-AML3 cells.
Gene Knockdown by shRNA
MCL1 was knocked down by lentiviral transduction using a MCL1-specific shRNA transfer vector targeting residues 2421-2440 on RefSeq NM_021960.4. Lentivirus was prepared by co-transfection of HEK293T cells (ATCC) with an equal molar mix of transfer vector and packaging plasmid (psPAX2 and pMD2.G, Addgene, Cambridge, MA) using JetPrime transfection reagent as directed by the manufacturer (Polyplus, Illkirch, France). Fresh lentiviral supernatants were passed through 0.45 μm surfactant-free cellulose acetate membranes; polybrene was added to 8 μg/mL, and the virus stock was used at once to spinoculate OCI-AML3 cells as described before (23). Infected cells were selected with 0.5 μg/mL puromycin. In parallel, control cells were transduced using lentivirus delivering a hairpin targeting GFP in pLKO.1 (Addgene). Knockdown was verified by Western blot analysis.
Selection of Resistant MOLM-13 Cells and Measurements of BCL-2 Protein by Intracellular Flow Cytometry
Resistant MOLM-13 cells were selected in RPMI 1640 medium supplemented with 10% FBS and 50 nM of ABT-199. Every two days, the cells were pelleted by centrifugation and resuspended in fresh medium with 50 nM ABT-199. Cell viability was monitored by Vi-CELL viability analyzer (Beckman Coulter, Irving, TX) until MOLM-13 cells reached a viability higher than 90%. The cellular content of BCL-2 protein was measured in both parental and resistant MOLM-13 cells by flow cytometry. Briefly, 1 million highly viable cells were washed twice with PBS and fixed in 1 mL of 4% paraformaldehyde for 15 minutes on ice, followed by washing with PBS and permeabilization with 1 mL of 0.1% Triton X-100 in PBS-buffered bovine serum albumin (BSA, 1% w/v). After incubation on ice for 10 minutes, cells were then washed with washing buffer (1% BSA in 1 x PBS), resuspended in 90 μL of washing buffer and stained with 10 μL of FITC-conjugated BCL-2 antibody or FITC-conjugated IgG1 isotype control (#F7053 and #X0927, DAKO, Carpinteria, CA). After incubation in dark at room temperature for 30 minutes, the cells were washed again with washing buffer and analyzed by flow cytometry using Gallios™ flow cytometer (Beckman Coulter). The intensity of BCL-2–associated fluorescence was measured on a logarithmic scale. For each sample, 20 000 cells were analyzed for Median Fluorescence Intensity (MFI) using Kaluza flow analysis software (Beckman Coulter).
BH3 Profiling of Cell Lines
AML cell lines were seeded at a density of 4 x 105 cells/mL in 10% FBS RPMI media supplemented with 10 mM L-glutamine, 100 U/ml penicillin and 10 mg/ml streptomycin 24 h before BH3 profiling. Two million cells of each cell line were pelleted at 400 xg for 5 minutes at RT and resuspended in 2 mL DTEB (135 mM Trehalose, 10 mM HEPES-KOH, 0.1% w/v BSA, 20 uM EDTA, 20 uM EGTA, 50 mM KCl, 5 mM succinate, final pH 7.4). Cell lines were profiled by using the plate-based JC-1 BH3 profiling assay previously described (16). Cells were permeabilized with digitonin, exposed to BH3 peptides, and mitochondrial transmembrane potential loss was monitoredusing the ratiometric dye JC-1.
Isolation and Treatment of Primary AML Cells with ABT-263, ABT-737 and ABT-199
Primary AML cells were obtained by informed consent from the Dana-Farber Cancer Institute, Leukemia Group, the Pasquarello Tissue Bank at the Dana-Farber Cancer Institute, the University of Texas MD Anderson Cancer Center, Leukemia Tissue Bank Shared Resource from the Ohio State University Comprehensive Cancer Center and the Germany-Austrian Study Group according to protocols approved by the Institute’s Institutional Review Board. Samples were Ficoll purified, used immediately or viably frozen in 90% FBS/10%DMSO.
Fresh (Figures 4A, 4D and 4E) or thawed (Figures 4B, 5) mononuclear cells were resuspended in culture medium supplemented with cytokines. Cells were treated with ABT-199, ABT-263 or ABT-737 for appropriate time. Cells were then washed with PBS and resuspended in Annexin binding buffer. Cell viability was assessed by FACS analysis following concurrent Annexin V and PI (or 7-AAD) staining. For more detailed methods, see Supplemental Methods.
Apoptosis of AML Stem/Progenitor Cells
AML mononuclear cells were isolated by Ficoll density centrifugation and cultured with 100 nM ABT-199 or ABT-737 as described above. After 24 h, AML cells were washed twice in Annexin binding buffer (ABB) and resuspended in 100 μL ABB containing 1:100 dilution of Annexin-V-APC (#550475), 1:50 dilution of CD45-APC-Cy7 (#557833), CD34-FITC (#555821), CD38-PE-Cy7 (#335790) and CD123-PercP-Cy5.5 (#58714) (all from BD Biosciences) for 20 minutes at room temperature in dark. Following staining, cells were washed with ABB and resuspended in 95 μL ABB containing 5 μL DAPI. Cells were analyzed by Gallios Flow Cytometer (Beckman Coulter). Results were expressed as percentage of specific apoptosis calculated as: .
In vivo Study of ABT-199 Efficacy in AML Mouse Models
All animal studies were conducted in accordance with the guidelines approved by the Institutional Animal Care and Use Committees at the University of Texas MD Anderson Cancer Center. Twenty female NOD SCID gamma (NSG) mice (6-wk old, Jackson Laboratory, Bar Harbor, MA) were intravenously injected with luciferase-labeled MOLM-13 cells (0.7 x 106 cells/100 μL) and randomly divided into two groups. Four days post injection, the mice were treated with vehicle or ABT-199 (100 mg/kg body weight) daily by oral gavage for 2 weeks. For oral dosing, ABT-199 (10 mg/mL) was formulated in 60% phosal 50 propylene glycol, 30% polyethyleneglycol-400, and 10% ethanol. Bioluminescence imaging (BLI) was used to monitor tumor burden on different time points. Briefly, mice were anaesthetized and injected intraperitoneally with firefly luciferase substrate D-luciferin and then imaged noninvasively using IVIS-200 in vivo imaging system (PerkinElmer, Waltham, MA). Three mice from each group were sacrificed by CO2 asphyxiation after 15 d. Bone marrow, spleen, and liver were collected for H&E and immunohistochemical staining. The remaining seven mice in each group were followed for survival.
For primary AML derived xenograft models, NSG mice were sub-lethally irradiated (250 cGy) the day prior to intravenous injection of 105 PDX21 patient-derived AML cells. Three weeks following injection and after confirmation of AML engraftment, the mice were randomly divided into two groups and treated with 100 mg/kg ABT-199 or vehicle via gavage daily for 2 weeks. All mice were then sacrificed and femur bone marrows were analyzed for leukemia burden by CD45 flow cytometry (using anti-human CD45-PE antibody #555483, BD Biosciences, San Jose, CA).
Immunohistochemistry Analysis
Immunohistochemistry was performed as described previously (24). Briefly, the tissue was formalin-fixed, paraffin-embedded, sectioned into 5-μm thickness and mounted onto microscope slides. Tissue sections were then deparaffinized and rehydrated using xylene and ethanol in decreasing concentration. Samples were stained with hematoxylin and eosin (H&E) for histopathological evaluation. For immunohistochemical staining, the tissue sections were incubated with primary antibody against human CD45 (#555480, BD Biosciences, San Jose, CA ), followed by sequential incubation with biotinylated secondary antibody, peroxidase labeled streptavidin and 3,3′ diaminobenzidine tetrahydrochloride/H2O2 (Dako), which resulted in a brown precipitate at the antigen site. Images were taken using an optical microscope under the same magnification.
Microarray-Based Gene Expression Profiling in AML
The expression of BCL-2 family genes was determined using oligonucleotide microarrays (HG-U133 Plus 2.0, Affymetrix) in 288 AML samples comprising all cytogenetic groups, and in 103 normal samples (healthy BM and non-leukemia conditions) as described in Haferlach et al (21). All samples in this study were obtained from untreated patients at the time of diagnosis. Cells used for microarray analysis were collected from the purified fraction of mononuclear cells after Ficoll density centrifugation. The study design adhered to the tenets of the Declaration of Helsinki and was approved by the ethics committees of the participating institutions before its initiation. The analysis is conducted at logarithm-2 transformed gene expression intensities. Correlation analysis based on Pearson correlation coefficient and Spearman’s rank correlation coefficient was performed to identify probe sets that have consistent expression pattern corresponding to a common gene. Two-sample t-test was performed for each two-group comparison, and the P value threshold of 0.005 was used to moderately control for multiple testing.
iBH3 of Primary AML Patient Cells
Thawed cells were washed 1x with PBS and stained with 1:100 Invitrogen Live/Dead – near IR stain (#10119, Life Technologies, Grand Island, NY) in FACS Buffer (2% FBS PBS, 1:100) for 20 min on ice, washed with FACS buffer and subsequently stained with CD45-V450 (#642275; BD Biosciences; San Jose, CA) 1:100 FACS buffer on ice for 20 min. Cells were pelleted at 400 x g for 5 min at RT and resuspended in DTEB. 100 uL of cells in DTEB was added to each tube containing twice the final concentration of each peptide treatment in 100 uL of DTEB with 0.002% w/v digitonin. Mitochondria in the permeabilized cells were exposed to peptides for 60 minutes at RT before the addition of 200 uL 4% v/v formaldehyde at RT for 15 minutes, quenched with 50 uL of 100 mM Tris / 2.5 M glycine pH 8.2 for 5 minutes at RT, and pelleted at 1500 xg for 5 minutes at RT. Cells were stained with anti-cytochrome c-Alexa488 (#560263, BD Bioscience) 1:100 in 0.1% Saponin/1%BSA/PBS overnight at 4°C and diluted 1:5 in PBS an hour before FACS on a LSR Fortessa flow cytometer (BD Bioscience) to quantify cytochrome c loss calculated from the median fluorescence intensity (MFI) as
AML blasts were identified by low-mid CD45/low SSC-A.
Statistical analysis
Statistical analyses were performed using GraphPad Prism software v6.0 (GraphPad, La Jolla, CA). Unless otherwise indicated, the results are expressed as the mean ± standard error of the mean (SEM) from at least three independent experiments. Differences with P values ≤ 0.05 were considered statistically significant.
Supplementary Material
Significance.
While targeting BCL-2 has largely been investigated in lymphoid cancers, we present preclinical results of targeting BCL-2 in AML. These results support clinical testing of the small molecule BCL-2 antagonist ABT-199 in AML, accompanied by testing of predictive biomarkers used in this study.
Acknowledgments
Financial Information
This work was supported by funds from the Leukemia and Lymphoma Society Translational Research Award 6387-13 (AL), LS2012-36558 and AML-P01 5 P01 CA055164-19 (MK and MA)
Footnotes
Conflict of Interest
A. Letai is an advisor for AbbVie and M. Konopleva has a sponsored research agreement from AbbVie. Dr. Andreeff serves on the Scientific Advisory Board of Eutropics Pharmaceuticals which once had a license for BH3 profiling.
References
- 1.Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuwana T, Newmeyer DD. Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr Opin Cell Biol. 2003;15:691–99. doi: 10.1016/j.ceb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 3.Letai A, Bassik C, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–92. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 4.Jabbour AM, Heraud JE, Daunt CP, Kaufmann TT, Sandow J, O’Reilly LA, et al. Puma indirectly activates Bax to cause apoptosis in the absence of Bid or Bim. Cell Death Differ. 2009;16:555–63. doi: 10.1038/cdd.2008.179. [DOI] [PubMed] [Google Scholar]
- 5.Ren D, Tu HC, Kim H, Wang GX, Bean GR, Takeuchi O, et al. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science. 2010;330:1390–93. doi: 10.1126/science.1190217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Certo M, Del Gaizo Moore V, Nishino N, Wei G, Korsmeyer S, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–65. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 7.Cheng EH, Wei MC, Weile, Flavell RA, Mak TTW, Lindsten T, et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8:705–711. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- 8.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen, Jin S, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–28. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 9.Gandhi L, Camidge DR, Ribeiro de Oliveira M, Bonomi P, Gandara G, Khaira D, et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol. 2011;29:909–16. doi: 10.1200/JCO.2010.31.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol. 2012;30:488–96. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schoenwaelder SM, Jarman KE, Gardiner EE, Hua M, Qiao J, White MJ, et al. Bcl-xL-inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood. 2011;118:1663–74. doi: 10.1182/blood-2011-04-347849. [DOI] [PubMed] [Google Scholar]
- 12.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–8. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 13.Vandenberg CJ, Cory S. ABT-199 a new Bcl-2-specific BH3 mimetic, has in vivo efficacy against aggressive Myc-driven mouse lymphomas without provoking thrombocytopenia. Blood. 2013;121:2285–8. doi: 10.1182/blood-2013-01-475855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vaillant F, Merino D, Lee L, Breslin K, Pal B, Ritchie ME, et al. Targeting BCL-2 with the BH3 Mimetic ABT-199 in Estrogen Receptor-Positive Breast Cancer. Cancer Cell. 2013;24:120–9. doi: 10.1016/j.ccr.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 15.Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–88. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 16.Vo TT, Ryan J, Carrasco R, Neuberg D, Rossi DJ, Stone RM, et al. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell. 2012;151:344–55. doi: 10.1016/j.cell.2012.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno JS, Akashi K, et al. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science. 2005;307:1101–4. doi: 10.1126/science.1106114. [DOI] [PubMed] [Google Scholar]
- 18.Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell. 2007;12:171–85. doi: 10.1016/j.ccr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 19.Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore VeG, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2001;334:1129–33. doi: 10.1126/science.1206727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brunelle JK, Ryan J, Yecies D, Opferman JT, Letai A. MCL-1-dependent leukemia cells are more sensitive to chemotherapy than BCL-2-dependent counterparts. J Cell Biol. 2009;187:429–42. doi: 10.1083/jcb.200904049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haferlach H, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Béné MC, et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Clin Oncol. 2010;28:2529–37. doi: 10.1200/JCO.2009.23.4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zeng Z, Shi YX, Samudio IJ, Wang RY, Ling X, Frolova O, et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood. 2009;113:6215–24. doi: 10.1182/blood-2008-05-158311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruvolo VR, Karanjeet KB, Schuster TF, Brown R, Deng Y, Hinchcliffe E, et al. Role for PKC δ in Fenretinide-Mediated Apoptosis in Lymphoid Leukemia Cells. J Signal Transduct. 2010;2010:584657. doi: 10.1155/2010/584657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Y, Jacamo R, Shi YX, Wang RY, Battula VL, Konoplev S, et al. Human extramedullary bone marrow in mice: a novel in vivo model of genetically controlled hematopoietic microenvironment. Blood. 2012;119:4971–80. doi: 10.1182/blood-2011-11-389957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seymore JF, Pagel JM, Kahl BS, Wierda WG, Miller TP, Gerecitano JF, et al. Updated results of a phase I first-in-human study of the BCL-2 inhibitor ABT-199 (GDC-0199) in patients with relapsed/refractory (R/R) chronic lymphocytic leukemia (CLL) J Clin Oncol. 2013;31:abstr 7018. [Google Scholar]
- 26.Vogler M, Furdas SD, Jung M, Kuwana T, Dyer MJ, Cohen GM. Diminished sensitivity of chronic lymphocytic leukemia cells to ABT-737 and ABT-263 due to albumin binding in blood. Clin Cancer Res. 2010;16:4217–25. doi: 10.1158/1078-0432.CCR-10-0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jordan CT, Upchurch D, Szilvassy SJ, Guzman ML, Howard DS, Pettigrew AL, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia. 2000;10:1777–84. doi: 10.1038/sj.leu.2401903. [DOI] [PubMed] [Google Scholar]
- 28.Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest. 2007;117:112–21. doi: 10.1172/JCI28281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Del Gaizo Moore V, Schlis KD, Sallan SE, Armstrong SA, Letai A. BCL-2 dependence and ABT-737 sensitivity in acute lymphoblastic leukemia. Blood. 2008;111:2300–9. doi: 10.1182/blood-2007-06-098012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Glaser SP, Lee EF, Trounson E, Bouillet P, Wei A, Fairlie WD, et al. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 2012;26:120–5. doi: 10.1101/gad.182980.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsujimoto Y, Finger LR, Yunis J, Nowell PC, Croce CM. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science. 1984;226:1097–9. doi: 10.1126/science.6093263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.