

Abstract
In bilaterians, which comprise most of extant animals, microRNAs (miRNAs) regulate the majority of messenger RNAs (mRNAs) via base-pairing of a short sequence (the miRNA “seed”) to the target, subsequently promoting translational inhibition and transcript instability. In plants, many miRNAs guide endonucleolytic cleavage of highly complementary targets. Because little is known about miRNA function in nonbilaterian animals, we investigated the repertoire and biological activity of miRNAs in the sea anemone Nematostella vectensis, a representative of Cnidaria, the sister phylum of Bilateria. Our work uncovers scores of novel miRNAs in Nematostella, increasing the total miRNA gene count to 87. Yet only a handful are conserved in corals and hydras, suggesting that microRNA gene turnover in Cnidaria greatly exceeds that of other metazoan groups. We further show that Nematostella miRNAs frequently direct the cleavage of their mRNA targets via nearly perfect complementarity. This mode of action resembles that of small interfering RNAs (siRNAs) and plant miRNAs. It appears to be common in Cnidaria, as several of the miRNA target sites are conserved among distantly related anemone species, and we also detected miRNA-directed cleavage in Hydra. Unlike in bilaterians, Nematostella miRNAs are commonly coexpressed with their target transcripts. In light of these findings, we propose that post-transcriptional regulation by miRNAs functions differently in Cnidaria and Bilateria. The similar, siRNA-like mode of action of miRNAs in Cnidaria and plants suggests that this may be an ancestral state.
MicroRNAs (miRNAs), small RNAs 20–24 nucleotides (nt) long, regulate messenger RNA (mRNA) expression in plants and animals (Ghildiyal and Zamore 2009). In animals, sequential processing of most primary miRNAs by the RNase III enzymes Drosha and Dicer produces miRNA/miRNA* duplexes (Grishok et al. 2001; Hutvagner et al. 2001; Ketting et al. 2001; Knight and Bass 2001; Lee et al. 2003). Subsequently, these duplexes are loaded into a member of the Argonaute (AGO) protein family and the miRNA* strand is ejected, producing an active RNA-induced silencing complex. miRNAs direct AGO proteins to repress expression of partially complementary mRNAs, thereby modulating diverse biological processes such as organogenesis, developmental timing, and cell proliferation (Bartel 2009). In bilaterian animals, miRNAs typically recognize their targets via a short nucleotide sequence, the “seed” (miRNA nucleotides 2–8), triggering translational inhibition and transcript decay by mechanisms such as deadenylation (Bartel 2009; Huntzinger and Izaurralde 2011). In plants, by contrast, most known targets exhibit near perfect complementarity to the miRNA, permitting AGO proteins to cleave the miRNA target (Baumberger and Baulcombe 2005; Axtell et al. 2011). Target cleavage directed by plant miRNAs resembles the action of small interfering RNAs (siRNAs), which are produced by Dicer alone and are found in most eukaryote lineages, suggesting that they represent ancestral small silencing RNAs (Ghildiyal and Zamore 2009).
The biogenesis and action of small silencing RNAs are well studied in Bilateria, the group that includes the majority of extant animals such as vertebrates, arthropods, and nematodes. In contrast, our knowledge about small silencing RNAs in early-branching, nonbilaterian lineages remains limited. Multiple phylogenomic studies position Cnidaria (sea anemones, corals, hydroids, and jellyfish) as the sister group of Bilateria (Hejnol et al. 2009; Philippe et al. 2011). Hence, comparison of cnidarians and bilaterians may shed light on the biology of their common ancestor that lived more than 600 million yr ago. A pioneering study on small RNAs in basally branching animals found that the model sea anemone, Nematostella vectensis, produces miRNAs and a second class of small RNAs, PIWI-interacting RNAs (piRNAs). Intriguingly, only a single miRNA, miR-100, appears to be conserved between bilaterians and cnidarians (Grimson et al. 2008). Here, we identify many new miRNAs in Nematostella and show by high-throughput sequencing of nine developmental stages that the majority of miRNAs are developmentally regulated. Nematostella miRNAs are frequently coexpressed with their targets and reveal extensive complementarity between most miRNAs and their targets. Further, this complementarity commonly leads to slicing of the target mRNA, like siRNAs and plant miRNAs. Our findings suggest that cnidarian miRNAs retain a potentially ancestral, RNAi-like mode of small RNA-mediated post-transcriptional regulation, unlike the vast majority of miRNAs in bilaterians.
Results
Discovery of Nematostella miRNAs and siRNAs
A large proportion (>93% in all libraries and developmental stages) (Table 1) of the sequenced Nematostella small RNAs exhibit sequence signatures typical of piRNAs: 23- and 24-mers displaying an adenosine bias at position 10 and longer reads exhibiting a uridine bias at position 1 (Aravin et al. 2007; Brennecke et al. 2007; Gunawardane et al. 2007). The high abundance of piRNAs in our libraries is in agreement with previous studies of Cnidaria (Grimson et al. 2008; Krishna et al. 2013). In addition to piRNAs, we found two types of small RNAs, which originate from predicted genomic inverted repeats. Small RNAs of the second type appeared to be processed from hairpins <100 nt long and had homogeneous 5′ ends, with one of the two arms of the hairpin typically generating more small RNAs than the other arm. These small RNAs likely correspond to miRNAs (Fig. 1A,B; Supplemental Figs. S1, S2; see below). The third type of small RNAs mapped to inverted repeats as long as 700 nt. These small RNAs were far more heterogeneous. The production of such complex populations of small RNA sequences from a single precursor is a hallmark of endogenous siRNAs (Fig. 1C; Czech et al. 2008; Ghildiyal et al. 2008; Okamura et al. 2008; Kim et al. 2009).
Table 1.
Small RNA abundance in deep-sequencing libraries of Nematostella
Figure 1.
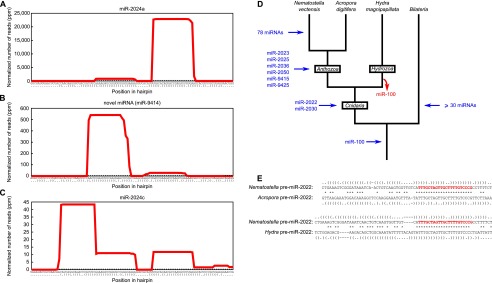
Discovery of novel Nematostella miRNAs. (A,B) Small RNA profiles along a pre-miRNA sequence (here exemplified by pre-miR-2024a and pre-nve-mir-9414). The x-axis indicates the nucleotide position along the hairpin sequence (predicted secondary structure is represented by dots and brackets, with dots indicating unpaired nucleotides and opening and closing brackets representing paired nucleotides). The y-axis indicates the number of reads covering each nucleotide in the pooled 18 deep-sequencing libraries. (C) A small RNA profile along endo-siRNA sequences (same conventions as in panels A and B). Several small RNAs (here exemplified by miR-2024c) were previously described as miRNAs, but the processing of their precursors into multiple small RNAs shows that these are actually siRNAs. (D) Proposed evolutionary scenario for the appearance of miRNA genes in the Nematostella vectensis lineage. The number of urbilaterian miRNAs (>30 in addition to miR-100) was estimated by comparing the known miRNA complement of a slowly evolving protostome (the annelid Capitella teleta) to the known miRNA complement of three deuterostomes (Strongylocentrotus purpuratus, Homo sapiens, and Tetraodon nigroviridis), requiring a perfectly conserved seed and an overall PHYLIP alignment score greater or equal than 0.3 (calculated by ClustalW). (E) Sequence and secondary structure alignment of Cnidarian pre-miR-2022. Mature miRNA sequences are shown in red. Conserved nucleotides are flagged with an asterisk. Secondary structures are represented by dots (for unpaired nucleotides) and brackets (for paired nucleotides). Dashes indicate alignment gaps.
We focused on those small RNAs with homogeneous 5′ ends that mapped to presumptive pre-miRNA structures (58-nt sequences folding into an unbranched hairpin, with a predicted folding free energy ≤ −14 kcal·mol−1). Among these, we identified 128 pre-miRNAs that produce 86 distinct mature miRNAs belonging to 84 seed-sequence families (Supplemental Table S1; Supplemental Fig. S1). In order to estimate the robustness of our miRNA annotations, we evaluated each of the 86 identified candidates by running the annotation program on random subsets of the 18 deep-sequencing libraries. When 10 subsets (each covering 90% of the actual library depth) were reanalyzed, 52 candidates were recovered in all 10 subsets, 65 candidates were recovered in at least half of the subsets, and 75 candidates were recovered in at least one subset. Supplemental Table S2 gives the confidence level (assessed by bootstrap score) for each candidate. Poorly recovered candidates tended to be those least expressed, and it is expected that they would have been missed by a shallower sequencing effort. Reciprocally, additional, yet-to-be-discovered miRNAs have probably escaped identification, but we expect that their expression levels would be very low (i.e., ∼10 reads per million). Our initial miRNA annotation included 28 of the 40 miRNAs that were annotated previously (Grimson et al. 2008). To assess why the remaining 12 had not been recovered, we reviewed each case and found that 11 miRNAs did not qualify as authentic miRNAs, either because their low abundance did not permit unambiguous annotation, or because they did not pass our precursor structure and sequence homogeneity criteria. Instead, they were represented by a complex set of slightly shifted small RNA sequences with heterogeneous 5′ ends, probably originating from imprecise nucleolytic processing (Fig. 1C; Supplemental Fig. S2). These small RNAs resemble both siRNAs and miRNAs derived from short hairpin precursors; we note that this is in contrast to the situation in bilaterians, where siRNAs and miRNAs are well-separated classes (Ghildiyal and Zamore 2009). In this respect, the case of the miR-2024 variants in Nematostella is of special interest: Our analysis suggests that four miR-2024 variants (miR-2024a, e, f, and g) are bona fide miRNAs, whereas the three remaining variants do not present all the features of canonical miRNAs. The overhangs of the miR-2024b/miR-2024b* duplex are unusually long, and miR-2024c is imperfectly processed, while the single-stranded flanks of the pre-miR-2024d hairpin liberate more reads than the miRNA*. The unusual features of pre-miRs 2024b, c, and d and their low abundance relative to other miR-2024 variants suggest that they may be degenerating, providing unprecedented examples of pre-miRNAs transitioning into sources of siRNAs. We found the conserved and experimentally confirmed miR-2022 (Chapman et al. 2010; Krishna et al. 2013; this study) to be a false negative in our initial annotation, because it did not pass one of the many stringent criteria (it folded less stably than another RNA hairpin located in close proximity). After the addition of miR-2022, the final set of 87 miRNAs includes 29 of the 40 previously annotated miRNAs (Grimson et al. 2008).
Of the 87 Nematostella miRNAs, miR-100 remains the only miRNA conserved between cnidarians and bilaterians (Fig. 1D). Thus, the previous observation indicating that the vast majority of miRNAs evolved independently in Cnidaria and Bilateria was not biased by sequencing depth or the samples used (Grimson et al. 2008). Comparing our annotated Nematostella miRNAs to the genome of the reef-building coral Acropora digitifera and to sequenced small RNAs from Hydra magnipapillata (Shinzato et al. 2011; Krishna et al. 2013) enabled us to reconstruct the gain and loss history of miRNAs in Cnidaria (Fig. 1D). Interestingly, only two miRNAs seem to be shared among the three cnidarian groups, compared to 31 ancestral miRNA families in bilaterians (Fig. 1D; Sperling et al. 2009). Notably, not only the mature sequences of these two miRNAs are conserved, but sequence homology can also be found in their hairpin structure (Fig. 1E).
Spatiotemporal regulation of Nematostella miRNAs
Most miRNAs (66 of 87) are barely detectable before the early planula stage (<10 ppm in untreated libraries) (Fig. 2A; Supplemental Table S1), and the expression of most peaks either at the primary polyp or adult stage (Fig. 2A). These temporal profiles indicate that the expression of the majority of Nematostella miRNAs is developmentally regulated. Eighteen miRNAs are male-specific and just one (miR-9459) is female-specific (enrichment in one sex > 10, with Fisher’s exact test P-value < 0.01). Sex-specific miRNAs have also been reported in mammals and flatworms and may exist in fish and birds (Wienholds et al. 2005; Ciaudo et al. 2009; Hao et al. 2010; Luo et al. 2012; Marco et al. 2013).
Figure 2.

Spatiotemporal expression of Nematostella miRNAs. (A) Heat map showing the normalized expression levels of distinct miRNAs derived from read counts in nonoxidized libraries from multiple developmental stages. The studied stages were unfertilized egg (UE), blastula (B; 8 h post-fertilization [hpf]), Gastrula (G; 22 hpf), early planula (EP; 72 hpf), late planula (LP; 120 hpf), metamorphosing planula (MP; 144 hpf), primary polyp (PP; 192–240 hpf), adult male (AM; older than 6 mo), and adult female (AF; older than 6 mo). The mean expression level of each miRNA as represented by the number of reads per million appears to the right of each row. Asterisk designates the mean expression of miR-2024a (2135 reads per million) and double asterisk designates the mean expression of miR-2023 (2249 reads per million). (B) Spatial expression of six miRNAs from panel A in late planulae (LP) and primary polyps (PP) as determined by in situ hybridization.
In situ hybridization experiments suggest that many Nematostella miRNAs have highly specific expression patterns (Fig. 2B) as documented for plant and bilaterian miRNAs (Flynt and Lai 2008; Axtell et al. 2011). For example, miR-100, miR-2022, and miR-2030 are expressed in groups of cells at the oral end; miR-2025 is expressed in the aboral endoderm; miR-2026 expression is confined to one side of the endoderm along the oral-aboral axis, and miR-2023 expression is restricted to cells in the aboral ectoderm. These data support a role for miRNAs in defining various cell or body region identities in Nematostella.
Nematostella miRNAs mediate target cleavage
In plants, miRNAs are 2′-O-methylated at their 3′ ends, whereas most animal miRNAs are not (Yu et al. 2005; Axtell et al. 2011). We assayed whether Nematostella miRNAs are methylated by sequencing oxidized small RNAs, a strategy that excludes unmethylated RNAs from the sequencing library (Ghildiyal et al. 2008). A substantial fraction of miRNAs in Nematostella is at least partially methylated, with similar normalized read counts in the untreated and oxidized libraries (Supplemental Table S1). This result agrees with our recent observation that the RNA methyltransferase Hen1 of Nematostella is expressed throughout the animal (Moran et al. 2013b), unlike in vertebrates where Hen1 expression is restricted to the germline where it methylates piRNAs (Kamminga et al. 2010). To estimate the efficiency of the oxidation reaction, we spiked eight of the libraries (oxidized and untreated libraries of unfertilized eggs, blastula, adult males, and adult females) with methylated and unmethylated RNA oligos. Unmethylated RNA oligos were depleted at >98% in the oxidized libraries, while methylated RNA oligos were detected at similar levels in untreated and oxidized libraries, demonstrating the high efficiency of the treatment (Supplemental Fig. S3; Supplemental Table S1). We classified miRNAs by their relative levels of methylation (see Methods): 43 miRNAs were overmethylated, 26 undermethylated, and 18 undetermined. Interestingly, overmethylated miRNAs were, on average, significantly longer than miRNAs with lower average methylation levels (P-value = 0.018, Wilcoxon test) (Fig. 3), whereas miRNAs whose methylation level could not be reliably determined displayed an intermediate size distribution. This may suggest that the methylation machinery in Nematostella has preference for longer small RNAs such as long miRNAs and piRNAs.
Figure 3.

Length distribution of the dominant mature sequence product for Nematostella miRNAs classified as overmethylated, undermethylated, and of undetermined methylation level. Overmethylated miRNAs were, on average, significantly longer than miRNAs with lower average methylation levels.
Methylation of small RNAs has been proposed to protect small RNAs—such as anti-viral siRNAs in insects—from degradation when they bind to extensively complementary target RNAs (Ameres et al. 2010). The presence of miRNAs with modified 3′ ends in Nematostella raised the possibility that, like siRNAs and plant miRNAs, these miRNAs might regulate highly complementary mRNAs. Indeed, we found the enrichment (fraction observed − fraction expected) of miRNAs with perfect and near-perfect mRNA matches (target prediction score ≤ 2.5; a weighted scoring of the alignment corresponding to a theoretical maximum of five mismatches; see Methods) to be significantly larger in Nematostella than in Drosophila melanogaster (P-value < 2.2 × 10−16, Fisher’s exact test) or in humans (P-value < 2.2 × 10−16) but similar to the enrichment in the plant Arabidopsis thaliana (Fig. 4A). Interestingly, this difference was more pronounced when miRNAs with relative expression levels above the mean of each set were considered; the fraction of high complementarity matches increased in plants and Cnidaria but decreased in human and fly (Supplemental Fig. S4).
Figure 4.

Complementarity of miRNAs to Nematostella transcripts and target cleavage. (A) Distribution of miRNA:mRNA alignment mismatch scores in Nematostella vectensis, Homo sapiens, Drosophila melanogaster, and Arabidopsis thaliana, shown as the difference in abundance between miRNAs and matching shuffled miRNAs, counting the fraction of sequences in each set having a best match with score 2.5 or lower. (B) The tag possession ratio (TPR) of the 75% most expressed (high), the 25% least expressed (low), and shuffled miRNAs as a function of the alignment mismatch score. (C) Fisher’s exact tests comparing the cumulative fraction of predicted cleavage sites supported by degradome reads of the top 75% expressed miRNAs to those with lower expression (bottom 25%), and to shuffled miRNAs, at alignment mismatch score thresholds ranging from 1 to 7. (D) Examples of degradome tag density along predicted miRNA target transcripts. Tags aligning to position 10 of the miRNA are indicated in orange and tags aligning to other positions in the miRNA are in gray. (E) RLM-RACE results for the miR-2022 targets shown in panel D. Cleavage positions are indicated by arrows and ratios of positive clones are shown above.
These results suggested that Nematostella miRNAs might direct cleavage of their mRNA targets just like siRNAs. To test this, we sequenced the degradome of Nematostella at the primary polyp, a stage which showed maximal mean miRNA expression among those sampled. Degradome sequencing captures RNA fragments bearing a 5′ monophosphate, the hallmark of AGO catalyzed target cleavage. Since AGO proteins slice their mRNA targets between the nucleotides paired with positions 10 and 11 of the small RNA guide, degradome reads starting across from position 10 of the miRNA indicate miRNA-mediated cleavage (Addo-Quaye et al. 2008; German et al. 2008; Karginov et al. 2010; Shin et al. 2010). Analysis of the primary polyp degradome identified 64 transcripts with more than one such cleavage supporting read in each of two biological replicates, suggesting a potential role in cleavage for at least 33 miRNAs (Supplemental Table S3). The fraction of predicted cleavage sites supported by the degradome (“tag possession ratio”) (Shin et al. 2010) increased with miRNA:target complementarity. When a few mismatches were tolerated, this was significantly higher for the 75% highest-expressed miRNAs, compared to miRNAs in the lowest quartile and to a random background with the same dinucleotide frequencies (2.53 × 10−117 < P-value < 2.04 × 10−2 for alignment mismatch score upper bounds ≥ 4) (Fig. 4B,C). While the maximum peak of degradome density was found at the cleavage position for some transcripts, other putative targets exhibited more complex degradation profiles (Fig. 4D).
To provide further support for miRNA-guided slicing in Nematostella, we assayed the cleavage of specific putative targets by gene-specific, 5′ RNA ligase-mediated rapid amplification of cDNA ends (RLM-RACE), a gene-specific and highly sensitive approach used previously in plants, green algae, and mammals (Llave et al. 2002; Zhao et al. 2007; Karginov et al. 2010). Among 25 potential miRNA targets assayed, seven could either not be detected or yielded shorter PCR products than expected, an inconclusive finding that might reflect further degradation of cleavage products. Of the 18 conclusive cases, 13 (>70%) supported miRNA-directed target cleavage (Table 2; Supplemental Table S4). Interestingly, we could validate a few cases of cleavage for moderately expressed miRNA targets, like the targets of miR-2022, which were barely detected by the degradome approach (Fig. 4D,E). This suggests that the degradome sequencing missed a portion of cleavage events, possibly due to downstream effects of exonucleases, as was shown in plants and mammals, and due to the use of polyA selection in the degradome sequencing protocol which hinders the detection of sliced products whose poly-A tails have been degraded (German et al. 2008; Shin et al. 2010). In sum, we demonstrated by RLM-RACE the specific cleavage of 13 different target mRNAs by 11 different miRNAs belonging to nine miRNA families. Together, the common occurrence of high complementarity of miRNAs to specific targets and our RLM-RACE and degradome data suggest that target slicing by miRNAs is a common mechanism in Nematostella.
Table 2.
Summary of RLM-RACE results in Nematostella
miRNA target sites and the slicing mechanism are conserved in Cnidaria
If the cleavage of targets by miRNAs in Nematostella is under selective pressure, we would expect some of the miRNA target sites to be conserved in other cnidarian species. Degradome analysis identified NvHoxD (also called Anthox8) as a target of miR-2026. NvHoxD, a homeobox-containing transcription factor localized to one side of the directive axis of Nematostella (Fig. 5A), is thought to be involved in the differentiation along this axis (Finnerty et al. 2004). HoxD has been reported to exist as two closely related paralogs (Chourrout et al. 2006), but our exhaustive cloning of HoxD transcripts as well as sequencing of the corresponding genomic region suggests that Nematostella possesses a single HoxD gene bearing duplicated 3′ exons that produce several mRNA isoforms via alternative splicing (Fig. 5B). Interestingly, the miR-2026 binding site is found in both copies of the exon containing the 3′ UTR of NvHoxD, despite otherwise low sequence conservation of this region, suggesting purifying selection to retain the miR-2026 binding site. RLM-RACE showed that both 3′ UTR copies are cleaved (Fig. 5B,C). Moreover, the miR-2026 binding site in the 3′ UTR of HoxD is conserved in the HoxD ortholog of the sea anemone Metridium senile, which is separated from Nematostella vectensis by about 250–450 million yr (Erwin et al. 2011; Park et al. 2012). This time range is comparable to that of the deepest miRNA target conservations known in plants (Addo-Quaye et al. 2009).
Figure 5.

Regulation of miRNA targets and target conservation. (A) Expression of microRNAs and their targets as determined by double in situ hybridization. (B) A scheme showing the structure of the partially duplicated HoxD gene and the position of the two binding sites of miR-2026. (C) The duplicated 3′ UTRs of HoxD in Nematostella (Nv) are very derived, yet the two binding sites of miR-2026 are highly conserved, and both transcript variants are cleaved. The site is also conserved in the sea anemone Metridium senile (Ms). Similarly, the miR-2025 binding site in the coding sequence of the Six3/6 transcript is conserved in Nematostella, Anemonia viridis (Av), and Anthopleura japonica (Aj). In panels B and C, cleavage positions are indicated by an arrow, and ratios of positive clones appear in brackets. (D) Schematic representation of coherent and incoherent circuits involving a transcription factor (TF) that regulates the expression of a miRNA and its targets and the degree of overlap between the expression domains of a miRNA and its targets that would indicate each of the two topologies. (E) miR-2030 and its host/target gene NVE19315 represent a clear case of incoherent regulatory circuit. The precursor of miR-2030 is located in an intron of NVE19315 as indicated by the green arrow. miR-2030 targets two sites in the NVE19315 transcript (indicated by magenta arrows) as supported by the degradome analysis (Site 1) and RLM-RACE (Site 2; cleavage position is indicated by an arrow, and ratios of positive clones appear in brackets).
Another unexpected finding regarding the targeting of HoxD by miR-2026 is the genomic location of the miRNA gene: In Bilateria, several miRNA genes reside in the Hox cluster upstream of the Hox gene they regulate (Mansfield and McGlinn 2012). This arrangement is thought to reinforce the posterior prevalence mediated by Hox genes (Yekta et al. 2008), a phenomenon that causes posterior phenotypes when both anterior and posterior genes are coexpressed. Nematostella lacks bilaterian-like Hox clusters, but several of its Hox genes are found in the same genomic region (Chourrout et al. 2006). Notably, the miR-2026 gene is located between the HoxC (Anthox7) and HoxD genes. Another unexpected similarity between miRNA-mediated regulation of Hox genes in Nematostella and bilaterians is the slicing of targets: One of the best-studied cases of miRNA-directed transcript slicing in bilaterians is the cleavage of HoxB8 mRNA by the Hox cluster-embedded miR-196 (Yekta et al. 2004). Since miR-196 and miR-2026 share no sequence motif, and because HoxD and HoxB8 are not orthologs, it is very likely that this reflects the convergent evolution of Hox gene regulation by neighboring miRNAs in Bilateria and Cnidaria.
Another example of a miRNA target of the homeobox transcription factor family in Nematostella is NvSix3/6. Six3/6 specifies the anterior-most part of the bilaterian brain and epidermis and has a role as an upstream regulator of the apical domain in the larva of Nematostella (Steinmetz et al. 2010; Sinigaglia et al. 2013). The Nematostella Six3/6 binding site for miR-2025 in the coding sequence (CDS) is conserved in the orthologous transcripts from the anemone species Anemonia viridis and Anthopleura japonica, despite poor overall sequence conservation of the mRNAs (Fig. 5C). The miRNA binding sites in the Six3/6 transcripts of A. viridis and A. japonica and the HoxD transcript of M. senile were the only conserved sites we detected between Nematostella and these three species. However, this might be explained by the limited transcriptome data sets available and the relatively large evolutionary distance between the species studied (≥250 million yr). The conservation of near-perfect binding sites for miRNAs in various species separated for several hundreds of millions of years strongly suggests that they are functional and that miRNA-mediated cleavage is also likely to take place in other cnidarians. To further test this idea, we used the same method employed in Nematostella to predict miRNA cleavage targets in H. magnipapillata, which is separated from Nematostella by more than 600 million yr (Erwin et al. 2011; Park et al. 2012). The data is currently limited; Hydra and Nematostella share only two miRNAs, their targets are not conserved, and the low coverage of the EST data set allowed prediction of only 28 putative targets for Hydra-specific miRNAs at a prediction score threshold of 2.5. Nevertheless, we detected specific cleavage products for two of the five mRNAs tested by RLM-RACE (Supplemental Table S4). This further supports the idea that miRNA-mediated target cleavage is widespread in the phylum Cnidaria.
Coherent and incoherent modes of regulation
miRNAs can be involved in either coherent or incoherent regulation (Shkumatava et al. 2009; Ebert and Sharp 2012). In coherent regulation, a transcription factor (TF) drives expression of a miRNA and concurrently inhibits, directly or indirectly, the expression of the miRNA target. Consequently, the miRNA reinforces the downstream effect of the upstream regulatory system. In contrast, in the incoherent regulation topology, the miRNA and its target are activated by the same regulators, and therefore the miRNA effect counteracts the effect of the upstream system. In coherent regulation, the expression domains of the miRNA and its target are nearly mutually exclusive in space or time, whereas in incoherent regulation they must overlap temporally and spatially (Fig. 5D; Shkumatava et al. 2009).
To reveal the logic by which Nematostella miRNAs regulate their targets, we performed double in situ hybridization. We localized the expression of miR-2022 and its target Nematogalectin-related 2 (NR2), miR-2026 and its target HoxD, miR-2030 and its target NVE19315, and miR-2025 and its targets Six3/6 and NVE8472 (Fig. 5A). NR2 encodes a protein found in the tubule of the nematocyst, the stinging organelle typifying Cnidaria (Hwang et al. 2007). Both NR2 and miR-2022 colocalized to nematocytes, the cells containing nematocysts (Fig. 5A). miR-2026 also colocalized with its target HoxD to one side of the endoderm. miR-2030 was colocalized with its target as well. All these examples are consistent with an incoherent topology. In contrast, miR-2025 is expressed in the endoderm of the aboral pole, whereas both of its targets are expressed in the adjacent, yet distinct, aboral ectoderm (Fig. 5A). This spatial distribution of miR-2025 and its targets indicates a coherent mode of regulation reminiscent of that reported in many cases for miRNAs and their targets in other organisms (Fig. 5D; Shkumatava et al. 2009; Ebert and Sharp 2012) and suggests that miR-2025 might help in setting sharper boundaries between cell layers.
Hence, we find evidence for both coherent and incoherent modes of regulation in Nematostella. miR-2030 and its target is a particularly interesting case of incoherent regulation: The precursor of this miRNA resides in an intron of NVE19315, a gene coding for an unknown protein (Fig. 5E). The transcript of this gene has two sites that are nearly perfectly complementary to miR-2030, and we show by RLM-RACE and degradome sequencing that both of them are active cleavage sites (Table 2; Supplemental Table S4; Fig. 5E). Since miR-2030 shows a similar expression pattern to its host/target gene, and there is no support in the transcriptome for independent transcription of the intronic region, it is clear that the expression of the miRNA and the hosting target gene are tightly linked (Fig. 5A; unpubl.). Thus, miR-2030 is part of a feed-forward incoherent circuit, a rare topology for miRNAs and their targets in bilaterians (Shkumatava et al. 2009). A similar case to miR-2030 and its host/target gene is miR-2028, as its precursor is also embedded in the intron of its own target (Supplemental Fig. S5).
Discussion
By deep sequencing of small RNAs from nine developmental stages of Nematostella, we increased the number of miRNAs to 87. Of these miRNAs, only two are conserved throughout Cnidaria (Fig. 1D). This low number could be due to acquisition of miRNAs after the divergence of these species from a common ancestor that evolved only a few miRNAs to begin with. Alternatively, this could be explained by a high turnover rate of miRNAs in the cnidarian lineage, in contrast to miRNA evolution in bilaterians, where losses of miRNA families are suggested to be rare (Wheeler et al. 2009). Interestingly, despite differences in sequencing depth, small RNA composition, and annotation methods that might affect the number of detected miRNAs, the numbers described here for Nematostella and recently for Hydra (126 miRNAs of 125 families) are comparable to those reported for several bilaterians such as annelids, echinoderms, planarians, and insects (Wheeler et al. 2009; Friedlander et al. 2012; Song et al. 2012). These findings call into question previous suggestions of a direct relation between body plan complexity and the number of miRNAs in an organism (Erwin et al. 2011). The low conservation of miRNAs among cnidarian lineages suggests that the birth and death of miRNA genes are more frequent in Cnidaria. Moreover, the minimal overlap of miRNA inventory between Cnidaria and Bilateria suggests that nearly all contemporary miRNAs appeared after the separation of these two groups.
In recent years, it was shown that Nematostella has gene families, gene structure, and genome architecture surprisingly reminiscent of those of vertebrates and slowly evolving protostomes (Kusserow et al. 2005; Technau et al. 2005; Putnam et al. 2007). Moreover, cis-regulation of transcription is very similar in Nematostella and bilaterians (Schwaiger et al. 2014). These findings are in striking contrast to the vastly different body plans and cell type compositions of Cnidaria and Bilateria.
An attractive hypothesis is that differences in post-transcriptional regulation may explain this apparent contradiction. Our findings suggest that miRNAs play substantially different regulatory roles in these two groups as the miRNA-directed target cleavage (slicing) appears far more common in cnidarians than in bilaterians, where only very few cases have been reported (Shin et al. 2010). For example, a recent comprehensive study employed degradome sequencing in the nematode C. elegans and, strikingly, found only a single case of miRNA-directed slicing (Park et al. 2013). Slicing requires extensive base-pairing between a miRNA and its target and generally has a stronger effect on target levels, but also greatly limits the number of possible targets. Indeed, each plant miRNA is known to have only a handful of targets compared to hundreds of potential targets modulated by a single animal miRNA, as well as several miRNAs targeting the same mRNA (Bartel 2009; Axtell et al. 2011). Moreover, translational inhibition is usually less potent than target slicing (e.g., Mullokandov et al. 2012). Such differences can considerably alter the role of miRNAs in regulatory gene networks. However, the finding of many miRNA-directed slicing events in Nematostella does not mean that bilaterian-like interactions between the miRNA seed and other transcript targets do not occur in parallel. Moreover, extended miRNA-mRNA matches in plants leading to target slicing were recently shown also to promote substantial translational inhibition, and it is possible that the same also occurs in Cnidaria (Brodersen et al. 2008; Yang et al. 2012; Li et al. 2013).
Another difference between cnidarian and bilaterian miRNA-based regulation is the topology of their circuits. In humans, zebrafish, and Drosophila, the vast majority of the miRNAs have very limited spatiotemporal expression overlap with their targets, pointing to the dominance of the coherent regulation (Stark et al. 2005; Sood et al. 2006; Shkumatava et al. 2009). Among the handful of cases for which we have spatial expression patterns, we find several examples of perfect overlap between the expression patterns of Nematostella miRNAs and their targets, suggesting incoherent regulatory topologies (Fig. 5A). Two miRNAs, miR-2028 and miR-2030, represent a topology where the miRNA is embedded within the intron of, and cotranscribed with, the gene encoding the target, which is sliced with the guidance of the miRNA (Fig. 5E; Supplemental Fig. S5). Strikingly, the exact same topology was shown experimentally to confer robustness of protein concentration to variation in transcription levels in synthetic genetic circuits (Bleris et al. 2011). These observations suggest that the incoherently regulated targets we detected in Nematostella are genes whose product amounts should be constant and resistant to fluctuation in transcription levels.
miRNAs are thought to have evolved independently in plants and animals because of the different biogenesis, the lack of miRNA sequence homology and, as discussed above, the different mode of action (Axtell et al. 2011). However, cnidarians and other nonbilaterians have recently been shown to possess a homolog of HYL1, previously considered to be a plant-specific protein required for the miRNA biogenesis (Moran et al. 2013b). Thus, HYL1 was present in the common ancestor of plants and animals and was lost in Bilateria. As in plants, a large fraction of miRNAs in Nematostella is methylated, presumably by HEN1. While Nematostella shares only one miRNA (miR-100) with Bilateria, another Nematostella miRNA has significant sequence identity to miR-156 (Fasta E-value = 0.0094), a miRNA conserved from mosses to higher plants (Arazi et al. 2005). The Nematostella miRNA is identical to Arabidopsis miR-156a in 16 of its positions, including the seed sequence (Fig. 6). While miRNAs are so short that such average sequence identity could arise by chance between any two miRNAs in the sets compared (Bonferroni corrected E-value = 0.8178), observing any alignment that spans the seed region or has an equal or higher number of consecutively matching bases to randomly shuffled Arabidopsis miRNAs with the same nucleotide distribution is very small (random sampling P-value < 0.01). Together, these findings suggest that miRNAs in plants and animals might have a common ancestry and that miRNAs in the common ancestor of the two kingdoms acted via slicing. miRNA-mediated slicing recalls the siRNA slicing mechanism, which is widespread among many eukaryotic lineages and this implies that slicing is the ancestral mechanism of miRNAs. However, the alternative hypothesis that the miRNA pathways and slicing originated convergently from siRNA mechanisms in the plant and cnidarian lineages cannot be rejected at this point.
Figure 6.

Nematostella miR-9422 exhibits significant sequence identity to the plant miR-156 family. Positions identical to the Nematostella miRNA are indicated by a black background. Abbreviations of species names are Ath, Arabidopsis thaliana (thale cress); Nve, Nematostella vectensis; Osa, Oryza sativa (rice); Ppt, Physcomitrella patens (moss); Smo, Selaginella moellendorffii (spikemoss).
In sum, our findings suggest that (1) the diversity of miRNAs in Cnidaria is comparable to that of some bilaterians, (2) miRNA composition is vastly different between different cnidarian groups, (3) miRNAs may play regulatory roles in the development of Cnidaria, (4) a large proportion of Nematostella miRNAs direct slicing of their mRNA targets, a mechanism with consequences different from the primary mode of action of bilaterian miRNAs, and (5) miRNAs participate in regulatory circuits of two opposite topologies in Cnidaria, including one that is rare in bilaterians. We propose that differences in post-transcriptional regulation like those reported here may contribute to the evident phenotypic differences between Cnidaria and Bilateria.
Methods
Sea anemone culture
N. vectensis was cultured under lab conditions (15 Promille sea water at 18°C) and spawning was induced as described (Genikhovich and Technau 2009b). The developmental stages studied were unfertilized eggs, blastula, gastrula, early planula, late planula, metamorphosing planula, primary polyps, adult male, and adult female.
High-throughput sequencing
Size-selection, oxidation, and sequencing were performed as described (Ghildiyal et al. 2008; Seitz et al. 2008). The resulting libraries were seeded at 2 pM on each Illumina lane (UMass Medical School Deep Sequencing Core). Read counts were normalized to the library sequencing depth, excluding abundant noncoding RNAs (rRNAs, tRNAs, and snRNAs). Abundant noncoding RNAs (rRNAs, tRNAs, snRNAs, and snoRNAs) were annotated to the N. vectensis genome using BLASTN (version 2.2.18) to identify homologs of (1) Drosophila melanogaster annotated rRNAs, tRNAs, snRNAs, and snoRNAs (downloaded from FlyBase: http://flybase.org/ and GenBank: http://www.ncbi.nlm.nih.gov/Genbank/index.html), (2) the most abundant rRNA variants found in previous studies involving sequencing of fly small RNAs (Ghildiyal et al. 2008; Seitz et al. 2008), (3) Homo sapiens rRNAs, snRNAs, and snoRNAs (downloaded from GenBank, and the LBME human snoRNA database: http://www-snorna.biotoul.fr/), and (4) the complete set of tRNAs found in the Genomic tRNA database (http://lowelab.ucsc.edu/GtRNAdb/). N. vectensis genomic sequences with a significant BLAST hit to those databases (E-value < 10−4) were flagged as “abundant noncoding RNAs,” and deep-sequencing reads showing perfect identity to one of those were discarded.
miRNA and siRNA annotation
Sequencing reads 20–23 nt long were mapped to the Nematostella genome using an exhaustive search allowing no mismatches and every 201-nt segment (centered on the first nucleotide of the read) was extracted. Stable RNA hairpins from these loci were identified using a sliding 58-nt window to scan the 201-nt segments, selecting those predicted to fold into unbranched hairpins with an RNAfold predicted ΔG ≤ −14 kcal·mol−1 (https://www.tbi.univie.ac.at/˜ronny/RNA/RNAfold.html), parameters that optimally identify known pre-miRNAs in bilateral animals. For each hairpin, we identified the nucleotide where read abundance reaches 25% of the maximal read abundance of that hairpin and the nucleotide where it reaches 95%. Small RNA 5′ ends were annotated as “homogeneous” if the distance between these two nucleotides was not larger than 1 nt. Small RNAs with homogeneous 5′ ends and derived from an arm rather than the loop or the flanks of the hairpin were retained. Conserved pre-miRNA genes were identified using BLAST with the 201-nt genomic segment as query, then screened for conserved secondary structure, conserved miRNA sequence, and perfectly conserved seed sequence. miRNAs exhibiting the same seed sequence (from nt 2 to nt 8) were grouped in families. To detect conserved miRNA genes in Cnidaria, Nematostella pre-miRNA sequences were aligned on the A. digitifera genome and the H. magnipapillata genome (both available Hydra genome assemblies, ABRM01 and ACZU01, gave the same result) using a low-stringency BLAST search (word size = 4, E-value cutoff = 10), followed by a stringent conservation screen for hairpin sequence and secondary structure. Identified orthologs of Nematostella pre-miRNAs had to fold in an unbranched hairpin, as predicted by RNAfold (with a predicted folding free energy not differing from that of the Nematostella hairpin by more than 10 kcal·mol−1). The miRNA seed had to align perfectly, and the whole miRNA sequence had to align on at least 90% of its sequence (alignments were performed using T-Coffee [Notredame et al. 2000]).
Methylation levels of miRNAs
For each miRNA, the ratio of normalized read count in oxidized and untreated libraries was calculated for each sample, requiring a minimum of 10 reads in each sample. If the abundance requirement was not met in any comparison, the miRNA was labeled “undetermined.” miRNAs for which the ratio was <1 in all libraries were classified as undermethylated, and miRNAs for which the ratio was ≥1 in at least one comparison were annotated as overmethylated.
Transcriptome data sets
We used the following data sets for all analyses. H. sapiens: All mRNA from NCBI RefSeq (accessed 01-08-2013), D. melanogaster: FlyBase release 5.46 (FB2012_04, dated 2012-07-06), A. thaliana: TAIR 10 gene models (ftp://ftp.arabidopsis.org/home/tair/Genes/TAIR10_genome_release/TAIR10_gff3), N. vectensis: NveGenes2.0 (http://www.cnidariangenomes.org/). Any precursor miRNA transcripts were removed from these sequence sets by intersection with miRNA annotation prior to target search.
Target site prediction
Mature miRNAs and shuffled sequences were mapped to the transcriptome and scored as described (Fahlgren et al. 2007). Briefly, we mapped sequences with FASTA v36 (Pearson and Lipman 1988) using the parameters -n -H -Q -f -16 -r +15/-10 -g -10 -w 100 -W 25 -E 100000 -i -U and scored the alignments using a weighted sum of the number of mismatches (scoremismatch = 1, scoreG:U = 0.5) with mismatches for guide RNA nucleotides g2–g13 counting double (scoremismatch = 2, scoreG:U = 1). While a perfect seed match is usually necessary for efficient target binding, recent reports in bilaterians show that it is not strictly required for target binding and slicing (Shin et al. 2010; Khorshid et al. 2013). The g2–g13 subsequence encompasses both the miRNA seed (g2–g7) and the scissile phosphate, and it was shown to be reasonably selective in an unbiased search for the most predictive miRNA subsequences. Predicted cleavage sites were annotated at the transcript coordinates corresponding to the position between miRNA nucleotides g10 and g11.
Comparative analysis of miRNA:target complementarity
We used published human, D. melanogaster, and A. thaliana miRNAs and associated data from miRBase release 19 (Kozomara and Griffiths-Jones 2011) and the Nematostella miRNAs described here. We calculated the relative expression value for each mature small RNA based on the number of supporting reads for each, and used this to classify each sequence as either miRNA or miRNA*, with the miRNA being the dominant product from each precursor; miRNA* sequences were not further analyzed. The resulting miRNAs for each species were aligned to the corresponding transcripts (with any miRNA precursors removed) from the same species, and the alignments scored as detailed above in the target prediction method to obtain the best scoring match for each miRNA. To control for differences in transcriptome size and dinucleotide composition between species, we generated the expected background distribution by repeating the analysis using 100 sets of dinucleotide composition-matched shuffled miRNAs for each species. We calculated the fraction of miRNAs and shuffled sequences at each 0.5 point interval for each species and calculated the enrichment as the fraction of miRNAs minus the fraction of shuffled miRNAs at each score level. We tested that these observations were robust to potential biases resulting from differences in miRNA annotation method and sequencing depth in the different species in two ways: first, by repeating the analysis using alternative miRNA sets for human (used for Fig. 4; Meunier et al. 2013) and Nematostella miRNAs from miRBase 19 (not shown); and second, by partitioning our Nematostella miRNAs and the miRBase miRNAs by relative abundance level in each species (Supplemental Fig. S4).
miRNA target site conservation
We downloaded ESTs from Metridium senile and Anemonia viridis from NCBI dbEST (accessed 11-15-2013), removed sequence redundancy by clustering using cd-hit-est-2d (Li and Godzik 2006) with default parameters, and obtained orthologous groups with Nematostella genes by pairwise bidirectional BLAST. We identified conserved miRNA target sites using the same Nematostella miRNA target prediction method as elsewhere to find matches to Nematostella miRNAs indicative of putative target sites in the nonredundant EST sets. Finally, we intersected ortholog information and predicted targets to obtain the final list of three conserved sites described in the text.
Degradome library construction, mapping, and analysis
Total RNA (150 μg) was extracted from primary polyps with TRIzol (Life Technologies), then purified using poly(A) selection (Dynabead mRNA Purification Kit; Life Technologies). Purified RNA was ligated to an RNA linker (5′-HO-GUUCAGAGUUCUACAGUCCGACGAUC-3′) with T4 RNA ligase (Life Technologies) at 20°C for 3 h. The ligated RNA was isolated using Agencourt RNAClean XP beads (Beckman Coulter) and reverse transcribed (SuperScript III; Invitrogen) using a degenerate primer (5′-GCACCCGAGAATTCCANNNNNNNN-3′). First-strand cDNA was purified with Agencourt RNAClean XP beads and amplified by PCR using primers 5′-CTACACGTTCAGAGTTCTACAGTCCGA-3′ and 5′-GCCTTGGCACCCGAGAATTCCA-3′ (five PCR cycles), then 5′-AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGA-3′ and 5′-CAAGCAGAAGACGGCATACGAGATN6GTGACTGGAGTTCCTTGGCACCCGAGAATTCCA-3′ (N6 indicates the 6-nt barcode for multiplexing; 13 PCR cycles). PCR products 250–500 bp were purified for sequencing on the HiSeq 2000 using the 50-nt read protocol (Illumina), and two biological replicates were sequenced.
Degradome reads were trimmed to remove the adapter sequence and then mapped to the nonredundant transcriptome using BWA 0.5.9 (Li and Durbin 2009). Two data sets were generated: reads aligning to the sense strand with no mismatches (52,301,385 and 49,642,119 reads in each library); and reads aligned to the sense strand with ≥6 consecutive 5′ end matches, a total alignment length ≥30 bp, and mapping quality ≥20 (8,761,317 and 8,440,491 reads, respectively). For each set, we compared the starting position of degradome reads on transcript sequences with the predicted miRNA-guided cleavage sites. For all statistical analyses, we required a minimum of two reads from each library starting at a predicted cleavage site to regard it as supported by the degradome. In addition, we mapped reads to the genome using the same procedure as detailed above for visualization purposes.
In situ hybridization
In situ hybridization (ISH) of RNA probes to detect pri-miRNA was performed as described (Genikhovich and Technau 2009a). Locked nucleic acid (LNA) probes labeled on both 5′ and 3′ ends with digoxygenin (Exiqon) were used to detect mature miRNAs. LNA ISH was performed as described except that the hybridization buffer contained 70% (v/v) formamide and 5% (w/v) buffered dextran sulfate (Sigma-Aldrich), and hybridization was at a temperature 30°C below the calculated Tm of that probe on a DNA substrate. LNA probe concentration (1–25 nM) was optimized experimentally. Simultaneous detection of pri-miRNAs and target mRNAs was as described (Genikhovich and Technau 2009a), whereas simultaneous detection of mature miRNAs and targets was performed under miRNA conditions to permit efficient hybridization of the LNA probe. Double ISH probes for mRNAs were labeled with fluorescein isothiocyanate (Roche), and miRNA probes were labeled with digoxygenin (Genikhovich and Technau 2009a). Probes were detected sequentially using alkaline phosphatase (AP) coupled to anti-fluorescein and anti-digoxygenin Fab antibody fragments (Roche) and nitro-blue tetrazolium (NBT) and 5-bromo-4-chloro-3′-indolyphosphate (BCIP; Roche) and Fast Red (Sigma-Aldridge) as described (Moran et al. 2013a). Staining was documented with an Eclipse 80i fluorescence microscope and Digital Sight DS-U2 camera (Nikon).
RLM-RACE
The RLM-RACE procedure was performed as described (German et al. 2008) using the FirstChoice RLM-RACE kit (Life Technologies) according to the manufacturer’s instructions. Ligated RNA was reverse transcribed (Superscript III; Life Technologies) according to the manufacturer’s instructions using gene-specific primers at 55°C for 60 min. First-strand cDNAs were used as template for PCR using a nested gene-specific primer, the outer 5′ RACE primer from the FirstChoice RLM-RACE kit, and Advantage 2 polymerase mix (Clontech) with five cycles (30 sec at 94°C, 12 sec at 72°C), five cycles (30 sec at 94°C, 20 sec at 70°C, 12 sec at 72°C), 27 cycles (30 sec at 94°C, 20 sec at 68°C, 12 sec at 72°C), and then 5 min at 72°C. The PCR product was diluted 1:1000 and used as template for a nested PCR reaction with a nested, gene-specific primer, the outer 5′ RACE primer from the FirstChoice RLM-RACE kit and Advantage 2 polymerase mix. The reaction conditions were as follows: 30 cycles (30 sec at 94°C, 20 sec at 68°C, 20 sec at 72°C), with a final elongation of 5 min at 72°C. PCR products were cloned into the pGEM-T Easy vector (Promega), and clones were sequenced (Macrogen). Clones were scored positively only when the gene-specific sequence following the adapter started in a position across position 10 of the miRNA.
Data access
Deep-sequencing libraries have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession number SRP000409.
Acknowledgments
This work was funded by a Marie Curie IEF fellowship and EMBO postdoctoral fellowship ALTF 1096-2009 to Y.M., FWF P22618-B17 grant to U.T., and HFSP Career Development Award #CDA00017/2010-C and a CNRS ATIP-Avenir grant to H.S.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.162503.113.
Freely available online through the Genome Research Open Access option.
References
- Addo-Quaye C, Eshoo TW, Bartel DP, Axtell MJ 2008. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr Biol 18: 758–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addo-Quaye C, Snyder JA, Park YB, Li YF, Sunkar R, Axtell MJ 2009. Sliced microRNA targets and precise loop-first processing of MIR319 hairpins revealed by analysis of the Physcomitrella patens degradome. RNA 15: 2112–2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameres SL, Horwich MD, Hung JH, Xu J, Ghildiyal M, Weng Z, Zamore PD 2010. Target RNA-directed trimming and tailing of small silencing RNAs. Science 328: 1534–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravin AA, Hannon GJ, Brennecke J 2007. The Piwi-piRNA pathway provides an adaptive defense in the transposon arms race. Science 318: 761–764 [DOI] [PubMed] [Google Scholar]
- Arazi T, Talmor-Neiman M, Stav R, Riese M, Huijser P, Baulcombe DC 2005. Cloning and characterization of micro-RNAs from moss. Plant J 43: 837–848 [DOI] [PubMed] [Google Scholar]
- Axtell MJ, Westholm JO, Lai EC 2011. Vive la difference: Biogenesis and evolution of microRNAs in plants and animals. Genome Biol 12: 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP 2009. MicroRNAs: Target recognition and regulatory functions. Cell 136: 215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumberger N, Baulcombe DC 2005. Arabidopsis ARGONAUTE1 is an RNA Slicer that selectively recruits microRNAs and short interfering RNAs. Proc Natl Acad Sci 102: 11928–11933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleris L, Xie Z, Glass D, Adadey A, Sontag E, Benenson Y 2011. Synthetic incoherent feedforward circuits show adaptation to the amount of their genetic template. Mol Syst Biol 7: 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ 2007. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128: 1089–1103 [DOI] [PubMed] [Google Scholar]
- Brodersen P, Sakvarelidze-Achard L, Bruun-Rasmussen M, Dunoyer P, Yamamoto YY, Sieburth L, Voinnet O 2008. Widespread translational inhibition by plant miRNAs and siRNAs. Science 320: 1185–1190 [DOI] [PubMed] [Google Scholar]
- Chapman JA, Kirkness EF, Simakov O, Hampson SE, Mitros T, Weinmaier T, Rattei T, Balasubramanian PG, Borman J, Busam D, et al. 2010. The dynamic genome of Hydra. Nature 464: 592–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chourrout D, Delsuc F, Chourrout P, Edvardsen RB, Rentzsch F, Renfer E, Jensen MF, Zhu B, de Jong P, Steele RE, et al. 2006. Minimal ProtoHox cluster inferred from bilaterian and cnidarian Hox complements. Nature 442: 684–687 [DOI] [PubMed] [Google Scholar]
- Ciaudo C, Servant N, Cognat V, Sarazin A, Kieffer E, Viville S, Colot V, Barillot E, Heard E, Voinnet O 2009. Highly dynamic and sex-specific expression of microRNAs during early ES cell differentiation. PLoS Genet 5: e1000620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech B, Malone CD, Zhou R, Stark A, Schlingeheyde C, Dus M, Perrimon N, Kellis M, Wohlschlegel JA, Sachidanandam R, et al. 2008. An endogenous small interfering RNA pathway in Drosophila. Nature 453: 798–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert MS, Sharp PA 2012. Roles for microRNAs in conferring robustness to biological processes. Cell 149: 515–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erwin DH, Laflamme M, Tweedt SM, Sperling EA, Pisani D, Peterson KJ 2011. The Cambrian conundrum: Early divergence and later ecological success in the early history of animals. Science 334: 1091–1097 [DOI] [PubMed] [Google Scholar]
- Fahlgren N, Howell MD, Kasschau KD, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA, Law TF, Grant SR, Dangl JL, et al. 2007. High-throughput sequencing of Arabidopsis microRNAs: Evidence for frequent birth and death of MIRNA genes. PLoS ONE 2: e219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnerty JR, Pang K, Burton P, Paulson D, Martindale MQ 2004. Origins of bilateral symmetry: Hox and dpp expression in a sea anemone. Science 304: 1335–1337 [DOI] [PubMed] [Google Scholar]
- Flynt AS, Lai EC 2008. Biological principles of microRNA-mediated regulation: Shared themes amid diversity. Nat Rev Genet 9: 831–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander MR, Mackowiak SD, Li N, Chen W, Rajewsky N 2012. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res 40: 37–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genikhovich G, Technau U 2009a. In situ hybridization of starlet sea anemone (Nematostella vectensis) embryos, larvae, and polyps. Cold Spring Harb Protoc 10.1101/pdb.prot5282 [DOI] [PubMed] [Google Scholar]
- Genikhovich G, Technau U 2009b. Induction of spawning in the starlet sea anemone Nematostella vectensis, in vitro fertilization of gametes, and dejellying of zygotes. Cold Spring Harb Protoc 10.1101/pdb prot5281 [DOI] [PubMed] [Google Scholar]
- German MA, Pillay M, Jeong DH, Hetawal A, Luo S, Janardhanan P, Kannan V, Rymarquis LA, Nobuta K, German R, et al. 2008. Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat Biotechnol 26: 941–946 [DOI] [PubMed] [Google Scholar]
- Ghildiyal M, Zamore PD 2009. Small silencing RNAs: An expanding universe. Nat Rev Genet 10: 94–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghildiyal M, Seitz H, Horwich MD, Li C, Du T, Lee S, Xu J, Kittler EL, Zapp ML, Weng Z, et al. 2008. Endogenous siRNAs derived from transposons and mRNAs in Drosophila somatic cells. Science 320: 1077–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A, Srivastava M, Fahey B, Woodcroft BJ, Chiang HR, King N, Degnan BM, Rokhsar DS, Bartel DP 2008. Early origins and evolution of microRNAs and Piwi-interacting RNAs in animals. Nature 455: 1193–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishok A, Pasquinelli AE, Conte D, Li N, Parrish S, Ha I, Baillie DL, Fire A, Ruvkun G, Mello CC 2001. Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell 106: 23–34 [DOI] [PubMed] [Google Scholar]
- Gunawardane LS, Saito K, Nishida KM, Miyoshi K, Kawamura Y, Nagami T, Siomi H, Siomi MC 2007. A slicer-mediated mechanism for repeat-associated siRNA 5′ end formation in Drosophila. Science 315: 1587–1590 [DOI] [PubMed] [Google Scholar]
- Hao L, Cai P, Jiang N, Wang H, Chen Q 2010. Identification and characterization of microRNAs and endogenous siRNAs in Schistosoma japonicum. BMC Genomics 11: 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hejnol A, Obst M, Stamatakis A, Ott M, Rouse GW, Edgecombe GD, Martinez P, Baguna J, Bailly X, Jondelius U, et al. 2009. Assessing the root of bilaterian animals with scalable phylogenomic methods. Proc Biol Sci 276: 4261–4270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntzinger E, Izaurralde E 2011. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat Rev Genet 12: 99–110 [DOI] [PubMed] [Google Scholar]
- Hutvagner G, McLachlan J, Pasquinelli AE, Balint E, Tuschl T, Zamore PD 2001. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 293: 834–838 [DOI] [PubMed] [Google Scholar]
- Hwang JS, Ohyanagi H, Hayakawa S, Osato N, Nishimiya-Fujisawa C, Ikeo K, David CN, Fujisawa T, Gojobori T 2007. The evolutionary emergence of cell type-specific genes inferred from the gene expression analysis of Hydra. Proc Natl Acad Sci 104: 14735–14740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamminga LM, Luteijn MJ, den Broeder MJ, Redl S, Kaaij LJ, Roovers EF, Ladurner P, Berezikov E, Ketting RF 2010. Hen1 is required for oocyte development and piRNA stability in zebrafish. EMBO J 29: 3688–3700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karginov FV, Cheloufi S, Chong MM, Stark A, Smith AD, Hannon GJ 2010. Diverse endonucleolytic cleavage sites in the mammalian transcriptome depend upon microRNAs, Drosha, and additional nucleases. Mol Cell 38: 781–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketting RF, Fischer SE, Bernstein E, Sijen T, Hannon GJ, Plasterk RH 2001. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev 15: 2654–2659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khorshid M, Hausser J, Zavolan M, van Nimwegen E 2013. A biophysical miRNA-mRNA interaction model infers canonical and noncanonical targets. Nat Methods 10: 253–255 [DOI] [PubMed] [Google Scholar]
- Kim VN, Han J, Siomi MC 2009. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 10: 126–139 [DOI] [PubMed] [Google Scholar]
- Knight SW, Bass BL 2001. A role for the RNase III enzyme DCR-1 in RNA interference and germ line development in Caenorhabditis elegans. Science 293: 2269–2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S 2011. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 39: D152–D157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna S, Nair A, Cheedipudi S, Poduval D, Dhawan J, Palakodeti D, Ghanekar Y 2013. Deep sequencing reveals unique small RNA repertoire that is regulated during head regeneration in Hydra magnipapillata. Nucleic Acids Res 41: 599–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusserow A, Pang K, Sturm C, Hrouda M, Lentfer J, Schmidt HA, Technau U, von Haeseler A, Hobmayer B, Martindale MQ, et al. 2005. Unexpected complexity of the Wnt gene family in a sea anemone. Nature 433: 156–160 [DOI] [PubMed] [Google Scholar]
- Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, et al. 2003. The nuclear RNase III Drosha initiates microRNA processing. Nature 425: 415–419 [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Godzik A 2006. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22: 1658–1659 [DOI] [PubMed] [Google Scholar]
- Li S, Liu L, Zhuang X, Yu Y, Liu X, Cui X, Ji L, Pan Z, Cao X, Mo B, et al. 2013. MicroRNAs inhibit the translation of target mRNAs on the endoplasmic reticulum in Arabidopsis. Cell 153: 562–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llave C, Xie Z, Kasschau KD, Carrington JC 2002. Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science 297: 2053–2056 [DOI] [PubMed] [Google Scholar]
- Luo GZ, Hafner M, Shi Z, Brown M, Feng GH, Tuschl T, Wang XJ, Li X 2012. Genome-wide annotation and analysis of zebra finch microRNA repertoire reveal sex-biased expression. BMC Genomics 13: 727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansfield JH, McGlinn E 2012. Evolution, expression, and developmental function of Hox-embedded miRNAs. Curr Top Dev Biol 99: 31–57 [DOI] [PubMed] [Google Scholar]
- Marco A, Kozomara A, Hui JH, Emery AM, Rollinson D, Griffiths-Jones S, Ronshaugen M 2013. Sex-biased expression of microRNAs in Schistosoma mansoni. PLoS Negl Trop Dis 7: e2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier J, Lemoine F, Soumillon M, Liechti A, Weier M, Guschanski K, Hu H, Khaitovich P, Kaessmann H 2013. Birth and expression evolution of mammalian microRNA genes. Genome Res 23: 34–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran Y, Praher D, Schlesinger A, Ayalon A, Tal Y, Technau U 2013a. Analysis of soluble protein contents from the nematocysts of a model sea anemone sheds light on venom evolution. Mar Biotechnol (NY) 15: 329–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran Y, Praher D, Fredman D, Technau U 2013b. The evolution of microRNA pathway protein components in Cnidaria. Mol Biol Evol 30: 2541–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullokandov G, Baccarini A, Ruzo A, Jayaprakash AD, Tung N, Israelow B, Evans MJ, Sachidanandam R, Brown BD 2012. High-throughput assessment of microRNA activity and function using microRNA sensor and decoy libraries. Nat Methods 9: 840–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J 2000. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol 302: 205–217 [DOI] [PubMed] [Google Scholar]
- Okamura K, Balla S, Martin R, Liu N, Lai EC 2008. Two distinct mechanisms generate endogenous siRNAs from bidirectional transcription in Drosophila melanogaster. Nat Struct Mol Biol 15: 581–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Hwang DS, Lee JS, Song JI, Seo TK, Won YJ 2012. Estimation of divergence times in cnidarian evolution based on mitochondrial protein-coding genes and the fossil record. Mol Phylogenet Evol 62: 329–345 [DOI] [PubMed] [Google Scholar]
- Park JH, Ahn S, Kim S, Lee J, Nam JW, Shin C 2013. Degradome sequencing reveals an endogenous microRNA target in C. elegans. FEBS Lett 587: 964–969 [DOI] [PubMed] [Google Scholar]
- Pearson WR, Lipman DJ 1988. Improved tools for biological sequence comparison. Proc Natl Acad Sci 85: 2444–2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe H, Brinkmann H, Lavrov DV, Littlewood DT, Manuel M, Worheide G, Baurain D 2011. Resolving difficult phylogenetic questions: Why more sequences are not enough. PLoS Biol 9: e1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam NH, Srivastava M, Hellsten U, Dirks B, Chapman J, Salamov A, Terry A, Shapiro H, Lindquist E, Kapitonov VV, et al. 2007. Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science 317: 86–94 [DOI] [PubMed] [Google Scholar]
- Schwaiger M, Schönauer A, Rendeiro AF, Pribitzer C, Schauer A, Gilles AF, Schinko JB, Renfer E, Fredman D, Technau U 2014. Evolutionary conservation of the eumetazoan gene regulatory landscape. Genome Res (this issue). doi: 10.1101/gr.162529.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz H, Ghildiyal M, Zamore PD 2008. Argonaute loading improves the 5′ precision of both microRNAs and their miRNA* strands in flies. Curr Biol 18: 147–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin C, Nam JW, Farh KK, Chiang HR, Shkumatava A, Bartel DP 2010. Expanding the microRNA targeting code: Functional sites with centered pairing. Mol Cell 38: 789–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinzato C, Shoguchi E, Kawashima T, Hamada M, Hisata K, Tanaka M, Fujie M, Fujiwara M, Koyanagi R, Ikuta T, et al. 2011. Using the Acropora digitifera genome to understand coral responses to environmental change. Nature 476: 320–323 [DOI] [PubMed] [Google Scholar]
- Shkumatava A, Stark A, Sive H, Bartel DP 2009. Coherent but overlapping expression of microRNAs and their targets during vertebrate development. Genes Dev 23: 466–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinigaglia C, Busengdal H, Leclère L, Technau U, Rentzsch F 2013. The bilaterian head patterning gene six3/6 controls aboral domain development in a cnidarian. PLoS Biol 11: e1001488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JL, Stoeckius M, Maaskola J, Friedlander M, Stepicheva N, Juliano C, Lebedeva S, Thompson W, Rajewsky N, Wessel GM 2012. Select microRNAs are essential for early development in the sea urchin. Dev Biol 362: 104–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood P, Krek A, Zavolan M, Macino G, Rajewsky N 2006. Cell-type-specific signatures of microRNAs on target mRNA expression. Proc Natl Acad Sci 103: 2746–2751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling EA, Vinther J, Moy VN, Wheeler BM, Semon M, Briggs DE, Peterson KJ 2009. MicroRNAs resolve an apparent conflict between annelid systematics and their fossil record. Proc Biol Sci 276: 4315–4322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark A, Brennecke J, Bushati N, Russell RB, Cohen SM 2005. Animal microRNAs confer robustness to gene expression and have a significant impact on 3′ UTR evolution. Cell 123: 1133–1146 [DOI] [PubMed] [Google Scholar]
- Steinmetz PR, Urbach R, Posnien N, Eriksson J, Kostyuchenko RP, Brena C, Guy K, Akam M, Bucher G, Arendt D 2010. Six3 demarcates the anterior-most developing brain region in bilaterian animals. Evodevo 1: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Technau U, Rudd S, Maxwell P, Gordon PM, Saina M, Grasso LC, Hayward DC, Sensen CW, Saint R, Holstein TW, et al. 2005. Maintenance of ancestral complexity and non-metazoan genes in two basal cnidarians. Trends Genet 21: 633–639 [DOI] [PubMed] [Google Scholar]
- Wheeler BM, Heimberg AM, Moy VN, Sperling EA, Holstein TW, Heber S, Peterson KJ 2009. The deep evolution of metazoan microRNAs. Evol Dev 11: 50–68 [DOI] [PubMed] [Google Scholar]
- Wienholds E, Kloosterman WP, Miska E, Alvarez-Saavedra E, Berezikov E, de Bruijn E, Horvitz HR, Kauppinen S, Plasterk RH 2005. MicroRNA expression in zebrafish embryonic development. Science 309: 310–311 [DOI] [PubMed] [Google Scholar]
- Yang L, Wu G, Poethig RS 2012. Mutations in the GW-repeat protein SUO reveal a developmental function for microRNA-mediated translational repression in Arabidopsis. Proc Natl Acad Sci 109: 315–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekta S, Shih IH, Bartel DP 2004. MicroRNA-directed cleavage of HOXB8 mRNA. Science 304: 594–596 [DOI] [PubMed] [Google Scholar]
- Yekta S, Tabin CJ, Bartel DP 2008. MicroRNAs in the Hox network: An apparent link to posterior prevalence. Nat Rev Genet 9: 789–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B, Yang Z, Li J, Minakhina S, Yang M, Padgett RW, Steward R, Chen X 2005. Methylation as a crucial step in plant microRNA biogenesis. Science 307: 932–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T, Li G, Mi S, Li S, Hannon GJ, Wang XJ, Qi Y 2007. A complex system of small RNAs in the unicellular green alga Chlamydomonas reinhardtii. Genes Dev 21: 1190–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]